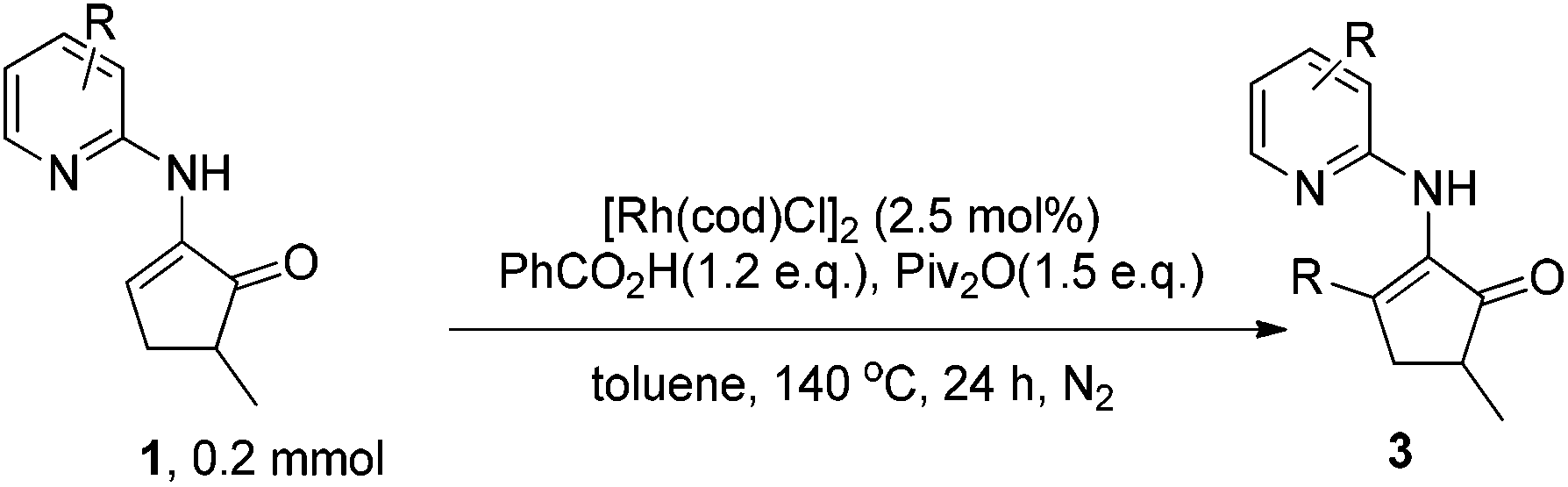
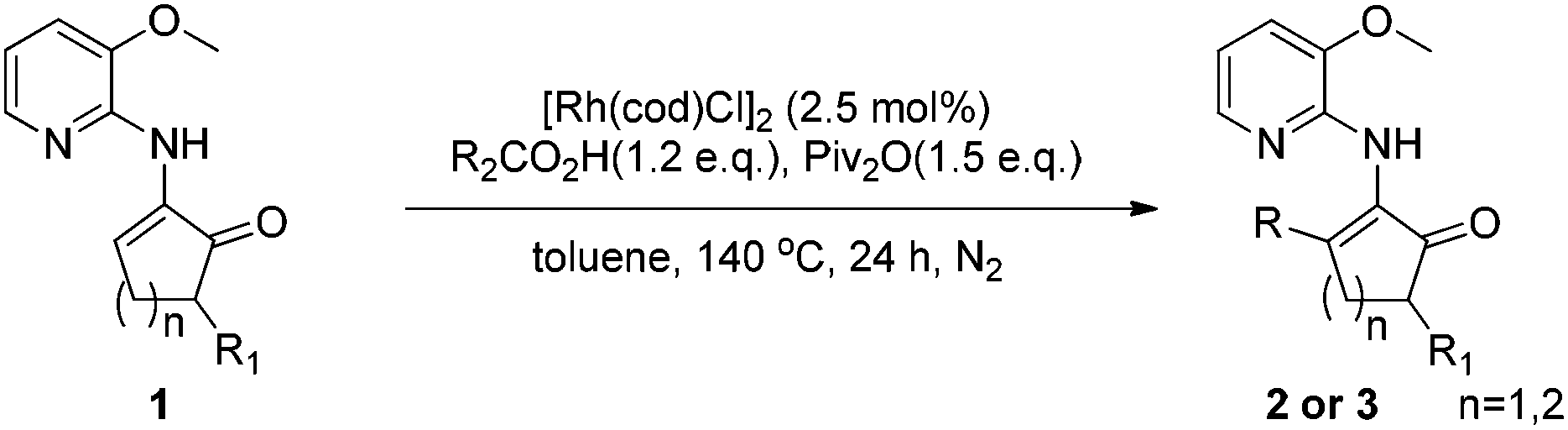
Direct alkenyl C–H functionalization of cyclic enamines with carboxylic acids via Rh catalysis assisted by hydrogen bonding†‡
Zhi-Quan
Lei
a,
Jian-Heng
Ye
a,
Jian
Sun
*a and
Zhang-Jie
Shi
*bc
aChengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, Sichuan 610041, China. E-mail: sunjian@cib.ac.cn
bBeijing National Laboratory of Molecular Sciences (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of the Ministry of Education, College of Chemistry and Green Chemistry Center, Peking University, Beijing 100871, China. E-mail: zshi@pku.edu.cn
cState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 21st April 2014
Abstract
Enamines and enamides are important synthetic intermediates. The transition metal catalyzed C–C coupling through direct β-C–H activation of enamines or enamides is an important method for their functionalization. But so far the effective coupling partners have been limited to organometallic reagents, arenes, olefins, and acrylates. In this study, a highly efficient method was developed to use carboxylic acids, an easily available and cheap carbon source, as coupling partners for the direct β-C–H functionalization of enamines in the presence of the Rh(I) catalyst and the aminopyridinyl directing group through decarbonylation coupling. The reaction was proved to be assisted by hydrogen bonding. The directing group was easily removed under acidic conditions. This method provides a useful alternative approach to synthesize C-alkylated and arylated cyclic diketones.
Enamines and enamides are important synthetic intermediates.1 They have broad utility in catalytic asymmetric C–C bond forming processes such as aza–ene,2 Michael,3 Friedel–Crafts,4 cycloaddition,5 and arylation.6 The transition metal catalyzed C–C coupling through direct β-C–H activation of enamines or enamides is an important method for their functionalization.7 The effective coupling partners include organometallic reagents,6c,d,g arenes,6f olefins7k and acrylates7i,j,8 (Scheme 1a).
 | ||
| Scheme 1 Transition metal catalyzed cross-couplings through direct β-H activation of enamides or enamines. | ||
In recent years, the development of decarboxylative coupling reaction of carboxylic acids has made significant progress.9 Its applications have extended to arylation,10 alkenylation,11 acylation,7h,12 and etherification.13 However, the decarbonylative coupling reactions of carboxylic acids were less developed.14 Recently, Yu15 and our group16 have independently developed a method for rhodium-catalyzed decarbonylative cross-coupling of acid derivatives with arenes. This method features a stable five-membered rhodium species as an intermediate. It, however, can only activate the aromatic C–H bond, and pyridine as the directing group is hardly removable. In our continuing efforts to extend the application scope of the decarbonylative system,16,17 we became interested in implementing decarbonylative coupling of acids with enamines installed with removable directing groups (DGs)18 through non-aromatic sp2 C–H activation (Scheme 1b),19 aiming at developing this transformation into a more general and economical method for the β-functionalization of enamines. The challenges facing this endeavour include: (1) the enamine may not be stable under acidic conditions at high temperatures; (2) the non-aromatic sp2 C–H bond may not be significantly active towards the previous catalyst system; (3) the six-membered rhodium species may not be as active as the previous five-membered one.
We first tested the reaction of enamine 1a, which was used by the Dong group,7k with benzoic acid under the standard conditions we previously used. Delightfully, the desired product 3a was obtained, albeit with only 30% isolated yield (80% conversion) (Scheme 2). The more cost-effective catalyst [Rh(cod)Cl]2 afforded a comparable yield. 3aa was observed as the main byproduct due to the amidation of the product. In order to suppress the side reaction, we installed a methyl group in the 3-position of the directing pyridinyl group (1b)20 hoping that the amidation could become less easy to occur due to steric hindrance (Table 1). But 1b turned out to be unstable and decomposed fairly fast under the reaction conditions. We then turned to the use of a methoxyl group (1c) at the same position considering that the methoxyl group is not only a steric hindrance provider, but also a hydrogen bond donor. If strong hydrogen bonds could be formed as depicted in Scheme 3,21,22 the substrate should not only have better stability under the reaction conditions, but also be able to facilitate the activation of the vinyl C–H bond since the directing group is fixed in the most favorable orientation towards the vinyl C–H bond. As expected, the reaction of 1c gave a dramatically increased yield. Notably, when the 3-methoxy group was replaced with a benzoxy group (1d), the yield was lowered to some degree. Moving the methoxy group to the 5-position of the pyridine ring (1f) or adding one more methoxy group at the 5-position (1e) also decreased the yield. On the other hand, if the aminopyridine DG is replaced by either an acetamido group (1g) or a 2-methoxyphenylamino group (1h), the reaction completely lost the reactivity, clearly indicating that the pyridinyl DG is crucial for the reactivity.
 | ||
| Scheme 2 The initial results obtained with the previous catalytic system. | ||
 | ||
| Scheme 3 Hydrogen bonding. | ||
|
|
|---|
| a Isolated yield. |

|
Thus, we chose 1c as the substrate for further studies (Table 2). A brief survey on the catalyst showed that [Rh(CO)2Cl]2 has similar catalytic activity to [Rh(cod)Cl]2 (entry 2 vs. 1), whereas Rh(CO)2(acac) is substantially less active (entry 3). In contrast, [Rh(cod)OH]2 exhibited almost no activity (entry 4). When the amount of acid was reduced from 1.5 to 1.2 equiv., the yield was further increased to 94% and 92% (entries 5 and 6, respectively). Lowering the catalyst amount from 2.5% to 1.25% caused some decrease in the yield (entries 7 and 8). O-xylene and chlorobenzene as solvents also gave excellent yields (entries 9 and 10). In contrast, the reaction completely lost the reactivity when other non-aromatic solvents such as acetonitrile and dichloroethane were used (entries 11 and 12). When the reaction temperature was lowered from 140 to 110 °C, the reaction was significantly slowed down and it only afforded a moderate yield (entry 13). If the reaction was initiated without degassing, again a moderate yield was obtained (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Acid (eq.) | Yieldb (%) |
| a The reactions were carried out with 0.2 mmol of 1c in the presence of 5.0 mol% Rh(I) catalysts and benzoic acid in 2.0 mL solvent, N2, 140 °C for 24 h. b Isolated yields in parenthesis. c 1.25 mol% of [Rh(cod)Cl]2 was added. d 1.25 mol% of [Rh(CO)2Cl]2 was added. e The reaction was carried out at 110 °C. f The reaction was carried out without degassing. | ||||
| 1 | [Rh(cod)Cl]2 | Toluene | 1.5 | 93(85) |
| 2 | [Rh(CO)2Cl]2 | Toluene | 1.5 | 91(81) |
| 3 | Rh(CO)2(acac) | Toluene | 1.5 | 77 |
| 4 | [Rh(cod)(OH)]2 | Toluene | 1.5 | <5 |
| 5 | [Rh(cod)Cl]2 | Toluene | 1.2 | 99(94) |
| 6 | [Rh(CO)2Cl]2 | Toluene | 1.2 | 98(92) |
| 7c | [Rh(cod)Cl]2 | Toluene | 1.2 | 73 |
| 8d | [Rh(CO)2Cl]2 | Toluene | 1.2 | 68 |
| 9 | [Rh(cod)Cl]2 | o-Xylene | 1.2 | 93 |
| 10 | [Rh(cod)Cl]2 | PhCl | 1.2 | 95 |
| 11 | [Rh(cod)Cl]2 | CH3CN | 1.2 | 0 |
| 12 | [Rh(cod)Cl]2 | DCE | 1.2 | 0 |
| 13e | [Rh(cod)Cl]2 | Toluene | 1.2 | 73 |
| 14f | [Rh(cod)Cl]2 | Toluene | 1.2 | 70 |

With the optimized reaction conditions in hand, various acids were subjected to reactions with 1c to explore the substrate scope (Table 3). In general, good to high yields were obtained for relatively electron-rich aromatic acids under condition A, including benzoic acids bearing various substituents (2a–f, 2i, 2j, 2m, 2n and 3i), naphthyl carboxylic acid (2s), and heteroaromatic thiophenyl and furanyl carboxylic acids (2o and 2p). Although sterically hindered and electron-deficient aromatic acids (2l, 2g, 2h, 2k, 2q and 2r) only afforded moderate yields under condition A, much better results could be attained with these substrates under condition B. Importantly, cinnamic acid (2u), crotonic acid (2t) and various aliphatic acids (2v–z) also proved to be good substrates for the reaction, furnishing the corresponding alkenyl and alkyl enamine products in good-to-excellent yields. It should be noted that no branched product due to isomerization was observed for long chain aliphatic acids, which partially ruled out the formation of cationic and radical intermediates during the course of reaction. It should be noted that enamine 1i devoid of the methyl group on the five-membered ring also proved to be an excellent substrate, affording the product 3i in 97% yield. In addition, the six-membered cyclic enamine 1j was found to be much less active. It reacted with benzoic acid to furnish the aromatization product 3j in 37% yield, which can be further transformed into other useful complexes.23
|
|
|---|
| a The reactions were carried out on the scale of 0.2 mmol 1, 1.2 equiv. acid, 140 °C, and N2. Condition A: in the presence of 2.5 mmol% [Rh(cod)Cl]2 as the catalyst, reacted for 24 h; condition B: in the presence of 2.5 mol% [Rh(CO)2Cl]2 as the catalyst, reacted for 36 h; isolated yields. b The isolated yield given under condition B. c The reaction was scaled up to 1.0 mmol under condition A. d The aromatic oligomer was exclusively formed under condition B. |

|
To demonstrate the practicability of the present reaction system, the reaction was conducted on a 1.0 mmol scale. Expectedly, the desired products (2a, 2u, 2w) were achieved with excellent yields. On the other hand, the 3-methoxypyridinyl amino DG of products 2a and 2w proved to be easily removable under modified conditions previously used by Dong and co-workers, affording the desired diketone products 4a and 4w in good yields (eqn (2) and (3)) (Scheme 4). Thus, this present method provides a convenient method for the preparation of C-alkylated7k and C-arylated24 products of 1,2-diketones.
 | ||
| Scheme 4 Removal of the directing group. | ||
Finally, a mechanism model16 was proposed for the present reaction system as follows (Scheme 5). The vinyl C–H bond was inserted by rhodium(I) with the assistance of the DG. The carboxylic acid reacts with (tBuCO)2O to form the anhydride 7, which then interacts with 4 to generate complex 5. The decarbonylation of 5 gives rise to the intermediate 6, which undergoes reductive elimination to produce the desired product 3 with the regeneration of the rhodium(I) catalyst.
 | ||
| Scheme 5 Proposed mechanism for decarbonylative coupling of enamines. | ||
In summary, we have successfully developed the first Rh-catalyzed decarbonylative coupling of cyclic enamines with simple carboxylic acids. The 3-methoxy-2-pyridinyl amino group proved to be a highly effective directing group for this transformation. A broad range of acids were subjected to the coupling to afford β-aryl, alkenyl and alkylation enamine products in high yields. The directing group proved to be easily removable, thus making the present reaction system a convenient and efficient approach to synthesize C-alkylated and C-arylated 1,2-diketone compounds. This work should have broad implications and serve as a seminal study toward catalytic ketone functionalization.
Experimental section
General procedure for the decarbonylative coupling of carboxylic acids with cyclic enamines
[Rh(cod)Cl]2 (0.005 mmol, 2.4 mg) or [Rh(CO)2Cl]2 (0.005 mmol, 1.9 mg), enamine 1 (0.2 mmol), and carboxylic acid (0.24 mmol) were added to a Schlenk flask, which was then degassed with N2 three times. (tBuCO)2O (0.3 mmol) and 2 mL of anhydrous toluene were added, and the reaction mixture was subsequently heated and kept at 140 °C in an oil bath for the indicated time with stirring. After cooling to room temperature, 1 mL of a concentrated ammonia solution was added. The mixture was directly subjected to column chromatography on silica gel with petroleum ether–EtOAc (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–5:1) as the eluent to afford the desired product 2 or 3.
:1–5:1) as the eluent to afford the desired product 2 or 3.
Acknowledgements
The authors gratefully acknowledge support for this work from the National Natural Science Foundation of China (Project No. 21272227) and SCST (Project No. 2012SZ0219).Notes and references
- (a) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292 CrossRef CAS PubMed; (b) D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455 RSC; (c) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599 CrossRef CAS PubMed.
- (a) R. Matsubara, Y. Nakamura and S. Kobayashi, Angew. Chem., Int. Ed., 2004, 43, 1679 CrossRef CAS PubMed; (b) M. Terada, K. Machioka and K. Sorimachi, Angew. Chem., Int. Ed., 2006, 45, 2254 CrossRef CAS PubMed; (c) K. Zheng, X. Liu, J. Zhao, Y. Yang, L. Lin and X. Feng, Chem. Commun., 2010, 46, 3771 RSC.
- (a) F. Berthiol, R. Matsubara, N. Kawai and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 7803 CrossRef CAS PubMed; (b) L. Zu, H. Xie, H. Li, J. Wang, X. Yu and W. Wang, Chem. – Eur. J., 2008, 14, 6333 CrossRef CAS PubMed.
- (a) Y.-X. Jia, J. Zhong, S.-F. Zhu, C.-M. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2007, 46, 5565 CrossRef CAS PubMed; (b) C. Baudequin, A. Zamfir and S. B. Tsogoeva, Chem. Commun., 2008, 4637 RSC; (c) Q.-X. Guo, Y.-G. Peng, J.-W. Zhang, L. Song, Z. Feng and L.-Z. Gong, Org. Lett., 2009, 11, 4620 CrossRef CAS PubMed.
- (a) R. A. Hughes, S. P. Thompson, L. Alcaraz and C. J. Moody, J. Am. Chem. Soc., 2005, 127, 15644 CrossRef CAS PubMed; (b) M. C. Bagley, C. Glover and E. A. Merritt, Synlett, 2007, 2459 CrossRef CAS PubMed; (c) H. Kim and C. Lee, J. Am. Chem. Soc., 2006, 128, 6336 CrossRef CAS PubMed; (d) Y. Hayashi, H. Gotoh, R. Masui and H. Ishikawa, Angew. Chem., Int. Ed., 2008, 47, 4012 CrossRef CAS PubMed; (e) W. Zhao, D. Zhao, A. A. Desai, R. O. M. A. de Souza, S. Guizzetti, L. Simon and J. Knight, Org. Process Res. Dev., 2012, 16, 1574 CrossRef CAS.
- (a) J. Mo, L. Xu and J. Xiao, J. Am. Chem. Soc., 2005, 127, 751 CrossRef CAS PubMed; (b) Z. Hyder, J. Ruan and J. Xiao, Chem. – Eur. J., 2008, 14, 5555 CrossRef CAS PubMed; (c) H. Zhou, Y.-H. Xu, W.-J. Chung and T.-P. Loh, Angew. Chem., Int. Ed., 2009, 48, 5355 CrossRef CAS PubMed; (d) H. Zhou, W.-J. Chung, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2009, 3472 RSC; (e) J. Ruan, J. A. Iggo, N. G. Berry and J. Xiao, J. Am. Chem. Soc., 2010, 132, 16689 CrossRef CAS PubMed; (f) S. Pankajakshan, Y.-H. Xu, J. K. Cheng, M. T. Low and T.-P. Loh, Angew. Chem., Int. Ed., 2012, 51, 5701 CrossRef CAS PubMed; (g) N. Gigant, L. Chausset-Boissarie, M.-C. Belhomme, T. Poisson, X. Pannecoucke and I. Gillaizeau, Org. Lett., 2013, 15, 278 CrossRef CAS PubMed; (h) N. Gigant, L. Chausset-Boissarie and I. Gillaizeau, Org. Lett., 2013, 15, 816 CrossRef CAS PubMed.
- (a) C. D. Gilmore, K. M. Allan and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 1558 CrossRef CAS PubMed; (b) S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585 CrossRef CAS PubMed; (c) D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326 CrossRef CAS PubMed; (d) J. Du, B. Zhou, Y. Yang and Y. Li, Chem. – Asian J., 2013, 8, 1386 CrossRef CAS PubMed; (e) B. Li, N. Wang, Y. Liang, S. Xu and B. Wang, Org. Lett., 2013, 15, 136 CrossRef CAS PubMed; (f) Y.-H. Xu, T. He, Q.-C. Zhang and T.-P. Loh, Chem. Commun., 2014, 50, 2784 RSC; (g) C. Feng and T.-P. Loh, Chem. Sci., 2012, 3, 3458 RSC; (h) H. Wang, L.-N. Guo and X.-H. Duan, Org. Lett., 2012, 14, 4358 CrossRef CAS PubMed; (i) N. Gigant and I. Gillaizeau, Org. Lett., 2012, 14, 3304 CrossRef CAS PubMed; (j) Z.-K. Wen, Y.-H. Xu and T.-P. Loh, Chem. Sci., 2013, 4, 4520 RSC; (k) Z. Wang, B. J. Reinus and G. Dong, J. Am. Chem. Soc., 2012, 134, 13954 CrossRef CAS PubMed; (l) C.-H. Lei, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 4708 CrossRef CAS PubMed; (m) S. Tong, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, Angew. Chem., Int. Ed., 2012, 51, 4417 CrossRef CAS PubMed; (n) L. Yang, C.-H. Lei, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Org. Lett., 2010, 12, 3918 CrossRef CAS PubMed; (o) L. Yang, D.-X. Wang, Z.-T. Huang and M.-X. Wang, J. Am. Chem. Soc., 2009, 131, 10390 CrossRef CAS PubMed; (p) Z. He, H. Li and Z. Li, J. Org. Chem., 2010, 75, 4636 CrossRef CAS PubMed; (q) W. Yu, Y. Du and K. Zhao, Org. Lett., 2009, 11, 2417 CrossRef CAS PubMed.
- (a) M. Li, L. Li and H. Ge, Adv. Synth. Catal., 2010, 352, 2445 CrossRef CAS; (b) T. Besset, N. Kuhl, F. W. Patureau and F. Glorius, Chem. – Eur. J., 2011, 17, 7167 CrossRef CAS PubMed; (c) Y.-Y. Yu, M. J. Niphakis and G. I. Georg, Org. Lett., 2011, 13, 5932 CrossRef CAS PubMed; (d) J. Zhang and T.-P. Loh, Chem. Commun., 2012, 48, 11232 RSC.
- (a) J. M. Anderson and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 1651 CrossRef CAS; (b) L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef CAS PubMed; (c) L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed; (d) L. J. Goossen, K. Goossen, N. Rodriguez, M. Blanchot, C. Linder and B. Zimmermann, Pure Appl. Chem., 2008, 80, 1725 CrossRef CAS; (e) L. J. Goossen and K. Goossen, Top. Organomet. Chem., 2013, 44, 121 CrossRef CAS; (f) W. I. Dzik, P. P. Lange and L. J. Goossen, Chem. Sci., 2012, 3, 2671 RSC.
- (a) L. J. Goossen, N. Rodriguez, B. Melzer, C. Linder, G. Deng and L. M. Levy, J. Am. Chem. Soc., 2007, 129, 4824 CrossRef CAS PubMed; (b) L. J. Goossen, N. Rodriguez and C. Linder, J. Am. Chem. Soc., 2008, 130, 15248 CrossRef CAS PubMed; (c) L. J. Goossen, B. Zimmermann and T. Knauber, Angew. Chem., Int. Ed., 2008, 47, 7103 CrossRef CAS PubMed; (d) J. Cornella, P. Lu and I. Larrosa, Org. Lett., 2009, 11, 5506 CrossRef CAS PubMed; (e) L. J. Goossen, N. Rodriguez, P. P. Lange and C. Linder, Angew. Chem., Int. Ed., 2010, 49, 1111 CrossRef CAS PubMed; (f) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef CAS PubMed; (g) J. Zhou, P. Hu, M. Zhang, S. Huang, M. Wang and W. Su, Chem. – Eur. J., 2010, 16, 5876 CrossRef CAS PubMed; (h) H. Zhao, Y. Wei, J. Xu, J. Kan, W. Su and M. Hong, J. Org. Chem., 2011, 76, 882 CrossRef CAS PubMed; (i) P. Hu, Y. Shang and W. Su, Angew. Chem., Int. Ed., 2012, 51, 5945 CrossRef CAS PubMed; (j) P. Hu, M. Zhang, X. Jie and W. Su, Angew. Chem., Int. Ed., 2012, 51, 227 CrossRef CAS PubMed.
- (a) A. G. Myers, D. Tanaka and M. R. Mannion, J. Am. Chem. Soc., 2002, 124, 11250 CrossRef CAS PubMed; (b) P. Hu, J. Kan, W. Su and M. Hong, Org. Lett., 2009, 11, 2341 CrossRef CAS PubMed; (c) M. Yamashita, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 592 CrossRef CAS PubMed; (d) Z.-M. Sun, J. Zhang and P. Zhao, Org. Lett., 2010, 12, 992 CrossRef CAS PubMed; (e) Z. Fu, S. Huang, W. Su and M. Hong, Org. Lett., 2010, 12, 4992 CrossRef CAS PubMed; (f) J. Zhou, G. Wu, M. Zhang, X. Jie and W. Su, Chem. – Eur. J., 2012, 18, 8032 CrossRef CAS PubMed; (g) M. Zhang, J. Zhou, J. Kan, M. Wang, W. Su and M. Hong, Chem. Commun., 2010, 46, 5455 RSC.
- (a) P. Fang, M. Li and H. Ge, J. Am. Chem. Soc., 2010, 132, 11898 CrossRef CAS PubMed; (b) M. Li and H. Ge, Org. Lett., 2010, 12, 3464 CrossRef CAS PubMed; (c) P. Mamone, G. Danoun and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 6704 CrossRef CAS PubMed.
- (a) S. Bhadra, W. I. Dzik and L. J. Goossen, J. Am. Chem. Soc., 2012, 134, 9938 CrossRef CAS PubMed; (b) S. Bhadra, W. I. Dzik and L. J. Goossen, Synthesis, 2013, 2387 CAS; (c) S. Bhadra, W. I. Dzik and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 2959 CrossRef CAS PubMed.
- (a) M. S. Stephan, A. J. J. M. Teunissen, G. K. M. Verzijl and J. G. De Vries, Angew. Chem., Int. Ed., 1998, 37, 662 CrossRef CAS; (b) L. J. Goossen and J. Paetzold, Angew. Chem., Int. Ed., 2002, 41, 1237 CrossRef CAS; (c) E. M. O'Brien, E. A. Bercot and T. Rovis, J. Am. Chem. Soc., 2003, 125, 10498 CAS; (d) T. Sugihara, T. Satoh, M. Miura and M. Nomura, Angew. Chem., Int. Ed., 2003, 42, 4672 CrossRef CAS PubMed; (e) L. J. Goossen and J. Paetzold, Angew. Chem., Int. Ed., 2004, 43, 1095 CrossRef CAS PubMed; (f) L. J. Goossen and N. Rodriguez, Chem. Commun., 2004, 724 RSC; (g) L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, J. Am. Chem. Soc., 2005, 127, 11102 CrossRef CAS PubMed; (h) S. E. Havlik, J. M. Simmons, V. J. Winton and J. B. Johnson, J. Org. Chem., 2011, 76, 3588 CrossRef CAS PubMed; (i) M. O. Miranda, A. Pietrangelo, M. A. Hillmyer and W. B. Tolman, Green Chem., 2012, 14, 490 RSC.
- (a) X. Zhao and Z. Yu, J. Am. Chem. Soc., 2008, 130, 8136 CrossRef CAS PubMed; (b) W. Ye, N. Luo and Z. Yu, Organometallics, 2010, 29, 1049 CrossRef CAS; (c) W. Jin, Z. Yu, W. He, W. Ye and W.-J. Xiao, Org. Lett., 2009, 11, 1317 CrossRef CAS PubMed.
- F. Pan, Z.-Q. Lei, H. Wang, H. Li, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2013, 52, 2063 CrossRef CAS PubMed.
- Z.-Q. Lei, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 2690 CrossRef CAS PubMed.
- (a) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS PubMed; (b) K. L. Tan, Nat. Chem., 2012, 4, 253 CrossRef CAS PubMed.
- (a) Y.-H. Xu, J. Lu and T.-P. Loh, J. Am. Chem. Soc., 2009, 131, 1372 CrossRef CAS PubMed; (b) K. D. Hesp, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 11430 CrossRef CAS PubMed; (c) Y. Li, X.-S. Zhang, Q.-L. Zhu and Z.-J. Shi, Org. Lett., 2012, 14, 4498 CrossRef CAS PubMed; (d) H. Wang, B. Beiring, D.-G. Yu, K. D. Collins and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12430 CrossRef CAS PubMed.
- X.-S. Zhang, Q.-L. Zhu, F.-X. Luo, G. Chen, X. Wang and Z.-J. Shi, Eur. J. Org. Chem., 2013, 6530–6534 CrossRef CAS.
- Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed.
- (a) S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812 CrossRef CAS PubMed; (b) H. J. Kim, M. J. Ajitha, Y. Lee, J. Ryu, J. Kim, Y. Lee, Y. Jung and S. Chang, J. Am. Chem. Soc., 2013, 136, 1132 CrossRef PubMed.
- X.-S. Zhang, Z.-W. Li and Z.-J. Shi, Org. Chem. Front., 2014, 1, 44–49 RSC.
- A. Jõgi, A. Paju, T. Pehk, T. Kailas, A.-M. Müürisepp, T. Kanger and M. Lopp, Synthesis, 2006, 3031 Search PubMed.
Footnotes |
| † In celebration of Max Malacria's 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, and HRMS data and copies of NMR spectra for all starting materials and products. See DOI: 10.1039/c4qo00074a |
| This journal is © the Partner Organisations 2014 |