
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intense red emitting monoclinic LaPO4:Eu3+ nanoparticles: host–dopant energy transfer dynamics and photoluminescence properties
Santosh K.
Gupta
*a,
P. S.
Ghosh
b,
M.
Sahu
c,
K.
Bhattacharyya
d,
R.
Tewari
b and
V.
Natarajan
a
aRadiochemistry Division, Bhabha Atomic Research Centre, Mumbai 400085, India. E-mail: santufrnd@gmail.com; Fax: +91-22-25505151; Tel: +91-22-25590636
bMaterials Science Division, Bhabha Atomic Research Centre, Mumbai 400085, India
cRadioanalytical Chemistry Division, Bhabha Atomic Research Centre, Mumbai 400085, India
dChemistry Division, Bhabha Atomic Research Centre, Mumbai 400085, India
First published on 18th June 2015
Abstract
LaPO4 nanoparticles were synthesized using complex polymerization method and characterized systematically using X-ray diffraction (XRD), transmission electron microscopy (TEM) and photoluminescence (PL) spectroscopy. Varied concentration of europium ion is doped in the LaPO4 lattice and optical properties and Judd–Ofelt analysis were investigated. It is observed that LaPO4 nanoparticles give violet–blue emission when irradiated with UV light. On doping europium ion the band gap of LaPO4 decreases. Based on DFT calculations it is proposed that energy range over which d and f states of Eu are distributed is coincident with the valence band of LaPO4 so causing an efficient energy transfer from LaPO4 to europium ion. The actual site symmetry for europium ion in lanthanum orthophosphate was also evaluated as D2d based on the Stark splitting pattern although it is C1 for La3+ in LaPO4. The critical energy-transfer distance for the Eu3+ ions was evaluated, based on which the quenching mechanism was verified to be an electric multipolar interaction. It is also observed that the red emission intensity of LaPO4:Eu3+ (2.0 mol%) is almost 85% of a commercial red phosphor, which clearly demonstrates that the as-prepared samples are promising red phosphors under near-UV for use in white-light emitting diodes.
1. Introduction
Lanthanide ion doped inorganic materials have attracted significant amount of attention in all areas of science and technology because of their interesting physico-chemical properties and potential applications in many new areas such as drug carriers,1 telecommunication,2 photovoltaics and photocatalysis,3 white light application,4 contrast agents for MRI and fluorescence imaging,5 nanothermometry6etc.Inorganic compounds bearing f0, f7 or f14 lanthanide ions have a negligible chance of undergoing quenching effect through cross relaxation from Ln3+ ion and hence they are the preferred choice as a host material.7 In that context LaPO4 is good luminescence host. Also because of its other desirable properties such as low toxicity, environmental stability, ability to accommodate large lanthanide ions, high refractive index and high thermal and chemical stability, it is extensively used as a host matrix.8,9 In fact, LaPO4 doped with cerium and terbium ion,10 has been a commercially applied green phosphor in fluorescent lamps. Lanthanum orthophosphate has three different polymorphs; hexagonal, monoclinic and tetragonal. Because the tetragonal phase is stable at high temperature and the hexagonal phase is stable at low temperature, they are not preferred choices for a luminescence host matrix, and it is the monoclinic form which is mainly explored for lanthanide-based photoluminescence (PL). Monoclinic LaPO4 has monazite structure; where lanthanum atoms have nine-fold coordination (LaO9). It is reported that out of nine bonds; four of the oxygen atoms are oriented in a distorted tetrahedron (PO4) that interpenetrates a planar pentagon formed by the other five other oxygen atoms.8 Europium (Eu3+) ions are notable among lanthanides for their characteristic red emission, which is most often excited by ultraviolet (UV) light. Eu3+ is also a special ion as it gives symmetry-sensitive emission because of the non-degenerate ground state 7F0 and non-overlapping 2S+1LJ multiplets and the most intense transition originates from the 5D0 state which is not split into sublevels due to the crystal field. Furthermore, in a crystal site with inversion symmetry the electric dipole transitions (EDT) are strictly forbidden and the magnetic dipole transitions (MDT) are usually the most intense emission peaks, while for a site without inversion symmetry EDT is usually the strongest emission line, because ΔJ = ±2 transitions are hypersensitive to small deviation from inversion symmetry. Eu3+ is a preferred structural probe and is used extensively for obtaining information about local symmetry, site occupancy etc.11–17
Substantial literature reports relate to the optical properties of europium-doped LaPO4. Chen et al.8 have investigated the effect of europium doping on the morphology and optical properties of LaPO4 crystal with varied dopant concentrations whereas Wang et al.18 has studied the influence of phosphate precursor on the PL properties of LaPO4:Eu nanoparticles. Another group19 has studied the influences of surfactants on PL properties of LaPO4:Eu nanoparticles. Yang et al. have studied the effects of the method of synthesis on the characteristics of the LaPO4:Eu phosphors.20 Another report on the optical properties of europium-doped LaPO4 nanorods and nanoparticles also exists in the literature.21 Lei et al.22 have explored the effect of lithium ion co-doping on the PL properties of LaPO4:Eu. There are also further reports on the PL properties of Eu3+-doped LaPO4.23–25
The most general method of synthesizing a phosphor material is doping different lanthanide ion into suitable inorganic hosts depending on the requirements. But in some cases even without doping any activator ion; hosts show intense self activated emission and subsequently can transfer their excitation energy to activator ions when doped. Therefore, energy transfer from dopant to host has become an effective route to enhance the emission intensity of activators. Some of the recent reports on application of host–dopant energy transfer, where defects play a prominent role, are in NIR phosphorescent phosphors26 and in deep tissue and cell imaging studies.27–29
None of these previous studies discuss about the origin of the violet-blue emission in lanthanum phosphate nanoparticles or explained the energy transfer mechanism on europium doping. Moreover none of these studies discuss about the local site symmetry around Eu3+ ion in LaPO4 and covalency of Eu–O bonds as a function of Eu3+ concentration.
Doping, which forms the basis of most photonic applications, is best achieved through a wet chemical method such as sol–gel, combustion, use of citrate precursor, microemulsion method. We have employed a citrate precursor route for the synthesis of LaPO4. This citrate precursor method scores over other methods because it is a relatively simple, easy and low-cost technique employing common chemicals with good stoichiometric and particle size control.
In this work we have presented a combined experimental and theoretical study on the optical properties of La1−xEuxPO4. Polymeric precursor derived La1−xEuxPO4 are characterized by X-ray diffraction (XRD), photoluminescence spectroscopy (PL) and ultraviolet–visible absorption analysis (UV-Vis). An effort was made to correlate the experimental and theoretical results for host–dopant energy transfer. We have studied the effect of europium ion concentration on photoluminescence properties and optical absorption spectroscopy of La1−xEuxPO4. We have also investigated the spectroscopic properties of La1−xEuxPO4 using Judd–Ofelt theory based on photoluminescence emission spectra as a function of Eu3+ concentration.
2. Experimental
2.1. Synthesis
La1−xEuxPO4 samples (x = 0, 0.005, 0.01, 0.02 and 0.05) were synthesized by a polymeric precursor route. The starting materials were (NH4)2HPO4 (99.99%, M/s Alfa Aesar, Lancaster), La2O3 (99.995%, Rare-earth development division, BARC, Mumbai) and Eu2O3 (99.999% purity supplied by Aldrich, USA). Citric acid (99.7%, M/s Chemco fine chemicals, Mumbai) was used as the complexing agent while ethylene glycol (99.0%, M/s Thomas Baker, Mumbai) was used for polyesterification to stabilize the complex. Initially the reactants La2O3 and Eu2O3 were preheated in a highly pure argon atmosphere at 1000 °C to remove moisture. Thereafter La2O3 was dissolved in concentrated HNO3 followed by addition of citric acid. Citric acid was added to maintain the fuel to oxidant ratio as unity. The solution was kept on the hot plate at 80 °C and the required amount of (NH4)2HPO4 was added. Ethylene glycol was then added and the mixture stirred. The citric acid and ethylene glycol were added in the mass ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The solution was kept overnight for stable polyester formation. For the Eu-doped samples, the required amount of Eu2O3 was also added in the nitric acid dissolution step. Upon evaporation of the reaction solution a transparent polymeric gel was formed. The gel was then burned at 500 °C in a preheated furnace for 1 h. The obtained foamy carbonaceous precursor was then ground, pelletised and kept at 800 °C for 6 h.
:4. The solution was kept overnight for stable polyester formation. For the Eu-doped samples, the required amount of Eu2O3 was also added in the nitric acid dissolution step. Upon evaporation of the reaction solution a transparent polymeric gel was formed. The gel was then burned at 500 °C in a preheated furnace for 1 h. The obtained foamy carbonaceous precursor was then ground, pelletised and kept at 800 °C for 6 h.
2.2. Instrumentation
X-Ray diffraction (XRD) patterns of the powdered undoped and europium-doped LaPO4 samples were recorded using a RIGAKU Miniflex-600 diffractometer operating in Bragg–Brentano focusing geometry. Cu-Kα radiation (λ = 1.5406 Å) was used as the X-ray source and the operating voltage and current of the instrument was kept at 40 kV and 30 mA, respectively. The XRD patterns were collected with scan rate of 1° min−1. PL data were recorded on an Edinburgh CD-920 unit equipped with M 300 monochromators. The data acquisition and analysis were done by F-900 software provided by Edinburgh Analytical Instruments, UK. A xenon flash lamp with frequency range of 10–100 Hz was used as the excitation source. Emission spectra for samples were recorded with a lamp frequency of 100 Hz. Multiple scans (at least five) were taken to minimize the fluctuations in peak intensity and to maximize S/N ratio. Fluorescence lifetime measurements were based on well established time-correlated single-photon counting (TCSPC) technique. Samples for TEM were prepared by suspending particles in a non-aqueous medium. To ensure uniform distribution samples were subjected to ultrasonication for 20 min and a few drops from the liquid were allowed to settle on 200 micron carbon coated copper grids. The grids were allowed to dry under vacuum for a few hours prior to loading in a transmission electron microscope (TEM). A Technai T-20 instrument of FEI was used for all TEM investigation. EPR spectra were recorded on a Bruker ESP-300 spectrometer operating at X-band microwave frequency (9.4186 GHz). Diphenyl picrylhydrazyl (DPPH) was used for the calibration of g-values of paramagnetic species. UV-Vis diffuse reflectance spectra (UV-Vis DRS) of the samples were obtained on an UV-Vis spectrophotometer (Hitachi U-3010) using BaSO4 as reference.2.3. Computational methodology
All calculations in this study are based on density functional theory (DFT) in conjunction with projector augmented wave (PAW) potentials, which is implemented in the plane wave based Vienna Ab initio Simulation Package (VASP).30,31 The generalized gradient approximation (GGA) parameterized by Perdew–Burke–Ernzerhof (PBE)32 was used as the exchange–correlation functional. The projector augmented wave (PAW) potentials33 were used for the ion–electron interactions including the valence states of La (5s, 5p, 5f – 11 valence electrons), P (3s, 3p – 5 valence electrons), Eu3+ (5p, 6s, 5d – 9 valence electrons) and O (2s, 2p – 6 valence electrons). In our calculations, the Kohn–Sham single-particle wave functions were expanded in a plane-wave basis with kinetic energy cutoff of 500 eV and it was shown that the results were well converged at this cut off. For monoclinic LaPO4 structure, optimization was carried out with respect to plane-wave cut-off energy and k-point meshes to ensure convergence of total energy to within a precision of 0.1 meV per atom. In order to study Eu3+-doped systems, a 2 × 2 × 1 supercell of LaPO4 unit cell containing 96 atoms was constructed. The Brillouin-zone (BZ) integrations were performed on an optimized Monkhorst–Pack34k-point grid of 10 × 10 × 8 for LaPO4 unit cell and 6 × 6 × 8 for 2 × 2 × 1 supercell. The total energy of LaPO4 and Eu3+-doped LaPO4 were optimized with respect to volume (or lattice parameter, b/a, c/a ratio), angle and atomic positions. The structural relaxations (b/a, c/a ratio and atomic positions) were performed for each structure using the conjugate gradient algorithm until the residual forces and stress in the equilibrium geometry were of the order of 0.005 eV Å−1 and 0.01 GPa, respectively. The final calculation of total electronic energy and density of states (DOS) were performed using the tetrahedron method with Blöchl corrections.353. Results and discussion
3.1. XRD study
Fig. 1 shows the XRD patterns of as-prepared samples of 0.5, 1.0, 2.0 and 5.0 mol% Eu3+-doped LaPO4 samples along with undoped LaPO4. XRD patterns of samples are in agreement with the monoclinic system of pure LaPO4 (JCPDS no. 84-0600) which reveals that all the products are monazite LaPO4 with monoclinic structure in P21/n space group. The XRD patterns shown in Fig. 1 do not show any diffraction peak related to pure oxide phase such as La2O3 or Eu2O3 phase. The fact that such phases are absent is an indication of a homogeneous solid solution of LaPO4 and Eu3+, which further confirms the occupancy of Eu3+ ions in the lattice sites of La3+ in LaPO4. Although three different polymorphs of LaPO4 exist – hexagonal, monoclinic and tetragonal, it is the monoclinic polymorph which is mostly used in commercial purposes.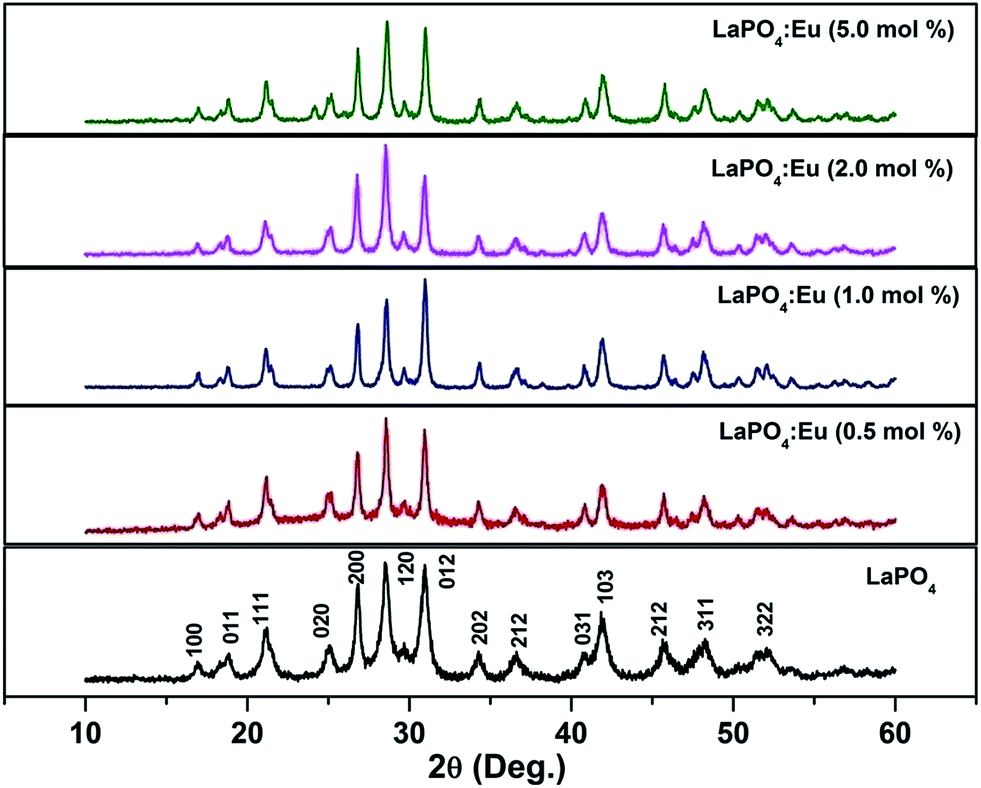 | ||
| Fig. 1 XRD pattern of undoped and europium-doped LaPO4 samples annealed at 800 °C. | ||
3.2. Transmission electron microscopy (TEM) investigations
Fig. 2 shows a representative TEM micrograph of LaPO4 samples. From the micrograph it can be seen that lanthanum phosphate particles formed are of nano-dimensions with irregular morphology. The average diameter of the LaPO4 particles was found to be approximately 15–30 nm. Selected area electron diffraction (SAED) pattern shows the presence of multiple sharp rings, confirming the nano-dimension and crystalline nature of the particles (inset of Fig. 2). The rings in the patterns could be successfully indexed in terms of the monoclinic phase of LaPO4 which is in agreement with XRD results. The formation of LaPO4 is also confirmed using the EDAX pattern shown in the left inset. The intense copper line arises from the copper-grid used in sample preparation while L and M lines of La and K lines of P and O are also present.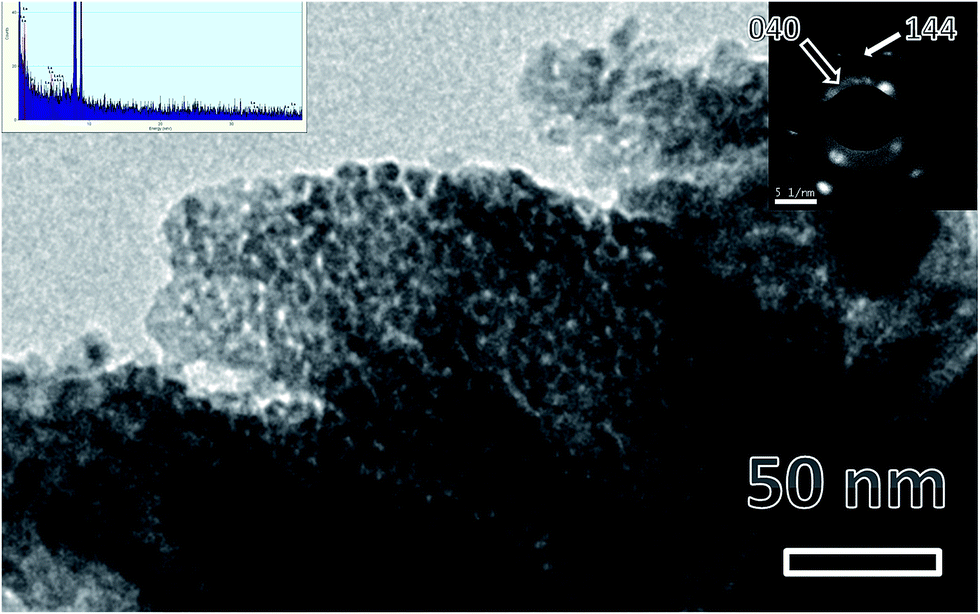 | ||
| Fig. 2 Representative TEM micrographs of LaPO4 nanoparticles synthesized using the citrate precursor route. Insets are EDS pattern (left top) and selected area electron diffraction (right top). | ||
3.3. Luminescence study
Fig. 3 shows the excitation spectra of 0.5 mol% doped Eu3+-doped LaPO4 nanoparticles by monitoring the emission at 593 nm corresponding to the 5D0 → 7F1 transition of europium ion. In the spectra a broad band centred at 256 nm is observed which is attributed to a charge transition state (CTS) arising due to transfer of an electron from the filled 2p shell of oxygen to a partially filled 4f shell of Eu3+ ion. Fig. 3 (inset) shows a magnified region of 300–450 nm in which the peaks at 322, 364, 384 and 397 nm are observed and correspond to 7F0,1 → 5H3,6, 7F0,1 → 5D4, 7F0,1 → 5G3 and 7F0 → 5L6 transitions of Eu3+, respectively. These peaks are relatively of weaker intensity compared to the Eu–O charge-transfer band absorption because f → f transitions are of forbidden nature.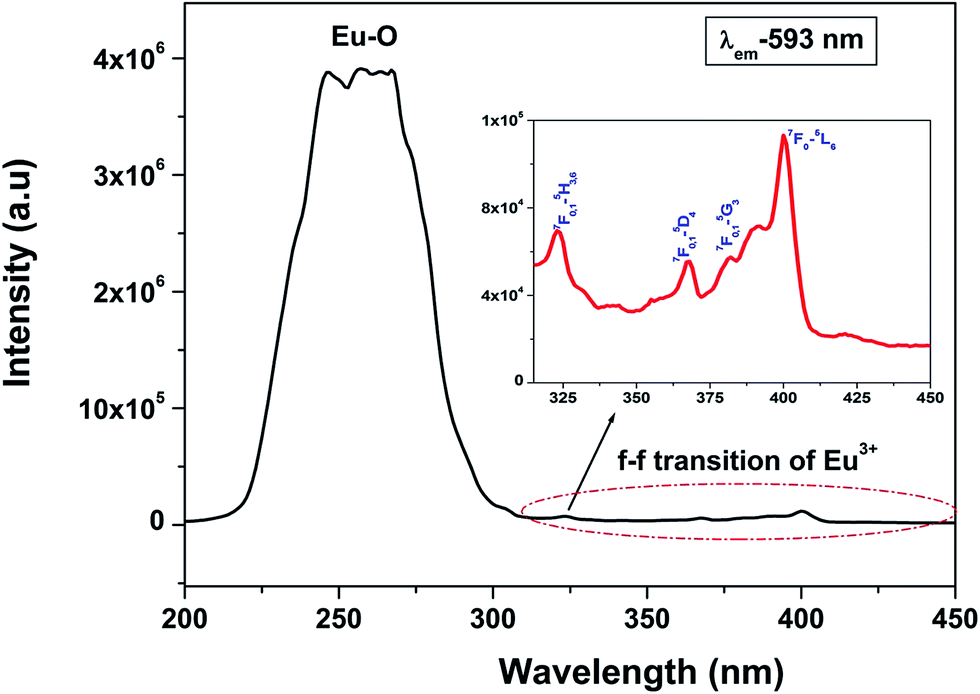 | ||
| Fig. 3 Excitation spectrum of LaPO4:Eu3+ nanoparticles at an emission wavelength of 593 nm. | ||
Fig. 4a shows the photoluminescent spectrum of undoped LaPO4 nanoparticles with excitation at 230 nm. It consists of a strong broad band in the range of 400–600 nm peaking at 455 nm. Such prominent emission in the case of pure LaPO4 nanoparticles at ∼455 nm is also reported by others.8,36
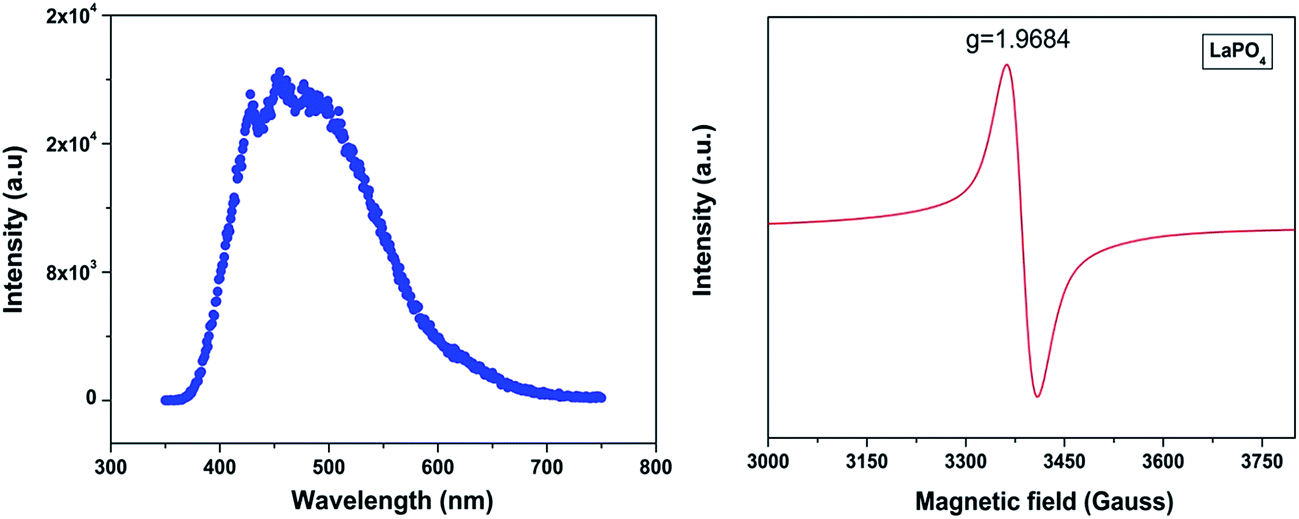 | ||
| Fig. 4 (a) Emission spectrum of undoped LaPO4 nanoparticles (λex – 230 nm) and (b) room-temperature EPR spectra of undoped LaPO4. | ||
The presence of photoluminescence in undoped LaPO4 reveals the presence of defects such as oxygen vacancies, lanthanum interstitials or dangling bonds which can be created during the synthesis procedure. Out of all these, oxygen vacancies are most likely candidate responsible for visible emission in LaPO4. Different types of oxygen vacancies (neutral, singly ionized or doubly ionized) could be responsible for such an intense emission without any activator ion. The presence of oxygen defects, which is responsible for visible emission in LaPO4, is confirmed using EPR. Fig. 4b shows the room-temperature EPR spectrum of LaPO4 which shows an intense peak at g ∼ 1.9684 which is typical of a singly ionized oxygen vacancy.37 Such intense violet-blue emission can arise in undoped LaPO4 from transitions of an electron which is formed during photon irradiation near to the conduction band (CB) to single ionized oxygen vacancy centres (VO+). Emission in such a case arises from a recombination process wherein an excited electron of the conduction band (CB) loses its energy and re-occupied the energy levels of an electron hole in the valence band (VB) through the localized defect levels. The schematic of defect induced emission is shown in Fig. 5.
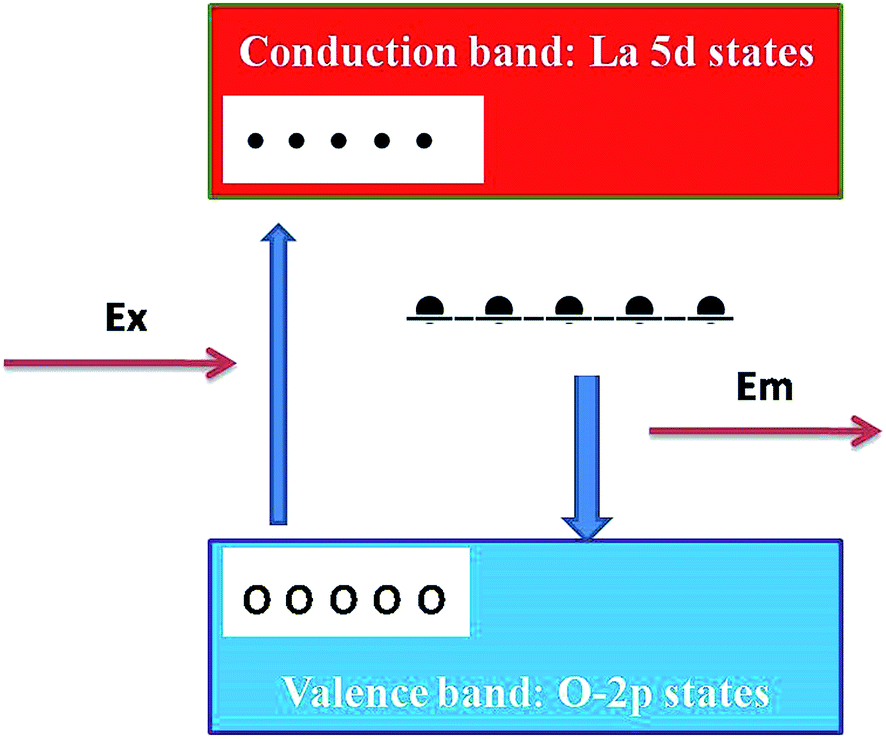 | ||
| Fig. 5 Schematic model of probable mechanisms responsible for observed emission in LaPO4. In the schematic model, the upper (red) and the lower (light blue) bands are La5d conduction bands and O2p valence bands, respectively. The black bar below the conduction band corresponds to the defect level created by the oxygen deficiency. White and black circles correspond to excited holes and electrons, respectively. | ||
Fig. 6 shows the emission spectra of 0.5 mol% Eu3+-doped LaPO4 nanoparticles when excited with 256 nm radiation corresponding to the charge-transfer state. As can be seen, it consists of various sharp lines due to the direct excitation of the Eu3+ ions from the ground level to higher levels of the 4f-manifold.
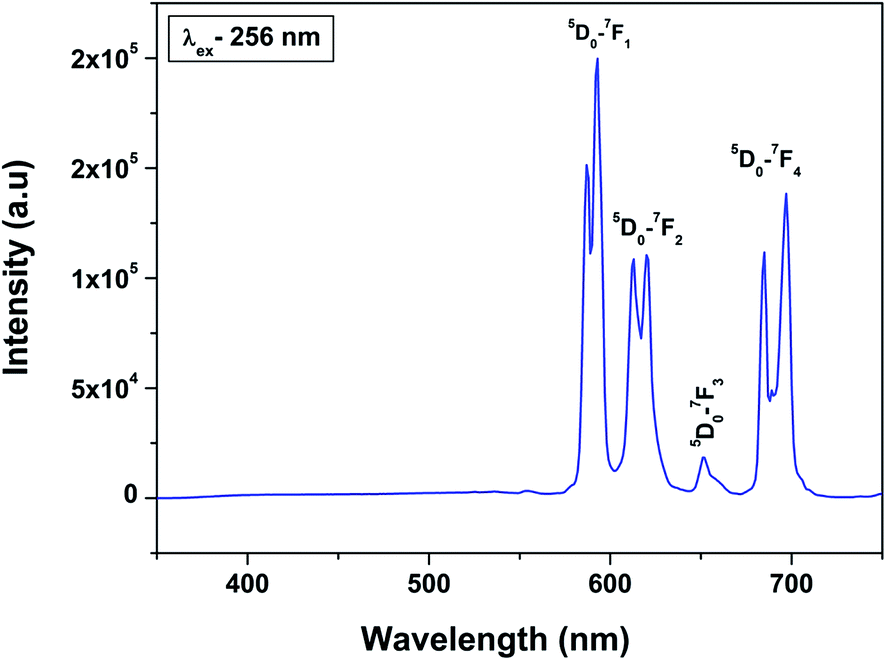 | ||
| Fig. 6 Emission spectrum of LaPO4:Eu3+ (0.5 mol%) at an excitation wavelength of 256 nm. | ||
The spectrum consists of main peaks at 593 and 614 nm corresponding to 5D0 → 7F1 and 5D0 → 7F2 transitions of Eu3+, respectively. Apart from these prominent peaks, we could also observe other weak emission peaks centered at 653 and 697 nm corresponding to 5D0 → 7F3 and 5D0 → 7F4 transitions, respectively.
The transition 5D0 → 7F1 is magnetic dipole (MD) allowed and is not sensitive to the local site symmetry around Eu3+ ion. Transition 5D0 → 7F1 is a hypersensitive forced electric dipole (ED) transition and it has electric dipole nature due to the mixing with 4fn−15d states of the opposite parity by the crystal field components. The intensity of this transition is strongly affected by local site symmetry around the Eu3+ ions and by the strength of electric field in the Eu3+ environment. The other transition 5D0–7F3 has a mixed ED and MD character while transition 5D0–7F4 is an ED transition.
The absence of broad host emission due to surface defects in emission spectra of the doped sample indicate incomplete energy transfer at 0.5 mol% of dopant ion concentration.
Among all the transitions observed, the 5D0 → 7F1 transition is the strongest and is characterized by an orange–red emission whereas the 5D0 → 7F2 transition is characterized by red emission. Generally in a crystal site with inversion symmetry the EDT are strictly forbidden and the MDT are usually the most intense emission peak whereas in a site without inversion symmetry EDT is usually the strongest emission line, because ΔJ = ±2 transitions are hypersensitive to small deviations from inversion symmetry. A sensitive parameter for understanding symmetry is the asymmetry ratio (I) which is described as ratio of integral intensities of EDT (5D0 → 7F2) to MDT (5D0 → 7F1). The fact that MDT is more intense than EDT (asymmetry ratio is less than unity), indicating that the majority of Eu3+ are at sites with inversion symmetry.
When the Eu3+ ion is inserted into any chemical/ligand environment (crystal field), the (2J + 1)-degenerate J-energy levels get split into the so-called Stark sub-levels by the ligand-field effect with the number of levels depending on the local site symmetry of the metal ion. It is reported that in LaPO4 systems, both La3+ as well as phosphate ions occupy C1 point group symmetry sites.38 The substitution of La3+ with Eu3+ may not result in significant lattice distortion because of identical charges, and any distortion would arise only due to size differences (103.2 pm for La3+ and 94.7 for Eu3+). From the Stark splitting pattern shown in Fig. 7, two lines for J = 0 → J = 1 transition, two lines for J = 0 → J = 2 transition, two lines for J = 0 → J = 3 transition and four lines for J = 0 → J = 4 transition of Eu3+ were resolved for LaPO4:Eu3+ (0.5 mol%). The fact that the 5D0 → 7F0 transition is absent rules out the possibility of the presence of C4v symmetry.39,40 Therefore the other possibility is D2d site symmetry of Eu3+ in LaPO4.
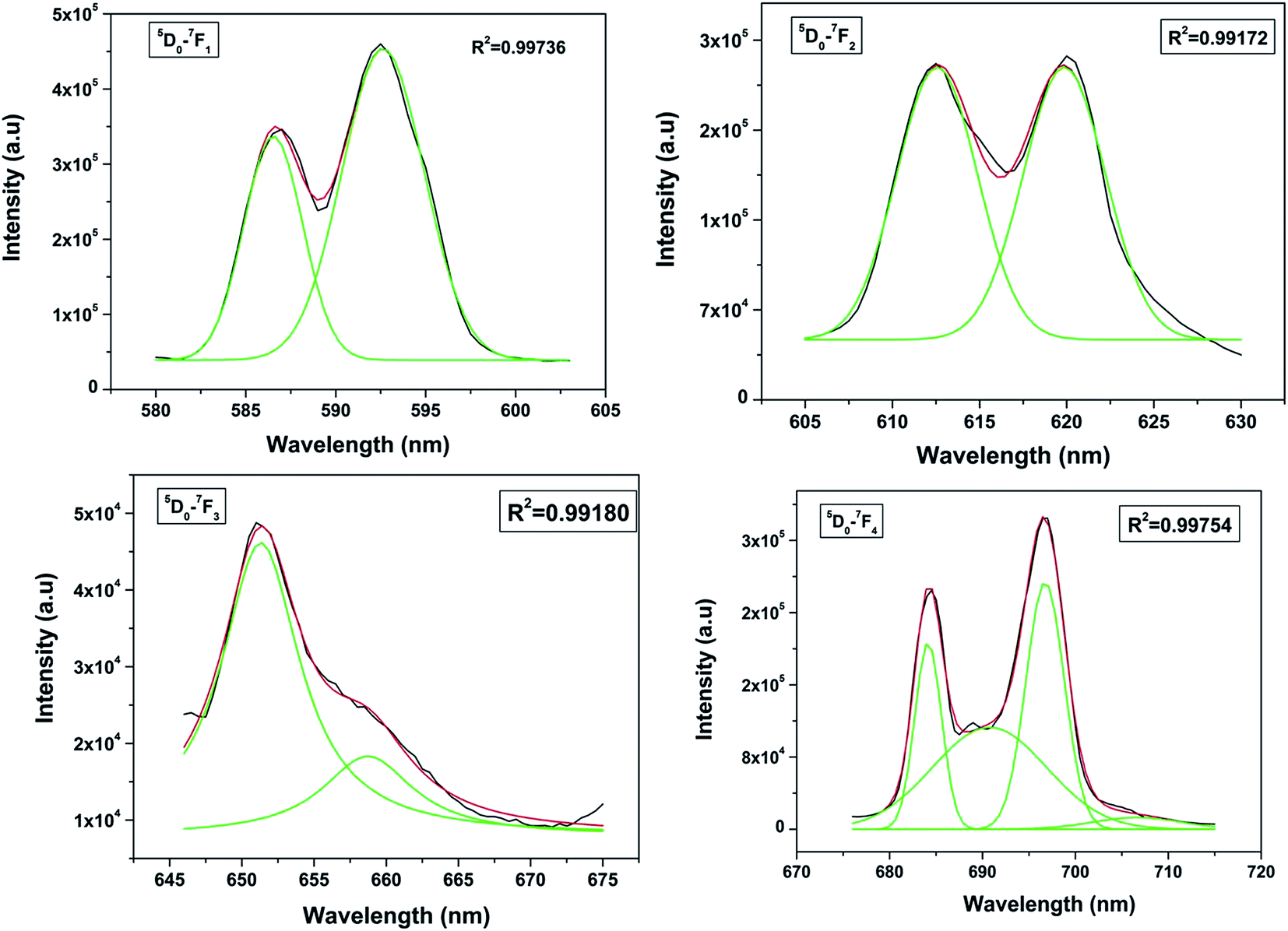 | ||
| Fig. 7 Stark splitting pattern of europium emission in lanthanum phosphate nanoparticles. | ||
As far as PL decay behavior is concerned, the 0.5 mol% doped sample shows monoexponential behavior (Fig. 8) which indicates that europium ion is uniformly distributed in the LaPO4 host lattice and occupies La3+ sites only. The fitting equation is given by eqn (1):
| I = I0 + Aexp(−t/τ) | (1) |
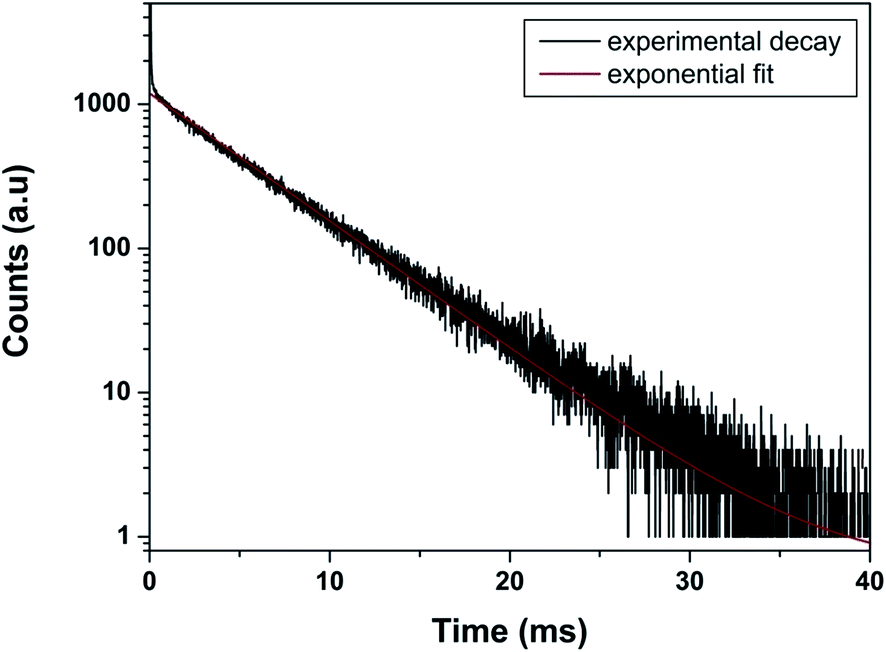 | ||
| Fig. 8 Photoluminescence decay profile of LaPO4:Eu3+ (0.5 mol%) at emission and excitation wavelength of 593 and 256 nm, respectively. | ||
3.4. Effect of dopant ion concentration on optical properties of LaPO4:Eu
Emission spectra of Eu3+ activated LaPO4 phosphors at different Eu3+ concentrations with excitation wavelength of 256 nm are shown in Fig. 9. It can be seen that there is not much change in the emission spectral features as a function of Eu3+ ion concentration. However, emission intensity increases with increase in Eu3+ ion concentration reaching a maximum at 2.0 mol%. Beyond 2.0% the intensity decreases due to concentration quenching, this resulting from resonant energy transfer between Eu3+ ions. To have a better understanding of the concentration quenching mechanism, the critical distance (Rc) needs to be evaluated using eqn (2):41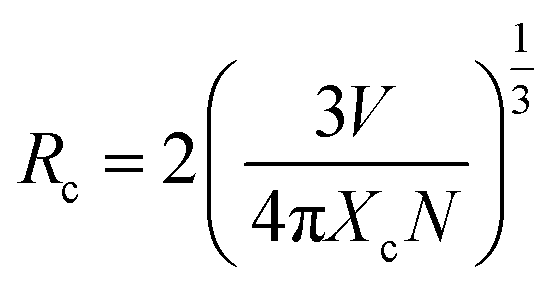 | (2) |
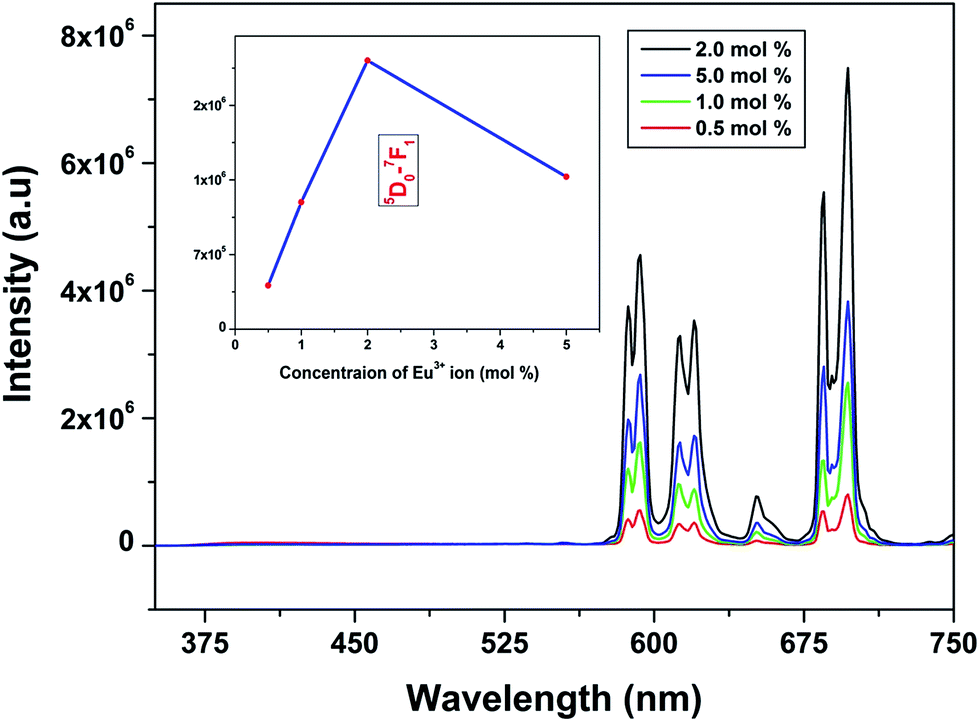 | ||
| Fig. 9 PL emission spectra (corrected w.r.t. source) of LaPO4:Eu3+ phosphors (mol% of Eu3+ = 0.5, 1.0, 2.0, 5.0) excited at 256 nm. The insets show the PL emission intensities of 5D0–7F1 as a function of Eu3+ ion concentration. | ||
For the LaPO4 host lattice, V = 304.86 Å3, N = 4 (four formula units per unit cell), and Xc = 0.02. Based on this value the critical distance is calculated to be 19.37 Å. In this case, the Eu3+–Eu3+ distance is larger than 10 Å. Thus exchange interactions can be ruled out. Therefore, the electric multipolar interaction is believed to be the only mode for the energy transfer among the Eu3+ ions in LaPO4 phosphor.
The effect of dopant ion concentration on lifetime value was also investigated and is shown in Fig. 10. It can be seen that all the samples exhibit monoexponential behavior indicating uniform distribution of europium at La3+ sites. The lifetime values increase up to 2.0 mol% and beyond that decreases due to concentration quenching. The lifetime values are 4.89, 5.77, 6.23, 6.08 ms for 0.5, 1.0, 2.0, 5.0 mol% europium, respectively. At higher concentration, when the critical distance reaches the value of 19.37 Å, the probability of cross-relaxation between Eu3+ becomes very high and thereby reduces the lifetime. There is a close correspondence between the effect of concentration on emission intensity and PL lifetimes.
 | ||
| Fig. 10 PL decay curves of LaPO4:Eu3+ samples (Eu3+ mol% = 0.5, 1.0, 2.0, 5.0) (monitored at 592 nm and excited at 256 nm). | ||
3.5. Band gap calculations: theory and experiment (DFT and DRS measurements)
LaPO4 has a monoclinic crystal structure (P21/n space group, no. 14) having four formula units per unit cell. The PAW-PBE calculated lattice parameters of this phase are a = 6.9372 Å, b = 7.1455 Å, c = 6.5411 Å (V0 = 314.98 Å3) and β = 103.73° (α = γ = 90°) (Fig. 11) which matches very well with the experimentally determined values, a = 6.8313 Å, b = 7.0705 Å, c = 6.5034 Å and β = 103.27°.42 The calculated relaxed atomic positions are compared with experimentally determined atomic positions in Table 1. All the calculated structural parameters of the LaPO4 unit cell are matching within 1.5% of experimentally measured room-temperature values42 and to within 0.2% of previous GGA calculations.43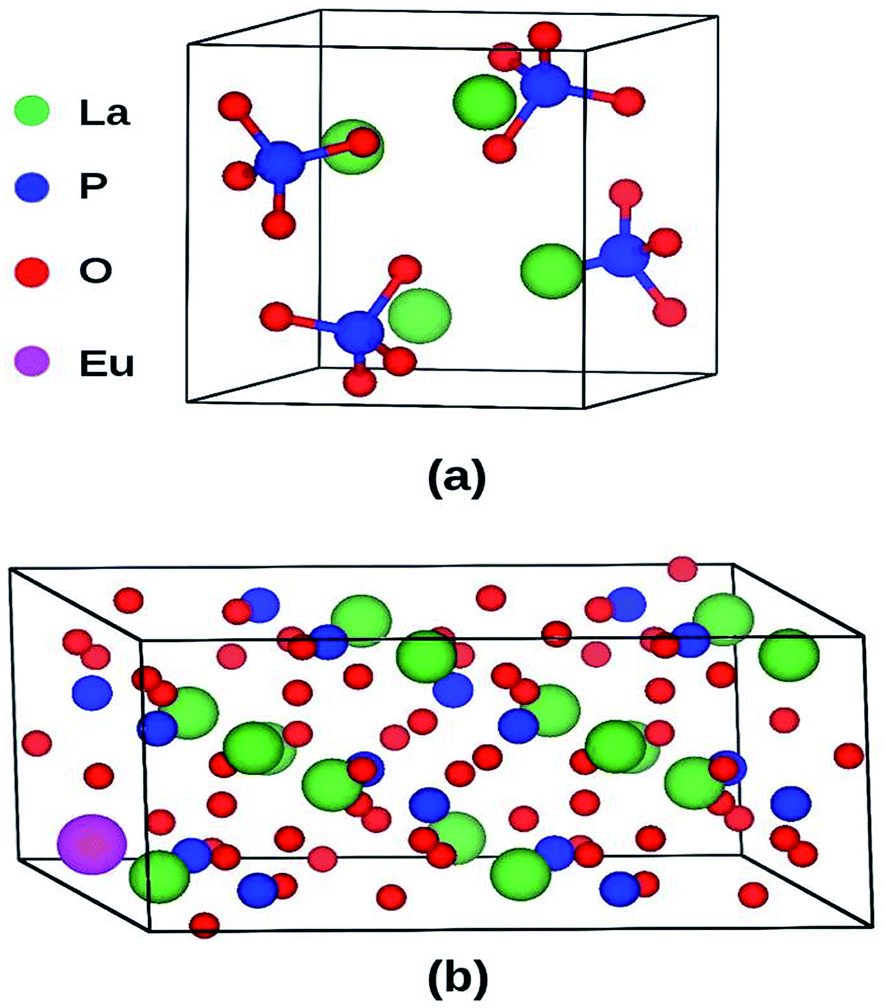 | ||
| Fig. 11 (a) Unit cell of LaPO4 (24 atoms) and (b) Eu3+-doped 2 × 2 × 1 supercell (96 atoms) of LaPO4. | ||
| Atomic positions (this study) | Atomic positions (ref. 42) | |||||
|---|---|---|---|---|---|---|
| x | y | z | x | y | z | |
| La (4e) | 0.2851 | 0.1589 | 0.0988 | 0.2815 | 0.1603 | 0.1006 |
| P (4e) | 0.3042 | 0.1618 | 0.6109 | 0.3047 | 0.1639 | 0.6121 |
| O1 (4e) | 0.2496 | 0.0064 | 0.4414 | 0.2503 | 0.0077 | 0.4477 |
| O2 (4e) | 0.3811 | 0.3293 | 0.4977 | 0.3799 | 0.3315 | 0.4964 |
| O3 (4e) | 0.4710 | 0.1035 | 0.8042 | 0.4748 | 0.1071 | 0.8018 |
| O4 (4e) | 0.1287 | 0.2129 | 0.7082 | 0.1277 | 0.2168 | 0.7101 |
The PAW-PBE calculated total and angular momentum projected electronic density of states (DOS) of LaPO4 is presented in Fig. 12. From Fig. 12 it can be noted that the conduction band from 4.5 to 4.8 eV is formed primarily by the La5d and f states. The valence band is located from −7.5 eV to 0. Besides the small contribution from La s, d and f states, the main contribution of the valence band arises from the O2p states and P3s, 3p states due to the strong hybridization effects. It is evident from Fig. 12 that the top of the valence band is formed predominantly by p states of oxygen with admixture of small amounts of La d and f states. The lower part of the valence band is mainly s and p state of P, p states of O along with very small contribution of La d and f states.
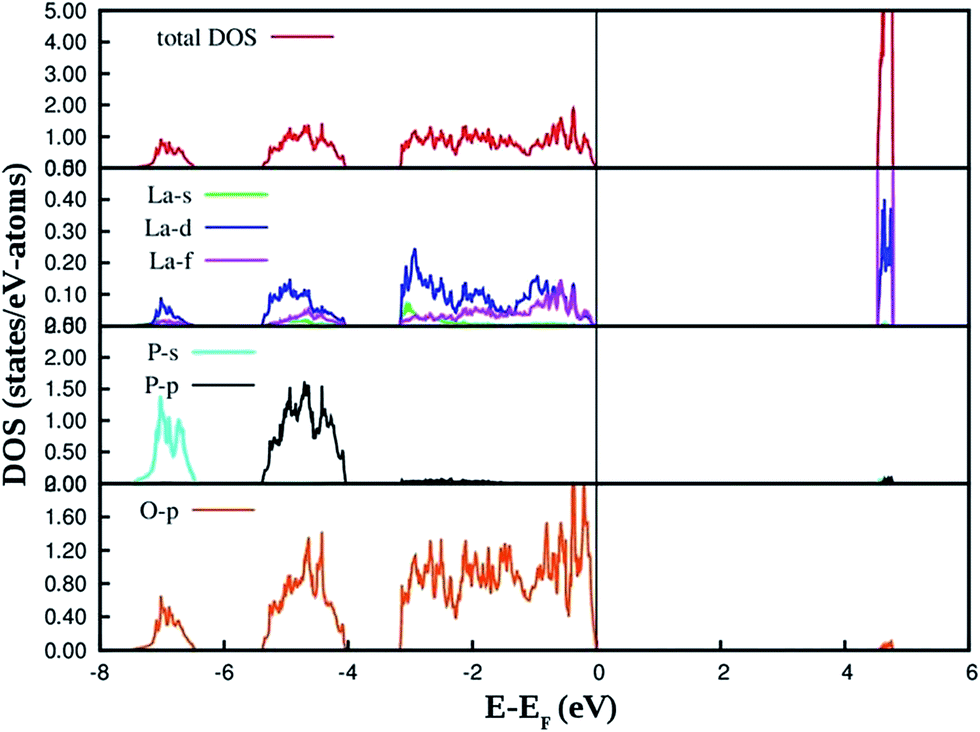 | ||
| Fig. 12 PAW-PBE calculated total and angular momentum decomposed DOS of LaPO4. | ||
The PAW-PBE calculated total and angular momentum projected electronic DOS of Eu3+-doped LaPO4 is presented in Fig. 13. The overall DOS of Eu3+-doped LaPO4 resembles LaPO4 DOS except for the impurity states very near to the Fermi level. The major contribution of Eu atom to the valence band is from d as well as f states and contributions to the conduction band are from solely Eu d states. Moreover, the energy range over which d and f states of Eu are distributed is exactly coincident with the valence band of LaPO4. Therefore, our DFT study predicts host (LaPO4) to dopant (Eu3+) energy transfer is preferable. Impurity energy states are contributed by Eu d and f states. An impurity state reduces the band gap of Eu3+-doped LaPO4 by ∼10% with respect to pure LaPO4.
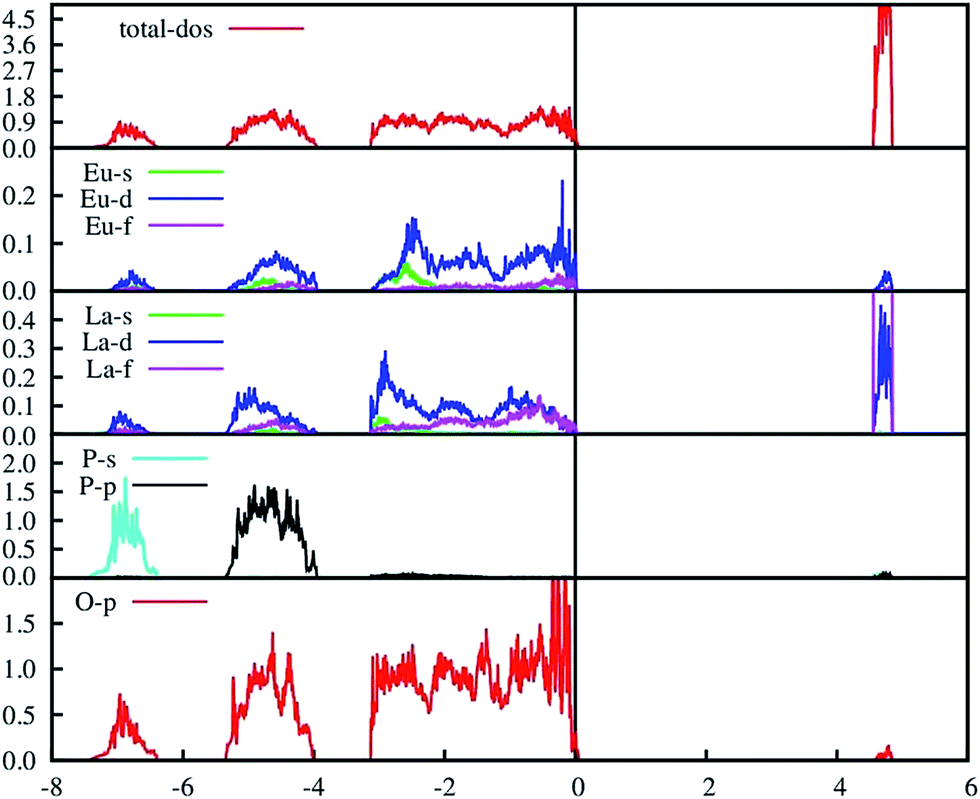 | ||
| Fig. 13 PAW-PBE calculated total and angular momentum decomposed DOS of Eu3+-doped LaPO4 (doping level is 6.25% at La3+ position). | ||
Fig. 14 and 15 illustrate the UV-Vis absorption spectra and absorption–energy curves (according to the Kubelka–Munk function), respectively, of undoped and europium-doped LaPO4 nanoparticles. It can be seen from the spectra that there is an absorption edge at around 252 nm which appears only on europium doping. Band gap values at different europium concentrations according to the KM spectra are listed in Table 2. It was observed that the band gap value of LaPO4 becomes narrower on europium doping and this phenomenon is known as band gap narrowing (BGN), which is in accordance with DFT calculations, and can be easily seen from Fig. 16. BGN is related to the change in the nature and strength of LaPO4 crystalline potential on europium doping (which has 4f valence electrons) which also influence its electronic level.44 Upon doping even at a low level of 0.5 mol% the impurity band broadens and merges with the bottom of the conduction band and thereby decreases the band gap of the material. With increase in europium concentration beyond 0.5% there is essentially no further change in band gap value.
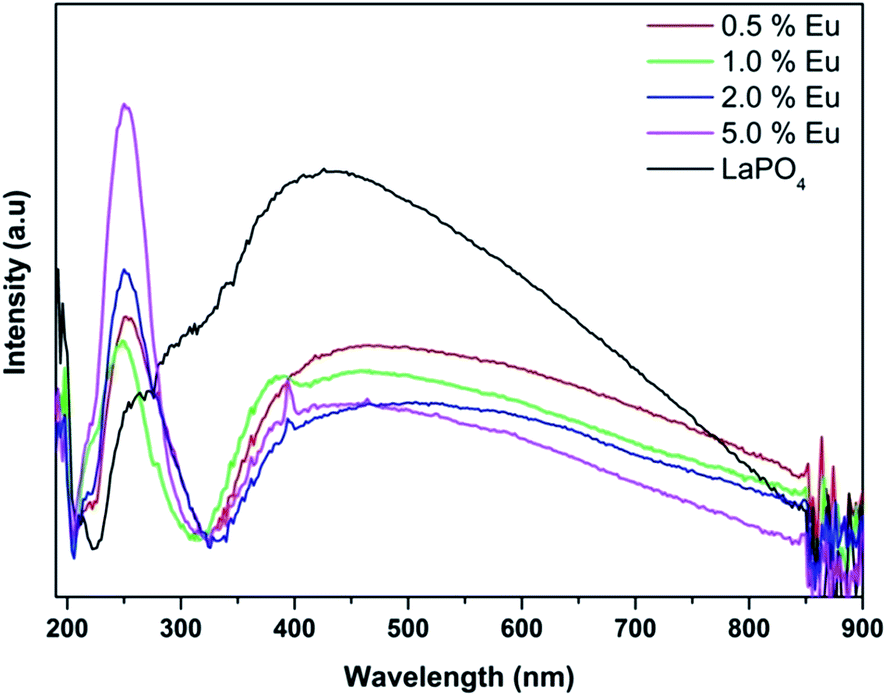 | ||
| Fig. 14 UV-Vis diffuse reflectance spectra of LaPO4 and La1−xEuxPO4. | ||
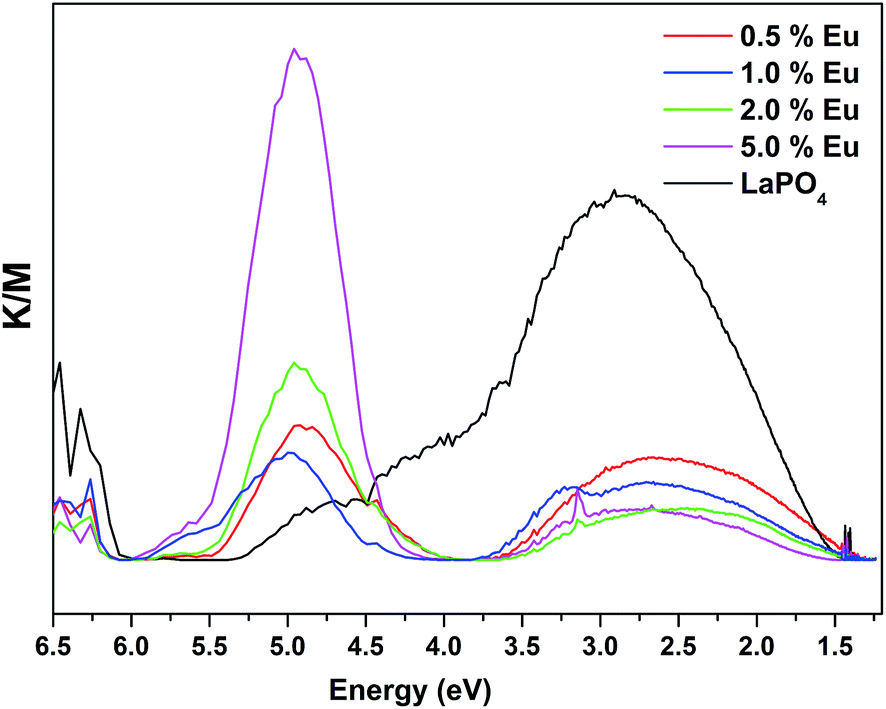 | ||
| Fig. 15 UV-Vis absorption–energy curves of LaPO4 and La1−xEuxPO4. | ||
| Sample | Band gap/eV |
|---|---|
| LaPO4 | 6.15 |
| LaPO4:Eu (0.5 mol%) | 4.13 |
| LaPO4:Eu (1.0 mol%) | 4.16 |
| LaPO4:Eu (2.0 mol%) | 4.25 |
| LaPO4:Eu (5.0 mol%) | 4.35 |
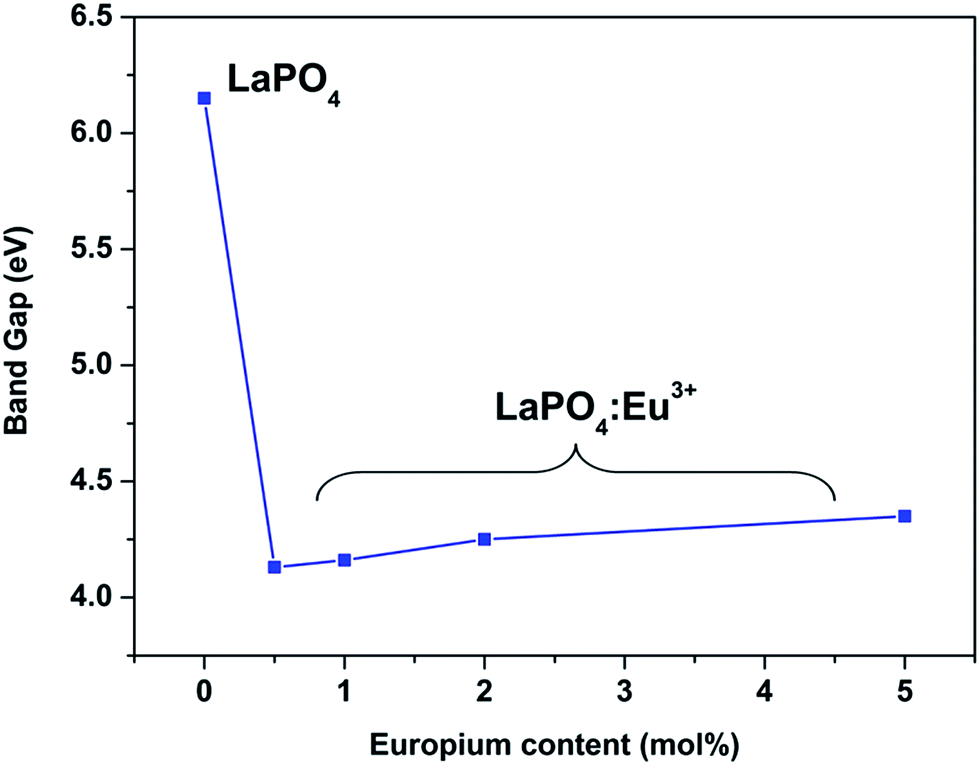 | ||
| Fig. 16 The energy gap dependence of LaPO4:Eu nanoparticles on the Eu doping level. | ||
3.6. Judd–Ofelt calculation: photophysical properties
Judd–Ofelt (JO) intensity parameters, ΩJ (J = 2, 4) calculated using Judd–Ofelt theory45,46 provide information about the local environment in the proximity of lanthanide ion. The application of JO theory to the quantitative analysis of Eu3+ emissive properties in a matrix is nicely presented by Werts et al.,47 and also by our group11,12,48Since the 5D0 → 7F1 transition, a magnetic dipole transitions is not affected much by environmental effects, its transition rate is constant with an approximate value of 50 s−1.49 For LaPO4 we have adopted a value for the refractive index of 1.78 for calculation. Emission quantum efficiency of the emitting 5D0 level is written as eqn (3):
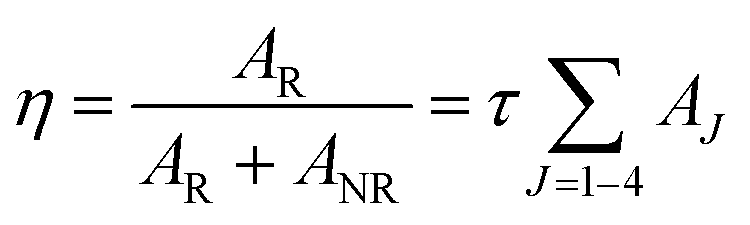 | (3) |
| Eu3+ mol% | A R/s−1 | A NR/s−1 | η (%) | Ω 2/10−20 cm2 | Ω 4/10−20 cm2 |
|---|---|---|---|---|---|
| 0.5 | 179 | 85 | 87.7 | 0.919 | 3.17 |
| 1.0 | 210 | 72 | 88.4 | 0.962 | 3.26 |
| 2.0 | 269 | 63 | 91.2 | 1.25 | 3.72 |
| 5.0 | 234 | 69 | 89.3 | 1.13 | 3.45 |
It is reported that the Ω2 parameter is related to the symmetry and the Ω4 parameter to long-range effects and electronic density surrounding the rare earth ion.50–52 It was observed that at all concentrations of Eu3+ the Ω4 value is greater than Ω2 indicating the existence of a symmetric environment around europium ions in LaPO4 which is highly probable because it occupies La3+ sites (no charge imbalance). This is also reflected in the intense MDT compared to EDT in our emission spectra. Also D2d point symmetry is observed for europium ion in LaPO4 which is relatively higher symmetry. It can also be seen from Table 3 that the Ω2 value increases with increase in Eu3+ concentration up to 2.0 mol% and thereafter decreases. This reveals that with increase in europium ion concentration the covalency of the M–L bond increases as does the distortion.
The value of Ω4 is not directly related to symmetry around Eu3+ but it does depend on electron density around the ligands. The fact that its value also increases with increase in dopant ion concentration indicates that the electron density on the ligand decreases.
Form Table 3 it can also be seen that, with increase in Eu3+ ion concentration up to 2.0 mol% that the radiative transition rate (AR) increases and non-radiative rate (ANR) decreases. This is matching with our emission spectral data where concentration quenching was observed beyond 2.0 mol% due to cross relaxation between europium ions. The ratio of intensity of electric dipole (5D0 → 7F2) to magnetic dipole (5D0 → 7F1), the asymmetry ratio (I) in Eu3+, is related to the degree of covalency between Eu3+ and O2−.53 The larger the difference in the intensities of these two lines, the larger is the asymmetry and covalency effects. For all the samples 5D0 → 7F1 is markedly more intense as compared with the 5D0 → 7F2 line i.e. R/O (red to orange ratio) shows a marked decrease, indicating high symmetry around the Eu3+ ion site, as also reflected by the JO parameters.
4. Potential application of Eu3+-doped LaPO4 as a luminescent material
To evaluate the material performance for phosphor applications, CIE chromaticity coordinates were evaluated for the LaPO4:Eu (2.0 mol%) sample adopting standard procedures. This is represented as the point ‘*’ in the CIE diagram shown in Fig. 17. It is clear from the values that LaPO4:Eu shows red emission with very high quantum efficiency of 91.2%. This is because of the fact that non-radiative losses are very low in this particular system. For the application of red phosphors in white LEDs with near UV light as the excitation source, the emission spectra of LaPO4:Eu3+ (2.0 mol%) and red phosphor (Y2O3:Eu from Nichia Corporation, Japan) were compared using the same excitation wavelength of 395 nm (near UV light) (Fig. 18). It is observed that the red emission intensity of LaPO4:Eu3+ (2.0 mol%) is almost 85% of the commercial red phosphor, which clearly demonstrates that the as-prepared samples are a promising red phosphor under near-UV radiation for white light emitting diodes.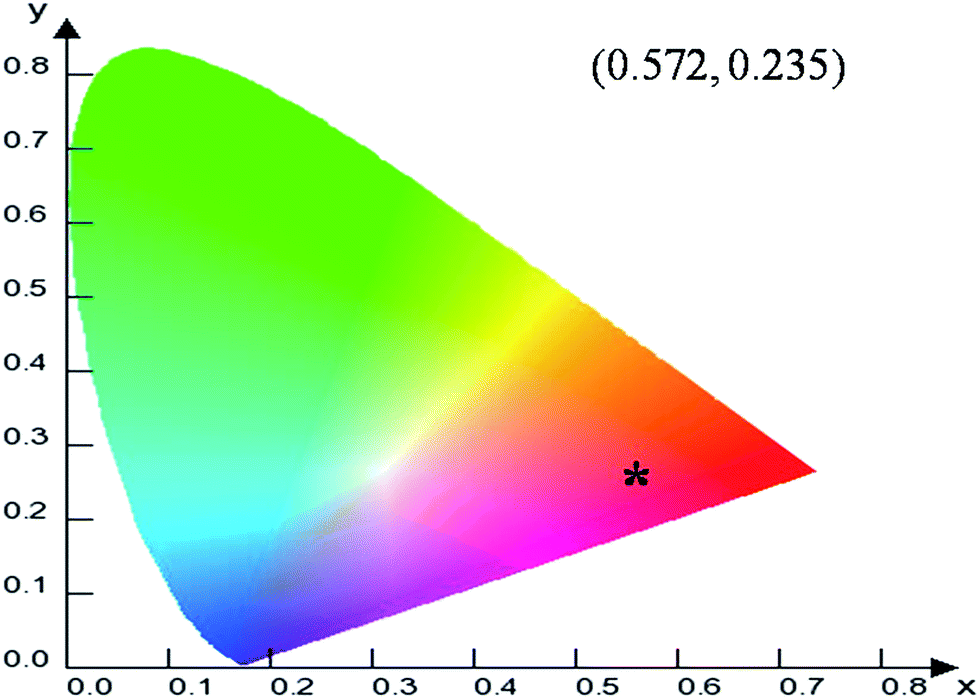 | ||
| Fig. 17 CIE diagram showing the co-ordinates and representing the color emitted by LaPO4:Eu3+. | ||
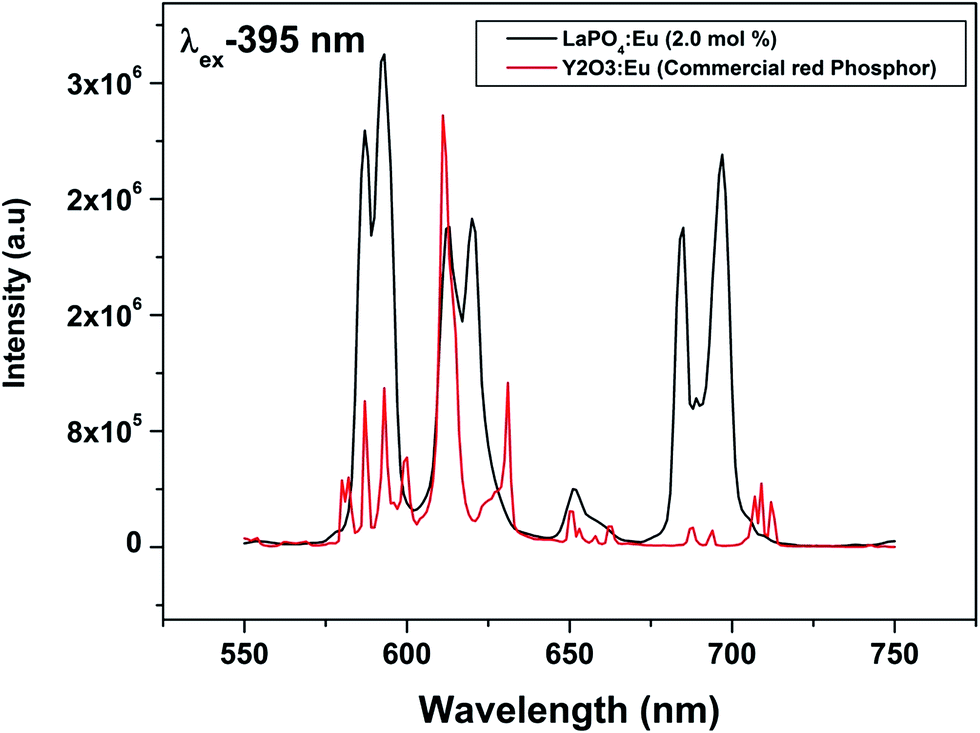 | ||
| Fig. 18 Comparison of emission spectra of commercial red phosphor (Y2O3:Eu from Nichia Corporation, Japan) and LaPO4:Eu3+ (2.0 mol%) under 395 nm excitation. The comparison is made on corrected spectra and integral intensity of all the peak is considered. | ||
5. Conclusions
Monoclinic LaPO4 nanoparticles of 20–30 nm size were synthesized using complex polymerization method and characterized systematically using X-ray diffraction (XRD), transmission electron microscopy (TEM) and photoluminescence (PL) spectroscopy. XRD shows the formation of the pure phase of LaPO4 and europium doping does not distort the crystal structure. The crystallinity and purity of the sample is further studied using EDAX and SAED. Based on PL and EPR it was inferred that intense violet–blue emission in the undoped sample is due to transitions of electrons which are formed during photon irradiation near to the conduction band (CB) to generate single ionized oxygen vacancy centres in LaPO4 nanoparticles. On europium doping, energy is completely transferred from the host to dopant ion, as also predicted using DFT calculations. Europium doping reduces the band gap of LaPO4 by almost 10%. Effects of europium ion on PL intensity, lifetime and photophysical properties were studied extensively. MDT is more intense than EDT in all samples indicating that the majority of Eu3+ are at relatively symmetric sites. This is also reflected in the JO parameters; at all concentrations Ω4 value is greater than Ω2 indicating the existence of low covalency between Eu3+ and ligand and symmetric environment around europium ion in LaPO4, which is highly probable because Eu3+ occupies La3+ sites (no charge imbalance). The point symmetry of Eu3+ in LaPO4 is D2d as calculated using the Stark splitting pattern. It was seen that LaPO4:Eu gives red emission with very high quantum efficiency of 91.2% for a 2.0 mol% doped sample. It is almost comparable to commercial red phosphor in terms of its efficiency indicating its potential for use as a red emitter in future LEDs.References
- C. Lv, W. Di, Z. Liu, K. Zheng and W. Qin, Dalton Trans., 2014, 43, 3681 RSC.
- D. Xu, Y. Zhang, D. Zhang and S. Yang, CrystEngComm, 2015, 17, 1106 RSC.
- W. Yang, X. Li, D. Chi, H. Zhang and X. Liu, Nanotechnology, 2014, 25, 482001 CrossRef PubMed.
- B. Mutelet, S. Boudin, O. Pérez, J. M. Rueff, C. Labbé and P. A. Jaff, Dalton Trans., 2015, 44, 1186 RSC.
- F. Chen, M. Chen, C. Yang, J. Liu, N. Luo, G. Yang, D. Chen and L. Li, Phys. Chem. Chem. Phys., 2015, 17, 1189 RSC.
- E. Hemmer, M. Quintanilla, F. Legare and F. Vetrone, Chem. Mater., 2015, 27, 235 CrossRef CAS.
- G. Phaomei, R. S. Ningthoujam, W. R. Singh, R. S. Loitongbam, N. S. Singh, A. Rath, R. R. Juluri and R. K. Vatsa, Dalton Trans., 2011, 40, 11571 RSC.
- H. Chen, Y. Ni, X. Ma and J. Hong, J. Colloid Interface Sci., 2014, 428, 141 CrossRef CAS PubMed.
- D. Che, X. Zhu, P. Liu, Y. Duan, H. Wang, Q. Zhang and Y. Li, J. Lumin., 2014, 153, 369 CrossRef CAS.
- J. Fang, M. Saunders, Y. Guo, G. Lu, C. L. Raston and K. S. Iyer, Chem. Commun., 2010, 46, 3074 RSC.
- R. Shukla, S. K. Gupta, V. Grover, V. Natarajan and A. K. Tyagi, Dalton Trans., 2015, 44, 10628 RSC.
- R. Gupta, S. K. Gupta, J. S. Gamre, K. V. Lohithakshan, V. Natarajan and S. K. Aggarwal, Eur. J. Inorg. Chem., 2015, 104 CrossRef CAS.
- S. K. Gupta, M. K. Bhide, S. V. Godbole and V. Natarajan, J. Am. Ceram. Soc., 2014, 97, 3694 CrossRef CAS.
- R. Phatak, S. K. Gupta, K. Krishnan, S. K. Sali, S. V. Godbole and A. Das, Dalton Trans., 2014, 43, 3306 RSC.
- S. K. Gupta, M. Mohapatra, V. Natarajan and S. V. Godbole, RSC Adv., 2013, 3, 20046 RSC.
- S. K. Gupta, M. Mohapatra, V. Natarajan and S. V. Godbole, J. Mater. Sci., 2012, 47, 3504 CrossRef CAS.
- S. K. Gupta, M. Mohapatra, S. Kaity, V. Natarajan and S. V. Godbole, J. Lumin., 2012, 132, 1329 CrossRef CAS.
- X. Z. Wang, J. Mater. Sci.: Mater. Electron., 2014, 25, 1264 CrossRef CAS.
- X. Y. Sun, X. D. Sun, X. G. Li, J. He and B. S. Wang, Nanosci. Nanotechnol. Lett., 2014, 6, 420 CrossRef CAS.
- S. H. Yang, C. K. Yang, J. H. Yan and C. M. Lin, J. Electron. Mater., 2014, 43, 3593 CrossRef CAS.
- P. Ghosh, A. Kar and A. Patra, J. Appl. Phys., 2010, 108, 113506 CrossRef.
- W. Lei and J. Lee, J. Phys. Chem. C, 2008, 112, 11679 Search PubMed.
- J. Li, X. Wu, Y. Fan, Y. Li, L. Hu and C. Tang, Mater. Chem. Phys., 2010, 124, 1172 CrossRef CAS.
- G. Phaomei, R. S. Ningthoujam, W. R. Singh, N. S. Singh, M. N. Luwang, R. Tewari and R. K. Vatsa, Opt. Mater., 2010, 32, 616 CrossRef CAS.
- M. J. Fisher, W. Wang, P. K. Dorhout and E. R. Fisher, J. Phys. Chem. C, 2008, 112, 1901 CAS.
- Y. Li, Y. Li, R. Chen, K. Sharafudeen, S. Zhou, M. Gecevicius, H. Wang, G. Dong, Y. Wu, X. Qin and J. Qiu, NPG Asia Mater., 2015, 7, e180 CrossRef.
- Y. Li, S. Zhou, Y. Li, K. Sharafudeen, Z. Ma, G. Dong, M. Peng and J. Qiu, J. Mater. Chem. C, 2014, 2, 2657 RSC.
- Y. Li, Y. Li, K. Sharafudeen, G. Dong, S. Zhou, Z. Ma, M. Peng and J. Qiu, J. Mater. Chem. C, 2014, 2, 2019 RSC.
- Y. Li, S. Zhou, G. Dong, M. Peng, L. Wondraczek and J. Qiu, Sci. Rep., 2013, 4, 4059 Search PubMed.
- G. Kresse and J. Furthmueller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmueller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Enzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- P. E. Blöchl, O. Jepsen and O. K. Andesen, Phys. Rev. B: Condens. Matter, 1994, 49, 16223 CrossRef.
- N. P. Shaik, N. V. P. Rao and K. V. R. Murthy, Adv. Mater. Lett., 2014, 5, 722 Search PubMed.
- C. Zhang and J. Lin, Chem. Soc. Rev., 2012, 41, 7938 RSC.
- A. Hezel and S. D. Ross, Spectrochim. Acta, 1966, 22, 1949 CrossRef CAS.
- S. V. Eliseevaa and J. C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- Q. Ju, Y. Liu, R. Li, L. Liu, W. Luo and X. Chen, J. Phys. Chem. C, 2009, 113, 2309 CAS.
- G. Blasse, Phys. Lett. A, 1968, 28, 444 CrossRef CAS.
- Y. X. Ni, J. M. Hughes and A. N. Mariano, Am. Mineral., 1995, 80, 21 CAS.
- K. Toyoura, N. Hatada, Y. Nose, T. Uda and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 184301 CrossRef.
- A. A. Dakhel, Opt. Mater., 2009, 31, 691 CrossRef CAS.
- B. R. Judd, Phys. Rev., 1962, 127, 750 CrossRef CAS.
- G. S. Ofelt, J. Chem. Phys., 1962, 37, 511 CrossRef CAS.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC.
- S. K. Gupta, M. Mohapatra, S. V. Godbole and V. Natarajan, RSC Adv., 2013, 3, 20046 RSC.
- G. F. de Sa, O. L. Malta, C. M. Donega, A. M. Simas, R. L. Longo, P. A. Santa-Cruz and E. F. da Silva, Coord. Chem. Rev., 2000, 196, 165 CrossRef CAS.
- E. M. Rodrigues, E. R. Souza, J. H. S. K. Monteiro, R. D. L. Gaspar, I. O. Mazali and F. A. Sigoli, J. Mater. Chem., 2012, 22, 24109 RSC.
- J. H. S. K. Monteiro, A. L. B. Formiga and F. A. Sigoli, J. Lumin., 2014, 154, 22 CrossRef CAS.
- R. A. Sá Ferreira, S. S. Nobre, C. M. Granadeiro, H. I. S. Nogueira, L. D. Carlos and O. L. Malta, J. Lumin., 2006, 121, 561 CrossRef.
- G. V. Prakash and R. Jagannathan, Spectrochim. Acta, Part A, 1999, 55, 1799 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |