 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biosynthesis of thiomarinol A and related metabolites of Pseudoalteromonas sp. SANK 73390†
Annabel C.
Murphy‡§
a,
Shu-Shan
Gao‡
a,
Li-Chen
Han
a,
Simon
Carobene
a,
Daisuke
Fukuda
bc,
Zhongshu
Song
a,
Joanne
Hothersall
b,
Russell J.
Cox
a,
John
Crosby
a,
Matthew P.
Crump
a,
Christopher M.
Thomas
b,
Christine L.
Willis
*a and
Thomas J.
Simpson
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: chris.willis@bristol.ac.uk; tom.simpson@bristol.ac.uk; Fax: +44 (0)117 925 1295; Tel: +44 (0)117 928 7660
bSchool of Biosiences, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
cDiscovery Science and Technology Department, Daiichi Sankyo RD Novare co., Ltd, 1-16-13, Kita-Kasai, Edogawa-ku, Tokyo 134-9630, Japan
First published on 1st November 2013
Abstract
The biosynthesis of the mixed PKS-FAS-NRPS hybrid antibiotic thiomarinol A was investigated using feeding studies to both wild type and mutant strains of the marine bacterium Pseudoalteromonas. Particularly interesting features of the pathway include assembly of the 8-hydroxyoctanoic acid side-chain via chain extension of a C4-precursor (4-hydroxybutyrate), and construction of the pyrrothine unit from cysteine via (HolA-D, F-H) prior to intact incorporation into thiomarinol (catalysed by TmlU). A series of thiomarinol-related and other minor metabolites have been isolated from wild-type and mutant strains. The results of these investigations are rationalised in terms of the overall biosynthetic pathway.
Introduction
Thiomarinol A 1 (Fig. 1), is the major metabolite produced by the marine bacterium Pseudoalteromonas sp. SANK 73390, along with minor analogues B to G (2 to 7).1–3 They display antibiotic activity against both Gram-negative and Gram-positive bacteria, and have particularly potent activity against methicillin-resistant Staphylococcus aureus (MRSA). They are unusual in being hybrid antibiotics, effectively two separate antibiotic classes in one molecule, and biosynthetically can be regarded as comprising three main elements: a highly functionalised polyketide-derived acid, which is esterified by a hydroxy fatty acid, which itself forms an amide with the bicyclic amino acid-derived pyrrothine. The main carbon skeleton of the thiomarinols is very closely related to that of mupirocin, an important antibiotic used clinically against MRSA.4 Mupirocin consists of a mixture of pseudomonic acids (PA A–C, 8 to 10),5–7 with PA-A comprising ca. 95% of the mixture. The major differences in thiomarinol A are the replacement of the 9-hydroxynonanoic acid moiety in the PAs with 8-hydroxyoctanoate in the thiomarinols, the presence of an extra 4-hydroxyl group, the lack of the 10,11-epoxide and the pyrrothine. Many pyrrothine natural products themselves display antibiotic activity, e.g. holomycin 11, N-propionylholothin 12, thiolutin 13, and aureothricin 14.8–10 Stable isotope labelling studies were used to confirm the origins of all of the atoms in pseudomonic acid A and were consistent with a polyketide/fatty acid origin.11,12 This was subsequently confirmed by isolation of the biosynthetic gene cluster13 which revealed that mupirocin was one of the first members of what is now known to comprise an increasingly large number of biologically active compounds. These are most often from diverse and unusual bacterial sources and are produced via trans-AT modular PKSs that have evolved independently from other modular systems.14 The main identifying feature of these systems is the lack of an integral acyl transferase (AT) domain in each chain extension module, this function being supplied “in trans” to each module by separately encoded mono- (or di-) functional ATs. In addition they show many other unusual features including extensive involvement of other trans acting enzymes during polyketide assembly. In particular β-branches are introduced onto β-keto–thiol ester intermediates by a cassette of enzymes that include an HMG CoA synthase (HMGS) analogue using chemistry similar to that involved in mevalonate formation in terpene biosynthesis.15 The biosynthesis of the pyrrothine class of natural products has not been studied in detail, although cysteine has been implicated as a precursor via radio-isotope labelling studies.16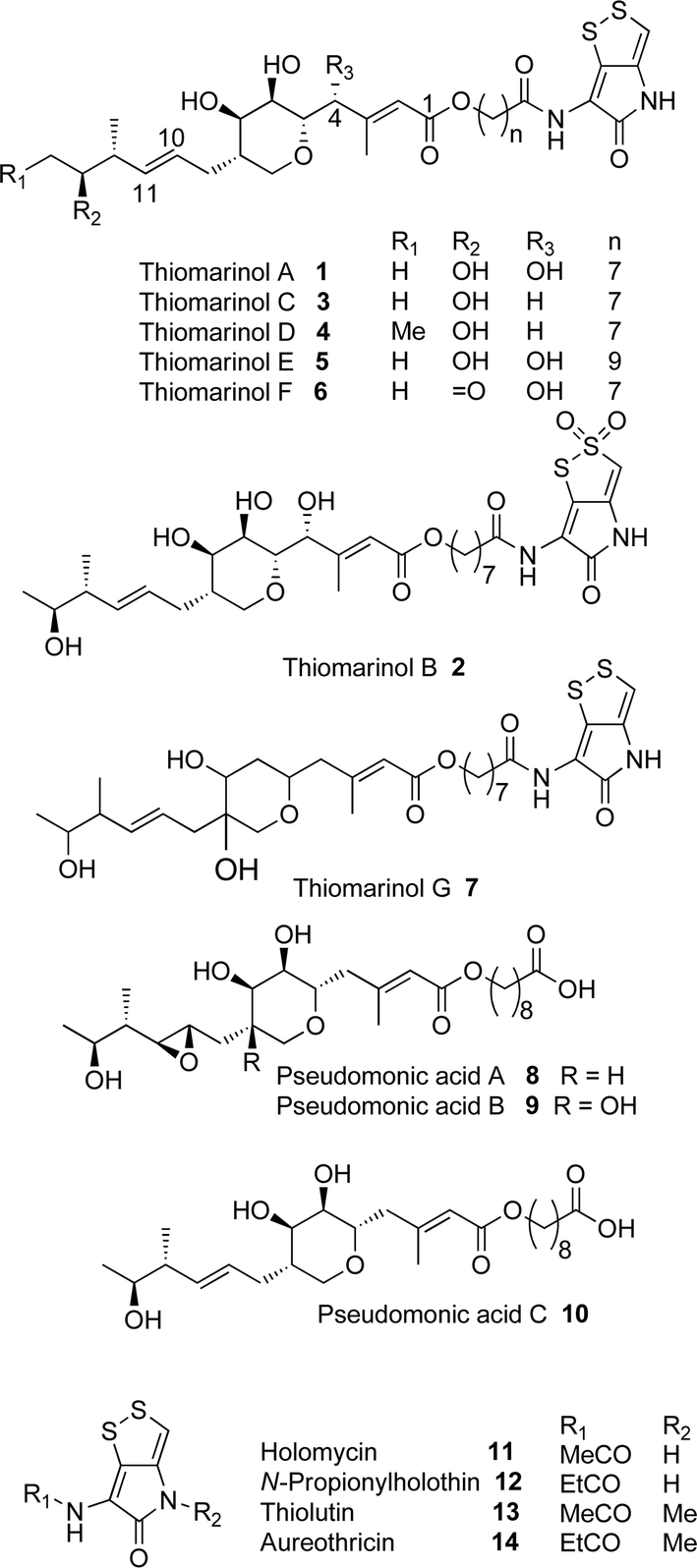 | ||
| Fig. 1 Thiomarinol, pseudomonic acid and acyl pyrrothine antibiotics. | ||
We have recently identified the thiomarinol (tml) biosynthetic gene cluster via full genome sequencing of SANK 73390. It is contained on a 97 kb plasmid consisting almost entirely of the thiomarinol biosynthetic genes.17 These consist of trans-AT PKSs (tmpA, C and D), a putative FAS (tmpB) and associated tailoring and resistance genes (tmlA-Z) with high homology to the mupirocin (mup) cluster (Fig. S1, ESI†). A non-ribosomal peptide synthetase (NRPS) linked to a set of tailoring enzymes (holA-H) is also present similar to that recently shown to control holomycin biosynthesis in Streptomyces clavuligerus.18,19 Analysis of the WT culture and mutant strains in which the PKS and NRPS parts of the cluster had been deactivated by in-frame deletions, resulted in isolation of a number of new thiomarinol related compounds.20 Analysis of extracts of the WT strain by reversed-phase HPLC-ESIMS led to the isolation of two additional polar metabolites which lacked the pyrrothine moiety. These were shown to be marinolic acid A 15 and the corresponding amide, marinolic amide 16. Similar analysis of the ΔNRPS mutant strain showed, as expected, that no pyrrothine containing compounds were produced, the major thiomarinol metabolite being marinolic acid A, but no amide 16. Two minor metabolites were isolated: marinolic acid A617 and A418 in which the octanoate moiety on marinolic acid A has been replaced by hexanoate and butanoate respectively. Re-examination of the WT extracts revealed trace amounts of 17. A number of new acyl-pyrrothine containing compounds were also isolated. On the basis of their similarity to metabolites previously isolated21–23 from Xenorhabdus spp, they were designated as xenorhabdins 8–13 (19–25) respectively (Fig. 2).
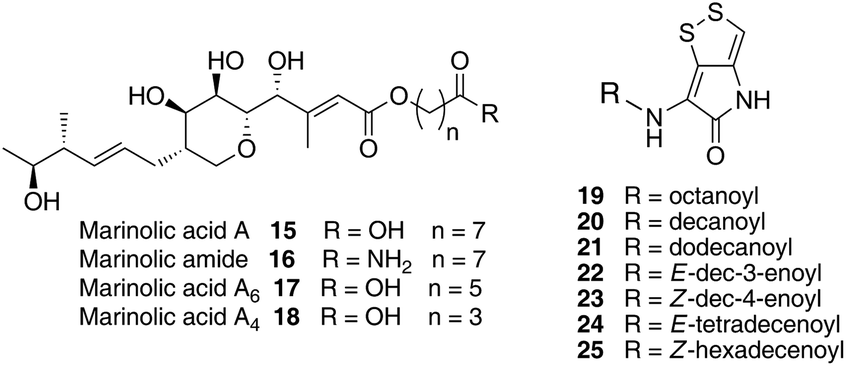 | ||
| Fig. 2 Marinolic acid and acyl-pyrrothine metabolites isolated from WT and mutant strains. | ||
We now report stable isotope labelling and other feeding experiments with WT and mutant strains of Pseudoalteromonas which confirm the origins of all of the atoms in thiomarinol, provide a rationalisation for the occurrence of the minor related metabolites, and for the timing of pyrrothine formation and its linkage to marinolic acid.
Results and discussion
The 13C NMR spectrum of thiomarinol A was rigorously assigned. This necessitated reassignment of two side-chain carbons (C4′ and C5′) compared to that made previously.1 This was achieved using a high resolution band-selective HMBC experiment with correlations observed via H3′ (to C4′ and C5′) and H7′ (to C6′ and C5′) allowing these two carbons, which are separated by 0.1 ppm, to be unambiguously assigned. In d6-DMSO, the 15- and 17-methyls are coincident, but they are resolved in d4-methanol. Prior to stable-isotope labelled precursor feeding, the production of thiomarinol A was monitored. This showed that thiomarinol production commences at around 7 hours after the start of fermentation, reaches a maximum around 24 hours and levels then slowly decrease thereafter. The labelled precursors were therefore fed to the fermentation media ca. 7 hours after inoculation, and the fermentation extracted after 24 hours. The crude extract obtained was purified to give thiomarinol A, which was then analysed by 13C NMR to determine the extent and positions of 13C and 18O incorporations. The results of feeding labelled acetates and [methyl-13C]-methionine to wild-type Pseudoalteromonas SANK 73390 are summarised in Scheme 1.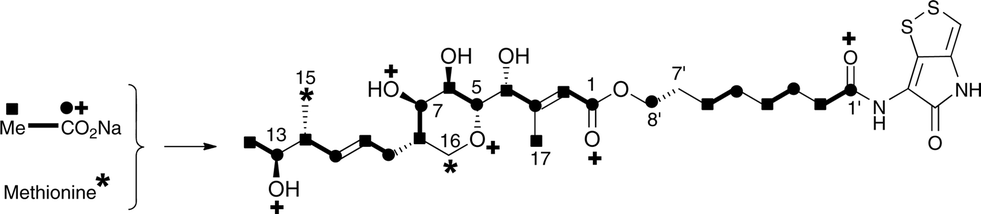 | ||
| Scheme 1 Incorporation of [13C]- and [18O]-labelled acetates and [methyl-13C]-methionine into thiomarinol A. | ||
The incorporation pattern confirms the similarity of the pseudomonic acid A11,12 and thiomarinol A biosynthetic pathways. A regular acetate incorporation pattern is observed in the C1–C14 fragment of thiomarinol. A high level of 13C incorporation (5–6%) is observed at all seven expected positions in the polyketide-derived moiety with high levels of 18O retention (ca. 80%) at carbons 1, 5, 7 and 13 (Table 1). The 4- and 6-hydroxyl groups are not labelled from 18O-acetate and are at positions consistent with their introduction being via tailoring enzymes using molecular oxygen. The 16-methylene (55% enriched from [methyl-13C]methionine) and 17-methyl carbons (40% enriched) are methionine derived consistent with the presence of C-MeT domains in the first and third extension modules, and the 15-methyl is derived from the methyl of a cleaved acetate unit consistent with it being derived via the HMGS β-branch pathway as in pseudomonic acid A.
| Carbon | δ C | [1,2-13C2]-acetatea % (JC–C Hz) | [2-13C]-acetatea % | [1-13C, 18O2]-acetatea % ΔδCb (ppm) |
|---|---|---|---|---|
a Percentage enrichment (>1 to nearest integer); significant differences from average values indicated in bold.
b Average ratio of enriched (13C) to enriched + isotopically shifted (13C18O) signals is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 except C-1′ which is ca. 1:1, all values to nearest integer.
c Spectra determined in d6-DMSO, and d4-MeOH in which the 15- and 17-methyls appear respectively at 14.9 and 15.2 ppm. :4 except C-1′ which is ca. 1:1, all values to nearest integer.
c Spectra determined in d6-DMSO, and d4-MeOH in which the 15- and 17-methyls appear respectively at 14.9 and 15.2 ppm.
|
||||
| 1 | 166.1 | 2 (76) | 5 (0.03) | |
| 2 | 114.3 | 2 (76) | 7 | |
| 3 | 160.8 | 2 (43) | 5 | |
| 4 | 72.4 | 2 (43) | 7 | |
| 5 | 76.2 | 2 (43) | 6 (0.02) | |
| 6 | 63.8 | 2 (43) | 7 | |
| 7 | 69.6 | 2 (35) | 6 (0.02) | |
| 8 | 42.2 | 2 (35) | 7 | |
| 9 | 31.8 | 2 (44) | 6 | |
| 10 | 127.7 | 2 (44) | 7 | |
| 11 | 134.1 | 2 (44) | 6 | |
| 12 | 43.1 | 2 (44) | 7 | |
| 13 | 69.2 | 2 (39) | 6 (0.02) | |
| 14 | 20.0 | 2 (39) | 7 | |
| 15 | 15.7 | 2 | 7 | |
| 16 | 64.2 | |||
| 17 | 15.7 | |||
| 1′ | 171.8 | 2 (50) | 6 (0.03) | |
| 2′ | 34.6 | 2 (50) | 7 | |
| 3′ | 24.9 | 2 (35) | 6 | |
| 4′ | 28.4 | 2 (35) | 7 | |
| 5′ | 28.3 | 1 (34) | 3 | |
| 6′ | 25.3 | 1 (34) | 4 | |
| 7′ | 28.2 | 0.3 (38) | 2 | |
| 8′ | 63.0 | 0.3 (38) | 2 | 1 |
| 1′′ | 167.9 | |||
| 2′′ | 115.3 | |||
| 3′′ | 133.9 | |||
| 4′′ | 133.6 | |||
| 5′′ | 110.3 | |||
Biosynthetic origin of the side-chain
Interestingly the labelling pattern in the octanoate side chain did not follow a standard polyketide labelling pattern. Whilst expected head-to-tail incorporation of intact acetate units from [1,2-13C2]acetate was observed for C1′ to C4′ (2% incorporation), a lower level of incorporation was observed in C5′ and C6′ (1%) and much lower into C7′ and C8′ (0.3%). [2-13C]-Acetate also gave a similar pattern with much lower incorporation of 13C into both C7′ and C8′ (∼2% compared to 7% in other acetate-derived positions), and an intermediate level of enrichment (4%) at C6′ (Table 1). [1-13C, 18O2]-Acetate gave incorporation levels of 13C (∼6%) at carbons 1′ and 3′ as was observed elsewhere, but incorporation into C5′ was half this level and at C7′ was negligible. A low incorporation, ca. 1%, into C8′ was observed. These results suggest that a C4 precursor is likely to be the starter unit for two rounds of FAS catalysed extension to give the octanoate side-chain. The observed scrambling of acetate incorporation at C7′ and C8′ suggested the possible involvement of a citric acid (Krebs) cycle intermediate.Feeding [2,3-13C2]succinate to cultures of wild-type Pseudoalteromonas SANK 73390 revealed an intact incorporation confirmed by observation of a 13C–13C coupling of 34 Hz between C6′ and C7′ (ca. 0.5% enrichment, Scheme 2, see insert expansion in Fig. S10†) in accord with the proposed C4 precursor. A lower level of incorporation (0.15%) was also observed elsewhere within the molecule resulting from degradation of [2,3-13C2]succinate to [1,2-13C2]acetate.
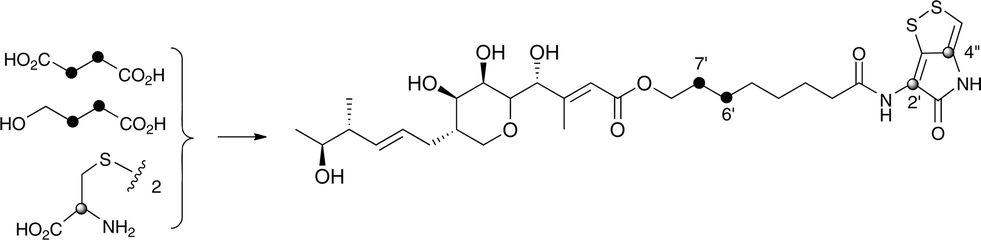 | ||
| Scheme 2 Incorporation of [13C]-labelled succinate, 4-hydroxybutyrate and cystine into thiomarinol A. | ||
It is known that 4-hydroxybutyrate can be formed via reduction of succinyl CoA.24 The overall results of acetate incorporation are also consistent with this: acetate is incorporated intact into only carbons 3 and 4 of succinyl CoA (Scheme S2, ESI†) which explains the significant but lower incorporation into C5′/C6′ of thiomarinol, presumably due to dilution by a pool of endogenously derived Krebs Cycle intermediate. Further cycling of labelled succinate would give single labels at carbons 1 and 2 of succinyl CoA which explains the lower levels of labelling from [2-13C]acetate into C7′ and C8′. To explain the low level of intact acetate labelling of C7′/C8′, the conversion of succinyl CoA to symmetrical succinate must be partially reversible so that ca. 30% of the succinate is incorporated via this route. The low level of labelling at C8′ from [1-13C]acetate is completely consistent with this hypothesis.
To further investigate the proposed C4 side-chain precursor, [2,3-13C2]-4-hydroxybutyrate was prepared from [2,3-13C2]succinate via cyclisation to succinic anhydride, reduction to the lactone with LiAlH4 followed by hydrolytic ring opening with aqueous NaOH (Scheme S1†). Feeding [2,3-13C2]-4-hydroxybutyrate to cultures of the wild-type Pseudoalteromonas SANK 73390 showed a site-specific labelling (0.2%) of thiomarinol A at C6′, C7′ confirming intact incorporation (Scheme 2, see insert expansion in Fig. S12†).
Thus the biosynthetic origin of C5′ to C8′ is somewhat similar to the situation presumed for 9-hydroxynonanoic in mupirocin biosynthesis, where labelling studies are consistent with a 3-hydroxypropionate starter unit, which then undergoes three reductive chain elongations. 3-Hydroxypropionate can be formed in bacteria by reduction of the thiolester of malonyl CoA25 and both the mup and tml clusters show the presence of genes encoding a malonyl/succinyl CoA dehydrogenase clustered with an acyl CoA synthetase and an ACP.
Interestingly, the biosynthetic gene cluster for difficidin,26 another trans-AT metabolite of Bacillus amyloliquefaciens, also contains an oxo-acyl CoA reductase encoded by difE which as in mupirocin/thiomarinol is grouped with an acyl CoA synthetase and an ACP (difD and difC). In addition there is an adjacent kinase (difB) which would be suitable for catalysing dehydration of 3-hydroxypropionate to produce the acryl thiol ester required to prime difficidin chain elongation.
The retention of 18O label at C1′ of thiomarinol A is slightly less than half that at the other acetate-oxygen bearing positions. This is consistent with C1′ existing as the free carboxylate, resulting in randomisation of the label between both carboxylate oxygens, before ligation via an amide bond to the pyrrothine. Analysis of the structure of the pyrrothine moiety of thiomarinol suggest that it is formed via two molecules of cysteine. To gain evidence for this proposal, [2-13C]cystine was prepared from ethyl [2-13C]pyruvate, (Scheme S2†) and fed to WT Pseudoalteromonas. A high level of incorporation of 13C into the C2′′ and C4′′ positions (16 and 20% respectively) confirmed that the pyrrothine is indeed cysteine, and therefore NRPS, derived (Scheme 2).
The biosynthetic gene cluster for holomycin biosynthesis in Streptomyces clavuligerus has been isolated and a pathway from cysteine proposed. This pathway involves initial dipeptide formation followed by a series of oxidative cyclisations, the final step being acetylation of the free pyrrothine.18 To prove that the pyrrothine moiety in thiomarinol is assembled and then incorporated as an intact unit rather than being assembled on marinolic acid, pyrrothine was synthesised as previously described27 and fed to the ΔNRPS mutant strain of SANK 73390. The mutant was prepared by a deletion in the holA gene encoding the amino acid activation domain which, from sequence analysis of the active site, is predicted to selectively activate cysteine. As indicated above, the ΔNRPS mutant normally produces mainly marinolic acid. When cultures were supplemented with pyrrothine, the normal WT phenotype was restored indicating that Pseudoalteromonas takes the intact pyrrothine as substrate (Scheme 3). Interestingly, the concentration of the feeds was critical, with maximum restoration of thiomarinol A production being observed at a pyrrothine concentration of 20 mg ml−1. At concentrations of >80 mg ml−1, no thiomarinol A was observed and production of marinolic acid was also completely inhibited (Fig. S3, ESI†).
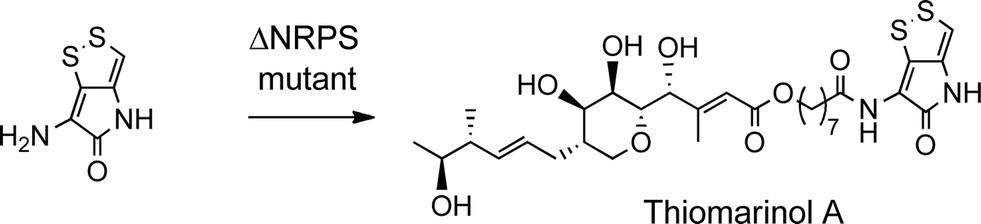 | ||
| Scheme 3 Feeding pyrrothine to the NRPS mutant gives thiomarinol A. | ||
Novel acyl pyrrothines via mutasynthesis
Previous studies have shown that when TmlU, a putative amide ligase is knocked out, the mutant strain produces no thiomarinol but does produce marinolic acid and the full set of xenorhabdins.20 The HolE analogue, ORF3483, from the holomycin cluster in S. clavuligerus has been expressed and purified and it has been shown18 to act as an acyl transferase capable of transferring acetyl and longer acyl groups to pyrrothine. Thus it appears that the thiomarinol cluster has two ligases, TmlU which forms the amide in thiomarinol, and HolE which links pyrrothine to a wide range of acyl CoAs available in Pseudoalteromonas to produce the xenorhabdins.To test if HolE can also transfer a range of non-endogenous substrates, various short, medium and long chain fatty acids and others with ω-functionalized side chains, e.g. hydroxyl, amino, carboxyl and phenyl were fed to the ΔPKS mutant (Scheme 4). Some of these were incorporated with varying efficiencies, presumably after conversion to their respective CoA esters, sometimes after catabolic chain shortening to give 19 and the novel acyl-pyrrothines 26–30.
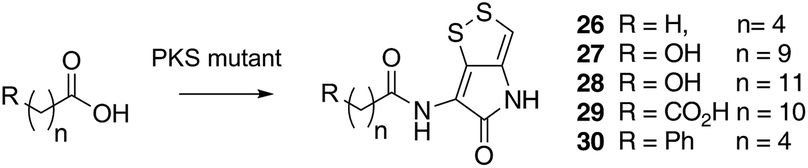 | ||
| Scheme 4 Novel acyl-pyrrothines generated using the ΔNRPS mutant. | ||
The biosynthesis of thiomarinol A and related metabolites
The isolation of the minor metabolites 15–18 from both WT and mutant strains and the incorporation of isotopically labelled substrates can be rationalised as shown in Scheme 5. Marinolic acid A 15 is produced by the ΔNRPS and ΔTmlU mutants, and is present in trace amounts in the WT which suggests that it is both a genuine biosynthetic product and intermediate in its own right and is not produced as an artefact, e.g. as a degradation product of thiomarinol A. The intermediacy of marinolic acid 15 was confirmed by feeding it to the ΔPKS mutant whereupon, thiomarinol production was restored (Fig. S19†). The amide 16 on the other hand was only seen in the WT cultures which suggests that it is formed by degradation of thiomarinol A, and is not formed by, e.g. amidation of marinolic acid. This was further evidenced by feeding thiomarinol A to the ΔPKS mutant when the presence of both marinolic acid and marinolic amide in the crude extract could be observed by LCMS. This suggests that the amide is indeed a degradation product of thiomarinol.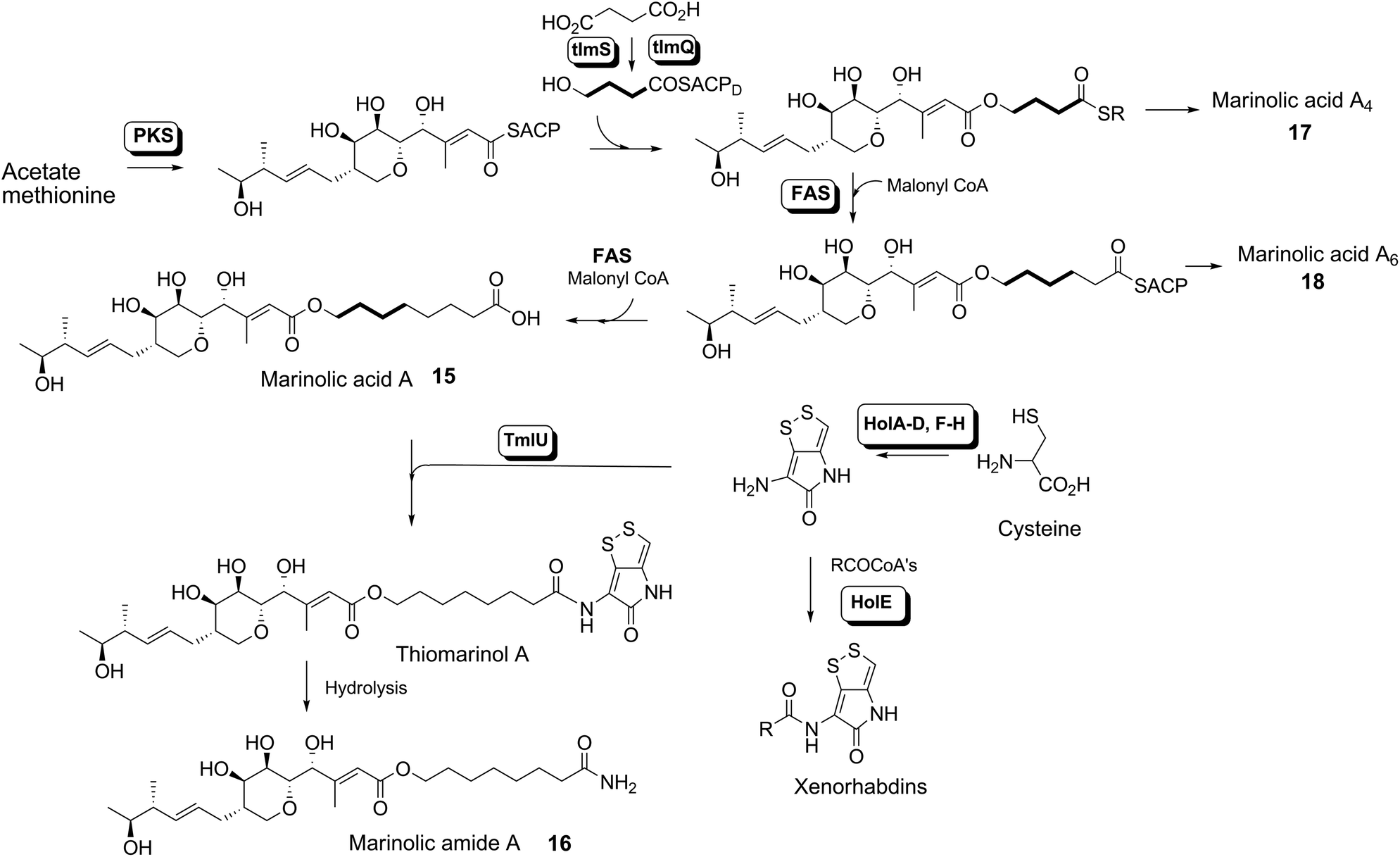 | ||
| Scheme 5 Overall biosynthesis of thiomarinol and related metabolites in Pseudoalteromonas. | ||
The isolation of truncated metabolites marinolic acids A4 and A6, is consistent with the isotope labelling studies which suggest that a C4 intermediate is added to the product of the PKS, e.g. by trans-esterification. This would then be elongated to give an enzyme bound hexanoate analogue which would be further elongated to give the completed octanoate side chain. In the absence of the pyrrothine in the ΔNRPS mutant they would be released either by enzymatic or spontaneous hydrolysis as marinolic acids. We have previously shown that release of assembly intermediates occurs when flux of intermediates along the normal mupirocin pathway is impaired by a range of targeted mutations of trans-acting and other tailoring genes.28 Similar mechanisms would allow leakage of marinolic acids A4 and A6 into the medium. It is significant that this appears to be more pronounced in the mutant than the WT. Truncated forms of mupirocin and related metabolites with C7 and C5 hydroxy acids have also been observed in both mutant strains and when the quorum sensing regulation mechanism has been manipulated to increase mupirocin production.29
Conclusions
Using a combination of genetic and isotopic labelling studies, evidence has been presented for a mixed PKS-NRPS biosynthetic pathway to the antibiotic thiomarinol A in Pseudoalteromonas sp. SANK 73390. Particularly interesting features of the pathway include assembly of the 8-hydroxyoctanoic acid side-chain, which is generated via chain extension of a C4 precursor (4-hydroxybutyrate), and both marinolic acids A418 and A617 have been isolated as minor metabolites from both wild-type and the ΔNRPS mutant. There was a good incorporation of [13C]cystine into thiomarinol A and feeding studies with synthetic pyrrothine to the ΔNRPS mutant are in accord with heterocycle assembly (HolA-D, F-H) prior to intact incorporation into thiomarinol A (TmlU). A further minor metabolite, marinolic amide 16, present in wild-type but not the ΔNRPS mutant, is a degradation product of thiomarinol. Isotopic labelling studies indicate that the biosynthesis of the C1–C17 fragment is analogous to that of pseudomonic acid, a trans-AT modular PKS pathway with many fascinating and, as yet, unexplained features and these studies lay the foundation for a further understanding of the pathways to these compounds.Acknowledgements
This work was supported by BBSRC grants BB/I014039, BB/I014737, BB/E021611 and BB/E022367. We thank Daiichi Sankyo Co. Ltd for funding (DF) and for strain SANK 73390. We are grateful to the Bristol Chemical Synthesis Centre for Doctoral Training, funded by EPSRC (EP/G036764/1), AstraZeneca, GlaxoSmithKline, Novartis, Pfizer, Syngenta and the University of Bristol for a studentship to SC.Notes and references
- H. Shiozawa, I. T. Kagasak, T. Kinoshita, H. Haruyama, H. Domon, Y. Utsui, K. Kodama and S. Takahashi, J. Antibiot., 1993, 46, 1834–1842 CrossRef CAS.
- H. Shiozawa, T. Kagasaki, A. Torikata, N. Tanaka, K. Fujimoto, T. Hata, Y. Furukawa and S. Takahashi, J. Antibiot., 1995, 48, 907–909 CrossRef CAS.
- H. Shiozawa, A. Shimada and S. Takahashi, J. Antibiot., 1997, 50, 449–452 CrossRef CAS.
- C. M. Thomas, J. Hothersall, C. L. Willis and T. J. Simpson, Nat. Rev. Microbiol., 2010, 8, 281–289 CrossRef CAS PubMed.
- A. T. Fuller, G. Mellows, M. Woolford, G. T. Banks, K. D. Barrow and E. B. Chain, Nature, 1971, 234, 416–417 CrossRef CAS.
- E. B. Chain and G. Mellows, J. Chem. Soc., Perkin Trans. 1, 1977, 294–309 RSC.
- E. B. Chain and G. Mellows, J. Chem. Soc., Perkin Trans. 1, 1977, 318–322 RSC.
- L. Ettlinger, E. Gäumann, R. Hütter, W. Keller-Schierlein, F. Kradolfer, L. Neipp, V. Prelog and H. Zähner, Helv. Chim. Acta, 1959, 42, 563–569 CrossRef CAS.
- W. D. Celmer and I. A. Solomons, J. Am. Chem. Soc., 1955, 77, 2861–2865 CrossRef CAS.
- K. Okamura, K. Soga, Y. Shimauchi, T. Ihikura and J. Lein, J. Antibiot., 1977, 30, 334–336 CrossRef CAS.
- T. C. Feline, R. B. Jones, G. Mellows and L. Phillips, J. Chem. Soc., Perkin Trans. 1, 1977, 309–318 RSC.
- F. M. Martin and T. J. Simpson, J. Chem. Soc., Perkin Trans. 1, 1989, 207–209 RSC.
- A. K. El-Sayed, J. Hothersall, S. M. Cooper, E. Stephens, T. J. Simpson and C. M. Thomas, Chem. Biol., 2003, 10, 419–430 CrossRef CAS.
- J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC.
- C. T. Calderone, Nat. Prod. Rep., 2008, 25, 845–853 RSC.
- M. Okanishi and H. Umezawa, Genetics of the Actinomycetales, Gustav Fischer, Stuttgart, New York, 1978 Search PubMed.
- D. Fukuda, A. S. Haines, Z. Song, A. Murphy, J. Hothersall, E. R. Stephens, R. Gurney, R. J. Cox, J. Crosby, C. L. Willis, T. J. Simpson and C. M. Thomas, PLoS One, 2011, 6, e18031 CAS.
- B. Li and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12629–12633 CrossRef PubMed.
- S. Huang, Y. Zhao, Z. Qin, X. Wang, M. Onega, L. Chen, J. He, Y. Yu and H. Deng, Process Biochem., 2011, 46, 811–816 CrossRef CAS PubMed.
- A. C. Murphy, D. Fukuda, Z. Song, J. Hothersall, R. J. Cox, C. L. Willis, C. M. Thomas and T. J. Simpson, Angew. Chem., Int. Ed., 2011, 123, 3271–3274 CrossRef PubMed.
- B. V. McInerney, R. P. Gregson, M. J. Lacey, R. J. Akhurst, G. R. Lyons, S. H. Rhodes, D. R. J. Smith, L. M. Engelhardt and A. H. White, J. Nat. Prod., 1991, 54, 774–784 CrossRef CAS.
- J. Li, G. Chen, J. M. Webster and E. Czyzewska, J. Nat. Prod., 1995, 58, 1081–1086 CrossRef CAS.
- S. Paik, Y. H. Park, S. I. Suh, H. S. Kim, I. S. Lee, M. K. Park, C. S. Lee and S. H. Park, Bull. Korean Chem. Soc., 2001, 22, 372–374 CAS.
- I. A. Berg, D. Kockelkorn, W. Buckel and G. Fuchs, Science, 2007, 318, 1782–1786 CrossRef CAS PubMed.
- M. Hügler, C. Menendez, H. Schägger and G. Fuchs, J. Bacteriol., 2002, 184, 2404–2410 CrossRef.
- X.-H. Chen, J. Vater, J. Piel, P. Franke, R. Scholz, K. Schneider, A. Koumoutsi, G. Hitzeroth, N. Grammel, A. W. Strittmatter, G. Gottschalk, R. D. Suessmuth and R. Borriss, J. Bacteriol., 2006, 188, 4024–4036 CrossRef CAS PubMed.
- T. Hjelmgaard, M. Givskovb and J. Nielsen, Org. Biomol. Chem., 2007, 5, 344–348 CAS.
- J.-e. Wu, J. Hothersall, C. Mazetti, Y. O'Connell, J. A. Shields, A. S. Rahman, R. J. Cox, J. Crosby, T. J. Simpson, C. M. Thomas and C. L. Willis, ChemBioChem, 2008, 9, 1500–1508 CrossRef CAS PubMed.
- J. Hothersall, A. C. Murphy, Z. Iqbal, G. Campbell, E. R. Stephens, J.-e. Wu, H. Cooper, S. Atkinson, P. Williams, J. Crosby, C. L. Willis, R. J. Cox, T. J. Simpson and C. M. Thomas, Appl. Microbiol. Biotechnol., 2011, 90, 1017–1026 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental details, feeding experiments, synthesis of labelled precursors, spectroscopic data and NMR spectra of labelled thiomarinol A. See DOI: 10.1039/c3sc52281d |
| ‡ These authors contributed equally to the work. |
| § Current address: Department of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge, CB2 1GA. |
| This journal is © The Royal Society of Chemistry 2014 |