Iridium(III) dipyridylamine complexes: synthesis, characterization and catalytic activities in photoredox reactions†‡
Elodie
Sauvageot
ab,
Ronan
Marion
ab,
Fabien
Sguerra
c,
Adèle
Grimault
ab,
Richard
Daniellou
de,
Matthieu
Hamel
c,
Sylvain
Gaillard
*ab and
Jean-Luc
Renaud
*ab
aNormandie University, University of Caen Basse Normandie, Laboratoire de Chimie Moléculaire et Thioorganique, 14050 Caen, France. E-mail: sylvain.gaillard@ensicaen.fr; jean-luc.renaud@ensicaen.fr
bCNRS, UMR 6507, 14050 Caen, France
cCEA, LIST, Laboratoire Capteurs et Architectures Électroniques, F-91191 Gif-sur-Yvette Cedex, France
dUniversity of Orléans, Institut de Chimie Organique et Analytique, 45067 Orléans, France
eCNRS, UMR 7311, 45067 Orléans, France
First published on 29th April 2014
Abstract
Two new series of cyclometalated iridium(III) complexes bearing a dipyridylamine as the N^N ligand have been prepared and fully characterized. After investigation of their photophysical properties, their catalytic activities have been evaluated in a model photoredox reaction and compared with the commercially available [Ir(ppy)2(dtbbpy)][PF6] complex.
Introduction
Nitrogen ligands are able to bind strongly to transition metal atoms and can stabilize both very low and high oxidation states.1–6 Among these nitrogen ligands, dipyridylamines (dpa) occupy a unique place in coordination chemistry because they are able to form six-membered chelates upon metal binding. They have found applications in transition metal-catalyzed polymerization, allylic oxidation, transesterification and magnetism and photomagnetism.7 Despite these interesting examples, the chemistry of 2,2′-dipyridylamines has mainly been limited to the study of their coordination behavior. Since several years, we have developed a research program devoted to the synthesis,8 the coordination,9 and the reactivity of metal complexes coordinated to dipyridylamine derivatives. This interest was directed by their easy functionalization and the possibility to have an anchoring site with the bridged nitrogen atom. These two reasons may give access to a broad range of applications such as supported catalysis,10 ion detection11 or Organic Light Emitting Diodes.12 Here, we present the synthesis and the full characterization of a series of neutral and cationic heteroleptic iridium(III) complexes bearing both C^N ligands and a dipyridylamine motif as the N^N ligand. An evaluation of their catalytic activities in a model photoredox reaction is also reported in order to demonstrate the potentiality of developing supported photoredox catalysis.Results and discussion
In order to have an orthogonal and homogeneous series of octahedral cationic and neutral iridium(III) complexes, 2-phenylpyridine (ppy), 2-p-tolylpyridine (4-Me-ppy), 2-(2,4-difluorophenyl)pyridine (2,4-F-ppy), benzoquinoline (bzq), N-phenylpyrazole (ppz) and 2-phenylisoquinoline (piq) were first selected as C^N cyclometalating ligands (1–6, Fig. 1). The corresponding iridium(III) dimer complexes were obtained in good to high yields (73 to 99% isolated yields) using a known procedure.13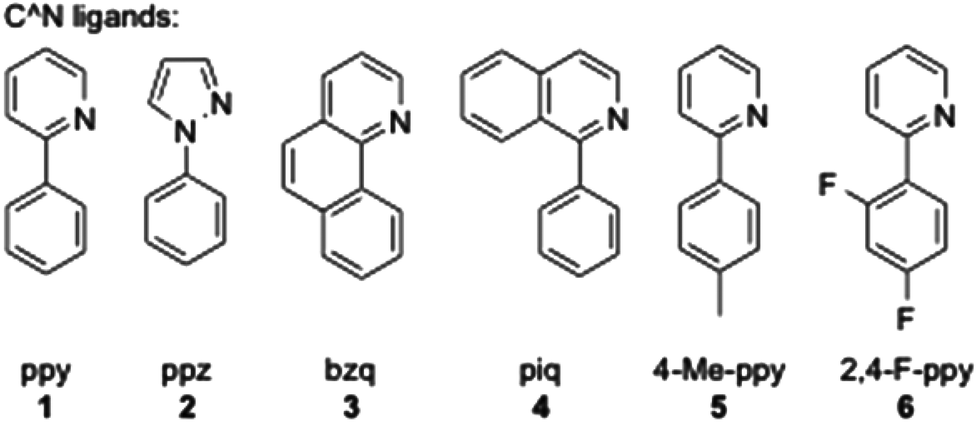 | ||
| Fig. 1 Cyclometalated ligands used in this study. | ||
Addition of various dipyridylamine derivatives14 (N^N ligands a–f, Fig. 2) to the mentioned iridium dimers followed by anion metathesis led to a library of cationic iridium complexes with yields ranging from 65 to 97%.15 It is noteworthy that the formation of the expected complexes between iridium(III) dimers and 6-Me-Hdpa was not observed. We assume that this non-coordination came from steric repulsions between the methyl substituent on the pyridine rings and the C^N ligands, already coordinated to the iridium centre.
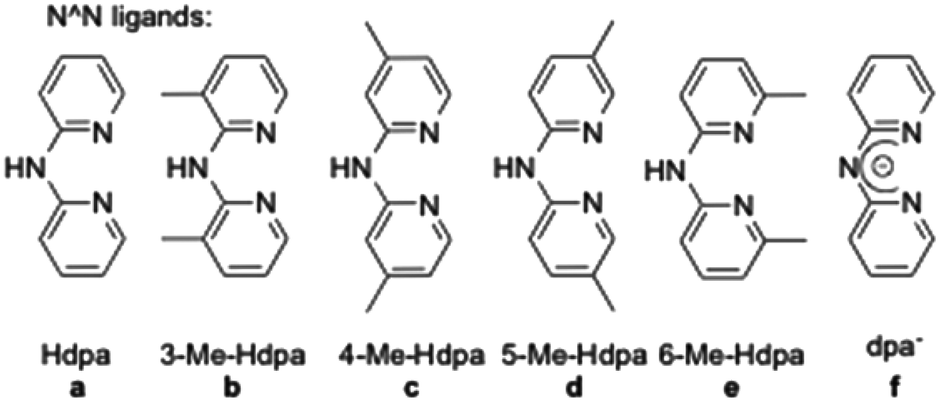 | ||
| Fig. 2 Dipyridylamine ligands used in this study. | ||
Finally, as summarized in Table 1, a series of 18 complexes was obtained in overall yields ranging from 39 to 80% (Scheme 1).
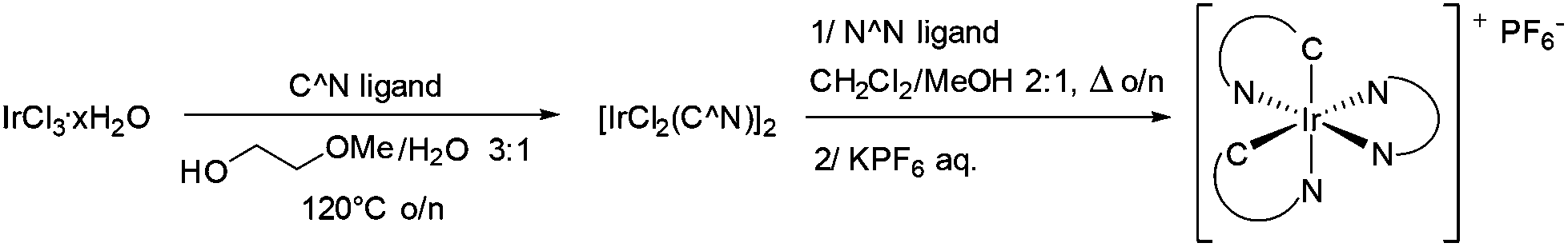 | ||
| Scheme 1 Synthesis of cationic Ir(III) complexes. | ||
| Entry | Complex | Yielda (%) |
|---|---|---|
| a Overall yields for the two steps, starting from IrCl3·xH2O. | ||
| 1 | [Ir(ppy)2(Hdpa)][PF6] 1a | 70 |
| 2 | [Ir(ppy)2(3-Me-Hdpa)][PF6] 1b | 80 |
| 3 | [Ir(ppy)2(4-Me-Hdpa)][PF6] 1c | 77 |
| 4 | [Ir(ppy)2(5-Me-Hdpa)][PF6] 1d | 77 |
| 5 | [Ir(ppz)2(Hdpa)][PF6] 2a | 67 |
| 6 | [Ir(ppz)2(3-Me-Hdpa)][PF6] 2b | 71 |
| 7 | [Ir(ppz)2(4-Me-Hdpa)][PF6] 2c | 79 |
| 8 | [Ir(ppz)2(5-Me-Hdpa)][PF6] 2d | 79 |
| 9 | [Ir(bzq)2(Hdpa)][PF6] 3a | 39 |
| 10 | [Ir(bzq)2(3-Me-Hdpa)][PF6] 3b | 42 |
| 11 | [Ir(bzq)2(4-Me-Hdpa)][PF6] 3c | 50 |
| 12 | [Ir(bzq)2(5-Me-Hdpa)][PF6] 3d | 44 |
| 13 | [Ir(piq)2(Hdpa)][PF6] 4a | 61 |
| 14 | [Ir(piq)2(3-Me-Hdpa)][PF6] 4b | 73 |
| 15 | [Ir(piq)2(4-Me-Hdpa)][PF6] 4c | 60 |
| 16 | [Ir(piq)2(5-Me-Hdpa)][PF6] 4d | 67 |
| 17 | [Ir(4-Me-ppy)2(Hdpa)][PF6] 5a | 77 |
| 18 | [Ir(2,4-F-ppy)2(Hdpa)][PF6] 6a | 53 |
Taking advantage of the acidic proton on the bridged nitrogen atom, we thought to prepare a series of neutral iridium complexes. The deprotonation was easily achieved in the presence of K2CO3 in dichloromethane at room temperature16 and yielded the corresponding “aza-acac” type ligands coordinated to the iridium(III) centre (54–70%). It is noteworthy that a complex mixture was observed by NMR analysis from [Ir(bzq)2(Hdpa)][PF6] 3a, and we were not able to isolate any complex (Scheme 2).
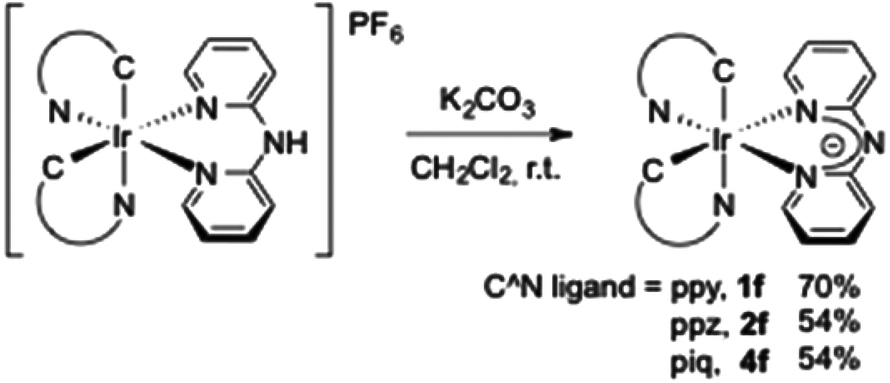 | ||
| Scheme 2 Synthesis of neutral iridium complexes bearing anionic dpa. | ||
To unambiguously establish the atom connectivity in the [Ir(C^N)2(N^N)][PF6] complexes, single crystals for X-ray diffraction (XRD) were grown by slow gas diffusion of pentane into a saturated dichloromethane or chloroform solution of [Ir(ppy)(4-Me-Hdpa)][PF6] complex 1c. Ball and stick representations of crystallized complexes [Ir(ppy)(4-Me-Hdpa)][PF6] 1c are presented in Fig. 3. These X-ray diffraction analyses confirmed the expected 6-membered ring chelate. It is worth mentioning that dipyridylamine ligands did not have a planar geometry due to the pyramidal nitrogen atom between the two pyridine rings.
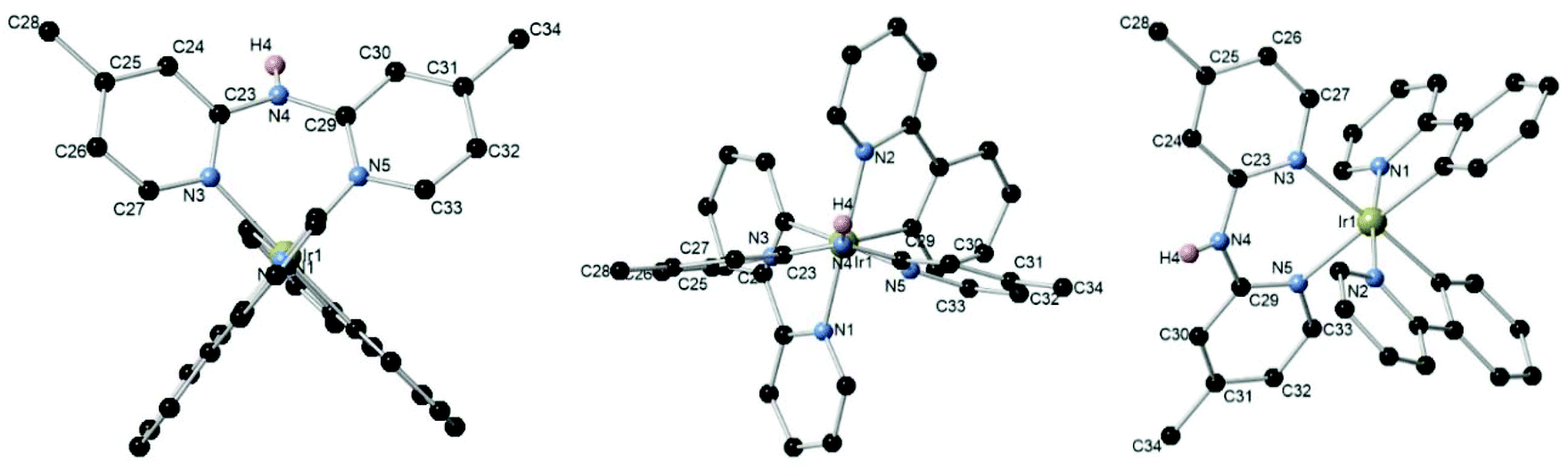 | ||
| Fig. 3 Different views of the ball and stick representation of [Ir(ppy)2(4-Me-Hdpa)][PF6] 1c. Some hydrogen atoms and counter anions were omitted for clarity. Selected bonds (Å) and angles (°): Ir1–N3 2.159(2), Ir1–N5 2.164(2), N3–Ir1–N5 86.21(9), C23–N4–C29 131.3(2). | ||
With these complexes in hand, their absorption properties were measured between 230 and 600 nm. In a general trend, the absorption spectra, recorded in CHCl3, show that the wavelength depends exclusively on the C^N ligands. For all complexes, the maximum of absorption, which can be assigned to a π–π* ligand-centred (LC) transition, was found between 238 and 277 nm (Table 2). The complexes bearing C^N ligands such as ppz (2a–d), bzq (3a–d), piq (4a–d) and 2,4-F-ppy (6a) presented a maximum of absorption which can be ascribed to a second π–π* ligand-centered transition, in a region between 292 and 323 nm (Table 2). It is noteworthy that concerning the complexes bearing bzq (3a–d) and ppy (1a–d), a third local maximum of absorption was found at 425 nm. This weak and broad band corresponds to a dπ–π* metal-to-ligand charge transfer (1MLCT) absorption, as also observed with other iridium complexes.18 More interestingly, for further applications in photoredox chemistry, the iridium complexes bearing a “piq” ligand exhibited this third local maximum at 440 nm and a more intense absorbance (Fig. 4). Then, if the position of the methyl substituent is considered, [Ir(piq)2(3-Me-Hdpa)][PF6] 4b showed a slightly lower absorbance than the Hdpa, 4-Me-Hdpa and 5-Me-Hdpa analogous complexes (4a, 4c–d, respectively, Fig. 4).
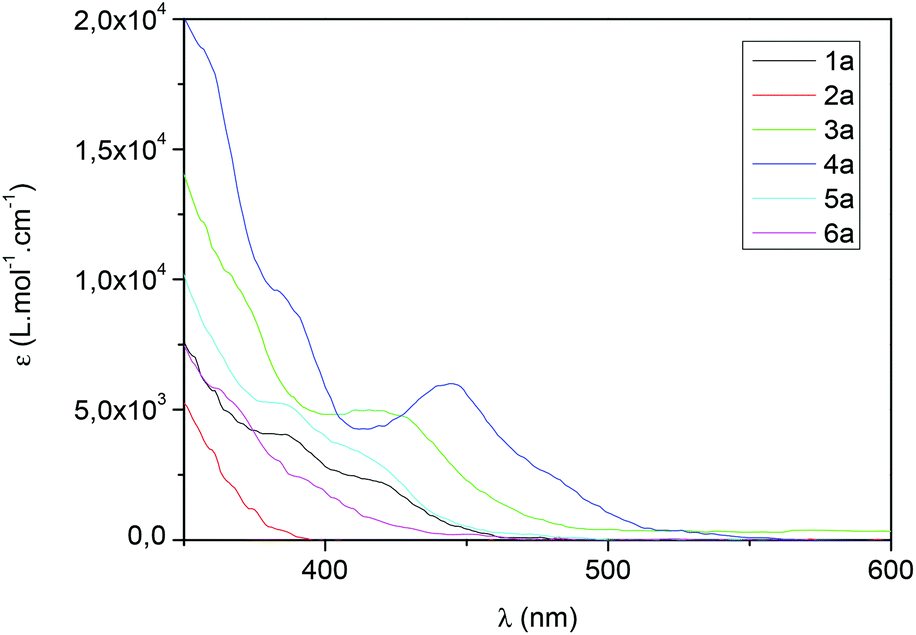 | ||
| Fig. 4 Absorption spectra of [Ir(C^N)2(Hdpa)][PF6] complexes. | ||
| Entry | Complex | λ/nm (ε/L mol−1 cm−1) | Emission max (nm) |
|---|---|---|---|
| 1 | [Ir(ppy)2(Hdpa)][PF6] 1a | 277 (3.60) | 486, 516 |
| 2 | [Ir(ppy)2(3-Me-Hdpa)][PF6] 1b | 277 (4.11) | 489, 517 |
| 3 | [Ir(ppy)2(4-Me-Hdpa)][PF6] 1c | 276 (4.02) | 489, 515 |
| 4 | [Ir(ppy)2(5-Me-Hdpa)][PF6] 1d | 277 (4.11) | 489, 516 |
| 5 | [Ir(ppy)2(dpa)] 1f | 277 (3.97), 398 (1.00) | 517 |
| 6 | [Ir(ppz)2(Hdpa)][PF6] 2a | 254 (4.35), 315 (1.77) sh | 395, 419, 439 |
| 7 | [Ir(ppz)2(3-Me-Hdpa)][PF6] 2b | 252 (3.83), 315 (1.64) sh | 395, 417, 441 |
| 8 | [Ir(ppz)2(4-Me-Hdpa)][PF6] 2c | 254 (4.36), 310 (1.85) sh | 398, 419, 447 |
| 9 | [Ir(ppz)2(5-Me-Hdpa)][PF6] 2d | 255 (3.87), 322 (1.57) | 396, 447 |
| 10 | [Ir(ppz)2(dpa)] 2f | 253 (3.45), 316 (1.45) sh | 427 |
| 11 | [Ir(bzq)2(Hdpa)][PF6] 3a | 248 (6.04), 323 (2.37), 410 (0.50) | 519, 547 |
| 12 | [Ir(bzq)2(3-Me-Hdpa)][PF6] 3b | 242 (5.70), 411 (0.46) | 523, 548 |
| 13 | [Ir(bzq)2(4-Me-Hdpa)][PF6] 3c | 260 (6.73), 315 (3.21), 410 (0.53) | 523, 553 |
| 14 | [Ir(bzq)2(5-Me-Hdpa)][PF6] 3d | 259 (6.31), 322 (2.75), 411 (0.51) | 521, 546 |
| 15 | [Ir(piq)2(Hdpa)][PF6] 4a | 239 (5.49), 292 (4.23), 444 (0.60) | 610, 639 |
| 16 | [Ir(piq)2(3-Me-Hdpa)][PF6] 4b | 238 (5.51), 293 (4.25), 445 (0.53) | 609, 640 |
| 17 | [Ir(piq)2(4-Me-Hdpa)][PF6] 4c | 239 (5.11), 293 (3.76), 445 (0.53) | 609, 635 |
| 18 | [Ir(piq)2(5-Me-Hdpa)][PF6] 4d | 239 (5.40), 292 (4.06), 446 (0.58) | 604, 637 |
| 19 | [Ir(piq)2(dpa)] 4f | 238 (3.96), 293 (2.92) | 628 |
| 20 | [Ir(4-Me-ppy)2(Hdpa)][PF6] 5a | 262 (5.31) | 482, 515 |
| 21 | [Ir(2,4-F-ppy)2(Hdpa)][PF6] 6a | 254 (5.21), 305 (2.52) | 457, 487 |
The emission spectra have also been recorded and weak emission was found between 388 and 416 nm for the iridium complexes bearing the ppz ligand (2a–d). More intense emissions in the visible area and wavelengths ranging from 457 to 639 nm were observed for the other C^N ligands. More precisely, complexes having a “ppy” type ligand have an emission in the blue region. For example, the emission wavelengths are in the blue region (457–487 nm) for the “2,4-F-ppy” ligand (6a), in the blue-green region (482–517 nm) for “ppy” (1a–d) and “4-Me-ppy” ligands (5a), in the green region (516–553 nm) for “bzq” ligands (3a–d), and finally in the red-orange domain for the “piq” ligand (4a–d, Table 2). All these emissions may be due to metal-to-ligand charge transfer (3MLCT). Moreover, for complexes 1a–d, 2a–c, 3a–d, 4a–db, 5a and 6a, a second maximum is also observed and is due to the vibronic transition.
The redox behaviour of these iridium complexes in acetonitrile was characterized by cyclic voltammetry (see ESI‡ for all the CVs). Due to the presence of a NH function in the N^N ligand, an irreversible oxidation of the nitrogen atom was observed around 1.25 VSCE. Such oxidation corresponds to previously described results for ruthenium Hdpa complexes.17 We assume that this irreversible oxidation might be due to water traces in our solution. As depicted in Table 3 (see ESI‡), redox processes are reversible for complexes 1a–f, 2a–f and 4a–d but not for complexes 3a–d and 6a. The oxidation wave (between +1.19 V and +1.30 V) corresponds to the metal centred IrIII/IV process whereas the reduction wave (between −1.30 V and −1.79 V) is assigned to the ligand-based (H)dpa0/− process.18 With complexes 3, in cyclic voltammetry, the oxidation wave was not reversible (see ESI‡).
| Complex | E 1/2 (M+/M*) (V) | E 1/2 (M*/M−) (V) | E 1/2 (M+/M) (V) | E 1/2 (M/M−) (V) | λ max em (nm) | Emission energy (eV) |
|---|---|---|---|---|---|---|
| a From ref. 18b. | ||||||
| [Ir(ppy)2(Hdpa)][PF6] 1a | −1.32 | 1.25 | 1.23 | −1.30 | 486 | 2.55 |
| [Ir(ppy)2(3-Me-Hdpa)][PF6] 1b | −1.32 | 1.13 | 1.21 | −1.40 | 489 | 2.53 |
| [Ir(ppy)2(4-Me-Hdpa)][PF6] 1c | −1.34 | 1.08 | 1.19 | −1.45 | 489 | 2.53 |
| [Ir(ppy)2(5-Me-Hdpa)][PF6] 1d | −1.33 | 1.06 | 1.20 | −1.47 | 489 | 2.53 |
| Ir(ppy)2(dpa) 1f | −1.18 | 1.05 | 1.22 | −1.34 | 517 | 2.39 |
| [Ir(ppz)2(Hdpa)][PF6] 2a | −1.83 | 1.77 | 1.28 | −1.34 | 399 | 3.11 |
| [Ir(ppz)2(3-Me-Hdpa)][PF6] 2b | −1.80 | 1.55 | 1.21 | −1.46 | 411 | 3.01 |
| [Ir(ppz)2(4-Me-Hdpa)][PF6] 2c | −1.81 | 1.53 | 1.25 | −1.53 | 405 | 3.06 |
| [Ir(ppz)2(5-Me-Hdpa)][PF6] 2d | −1.95 | 1.79 | 1.24 | −1.40 | 388 | 3.19 |
| Ir(ppz)2(dpa) 2f | −1.60 | 1.58 | 1.30 | −1.32 | 427 | 2.90 |
| [Ir(bzq)2(Hdpa)][PF6] 3a | 0.81 | −1.60 | 515 | 2.41 | ||
| [Ir(bzq)2(3-Me-Hdpa)][PF6] 3b | 0.86 | −1.54 | 516 | 2.40 | ||
| [Ir(bzq)2(4-Me-Hdpa)][PF6] 3c | 0.64 | −1.62 | 549 | 2.26 | ||
| [Ir(bzq)2(5-Me-Hdpa)][PF6] 3d | 0.59 | −1.79 | 521 | 2.38 | ||
| [Ir(piq)2(Hdpa)][PF6] 4a | −0.83 | 0.50 | 1.20 | −1.53 | 610 | 2.03 |
| [Ir(piq)2(3-Me-Hdpa)][PF6] 4b | −0.85 | 0.43 | 1.19 | −1.61 | 609 | 2.04 |
| [Ir(piq)2(4-Me-Hdpa)][PF6] 4c | −0.84 | 0.48 | 1.19 | −1.55 | 609 | 2.03 |
| [Ir(piq)2(5-Me-Hdpa)][PF6] 4d | −0.88 | 0.60 | 1.17 | −1.45 | 604 | 2.05 |
| [Ir(4-Me-ppy)2(Hdpa)][PF6] 5a | −1.42 | 1.10 | 1.15 | −1.47 | 482 | 2.57 |
| [Ir(2,4-F-ppy)2(Hdpa)][PF6] 6a | 1.39 | −1.32 | 457 | 2.71 | ||
| [Ir(ppy)2(dtbbpy)][PF6] | −0.96 | 0.66 | 1.21 | −1.51 | 581 | 2.14a |
| [Ir(2,4-F-ppy)2(dtbbpy)][PF6] | −0.92 | 0.97 | 1.49 | −1.44 | 2.41a | |
As the excited state for the iridium complexes is mainly a triplet state, the potential in the excited state can be approximated to the optic gap (energy gap) and then can be calculated from the emission wavelength (Table 3). This energy gap gives also access to the corresponding half potential E1/2 (M+/M*) and E1/2 (M*/M−).
Taking advantage of the photophysical properties of these dipyridylamine iridium complexes, we anticipated that our new iridium complexes could also have interesting photoredox catalytic activities. Photoredox chemistry is a flourishing research area and photoinduced redox processes using visible light offer a great variety of catalytic transformations useful in organic synthesis.19 As a model reaction we chose the aza-Henry reactions between N-phenyl tetrahydroisoquinoline, which will be in situ oxidized to the corresponding iminium, and nitromethane.20 This reaction is formally an α-alkylation of amines via a CH-functionalization. Catalytic activities of all our complexes will be compared with the commercially available [Ir(ppy)2(dtbbpy)][PF6] (dtbbpy = 4,4′-di-tert-butylbipyridine).18a
No conversion was detected in the absence of light and low conversions were observed in the absence of catalysts as already reported by Stephenson et al.20 After a rapid screening of the reaction conditions with the commercially available complex (see ESI‡ for a picture of the irradiation system used in this work), we found that a 0.25 M solution of N-phenyl tetrahydroisoquinoline (0.5 mmol) in nitromethane, in the presence of 1 mol% of [Ir(ppy)2(dtbbpy)][PF6], under air and blue LED irradiation led to the best results and provided the alkylated product with a complete conversion within 11 h (Scheme 3). We have also highlighted practical problems encountered during the scale up. Indeed, the conversion decreased proportionately to the increase of the starting material quantities, namely conversion was twice as low starting from 1 mmol of N-phenyl tetrahydroisoquinoline 7. We assume that, in batch reactions, the power of the LEDs is crucial to allow efficient processing and to retain catalytic activities more LED strips have to be used.
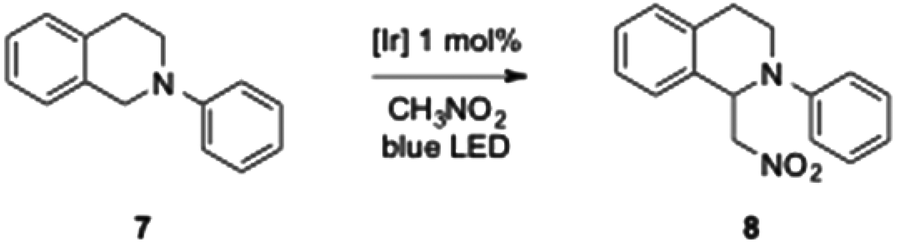 | ||
| Scheme 3 Iridium(III)-catalysed CH-functionalization of 7. | ||
We evaluated the photoredox activities of all our complexes under the mentioned optimized reaction conditions and followed the conversion rate by GC analysis (see ESI‡ for the conversion of all complexes). First, we noted that, whatever the dipyridylamine derivatives used, these N^N ligands have a marginal impact on the reactivity, and thus slight variations of the conversion were observed (see ESI‡). The essential feature for the redox activity is the nature of the C^N ligand (1–6, Fig. 1). Low conversions (5 to 13%) were obtained with complexes 2 having a “ppz” type ligand. Such activity is in good agreement with their photophysical properties. Good to excellent catalytic activities were achieved with the other C^N ligand families, the “piq” ligand 4 providing the best results. As described in Fig. 5, [Ir(ppy)2(dtbbpy)][PF6] has a higher TOF than our dipyridyl–iridium complexes and the reaction is complete within 9 h. However, full conversion can be achieved with [Ir(piq)2(Hdpa)][PF6] 4a within 11 h.
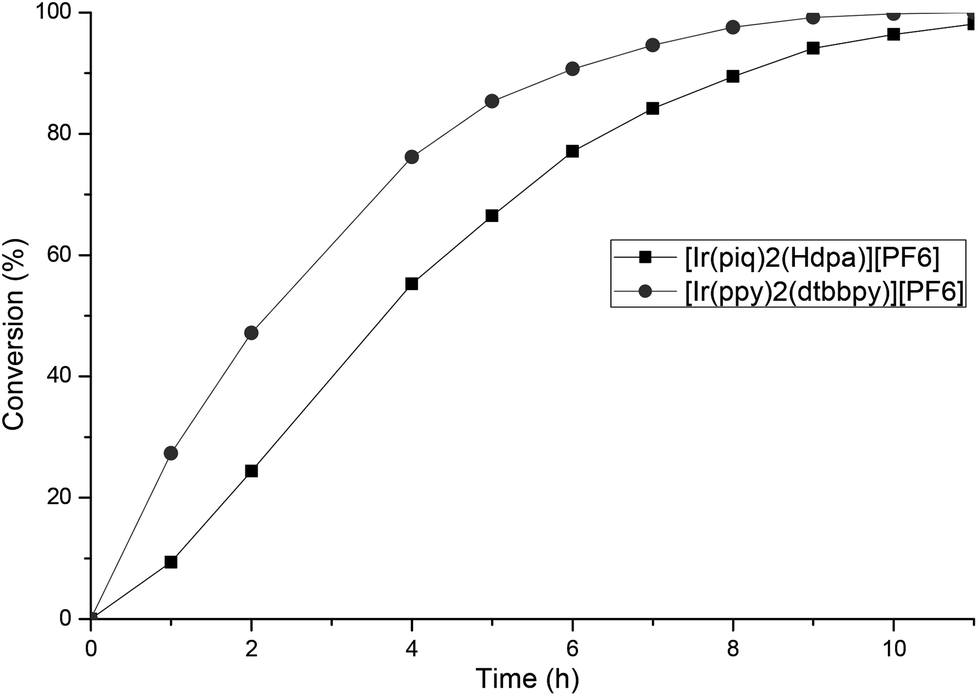 | ||
| Fig. 5 Conversion of 7 into 8 followed by GC. | ||
Finally, results of the aza-Henry reaction with seven more active complexes and [Ir(ppy)2(dtbbpy)][PF6] are summarized in Table 4 and it is validated that these iridium complexes are active in photoredox chemistry. The amines were isolated in moderate to high yields (49–79%). Thus, under these reaction conditions, the alkylated N-phenyl tetrahydroisoquinoline was isolated in 79% yield in the presence of [Ir(ppy)2(dtbbpy)][PF6] and in 69% yield in the presence of [Ir(piq)2(Hdpa)][PF6].
| Entry | Complex | Yieldb (%) |
|---|---|---|
| a General conditions: [Ir] (1 mol%) and 7 (52 mg, 0.25 mmol, 1 equiv.) in 1 mL of CH3NO2 at r.t. for 11 h. b Isolated yields. | ||
| 1 | [Ir(bzq)2(Hdpa)][PF6] 3a | 49 |
| 2 | [Ir(bzq)2(5-Me-Hdpa)][PF6] 3d | 56 |
| 3 | [Ir(piq)2(Hdpa)][PF6] 4a | 69 |
| 4 | [Ir(piq)2(3-Me-Hdpa)][PF6] 4b | 62 |
| 5 | [Ir(piq)2(4-Me-Hdpa)][PF6] 4c | 60 |
| 6 | [Ir(piq)2(5-Me-Hdpa)][PF6] 4d | 57 |
| 7 | [Ir(piq)2(dpa)] 4f | 50 |
| 8 | [Ir(ppy)2(dtbbpy)][PF6] | 79 |
Stephenson et al. have proposed a catalytic cycle for this transformation (Scheme 4).20 Reductive quenching of the excited state of the iridium complex forms the amine radical cation. The powerful reducing iridium(II) complex may then reduce oxygen to the corresponding radical anion. Finally, the iminium ion is produced by hydrogen abstraction from the ammonium radical. Addition of nitromethane to the iminium led to the alkylated amine.
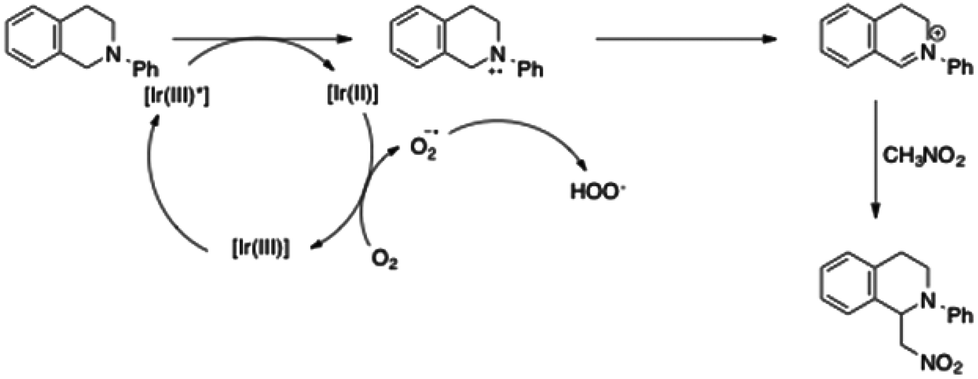 | ||
| Scheme 4 Mechanism proposed by Stephenson for the iridium(III)-catalysed CH-functionalization of 7. | ||
Oxidation of the amine (or reduction of the excited iridium complex) is the crucial step in this catalytic cycle. As a consequence, the half potential E1/2 (M*/M−) must be lower than E1/2 (R3N˙+/R3N) (for the tetrahydroisoquinoline, E1/2 = 0.93 V) to enable the overall amine alkylation. Then, the mechanism and the potentials in Table 3 clearly explain the absence of reactivity when iridium complexes 3, bearing a phenylpyrazole ligand, are used. Conversely, complexes 3d and 4, which have a half potential in reduction in the same range as the half potential of [Ir(ppy)2(dtbbpy)][PF6], led to similar catalytic activities and afforded the amine with the highest yields. These simple results clearly demonstrate the correlations between the photophysical properties of the iridium complexes and their catalytic activities in photoredox reactions.
Conclusion
In conclusion, two new series of neutral and cationic iridium complexes bearing dipyridylamine derivatives as N^N ligands have been synthesized and fully characterized. Almost 20 different complexes have been prepared with generally good yields. Absorption and emission properties of this series of complexes have also been reported and shown that some complexes exhibit absorption in the visible region corresponding to the blue area, depending on the C^N ligand. Their evaluation for photoredox reaction has been performed and compared with commercially available [Ir(ppy)2(dtbbpy)][PF6]. These new complexes bearing Hdpa motifs presented interesting catalytic activities and may compete with known iridium complexes. Furthermore, these results open now some new perspectives in photoredox organic transformations using supported iridium catalysts. These studies are now ongoing in our laboratories.Acknowledgements
This work was supported by the “Ministère de la Recherche et des Nouvelles Technologies”, CNRS (Centre National de la Recherche Scientifique). The LABEX SynOrg (ANR-11-LABX-0029) is acknowledged for a PhD grant (E.S.). The authors thank the “Agence Nationale de la Recherche”, within the CSOSG program (ANR-12-SECU-0002), and the “Région Basse-Normandie” for their funding (R.M., F.S. and A.G.).Notes and references
- R. Guilard and K. M. Kadish, Chem. Rev., 1988, 88, 1121 CrossRef CAS.
- R. van Asselt, E. Rijnberg and C. J. Elsevier, Organometallics, 1994, 13, 706 CrossRef CAS.
- P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1986, 1722 RSC.
- W. de Graaf, J. Boersma, W. J. J. Smeets, A. L. Spek and G. van Koten, Organometallics, 1989, 8, 2907 CrossRef CAS.
- A. J. Canty, Acc. Chem. Res., 1992, 25, 83 CrossRef CAS.
- D. G. Brown, P. K. Byers and A. J. Canty, Organometallics, 1990, 9, 1231 CrossRef CAS.
- (a) M. E. Wright, S. A. Svejda, M. J. Jin and M. A. Peterson, Organometallics, 1990, 9, 136 CrossRef CAS; (b) G. Chelucci, S. Chessa and G. Orrù, J. Mol. Catal. A, 2004, 220, 145 CrossRef CAS PubMed and references cited therein; (c) J. Gil-Moltó and C. Nájera, Eur. J. Org. Chem., 2005, 4073 CrossRef and references cited therein; (d) H.-L. Kwong, L.-S. Cheng, W.-S. Lee, W.-L. Wong and W.-T. Wing, Eur. J. Inorg. Chem., 2000, 1997 CrossRef CAS; (e) H. S. Scott, T. M. Ross, N. F. Chilton, I. A. Gass, B. Moubaraki, G. Chastanet, N. Paradis, J.-F. Lètard, K. R. Vignesh, G. Rajaraman, S. R. Batten and K. S. Murray, Dalton Trans., 2013, 42, 16494 RSC.
- S. Gaillard, M. K. Elmkaddem, C. Fischmeister, C. M. Thomas and J.-L. Renaud, Tetrahedron Lett., 2008, 49, 3471 CrossRef CAS PubMed.
- (a) Z. Zheng, M. K. Elmkaddem, C. Fischmeister, T. Roisnel, C. M. Thomas, J.-F. Carpentier and J.-L. Renaud, New J. Chem., 2008, 32, 2150 RSC; (b) C. Romain, S. Gaillard, M. K. Elmkaddem, L. Toupet, C. Fischmeister, C. M. Thomas and J.-L. Renaud, Organometallics, 2010, 29, 1992 CrossRef CAS.
- (a) F. Sinner, M. R. Buchmeiser, R. Tessadri, M. Mupa, K. Wurst and G. K. Bonn, J. Am. Chem. Soc., 1998, 120, 2790 CrossRef CAS; (b) R. Kröll, C. Eschbaumer, U. S. Schuber, M. R. Buchmeiser and K. Wurst, Macromol. Chem. Phys., 2001, 202, 645 CrossRef.
- (a) J. Ni, K.-J. Wei, Y. Min, Y. Chen, S. Zhan, D. Li and Y. Liu, Dalton Trans., 2012, 41, 5280 RSC; (b) Y. Xie, Y. Ding, C. Wang, J. P. Hill, K. Ariga, W. Zhang and W. Zhu, Chem. Commun., 2012, 48, 11513 RSC; (c) S. Martić, G. Wu and S. Wang, Inorg. Chem., 2008, 47, 8315 CrossRef PubMed.
- (a) Q.-D. Liu, W.-L. Jia, G. Wu and S. Wang, Organometallics, 2003, 22, 3781 CrossRef CAS; (b) J. Lee, Q.-D. Liu, M. Motala, J. Dane, J. Gao, Y. Kang and S. Wang, Chem. Mater., 2004, 16, 1869 CrossRef CAS; (c) D.-R. Bai and S. Wang, Organometallics, 2006, 25, 1517 CrossRef CAS; (d) R. Tan, Z.-B. Wang, Y. Li, D. J. Kozera, Z.-H. Lu and D. Song, Inorg. Chem., 2012, 51, 7039 CrossRef CAS PubMed.
- K. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 467 CrossRef.
- (a) S. Wagaw and S. L. Buchwald, J. Org. Chem., 1996, 61, 7240 CrossRef CAS; (b) M.-H. Yang, C.-C. Chou, H.-C. Lee, G.-H. Lee, M.-K. Leung and S.-M. Peng, Chem. Commun., 1997, 2279 RSC.
- Q. Zhao, S. Liu, M. Shi, C. Wang, M. Yu, L. Li, T. Yi and C. Huang, Inorg. Chem., 2006, 45, 6152 CrossRef CAS PubMed.
- H. Ben Ammar, J. Le Nôtre, M. Salem, M. T. Kaddachi and P. H. Dixneuf, J. Organomet. Chem., 2002, 662, 63 CrossRef CAS.
- D. E. Morris, Y. Ohsawa, D. P. Segers, M. K. DeArmond and K. W. Hanck, Inorg. Chem., 1984, 23, 3010 CrossRef CAS.
- (a) J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763 CrossRef CAS PubMed; (b) M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal Jr., G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS.
- For recent reviews, see: (a) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. Mac Millan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (c) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727 CrossRef; (d) J. Hu, J. Wang, T. H. Nguyen and N. Zheng Beilstein, J. Org. Chem., 2013, 9, 1977 Search PubMed.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 988580 (1c) and 988579 (2c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00059e |
| This journal is © the Partner Organisations 2014 |