Dispersing Pt atoms onto nanoporous gold for high performance direct formic acid fuel cells†
Rongyue
Wang
b,
Jianguo
Liu
*a,
Pan
Liu
c,
Xuanxuan
Bi
b,
Xiuling
Yan
b,
Wenxin
Wang
b,
Xingbo
Ge
bc,
Mingwei
Chen
*c and
Yi
Ding
*bd
aEco-Materials and Renewable Energy Research Center, Department of Materials Science and Engineering, and National Laboratory of Solid State Microstructures, Nanjing University, Nanjing 210093, China. E-mail: jianguoliu@nju.edu.cn; Fax: +86-25-83686632; Tel: +86-25-83621219
bCenter for Advanced Energy Materials & Technology Research (AEMT), and School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China. E-mail: yding@sdu.edu.cn; Fax: +86-531-88366269; Tel: +86-531-88366513
cWPI Advanced Institute for Materials Research, Tohoku University, Sendai 980-8577, Japan. E-mail: mwchen@wpi-aimr.tohoku.ac.jp; Fax: +81-22-217-5955; Tel: +81-22-217-5990
dShandong Applied Research Center for Gold Technology (Au-SDARC), Yantai 264005, China
First published on 14th October 2013
Abstract
Direct formic acid fuel cells (DFAFCs) are promising portable energy conversion devices for supplying our off-grid energy demands. However, traditional Pt-based catalysts suffer from poor performance; consequently the precious metal loading in an actual fuel cell has to be maintained at a very high value, typically orders of magnitude higher than the acceptable level. Through a molecular self-assembly/electro-deposition process, Pt atoms are effectively dispersed onto the surface of a nanoporous gold substrate, and the resulting nanocomposites demonstrate superior electrocatalytic performance toward formic acid electro-oxidation, which can be attributed to a nearly ideal catalyst configuration where all the Pt atoms are involved in a highly desired direct reaction path. In both half-cell electrochemical testing and actual DFAFCs, these rationally designed electrodes show over two orders of magnitude improvement in Pt efficiency, as compared with the state-of-the-art Pt/C catalyst. This design strategy allows customized development of new generation electrocatalysts for high performance energy saving technologies.
Introduction
The development of highly efficient power sources has a tremendous impact on our sustainable societies and economies.1,2 Among various energy management systems, direct liquid fuel cells show great potential for powering portable electronic devices such as laptop computers and cell phones.3,4 Compared with methanol, formic acid has become increasingly attractive as an alternative fuel option in recent years due to its advantages of being less toxic, having a higher theoretical open circuit potential and lower fuel crossover.5–7 However, before the real application of direct formic acid fuel cells (DFAFCs), the usage of precious metal catalysts such as Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 should be drastically decreased in both the anode and the cathode. Recent years have seen a surge in the development of non-Pt oxygen reduction reaction catalysts, aimed at eliminating the use of Pt at the fuel cell cathode.9–11 However, at anode there are not yet any choices to replace Pt or Pd for formic acid electro-oxidation. Pd possesses high initial activity for this reaction, but is much less stable.5 Pt is more resistant to corrosion, but pure Pt can be readily poisoned by CO intermediates12 which inhibit the direct oxidation of formic acid. Although the exact mechanism of formic acid electro-oxidation on Pt is still under debate,13–15 it is generally agreed that the unwanted formation of the CO intermediates requires continuous surface Pt lattices, while the desired direct oxidation path needs smaller ensembles. This has been proven using DFT calculations16 and also demonstrated by experimental work which showed that covering the Pt surface with adatoms17–21 or alloying Pt with other metals22,23 could greatly inhibit the formation of CO, and result in improved catalytic activity.
8 should be drastically decreased in both the anode and the cathode. Recent years have seen a surge in the development of non-Pt oxygen reduction reaction catalysts, aimed at eliminating the use of Pt at the fuel cell cathode.9–11 However, at anode there are not yet any choices to replace Pt or Pd for formic acid electro-oxidation. Pd possesses high initial activity for this reaction, but is much less stable.5 Pt is more resistant to corrosion, but pure Pt can be readily poisoned by CO intermediates12 which inhibit the direct oxidation of formic acid. Although the exact mechanism of formic acid electro-oxidation on Pt is still under debate,13–15 it is generally agreed that the unwanted formation of the CO intermediates requires continuous surface Pt lattices, while the desired direct oxidation path needs smaller ensembles. This has been proven using DFT calculations16 and also demonstrated by experimental work which showed that covering the Pt surface with adatoms17–21 or alloying Pt with other metals22,23 could greatly inhibit the formation of CO, and result in improved catalytic activity.
The ultimate goal of catalyst development is to realize 100% utilization of the catalytically active species while maintaining good enough durability against dissolution and poisoning. This is particularly challenging for formic acid oxidation catalysts as the target molecules are inherently corrosive and formation of the CO intermediates is almost spontaneous on the Pt surface. A common strategy to achieve high Pt utilization is to make the Pt or Pt-based alloy nanoparticles as small as possible, which are in turn adsorbed onto conductive substrates such as carbon black. For example, Ji and co-workers23 recently prepared sulfur modified ordered mesoporous carbon (OMC) supported Pt intermetallic nanoparticles, the highest mass specific activity toward formic acid electro-oxidation was realized on OMC supported 3 nm PtBi nanoparticles (∼770 mA mg−1). Core–shell nanostructures, especially those with a monolayer-type Pt shell, have attracted great interest in recent years, and enhanced mass specific catalytic activity toward the oxygen reduction reaction is often observed.24–26 Although these nanostructures could, to some extent, approach 100% Pt utilization, they are not favorable for formic acid electro-oxidation as the continuous Pt surface lattices would still facilitate the undesired indirect path which forms the poisoning CO intermediates. In this context, the ideal catalyst structure for this purpose should be a highly conductive high-surface-area electrode on which discrete Pt atoms are uniformly dispersed and tethered to form a stable sub-monolayer type catalyst, so that formic acid electro-oxidation would proceed via the highly efficient direct oxidation path. Here we report on the fabrication and application of this kind of nanoengineered electrocatalyst using a precise molecular self-assembly/electro-deposition method. By dispersing Pt atoms onto a highly conductive nanoporous metal support, these novel electrodes show unparalleled electrocatalytic performance toward formic acid electro-oxidation in both half-cell electrochemical testing (with mass specific activity up to 64 A mg−1 Pt under the noted conditions) and actual DFAFCs.
Experimental section
Preparation of NPG and NPG–Ptx
All electrochemical experiments were carried out at room temperature (approximately 25 °C) in a standard three-electrode cell using a CHI 760 electrochemical workstation (CH instruments, Inc.). Before each electrochemical measurement, the solution was purged with ultra-pure nitrogen for at least 20 min to remove dissolved oxygen. A mercury/mercurous sulfate electrode (MMSE) and a reversible hydrogen electrode (RHE) were used as reference electrodes under different electrochemical conditions. The electrode potentials were reported with respect to RHE. Platinum foil with dimensions of 1 × 1 cm2 was used as the counter electrode.NPG membranes were prepared by dealloying silver–gold alloy leaves (Sepp Leaf Products Inc.) on the surface of concentrated nitric acid at 20 °C for 2 hours. NPG membranes were mounted onto glassy carbon (GC, Ø 3 or Ø 4 mm) electrodes for electrochemical characterization. The electrochemical surface area (ECSA, m2 g−1) of an NPG leaf was measured to be approximately 9.3 m2 g−1 by integrating the reduction peak of the Au oxidant in 0.1 M HClO4 using Brummer's method (Fig. S1†). The electrode was then immersed in 1 mM H2PtCl6 solution to allow adsorption of PtCl62− onto the ligament surface of the NPG leaf (immersion step). The PtCl62− ions in solution were washed away by immersing the electrode in ultra-pure water (18.23 MΩ) for five minutes (cleaning step), the cleaning step was repeated 6 times to ensure that the ions in solution were totally removed. The adsorbed PtCl62− ions were reduced by sweeping the potential to 0 V from an open circuit potential of approximately 0.93 V (reduction step). The amount and structure of Pt deposited onto the ligament surface of the NPG could be adjusted by repeating the immersion, cleaning, and reduction steps.
Inductively coupled plasma atomic emission spectroscopy (ICP-AES) results indicated that the freshly prepared NPG membranes were composed almost entirely of gold (1.04 at.% Ag) and the surface Ag amount could be even lower because the surface Ag was preferentially dissolved away by concentrated nitric acid, which was further confirmed by the X-ray photoelectron spectroscopy (XPS) results (0.5 at.% Ag). These results confirm that the effect of residual Ag atoms could be neglected and the deposited amount of Pt can be obtained by integrating the reduction peak of the PtCl62− ions on the assumption that 100% current efficiency was achieved (see Fig. S1†). Considering the valence state of Pt in PtCl62− and the one-to-one correspondence in the chemisorbed oxygen monolayer on the Au surface (see Ref. S2†), Pt coverage (θPt) could be obtained by dividing half of the PtCl62− reduction charge (QPt) by the Au oxidant reduction charge (QAu).
| θPt = 0.5QPt/QAu |
The morphology of NPG was examined using high-resolution transmission electron microscopy (HRTEM, JEOL JEM-2100 at 200 kV). The actual compositions of the NPG and NPG–Pt samples were determined using ICP-AES (IRIS Advantage). The HAADF STEM characterization was performed with a 200 kV JEM-2100F electron microscope (JEOL) equipped with two aberration correctors (CEOS GmbH) for the image- and probe-forming lens systems and X-ray energy-dispersive spectroscopy (JED-2300T, JEOL) for chemical composition analysis. Both Cs correctors were optimized for image observations and the point-to-point resolutions of TEM and STEM were 1.2 and 1.0 Å, respectively.
Preparation of the working electrode using commercial Pt/C catalysts
A catalyst slurry was prepared by ultrasonically dispersing 5 mg Pt/C (20 wt%, Johnson Matthey) into a 0.5 ml Nafion® alcohol (5 wt%) and 0.5 ml ultra-pure water mixed solution. Then 5 μl of the suspension was pipetted onto a GC electrode (Ø 4 mm) to prepare the working electrode. Finally the catalyst coated GC electrode was allowed to dry in air at room temperature overnight.Electrochemical characterization and catalytic activity of Pt/C and NPG–Ptx catalysts
The ECSA of the Pt/C and NPG–Ptx catalysts was measured using a hydrogen adsorption–desorption method (H-UPD) on platinum in acidic solution (the electric charge density for hydrogen adsorption is known to be 210 μC cm−2) (Fig. S2†). The charges for H-UPD (QH) were obtained in 0.1 M HClO4 by integrating the peaks between 0.05 and 0.4 V as shown in Fig. S2.† The charge associated with hydrogen under-potential adsorption and desorption were averaged to decrease the possible influence of GC.| ECSA = QH/210 |
The Pt utilization (UPt) was obtained by dividing the H-UPD charge (QH), multiplied by four, by reduction charge for PtCl62− (QPt).
| UPt = 4QH/QPt |
The Pt loading (mPt) of the NPG–Pt catalysts could be obtained by integrating the PtCl62− reduction peaks as shown in Fig. S1.†
| mPt = MPtQPt/(nF) |
485.
The catalytic activities of Pt/C and NPG–Ptx were measured in 0.1 M HClO4 containing 0.05 M HCOOH or 0.1 M CH3CH2OH by cyclic voltammetry (CV) using MMSE as the reference electrode with a salt bridge connecting it to the electrochemical cell. The catalytic activity of the different catalysts was normalized to ECSA or Pt loading by directly dividing the data in the CV curves by ECSA or Pt mass, respectively.
Preparation of MEA and fuel cell testing
NPG was floated on the solution surface and attached with one edge to a gold foil for the preparation of samples for the fuel cell test. NPG–Pt catalyst (with a thickness of about 500 nm and an area of 1 × 1 cm2) was loaded onto a piece of Nafion® (115) membrane as an anode. The membrane electrode assembly (MEA) was prepared by pressing carbon paper (anode side) and carbon paper supported Pt/C catalyst (cathode, 60% Pt loading, 2.2 mg Pt per cm2) onto each side of the catalyst loaded Nafion® (115) membrane at 110 °C for 195 s at a pressure of 0.3 MPa cm−2. The carbon paper supported Pt/C catalysts were fabricated by coating the catalyst ink (the mass ratio of dry Nafion to Pt was 1:5) on carbon paper with a brush. The loading of Pt/C on carbon paper was monitored by weighing on an electronic balance. The actual Pt loading on the NPG–Pt anodes was measured using ICP-AES.
The MEA mounted into the fuel cell was firstly conditioned in flowing hot water (90 °C) for 2 h. Then the fuel cell was activated by CV (0–1.3 V) and tested at 40 °C by supplying 3 M formic acid to the anode (about 2 ml min−1) and dry air (about 120 ml min−1) to the cathode. Cell discharging and polarization tests were conducted on a fuel cell station equipped with a Kikusui PLZ30 (Japan) electronic load.
To evaluate the durability of the catalysts in DFAFC, accelerated stability tests (AST) were performed. After MEA activation, the maximum power density of the fuel cell was collected. Hydrogen gas was then supplied to the cathode and potential cycling between 0.5 and 1 V was applied to the anode with a scan rate of 0.5 V s−1 for thousands of cycles. It should be pointed out that under the operation conditions, the potential the of anode is usually between 0.2 and 0.5 V. However, under storage conditions, the fuel cell anode would be filled with air (containing oxygen), which shifts the potential of anode to an even higher value. We therefore chose a potential range between 0.5 and 1 V to monitor both the operation and storage conditions. After cleaning with ultra-pure water for approximately 10 min, the fuel cell was discharged again and the maximum power density was collected.
Results and discussion
As a high surface area nanomaterial, nanoporous gold (NPG) leaf is corrosion resistant, highly conductive, has tunable porosity and can be readily employed as a cost-effective electrode support.27–29 Its inter-connected open porous structure facilitates the collection and transportation of electrons from the surface to current collectors, as well as providing easily accessible electrode surfaces for reactants and products.28,29 More importantly, the well developed surface chemistry of Au provides rich opportunities to manage the surface structures and functions of these novel electrodes with sub-nanometer precision. However, previously reported plating methods can only deposit a monolayer or several layers of Pt which is not favorable for formic acid electro-oxidation as mentioned above.28,29We have developed a molecular self-assembly/electro-deposition (MSA-ED) method to disperse Pt atoms onto the NPG surface, which can be used to deposit catalytic Pt atoms onto the substrate with sub-mono-atomic-layer precision (Scheme 1). We found that PtCl62− ions could form a stable monolayer structure on the ligament surface of the NPG leaf, a phenomenon very similar to those of platinate complex ions adsorbed on the single crystalline Au (111) electrode.30 These complex ions do not de-sorb upon repeated rinsing in pure water. Upon electro-reduction, these self-assembled PtCl62− ions can be reduced and deposited onto the surface of the NPG. The deposited Pt could be measured by integrating the reduction peak of the hexachloroplatinate complexes (Fig. S1†). Experimental investigations showed that PtCl62− ions could form a monolayer structure on the NPG surfaces in approximately 5 min, and further prolonging the immersion time did not increase the quantity of deposited Pt (Fig. 1a). The calculated Pt coverage was approximately 14% which was, surprisingly, in agreement with the reported results acquired from single crystal surfaces, where one PtCl62− ion was found to occupy the surface area of seven sub-layer Au atoms (Scheme 1).30
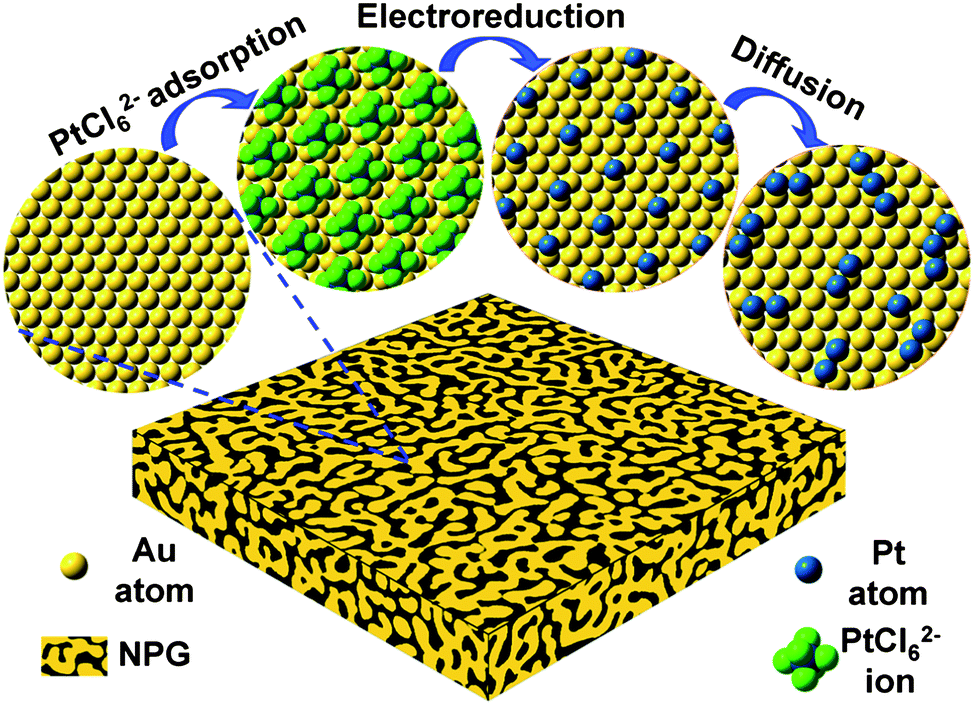 | ||
| Scheme 1 Schematic illustration of the fabrication procedure. Note the actual ligament surfaces of NPG leaves are highly curved. | ||
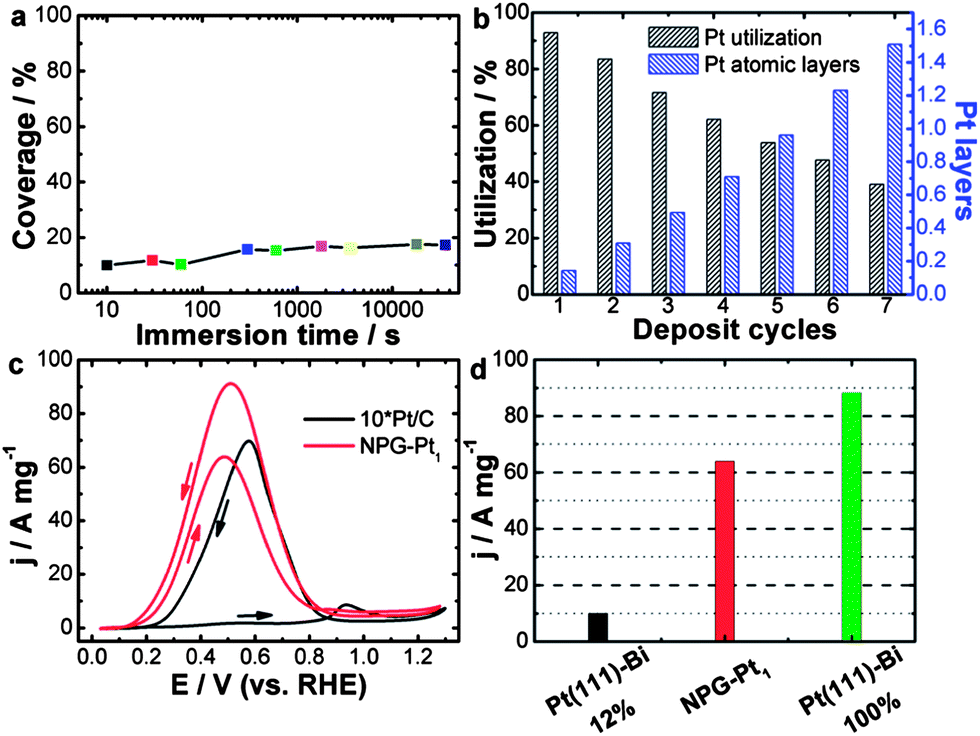 | ||
| Fig. 1 (a) Calculated Pt coverage of NPG–Pt1 for different immersion time periods. (b) Calculated Pt layer and utilization of NPG–Pt at different deposition cycles. (c) Pt mass specific catalytic activities of NPG–Pt1 and Pt/C (magnified by 10-fold for clarity) in a H2SO4/HCOOH mixed solution, 0.5 M and 1 M respectively. Sweep rate: 20 mV s−1. (d) Pt mass specific current peak value (forward scan) of NPG–Pt1 and theoretical values calculated based on experimental results of Bi covered Pt (111) surface.18 | ||
With this method, essentially all deposited Pt atoms were exposed on the electrode surface, indicating perfect Pt utilization at ∼100%. Indeed, using a carefully calibrated hydrogen adsorption–desorption electrochemically active surface area measurement (Fig. S2†), this process generated NPG–Pt1 nanocomposites with Pt utilization as high as 93% (Fig. 1b), which is much higher than the state-of-the-art Pt/C catalyst.11 To deposit more Pt, this MSA-ED process was simply repeated (Fig. 1b, S3 and S4†), and after 5 cycles when the overall amount of deposited Pt was approximately equivalent to monolayer coverage on all NPG surfaces, the calculated Pt utilization was still over 54%, which indicated that most Pt atoms existed as two-dimensional clusters with mono-atomic or bi-atomic heights (Fig. 1b).
The catalytic activity of NPG–Pt1 was tested by cyclic voltammetry (CV) in a H2SO4/HCOOH mixed solution, 0.5 M and 1 M respectively, at room temperature and the result is presented in Fig. 1c, where the data acquired from the Pt/C catalyst (magnified by 10-fold for clarity) is also plotted for comparison. On the Pt/C catalyst, in the forward scan, there was only a small oxidation current at potentials below 0.8 V, as the surface was almost totally passivated by CO. When the poisoning CO molecules started to be oxidized, there was a steep increase in the anodic current at about 0.8 V.13 In contrast, NPG–Pt1 exhibited completely different electrochemical responses in this potential range, which are characterized by a dramatic negative-shift in onset potential to below 0.2 V and huge anodic peaks at around 0.5 V, indicative of successful inhibition of the formation of the poisoning CO intermediates due to the selection of the direct reaction path.19 To date, the highest electrochemical surface area (ECSA) specific peak current of Pt-based catalysts is reported to be 42 mA cm−2 on Bi covered preferential (111) Pt nanoparticles with a Bi coverage of 0.88.18 If we propose that this performance can be realized on a Bi covered monolayer Pt (111) catalyst, the highest Pt mass specific activity could then be calculated to be 10.6 A mg−1 (42 mA cm−2 × 210 m2 g−1 × 0.12). If we further assume that the Pt atoms covered by Bi are not counted (88%), this would indicate that the highest possible Pt mass specific activity is 88.2 A mg−1 under the same testing conditions as in the present work. It is then striking to see that the Pt mass specific activity of the NPG–Pt1 catalyst (64 A mg−1, at a sweep rate of 20 mV s−1) comes very close to this ideal value (Fig. 1d), which to our knowledge is about 2 orders of magnitude higher than most literature results reported to date.23
The catalytic activities of NPG–Pt specimens with different deposition cycles were further tested in a HClO4/HCOOH mixed solution, 0.1 M and 0.05 M respectively, the results are shown in Fig. S5 and S6.† A comparison of the NPG–Ptx catalysts with Pt/C is summarized in Fig. S7 and S8.† It was expected that the Pt mass specific activity would decrease with increasing deposition cycles (Fig. S5 and S7†). However, the decrease in Pt mass specific activity was not solely due to a decrease in Pt utilization. Rather, it was associated with the ensemble effect. With more Pt atoms deposited onto the ligament surface of the NPG, these two-dimensional Pt clusters tended to grow larger, which is less desirable for direct oxidation of formic acid. This hypothesis was further demonstrated by the decreased ECSA specific activities and the positive shifts of the onset and peak potentials (Fig. S6 and S8†) with the increasing number of deposition cycles. With the highest catalytic efficiency, the NPG–Pt1 catalyst showed a peak current density of 21.7 A mg−1 at a sweep rate of 50 mV s−1, which was approximately 380 times higher than that of the Pt/C catalyst (0.057 A mg−1) at the same potential.
It is well known that at lower potential the surface of the Pt-based electrode would be easily covered by CO poisoning intermediates which inhibit further formic acid oxidation. To prove that our nanoengineered electrocatalysts adopted a nearly ideal surface configuration, avoiding the undesired indirect path completely, the following experiment was carried out. Pt/C and NPG–Pt1 electrodes were first dipped into a solution of HClO4/HCOOH, 0.1 M and 0.2 mM respectively, for 5 min to allow the formation (if any) of CO poisoning intermediates, after which the adsorbed poisoning intermediates were electrochemically stripped off in 0.1 M HClO4, the recorded I–V curves are shown in Fig. S9.† It was quite clear that during this process the surface of the Pt/C electrode was covered by CO-like intermediates which caused the decrease in the H-UPD peaks and a dominant CO stripping peak could be seen at ∼0.7 V. In sharp contrast, on NPG–Pt1 the first and second cycles of the CV curves coincided nicely which implied nearly complete suppression of the formation of poisoning intermediates (indirect path). These results provide strong evidence for the hypothesis that the Pt ensembles on NPG–Pt1 are small enough to allow only the direct path for formic acid electro-oxidation to occur.
As the Pt coverage after one deposition cycle was only around 1/7, the Pt atoms should have to diffuse a relatively long distance to meet each other and aggregate into atomic ensembles. The structure of the NPG was examined using high resolution transmission electron microscopy (HRTEM). As shown in Fig. 2a and b, the NPG is composed of well crystallized interconnected ligaments with diameters of ∼25 nm. Interestingly, the ligament surface was found to be mostly terminated with {111} facets, showing very small steps in size, less than 1 nm (Fig. 2b). The highly stepped nature of the NPG surface was also demonstrated by T. Fujita and co-workers recently.31 The dispersion of Pt atoms on these high density surface steps should be quite different from that on smooth single crystal Au (111) or bulk Au surfaces (Fig. S10†), where Pt atoms are expected to be more mobile forming larger ensembles or clusters.32,33 The presence of small steps on the NPG surface is believed to be beneficial to inhibiting the diffusion of Pt atoms and trapping them on the surface of the NPG forming very small ensembles. While it is less practical to identify the exact ensemble structure of Pt supported on high surface area gold substrates such as NPG, we used a scanning transmission electron microscope (STEM) to characterize the surface structure of NPG–Pt1 to give some clues. As shown in Fig. 2c–f, no Pt cluster was observed on the ligament surface from the high-angle annular dark-field (HAADF) TEM and HRTEM images of the NPG–Pt1 sample. Fig. 3a1–a4 show the element mappings for Pt and Au on the edge of a ligament. Although the Pt content was too low to be determined quantitatively through the whole sample, the EDS mapping gave ∼6.3% atomic content for Pt at the edge of the ligament. Moreover, the uniform distribution of Pt on Au clearly demonstrated the highly dispersed state of the Pt atoms on the ligament surface of NPG. The line scan results shown in Fig. 3b and c further demonstrate the core–shell type structure of the NPG–Pt1 sample. Again, the Pt loading was too low to be determined quantitatively in the center part of the ligaments. However, the EDS line scan shown in Fig. 3c2 gave ∼16.2% Pt contents, as the electron beam collected fewer gold signals on the edge. To give more evidence, Fig. S11† shows HAADF TEM and HRTEM images and the corresponding element mappings and line scan results for the NPG–Pt5 sample. With higher Pt loading, one still sees continuous and smooth lattices for the entire ligament, the lack of Pt nano-islands and the good distribution of Pt element provide further evidence of the well dispersed nature of Pt on NPG. As the coverage of Pt on NPG is only approximately 1/7 of a monolayer for NPG–Pt1, it was thus reasonable to postulate that the Pt atoms existed as well separated atomic ensembles which were small enough to facilitate the direct reaction path for formic acid electro-oxidation (Scheme 1, top right panel).
 | ||
| Fig. 2 TEM images of NPG and NPG–Pt1. (a) TEM image of NPG. (b) Fourier filtered HRTEM image of one ligament. The surface steps are illustrated by dashed lines. (c) HAADF TEM image of NPG–Pt1. (d–f) HAADF HRTEM images of NPG–Pt1. | ||
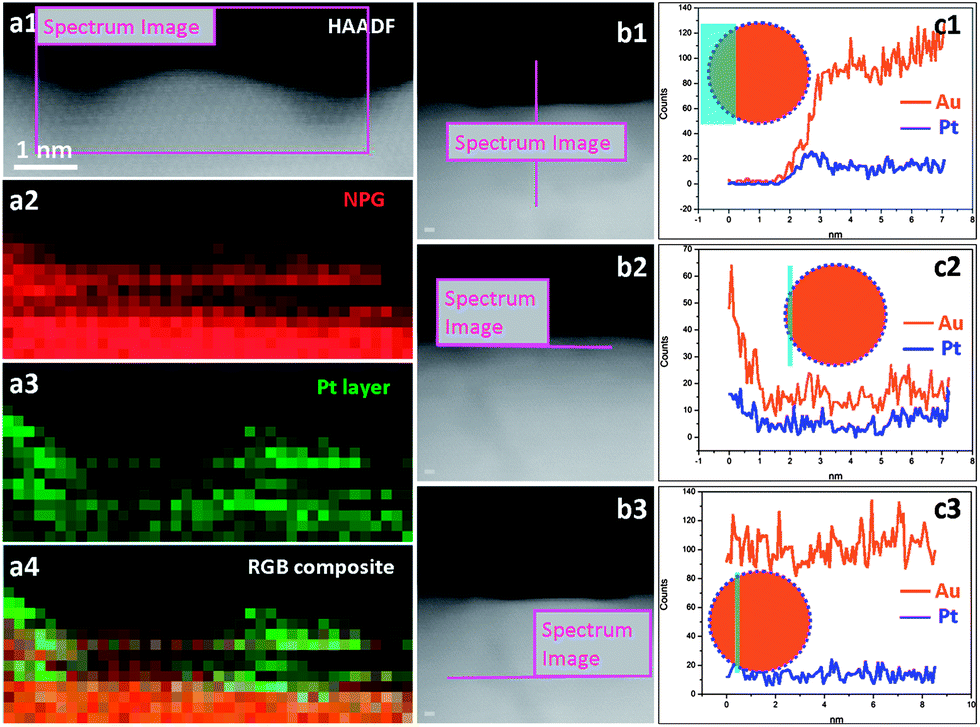 | ||
| Fig. 3 HAADF TEM images of NPG–Pt1 and the corresponding EDS mappings and line scans. (a1) HAADF TEM image. (a2) EDS mapping for Au. (a3) EDS mapping for Pt. (a4) EDS mapping overlay for Au and Pt. (b1–b3) HAADF TEM images. (c1–c3) EDS line scans for Au and Pt. Inserts in c1–c3 illustrate schematic cross-section views for the configuration of the electron beam and the NPG–Pt1 ligament. | ||
We also investigated the oxidation state of Pt in the NPG–Pt1 sample using XPS. As shown in Fig. S12a,† the 4f7/2 peak of Pt in the NPG–Pt1 sample is located at 72.9 eV which is significantly higher than that of metallic Pt (71.1 eV) and reveals a higher oxidation state. As there is no Cl signal observed (see Fig. S12b†), we can rule out the possibility of unreduced PtCl62− (75.5 eV) and PtCl42− (73 eV). Higher XPS peaks for Pt have been observed for nanoparticles smaller than 1 nm and were attributed to platinum oxidants.34 As the Pt ensembles in the NPG–Pt1 sample are very small, it is quite possible that the Pt atoms could be oxidized during the preparation of the XPS samples. It was reported that oxygen containing groups could help to stabilize the high-index surface formed during the synthesis of tetrahexahedral platinum nanocrystals using a potential cycling method.35 We believe that the adsorption of oxygen containing groups on Pt together with the high density atomic steps on NPG contribute to stabilizing the small Pt ensembles in the NPG–Pt1 sample.
With high catalytic activity, these high surface area NPG–Pt membrane catalysts were designed for real fuel cells. To make the MEA, NPG–Pt catalysts were attached to carbon paper to form anodes, while on the cathode side was Pt/C (2.2 mg Pt per cm2) loaded carbon paper prepared using the conventional brushing method (Fig. S13†). These two electrodes were then hot-pressed onto each side of a Nafion® 115 membrane. Considering that these fuel cells are most likely to function at lower temperatures for portable electronic devices, testing was carried out at 40 °C. Fig. 4a shows the current–voltage and current–power polarization curves for the NPG–Pt catalysts with different Pt loadings. A control sample using the commercial Pt/C catalysts (2.2 mg Pt per cm2) is also presented for comparison. Although the Pt loading is typically orders of magnitude lower, the NPG–Pt 0.013 mg catalyst shows both a higher open circuit voltage and a higher maximum power output (Fig. 4a) with impressive stability (Fig. 4b). Specifically, the maximum power output for the NPG–Pt 0.013 mg catalyst is 61 mW cm−2, which is 1.5 times that of the Pt/C 2.2 mg catalyst (40 mW cm−2). If we normalize the apparent maximum power to the amount of Pt used in the anode, the NPG–Pt 0.013 mg sample exhibits a 260-fold enhancement in specific power efficiency (4.7 vs. 0.018 W mg−1, Fig. 4c & S14†). This specific power efficiency is also at least one order of magnitude higher than the highest value reported in the literature for PtBi nanocatalysts (∼0.22 W mg−1 at 60 °C using 6 M formic acid).36 Once again, the tremendous performance enhancement should be mainly attributed to the nearly optimized Pt ensemble structure involved in the NPG–Pt structure. This was proved by the observation that upon deposition of more Pt on the NPG, one actually saw a decrease in the performance of the DFAFC. Also profiled in Fig. 4a & c is a NPG–Pt sample with a 0.065 mg Pt loading, with 4 times more Pt, this sample exhibited a lower maximum power density (44 mW cm−2) and Pt mass specific power efficiency (0.68 W mg−1) as compared with the NPG–Pt 0.013 mg sample. The durability of the NPG–Pt catalyst in a real fuel cell was further evaluated using an accelerated stability test (AST). After a potential excursion between 0.5 and 1 V, the fuel cell was tested and no apparent performance decay was observed (Fig. 4d). The excellent durability may be attributed to: (1) the strong bonding between the Pt surface ensembles and the Au substrate;28 and (2) an electric stabilization effect by Au which could inhibit the oxidation of Pt.19
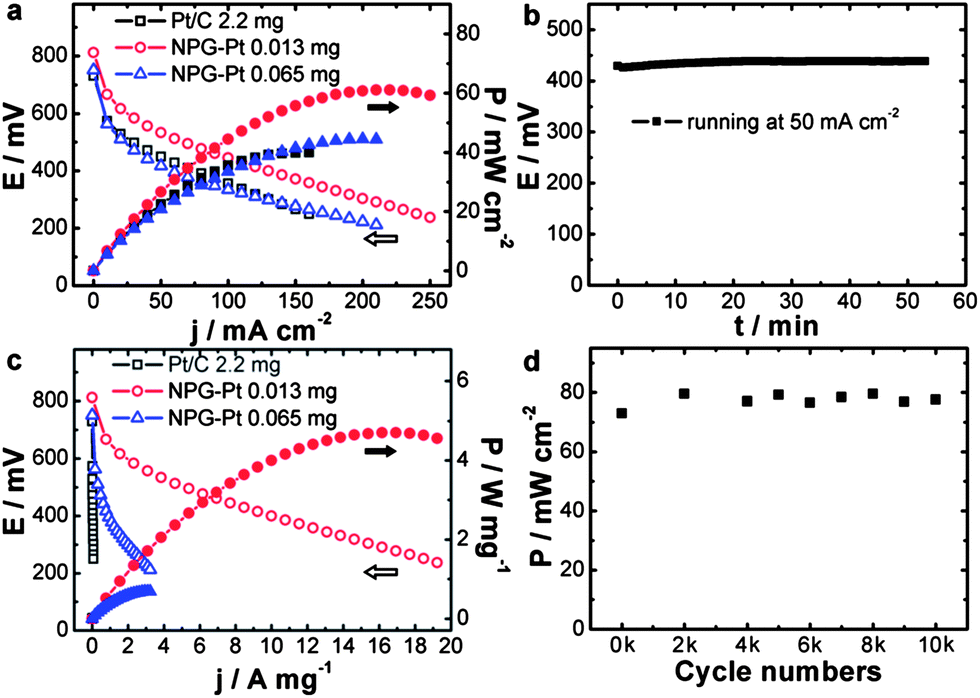 | ||
| Fig. 4 (a) Current–voltage and current–power polarization curves and (c) Pt mass specific current–voltage and current–power polarization curves for Pt/C 2.2 mg, NPG–Pt 0.013 mg and NPG–Pt 0.065 mg catalysts. (b) Voltage–time curve for NPG–Pt 0.013 mg sample at a constant current density of 50 mA cm−2. (d) Maximum power densities of NPG–Pt 0.014 mg catalyst after different cycle numbers. All fuel cells were operated at 40 °C, using the same Pt/C (2.2 mg Pt per cm2) cathode, with formic acid (3 M) as the fuel and dry air as the oxidant. | ||
Conclusions
In conclusion, we describe a rational design strategy for the construction of very high performance DFAFC anode catalysts. Using a molecular self-assembly/electro-deposition process, Pt atoms were effectively dispersed onto the surfaces of a highly conductive high surface area nanoporous substrate with sub-monolayer precision. With nearly perfect Pt utilization and the desired surface ensemble structure, these nanocatalysts showed orders of magnitude improvement in performance in both formic acid electro-oxidation and the actual DFAFC. Preliminary investigations demonstrated that this method could be extended to decorate other catalytically active metals such as Pd in a highly controllable manner, and these nanomaterials also showed superior performance in catalyzing other electrode reactions such as ethanol oxidation (Fig. S15†). These functional hybrid nanostructures thus possess great potential for the development of new generation clean energy technologies.Acknowledgements
Financial support from the National 973 (2012CB932800) Program Project of China and the National Science Foundation of China (51171092) is acknowledged.Notes and references
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- M. Z. Jacobson, W. G. Colella and D. M. Golden, Science, 2005, 308, 1901–1905 CrossRef CAS PubMed.
- R. F. Service, Science, 2002, 296, 1222–1224 CrossRef CAS PubMed.
- C. M. Miesse, W. S. Jung, K. J. Jeong, J. K. Lee, J. Lee, J. Han, S. P. Yoon, S. W. Nam, T. H. Lim and S. A. Hong, J. Power Sources, 2006, 162, 532–540 CrossRef CAS PubMed.
- X. W. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef CAS PubMed.
- C. Rice, R. I. Ha, R. I. Masel, P. Waszczuk, A. Wieckowski and T. Barnard, J. Power Sources, 2002, 111, 83–89 CrossRef CAS.
- K. J. Jeong, C. A. Miesse, J. H. Choi, J. Lee, J. Han, S. P. Yoon, S. W. Nam, T. H. Lim and T. G. Lee, J. Power Sources, 2007, 168, 119–125 CrossRef CAS PubMed.
- C. Sealy, Mater. Today, 2008, 11, 65–68 CrossRef CAS.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- B. Beden, A. Bewick and C. Lamy, J. Electroanal. Chem., 1983, 148, 147–160 CrossRef CAS.
- G. Samjeske, A. Miki, S. Ye and M. Osawa, J. Phys. Chem. B, 2006, 110, 16559–16566 CrossRef CAS PubMed.
- Y. X. Chen, M. Heinen, Z. Jusys and R. J. Behm, Angew. Chem., Int. Ed., 2006, 45, 981–985 CrossRef CAS PubMed.
- M. Osawa, K. Komatsu, G. Samjeske, T. Uchida, T. Ikeshoji, A. Cuesta and C. Gutierrez, Angew. Chem., Int. Ed., 2011, 50, 1159–1163 CrossRef CAS PubMed.
- M. Neurock, M. Janik and A. Wieckowski, Faraday Discuss., 2008, 140, 363–378 RSC.
- J. Clavilier, A. Fernandez-Vega, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1989, 258, 89–100 CrossRef CAS.
- A. Lopez-Cudero, F. J. Vidal-Iglesias, J. Solla-Gullon, E. Herrero, A. Aldaz and J. M. Feliu, Phys. Chem. Chem. Phys., 2009, 11, 416–424 RSC.
- R. Y. Wang, C. Wang, W. B. Cai and Y. Ding, Adv. Mater., 2010, 22, 1845–1848 CrossRef CAS PubMed.
- E. Leiva, T. Iwasita, E. Herrero and J. M. Feliu, Langmuir, 1997, 13, 6287–6293 CrossRef CAS.
- A. Cuesta, M. Escudero, B. Lanova and H. Baltruschat, Langmuir, 2009, 25, 6500–6507 CrossRef CAS PubMed.
- E. Casado-Rivera, D. J. Volpe, L. Alden, C. Lind, C. Downie, T. Vazquez-Alvarez, A. C. D. Angelo, F. J. DiSalvo and H. D. Abruna, J. Am. Chem. Soc., 2004, 126, 4043–4049 CrossRef CAS PubMed.
- X. L. Ji, K. T. Lee, R. Holden, L. Zhang, J. J. Zhang, G. A. Botton, M. Couillard and L. F. Nazar, Nat. Chem., 2010, 2, 286–293 CrossRef CAS PubMed.
- K. Sasaki, H. Naohara, Y. Cai, Y. M. Choi, P. Liu, M. B. Vukmirovic, J. X. Wang and R. R. Adzic, Angew. Chem., Int. Ed., 2010, 49, 8602–8607 CrossRef CAS PubMed.
- D. L. Wang, H. L. Xin, Y. C. Yu, H. S. Wang, E. Rus, D. A. Muller and H. D. Abruna, J. Am. Chem. Soc., 2010, 132, 17664–17666 CrossRef CAS PubMed.
- M. H. Shao, K. Shoemaker, A. Peles, K. Kaneko and L. Protsailo, J. Am. Chem. Soc., 2010, 132, 9253–9255 CrossRef CAS PubMed.
- Y. Ding, Y. J. Kim and J. Erlebacher, Adv. Mater., 2004, 16, 1897–1900 CrossRef CAS.
- Y. Ding, M. W. Chen and J. Erlebacher, J. Am. Chem. Soc., 2004, 126, 6876–6877 CrossRef CAS PubMed.
- P. P. Liu, X. B. Ge, R. Y. Wang, H. Y. Ma and Y. Ding, Langmuir, 2009, 25, 561–567 CrossRef CAS PubMed.
- K. Uosaki, S. Ye, Y. Oda, T. Haba and K. Hamada, Langmuir, 1997, 13, 594–596 CrossRef CAS.
- T. Fujita, P. Guan, K. McKenna, X. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed.
- Y. Nagahara, M. Hara, S. Yoshimoto, J. Inukai, S. L. Yau and K. Itaya, J. Phys. Chem. B, 2004, 108, 3224–3230 CrossRef CAS.
- J. Kim, C. Jung, C. K. Rhee and T. Lim, Langmuir, 2007, 23, 10831–10836 CrossRef CAS PubMed.
- H. L. Wang, Y. H. Wang, Z. W. Zhu, A. Sapi, K. J. An, G. Kennedy, W. D. Michalak and G. A. Somorjai, Nano Lett., 2013, 13, 2976–2979 CrossRef CAS PubMed.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- S. Uhm, H. J. Lee, Y. Kwon and J. Lee, Angew. Chem., Int. Ed., 2008, 47, 10163–10166 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures S1–S15. See DOI: 10.1039/c3sc52792a |
| This journal is © The Royal Society of Chemistry 2014 |