 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetrathiafulvalene mono- and bis-1,2,3-triazole precursors by click chemistry: structural diversity and reactivity†
Thomas
Biet
and
Narcis
Avarvari
*
Laboratoire MOLTECH-Anjou, Université d'Angers, CNRS, UMR 6200, UFR Sciences, Bât. K, 2 Bd. Lavoisier, 49045 Angers, France. E-mail: narcis.avarvari@univ-angers.fr; Fax: (+33)02 41 73 54 05; Tel: (+33)0241 73 50 84
First published on 14th February 2014
Abstract
The donor ortho-dimethyl-TTF-(N-n-Bu-1,2,3-triazole) 1,5-isomer has been synthesized by click chemistry following a ruthenium-catalyzed azide–alkyne cycloaddition procedure. The single crystal X-ray analysis showed a planar conformation between the TTF and triazole units and a set of intermolecular interactions at the supramolecular level in the solid state. The same procedure allowed the preparation of the corresponding ortho-dimethyl-TTF-bis(triazole) which was also structurally characterized. Because of the steric hindrance, the triazole units are no longer planar with the TTF backbone. The reactivity of the triazole ring has been investigated in protonation and alkylation reactions, monitored by UV-visible spectroscopy, which clearly showed the red shift of the intramolecular charge transfer band. A TTF-methyl-triazolium salt has been isolated and analyzed by single crystal X-ray analysis. All of the TTF-triazoles and triazolium salts are valuable precursors for radical cation salts due to their oxidation potentials and variety of possible intermolecular interactions.
Introduction
Synthetic methods based on click chemistry strategies have developed increasingly over the last decade,1 with one of the most useful reactions being the copper-catalyzed azide–alkyne cycloaddition (CuAAC) that allows the straightforward preparation of 1,2,3-triazoles (1,4-isomers).2 These heterocycles have proved to be useful units in chemical sensing3 and peptidomimetic research,4 and are also interesting ligands towards transition metals.5 Alternatively, the 1,5-isomers of 1,2,3-triazoles have been efficiently prepared by the azide–alkyne cycloaddition mediated by a ruthenium(II)-based catalyst.6 We have recently employed both strategies for the synthesis of the new electroactive ligands TTF-1,2,3-triazoles as 1,4- and 1,5-isomers (Scheme 1),7 respectively, where the electroactive part is represented by the TTF (tetrathiafulvalene) unit, and in which TTF and triazoles are directly connected. A large difference in their electrochemical behavior was observed, e.g. much easier oxidation for the 1,4-isomers than for the 1,5-isomers, and, accordingly, in their relative kinetic stability. Moreover, coordination of the more stable 1,5-isomers to Cu(hfac)2 (hfac = hexafluoroacetylacetonate) fragments induced an unusual cathodic shift of the oxidation potentials of TTF. | ||
| Scheme 1 TTF-1,2,3-triazoles. | ||
In the other few published examples of TTF-triazole systems, with linkers of various lengths between TTF and triazole,8 the latter serves mainly to connect other electroactive moieties to TTF, such as perylene diimide9 or ferrocene.10 From a general point of view, the association of TTF with coordinating groups within electroactive ligands11 represents a powerful strategy to access either multifunctional molecular materials, in which a combination of different physical properties can be achieved,12 or electroactive catalysts.13 In this respect, nitrogen-containing five-membered rings, such as oxazolines,14 pyrazoles15 or imidazoles16 have been attached to TTF, while the synthesis and coordination chemistry of TTF-triazole ligands has been reported much more recently.7 Beside their use as ligands, the basicity and nucleophilicity of the triazole ring should in principle allow for the modulation of the intramolecular charge transfer and the electrodonating character of TTF upon protonation or reaction with electrophiles.16b Moreover, the versatility of the two “click” strategies, i.e. Cu versus Ru catalysis, guarantees the preparation of a whole library of TTF-triazoles by the appropriate choice of the acetylene and azide units. The substitution pattern, type of isomers, i.e. 1,4-versus 1,5-, and number of triazole units should have an important influence not only on the electrochemical properties and coordination ability, but also on the supramolecular architectures in crystals, which ultimately determines the solid state properties of the derived materials. We report herein the synthesis, reactivity and solid state structures of TTF-mono and -bis(1,2,3-triazoles), together with those of a TTF-triazolium salt.
Results and discussion
The TTF-triazole 1 (Schemes 1 and 2), with Me groups on TTF and a benzyl (Bn) substituent on the nitrogen atom N1, was synthesized as previously described by a Ru-catalyzed azide–alkyne cycloaddition (RuAAC) reaction between the dimethyl-TTF-acetylene and benzyl azide.7 In order to evaluate the influence of different substituents on the triazole ring, the ligand TTF-triazole 2 with an n-butyl (n-Bu) group instead of Bn was prepared following the same procedure by using n-butyl azide (Scheme 2). | ||
| Scheme 2 Synthesis of the TTF-mono(triazoles) 1 and 2. | ||
The donor ortho-dimethyl-TTF 3 is well suited for a selective bis-functionalization, allowing us to attempt the preparation of TTF-bis(triazoles) through both strategies. Accordingly, the diiodo derivative 4 has been prepared upon the double lithiation of 3 followed by treatment with perfluorohexyl iodide. Then, a double Sonogashira coupling under classical conditions with trimethylsilyl acetylene afforded the bis(acetylenic) compound 5 which was further deprotected in the presence of (n-Bu)4NF. The resulting TTF-bis(acetylene) 6 was then rapidly engaged in a double cycloaddition reaction with benzyl azide following either the copper or the ruthenium-based catalysis (Scheme 3).
 | ||
Scheme 3 Synthesis of 7 and 8. Reaction conditions: (i) (n-Bu)4NF, THF–MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); (ii) CuI, PhCH2N3, CHCl3–DIPEA (1:1), 65 °C; (iii) Cp*RuCl(PPh3)2, PhCH2N3, THF, 65 °C. :1); (ii) CuI, PhCH2N3, CHCl3–DIPEA (1:1), 65 °C; (iii) Cp*RuCl(PPh3)2, PhCH2N3, THF, 65 °C. | ||
While in the case of the Ru-catalyzed reaction the TTF-bis(triazole) 8 was obtained in low yield (15%) as the bis(1,5-isomer), the Cu-based catalysis afforded only the TTF-triazole-acetylene 7 as the 1,4-isomer in even lower yield (10%) with no trace of the bis(triazole). The low yields are very likely attributed to the sensitivity of the TTF-diacetylene 6 which partially degrades under the reaction conditions. Moreover, as previously mentioned, the 1,4-isomers are less stable than the 1,5-isomers,7 therefore compound 7, which is an intermediate towards the desired bis(triazole), degrades before the second acetylene unit reacts in the cycloaddition reaction. Nevertheless, a small amount of 7 could be purified by column chromatography and characterized by 1H NMR and mass spectrometry, yet the compound has a limited stability in air even in the solid state.
Cyclic voltammetry measurements (see ESI†) show that the oxidation potentials for the donor 2, of 0.45 and 0.85 V versus SCE for E11/2 and E21/2 respectively, are identical to those for the benzyl derivative 1. In the case of the bis(triazole) 8, anodic shifts of 0.13 and 0.11 V for the first and second oxidation processes are observed when compared to the mono(triazole) 1 (e.g. E11/2 = 0.44 V (1) and 0.57 V (8), E21/2 = 0.85 V (1) and 0.96 V (8)), thus evidencing the withdrawing effect of the triazole ring when connected to TTF as the 1,5-isomer.
The solid state structures of the mono(triazole) 2, bis(triazole) 8, as well as the intermediate 5 (see ESI†), have been determined by single crystal X-ray diffraction analysis.
The crystal structure of 1-butyl-5-(ortho-dimethyl-TTF)-1,2,3-triazole (2)
The solid state structure of the benzyl derivative 1, previously described,7 shows quasi-planarity between the TTF and triazole units, with a dihedral angle of 6.0° and the s-trans conformation, and the existence of short intermolecular S⋯S contacts. In the case of 2, suitable crystals, as orange needles, for X-ray analysis were obtained by the slow evaporation of the solvent from a 1:1 MeOH–CH2Cl2 solution. The compound crystallizes in the monoclinic system, space group P21/c, with one independent molecule in the unit cell. The conformation between the TTF and triazole is s-trans when considering the double bonds C5![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C6 and C7C8, with a very small value of 7.4° for the TTF⋯triazole dihedral angle (Fig. 1). The central C3C4 bond measures 1.260(4) Å, in agreement with the neutral state of the donor.
C6 and C7C8, with a very small value of 7.4° for the TTF⋯triazole dihedral angle (Fig. 1). The central C3C4 bond measures 1.260(4) Å, in agreement with the neutral state of the donor.
 | ||
| Fig. 1 Molecular structure of 2 along with the numbering scheme. | ||
At the supramolecular level the formation of columns is observed, in which the donors are shifted with respect to each other along the longitudinal axis in such a way that a triazole ring stacks with the TTF unit below, with N⋯S and S⋯S contacts of 3.55 and 3.72 Å, respectively (Fig. 2).
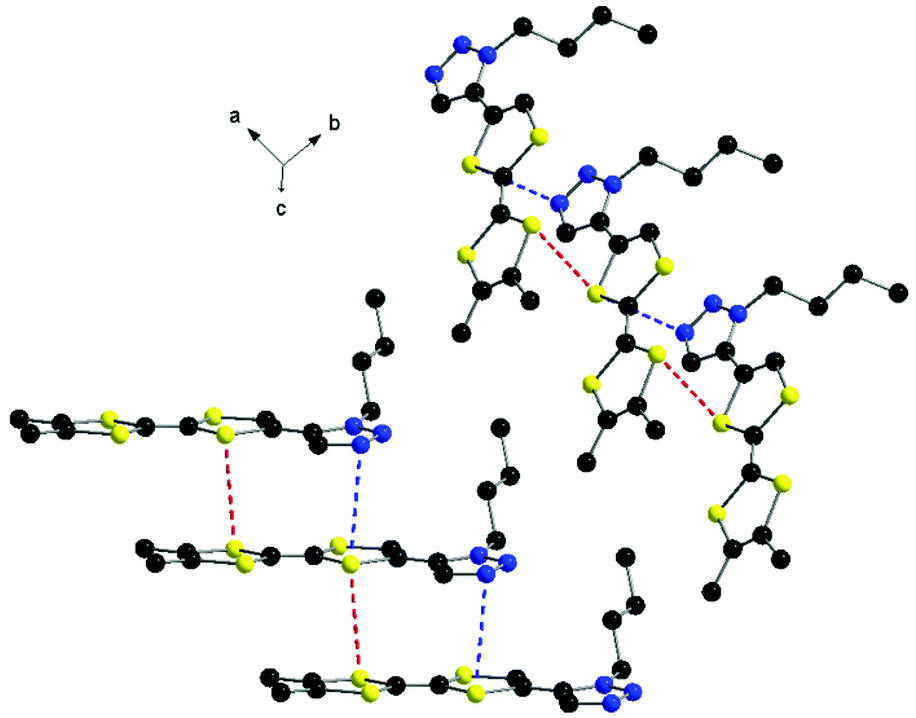 | ||
| Fig. 2 Columns of donors 2 with an emphasis on the N⋯S and S⋯S intermolecular contacts. Hydrogen atoms have been omitted. | ||
This arrangement is different to that encountered in the structure of 1, where the formation of head-to-tail dimers was observed. The columns of 2 further interact laterally along the c axis through short N⋯S contacts and N⋯Hvinyl hydrogen bonds of 2.87 and 2.83 Å, respectively (Fig. 3). Thus, the overall supramolecular architecture results from the interplay of S⋯S, N⋯S and N⋯H intermolecular interactions. As a general feature of the TTF-mono(triazole) donors, when comparing the structures of 17 and 2, one can disclose the planarity of the system, suggesting conjugation between the two units.
 | ||
| Fig. 3 Packing of 2 along the c axis with an emphasis on the N⋯S and N⋯Hvinyl interactions. | ||
The crystal structure of ortho-dimethyl-TTF-bis(1-benzyl-1,2,3-triazol-5-yl) (8)
Single crystals of 8 as orange plates were obtained by vapour diffusion of pentane into a solution of the compound in methylene chloride. The donor crystallizes in the orthorhombic system, with the non-centrosymmetric space group P212121, with one independent molecule in the asymmetric unit. The most striking peculiarity in the structure is the non-planarity of the two triazole rings with respect to the TTF backbone, with dihedral angles of 87.9° and 37.0° for TTF⋯triazoleN1N2N3 and TTF⋯triazoleN4N5N6, respectively (Fig. 4). This feature is very likely due to the steric hindrance caused by the vicinity of the two heterocycles, which is comparable to the situation observed in the solid state structures of TTF-mono(oxazolines)14a and TTF-bis(oxazolines).17 Moreover, the intramolecular π-stacking interaction established between the triazole N4N5N6 and the Ph ring of the other triazole unit, which are parallel at a distance of 3.4 Å, probably also contributes to the stabilization of this conformation. Bond lengths, such as the central C3C4 bond and the internal C–S bonds, have typical values for a neutral donor (see ESI†).
 | ||
| Fig. 4 Molecular structure of 8 along with the numbering scheme. | ||
At the supramolecular level there is no short S⋯S intermolecular contact, very likely because of the presence of the bulky benzyl groups on TTF. However, a network of intermolecular N⋯H interactions develops, particularly involving the benzylic protons (see ESI†).
For the use of precursors such as the TTF-bis(triazole) 8 in electrocrystallization experiments for the preparation of radical cation salts, the replacement of the benzyl groups by less bulky substituents, e.g. n-Bu as in 2, can be envisaged in order to allow the occurrence of TTF⋯TTF intermolecular interactions which are crucial for the establishment of conductivity paths. It is clear that, when analysing the structures of 1,72 and 8, and also the other TTF-triazole derivatives,7 the presence of the triazole ring on TTF provides additional possibilities for intermolecular interactions such as hydrogen bonding, π–π stacking and donor–acceptor, and thus results in the emergence of original architectures. Moreover, the unsubstituted nitrogen atoms can in principle be protonated or alkylated, which should allow for the modulation of the electron donor character of TTF, the intra- and intermolecular charge transfer, and the geometry of the compound.
Protonation and alkylation studies with 1 and 8
The protonation of the mono(triazole) 1 and bis(triazole) 8, both possessing benzyl substituents, has been monitored by UV-vis spectroscopy. As previously described, in the UV-vis spectra of TTF-triazoles, an absorption band is observed at around λmax = 388–394 nm, corresponding to a partial charge transfer from TTF to triazole, as determined by TD DFT calculations.7 In the bis(triazole) 8, this band is centered at 415 nm (Fig. 5), with the red shift with respect to the mono(triazole) 1 being in agreement with a smaller HOMO–LUMO gap when compared to 1. Since the oxidation of 8 occurs at a more anodic potential than the oxidation of 1, indicating a lower energy HOMO level for 8 than for 1, it can be concluded that the variation ΔELUMO is larger than ΔEHOMO when going from 1 to 8. The protonation or alkylation of the triazole ring is expected to increase its withdrawing character, and, consequently, result in a red shift of the charge transfer band. The stepwise addition of up to 1 equivalent of HBF4 in a CH2Cl2 solution of 1 indeed leads to a red shift of the charge transfer band from 394 nm (1) to 422 nm ([1–H]+BF4−), with the existence of a clear isosbestic point at 416 nm (see ESI†). In the same time, practically no variation of the position of the high energy bands ranging between 250–350 nm is observed. Further addition of HBF4 produces no further change to the spectrum, in agreement with the presence of only one basic N atom. When the same type of experiment was performed with the bis(triazole) 8, a first bathochromic shift of 14 nm was observed by adding up to 1 equivalent of HBF4, followed by a second shift of 22 nm between 1 and 2 equivalents of HBF4 (Fig. 5). | ||
| Fig. 5 UV-vis absorption spectra of 8 (10−4 M in CH2Cl2) during the stepwise addition of HBF4; the insets magnify the charge transfer band. | ||
In order to isolate and characterize in detail a triazolium salt, the precursor 1 was reacted with trimethyloxonium tetrafluoroborate (Me3O)BF4 to afford 9 (Scheme 4).
 | ||
| Scheme 4 Alkylation of 1. | ||
The triazolium salt 9 was obtained as an analytically pure brown solid, and suitable single crystals, as red blocks, were grown by the slow diffusion of diethyl ether into a solution of 9 in acetonitrile. The cyclic voltammetry measurements show anodic shifts of 0.07 and 0.06 V for the first and second oxidation potentials respectively, when compared to the neutral precursor 1 (see ESI†). In line with the observations from the protonation experiment, a large red shift of 57 nm in the UV-vis spectrum is observed for the charge transfer band which is now centred at 451 nm (see ESI†).
The final proof for the cationic structure of 9 was provided by the single crystal X-ray analysis. The compound crystallizes in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with one independent TTF-triazolium cation and one BF4 anion in the asymmetric unit. As clearly observed (Fig. 6), the methylation took place at the N3 atom, which is the most nucleophilic. There is now perfect planarity between the TTF and triazolium units, while the Ph ring is perpendicular to the rest of the molecule.
, with one independent TTF-triazolium cation and one BF4 anion in the asymmetric unit. As clearly observed (Fig. 6), the methylation took place at the N3 atom, which is the most nucleophilic. There is now perfect planarity between the TTF and triazolium units, while the Ph ring is perpendicular to the rest of the molecule.
 | ||
| Fig. 6 Molecular structure of 9 along with the numbering scheme (up) and a lateral view of the donor molecule (bottom). | ||
The central C3C4 bond length of 1.351(4) Å is in the normal range for a neutral donor and compares very well with the value of 1.350(5) Å in the neutral precursor 1. Nevertheless, the most important structural effect of the methylation concerns, as expected, the triazole ring, since in 9 the N–N distances have closer values, i.e. 1.326(4) Å for N1–N2 and 1.306(4) Å for N2–N3, as an effect of the conjugation, and, moreover, are shorter than the distances of 1.353(4) and 1.316(4) Å, respectively, observed in the neutral triazole. In the packing there is no short intermolecular S⋯S contact (see ESI†), yet hydrogen bonds of F⋯H-type are established between the BF4 anions and vinylic, benzylic and NMe protons (see ESI†).
Conclusions
The synthesis of TTF-bis(triazoles) as 1,4- and 1,5-isomers was attempted following copper and ruthenium-catalyzed azide–alkyne cycloaddition reactions, respectively. The access to these bis(triazole) precursors proved to be much more problematic than the preparation of TTF-mono(triazoles), very likely because of the sensitivity of the TTF-bis(acetylene) precursor and the TTF-triazole-acetylene intermediate, since only the 1,5-isomer of the ortho-dimethyl-TTF-bis(triazole) could be prepared by ruthenium-based catalysis. The same reaction allowed the preparation of TTF-mono(triazoles) with benzyl or n-butyl substituents on the triazole ring. The substitution patterns and number of triazole units greatly influences the solid state structures of these electroactive precursors. As a general structural feature, the triazole units engage in the solid state in intermolecular π–π interactions and hydrogen bonding, thus competing with the classical TTF⋯TTF interactions. Modulation of the intramolecular charge transfer, TTF → triazole, was achieved by protonation of the triazole units with HBF4, monitored by UV-visible spectroscopy. Moreover, a triazolium salt was isolated and structurally characterized by methylation with the strong electrophilic agent (Me3O)BF4. All of these TTF-triazoles and derivatives constitute valuable precursors for radical cation salts since they all possess good electron donating properties. The coordinating properties of triazoles can be also exploited, especially when associated to pyridines within chelating ligands.18 These possibilities are actively being investigated in our laboratory.Experimental
General
Dry THF and diethyl ether were obtained from a solvent purification system (LC Technology Solutions Incorporated). Nuclear magnetic resonance spectra were recorded on a Bruker Advance DRX 300 spectrometer operating at 300 MHz for 1H and 75 MHz for 13C. Chemical shifts are expressed in parts per million (ppm) downfield from external TMS. The following abbreviations are used: s, singlet; d, doublet; t, triplet; m, multiplet. MALDI-TOF MS spectra were recorded on a Bruker Biflex-IIITM apparatus, equipped with a 337 nm N2 laser. Elemental analyses were recorded using a Flash 2000 Fisher Scientific Thermo Electron analyzer. IR spectra were recorded on a Bruker FT-IR Vertex 70 spectrometer equipped with a platinum diamond ATR accessory.The compounds ortho-dimethyl-TTF 319 and ortho-dimethyl-ethynyl-TTF20 were prepared according to the published procedures.
1-Butyl-5-(ortho-dimethyl-tetrathiafulvalenyl)-1,2,3-triazole 2
In a Schlenk tube, ortho-dimethyl-ethynyl-TTF (120 mg, 0.47 mmol), n-butyl azide (70 mg, 0.71 mmol) and Cp*RuCl(PPh3)2 (22 mg, 6 mol%) were dissolved in dry THF (8 mL). The reaction mixture was heated at 65 °C overnight and concentrated under vacuum. The crude product was then purified by chromatography over SiO2 (CH2Cl2–AcOEt 3:1 as the eluent, Rf = 0.3) to yield 2 as an orange solid (130 mg, 78%). Suitable single crystals for X-ray analysis were obtained by slow evaporation of the solvent from a MeOH–CH2Cl2 (1:1) solution of 2.
1H NMR (CDCl3 + NEt3, 300 MHz) δ (ppm) 7.70 (s, 1H), 6.44 (s, 1H), 4.42 (t, 2H, J = 7.3 Hz), 1.99 (s, 6H), 1.91 (qt, 2H, J = 7.3 Hz), 1.37 (sext, 2H, J = 7.3 Hz), 0.98 (t, 3H, J = 7.3 Hz); {1H}13C NMR (CDCl3 + NEt3, 75 MHz) δ (ppm) 134.2, 129.4, 123.0, 122.9, 121.1, 119.8, 113.2, 106.5, 46.2, 32.13, 19.8, 13.8, 13.5; MS (MALDI-TOF) m/z = 354.7 (M+); Anal. calcd for C14H17N3S4: C, 47.29; H, 4.82; N, 11.82; S, 36.07%, found: C, 47.58; H, 4.75; N, 11.44; S, 35.83%.
2,3-Diiodo-6,7-dimethyltetrathiafulvalene 4
In a Schlenk tube, ortho-dimethyl-TTF 3 (600 mg, 2.58 mmol) was dissolved in dry diethyl ether (80 mL) under argon at −78 °C. Diisopropylethylamine (798 μL, 5.68 mmol), followed by butyllithium (3.55 mL, 5.68 mmol) (1.6 M solution in hexane) were then added to the solution. The reaction mixture was stirred at −78 °C for one hour and a yellow precipitate appeared. Perfluorohexyl iodide (1.45 mL, 6.71 mmol) was added and the reaction mixture was allowed to warm up slowly to room temperature and was stirred overnight. After evaporation of the solvent, the crude product was purified by chromatography over SiO2 (cyclohexane–CS2 1:1 as the eluent, Rf = 0.8), yielding 4 as a light red solid (662 mg, 53%).
1H NMR (CDCl3 + NEt3, 300 MHz) δ (ppm) 2.00 (s, 6H); {1H}13C NMR (CDCl3 + NEt3, 75 MHz) δ (ppm) 128.4, 128.3, 122.9, 114.5, 111.4, 77.2, 13.8; MS (MALDI-TOF) m/z = 483.7 (M+); Anal. calcd for C8H6I2S4: C, 19.84; H, 1.25; S, 26.49%, found: C, 20.66; H, 1.30; S, 26.54%.
2,3-Di(trimethylsilylethynyl)-6,7-dimethyltetrathiafulvalene 5
In a Schlenk tube, 4 (500 mg, 1.03 mmol), Pd(PPh3)4 (60 mg, 5 mol%) and CuI (20 mg, 10 mol%) were dissolved in dry THF (15 mL). Diisopropylamine (871 μL, 6.20 mmol) and trimethylsilylacetylene (882 μL, 6.20 mmol) were then added to the solution. The reaction mixture was heated at 60 °C overnight under argon. After evaporation of the solvent, the crude product was purified by chromatography over SiO2 (cyclohexane as the eluent), yielding 5 as a purple solid (377 mg, 86%). Suitable single crystals for X-ray analysis were obtained by the recrystallization of 5 in acetonitrile.1H NMR (CDCl3 + NEt3, 300 MHz) δ (ppm) 1.97 (s, 6H), 0.25 (s, 18H); {1H}13C NMR (CDCl3 + NEt3, 75 MHz) δ (ppm) 125.8, 122.7, 122.0, 114.0, 106.5, 99.7, 94.9, 13.8, −0.29; MS (MALDI-TOF) m/z = 424.0 (M+); Anal. calcd for C18H24S4Si2: C, 50.89; H, 5.69; S, 30.19%, found: C, 51.21; H, 5.75; S, 30.05%.
2,3-Di(ethynyl)-6,7-dimethyltetrathiafulvalene 6
To a degassed solution of 5 (150 mg, 0.35 mmol) in THF–methanol (20 mL, 1:1 v/v), tetrabutylammonium fluoride (850 μL, 0.85 mmol) (1 M solution in THF) was added. The reaction mixture was stirred at RT for 45 min and then the solvents were removed under vacuum. The crude product was then purified by flash chromatography over neutral alumina (CH2Cl2 as the eluent) to yield 6 as a light purple solid (70 mg, 71%) which was directly engaged in the next step.
1H NMR (CDCl3 + NEt3, 300 MHz) δ (ppm) 3.44 (s, 2H), 2.00 (s, 6H).
ortho-Dimethyl-TTF-2′-(N-benzyl-1,2,3-triazol-5-yl)-3′-ethynyl 7
In a Schlenk tube, 6 (70 mg, 0.25 mmol), benzyl azide (90 mg, 0.75 mmol) and CuI (5.7 mg, 6 mol%) were dissolved in CHCl3 (5 mL) and N,N-diisopropylethylamine (3 mL). The reaction mixture was heated at 65 °C overnight and concentrated under vacuum. The crude product was then purified by chromatography over SiO2 (CH2Cl2–AcOEt 3:1, and a few drops of NEt3 as the eluent, Rf = 0.4) to yield 7 as an orange solid (10 mg, 10%).
1H NMR (CDCl3 + NEt3, 300 MHz) δ (ppm) 8.16 (s, 1H), 7.44–7.39 (m, 3H), 7.30 (m, 2H), 5.60 (s, 2H), 3.61 (s, 1H), 1.98 (s, 6H); MS (MALDI-TOF) m/z = 413.2 (M+).
ortho-Dimethyl-TTF-bis(1-benzyl-1,2,3-triazol-5-yl) 8
In a Schlenk tube, 6 (120 mg, 0.28 mmol), benzyl azide (112 mg, 0.85 mmol) and Cp*RuCl(PPh3)2 (14 mg, 6 mol%) were dissolved in dry THF (8 mL). The reaction mixture was heated for 24 h at 65 °C and concentrated under vacuum. The crude product was then purified by chromatography over SiO2 (CH2Cl2–AcOEt 1:1 as the eluent) to yield 8 as a beige solid (35 mg, 15%). Suitable single crystals for X-ray analysis were grown by vapour diffusion of pentane onto a CH2Cl2 solution of 8.
1H NMR (CDCl3 + NEt3, 300 MHz) δ (ppm) 7.43–7.37 (m, 6H), 7.30–7.26 (m, 4H), 6.54 (s, 2H), 5.41 (s, 4H), 2.08 (s, 6H); {1H}13C NMR (CDCl3 + NEt3, 75 MHz) δ (ppm) 134.6, 134.5, 129.0, 128.9, 128.2, 127.2, 123.1, 121.7, 117.7, 65.7, 13.6; MS (MALDI-TOF) m/z = 545.2 (M+); Anal. calcd for C26H22N6S4: C, 57.12; H, 4.06; N, 15.37; S, 23.46%, found: C, 56.86; H, 4.20; N, 14.43; S, 21.96%.
1-Benzyl-3-methyl-5-(6′,7′-dimethyltetrathiafulvalenyl)-1,2,3-triazolium·BF49
In a Schlenk tube, 1 (35 mg, 0.09 mmol) was dissolved in a 6 mL mixture of acetonitrile–dichloromethane (1:1). Then, trimethyloxonium tetrafluoroborate (13.3 mg, 0.09 mmol) dissolved in acetonitrile (3 mL) was added and the solution, which turned darker, was stirred for 1 h at room temperature. Diethyl ether (10 mL) was added to the reaction mixture, and the precipitate formed was filtered and washed with cold dichloromethane and diethyl ether to yield 9 as a brown solid (31 mg, 77%). Suitable single crystals for X-ray analysis were grown by the slow diffusion of diethyl ether into an acetonitrile solution of 9.
1H NMR (DMSO, 300 MHz) δ (ppm) 9.19 (s, 1H), 7.48–7.40 (m, 4H), 7.38–7.31 (m, 2H), 5.93 (s, 2H), 4.32 (s, 3H), 1.99 (s, 6H); {1H}13C NMR (DMSO, 75 MHz) δ (ppm) 134.1, 132.5, 132.3, 130.6, 129.6, 129.5, 128.9, 123.5, 113.9, 113.1, 104.7, 55.5, 13.9; MS (MALDI-TOF) m/z = 403.8 (M+); Anal. calcd for C18H18BF4N3S4: C, 43.99; H, 3.69; N, 8.55; S, 26.10%, found: C, 44.38; H, 3.96; N, 8.26; S, 25.68%.
X-Ray structure determination
Details of the data collection and solution refinement are given in Table 1 (see also ESI† for 5). X-ray diffraction measurements were performed on a Bruker Kappa CCD diffractometer, operating with a Mo Kα (λ = 0.71073 Å) X-ray tube with a graphite monochromator. The structures were solved (SHELXS-97) by direct methods and refined (SHELXL-97) by full matrix least-square procedures on F2.21 All of the non-hydrogen atoms were refined anisotropically. Hydrogen atoms were introduced at calculated positions (riding model) and included in structure factor calculations, but not refined. Crystallographic data for the four structures have been deposited with the Cambridge Crystallographic Data Centre, with the deposition numbers CCDC 982420 (2), CCDC 982421 (5), CCDC 982422 (8) and CCDC 982423 (9).| Compound | 2 | 8 | 9 |
|---|---|---|---|
| Empirical formula | C14H17N3S4 | C26H22N6S4 | C18H18BF4N3S4 |
| fw | 355.55 | 546.74 | 491.40 |
| T (K) | 293(2) | 293(2) | 293(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Orthorhombic | Triclinic |
| Space group | P21/c | P212121 |
P |
| Unit cell dimensions | |||
| a (Å) | 5.3381 (5) | 6.7025 (5) | 9.7854 (6) |
| b (Å) | 25.844 (19) | 19.621 (2) | 11.173 (7) |
| c (Å) | 11.069 (10) | 20.267 (3) | 11.332 (5) |
| α (°) | 90.00 | 90.00 | 70.909 (4) |
| β (°) | 99.56 (7) | 90.00 | 81.108 (4) |
| γ (°) | 90.00 | 90.00 | 68.833 (4) |
| V (Å3) | 1505.9 (2) | 2665.3 (6) | 1090.88 (11) |
| Z | 4 | 4 | 2 |
| D c (g cm−3) | 1.568 | 1.363 | 1.496 |
| Abs coeff (mm−1) | 0.626 | 0.384 | 0.480 |
| F(000) | 744 | 1136 | 504 |
| Crystal size (mm3) | 0.6 × 0.1 × 0.1 | 0.6 × 0.3 × 0.08 | 0.8 × 0.4 × 0.4 |
| θ range for data collection (°) | 3.15–27.50 | 2.71–24.03 | 2.35–27.00 |
| Limiting indices | −6 ≤ h ≤ 6, | −7 ≤ h ≤ 7, | −12 ≤ h ≤ 12, |
| −33 ≤ k ≤ 32, | −22 ≤ k ≤ 21, | −14 ≤ k ≤ 14, | |
| −14 ≤ l ≤ 14 | −23 ≤ l ≤ 20 | −14 ≤ l ≤ 14 | |
| Reflections collected | 17019 |
13973 |
21173 |
| Independent reflections | 3380 | 4058 | 4719 |
| Completeness (%) to θ = 25.59° | 98.1 | 98.1 | 98.9 |
| Abs correction | Multi-scan | Multi-scan | Multi-scan |
| Refinement method | Full-matrix least squares on F2 | Full-matrix least squares on F2 | Full-matrix least squares on F2 |
| Data/restraints/parameters | 3380/0/190 | 4058/0/325 | 4719/0/271 |
| GOF on F2 | 1.011 | 1.125 | 1.132 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0579, wR2 = 0.1453 | R 1 = 0.0482, wR2 = 0.0789 | R 1 = 0.0657, wR2 = 0.2094 |
| R indices (all data) | R 1 = 0.1176, wR2 = 0.1640 | R 1 = 0.0876, wR2 = 0.0923 | R 1 = 0.0804, wR2 = 0.2255 |
| Largest diff. peak and hole (e Å−3) | 0.393 and −0.302 | 0.196 and −0.172 | 1.328 and −0.768 |
Acknowledgements
This work was supported by the CNRS, University of Angers and the Ministry of Education and Research (grant to T.B.). I. Freuze and C. Mézière are thanked for the MS analyses.References
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldalb, J. Org. Chem., 2002, 67, 3057 CrossRef; (c) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC.
- (a) Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848 RSC; (b) J. J. Bryant and U. H. F. Bunz, Chem. - Asian J., 2013, 8, 1354 CrossRef CAS PubMed.
- D. S. Pedersen and A. Abell, Eur. J. Org. Chem., 2011, 2399 CrossRef CAS.
- (a) R. Bronisz, Inorg. Chem., 2005, 44, 4463 CrossRef CAS PubMed; (b) B. M. J. M. Suijkerbuijk, B. N. H. Aerts, H. P. Dijkstra, M. Lutz, A. L. Spek, G. van Koten and R. J. M. K. Gebbink, Dalton Trans., 2007, 1273 RSC; (c) M. L. Gower and J. D. Crowley, Dalton Trans., 2010, 39, 2371 RSC; (d) J. D. Crowley and E. L. Gavey, Dalton Trans., 2010, 39, 4035 RSC; (e) H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675 RSC; (f) D. Schweinfurth, J. Krzystek, I. Schapiro, S. Demeshko, J. Klein, J. Telser, A. Ozarowski, C.-Y. Su, F. Meyer, M. Atanasov, F. Neese and B. Sarkar, Inorg. Chem., 2013, 52, 6880 CrossRef CAS PubMed.
- (a) L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998 CrossRef CAS PubMed; (b) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923 CrossRef CAS PubMed.
- T. Biet, T. Cauchy and N. Avarvari, Chem. - Eur. J., 2012, 18, 16097 CrossRef CAS PubMed.
- Y.-L. Zhao, W. R. Dichtel, A. Trabolsi, S. Saha, I. Aprahamian and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 11294 CrossRef CAS PubMed.
- K. Qvortrup, M. Å. Petersen, T. Hassenkam and M. B. Nielsen, Tetrahedron Lett., 2009, 50, 5613 CrossRef CAS.
- B.-T. Zhao, L.-W. Liu, X.-C. Li, G.-R. Qu, E. Belhadj, F. Le Derf and M. Sallé, Tetrahedron Lett., 2013, 54, 23 CrossRef CAS.
- (a) D. Lorcy, N. Bellec, M. Fourmigué and N. Avarvari, Coord. Chem. Rev., 2009, 253, 1398 CrossRef CAS; (b) M. Shatruk and L. Ray, Dalton Trans., 2010, 39, 11105 RSC.
- (a) E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419 CrossRef CAS PubMed; (b) L. Ouahab and T. Enoki, Eur. J. Inorg. Chem., 2004, 933 CrossRef CAS; (c) F. Riobé and N. Avarvari, Coord. Chem. Rev., 2010, 254, 1523 CrossRef.
- (a) C. Réthoré, I. Suisse, F. Agbossou-Niedercorn, E. Guillamón, R. Llusar, M. Fourmigué and N. Avarvari, Tetrahedron, 2006, 62, 11942 CrossRef; (b) C. Réthoré, F. Riobé, M. Fourmigué, N. Avarvari, I. Suisse and F. Agbossou-Niedercorn, Tetrahedron: Asymmetry, 2007, 18, 1877 CrossRef.
- (a) C. Réthoré, M. Fourmigué and N. Avarvari, Chem. Commun., 2004, 1384 RSC; (b) C. Réthoré, M. Fourmigué and N. Avarvari, Tetrahedron, 2005, 61, 10935 CrossRef; (c) A. M. Madalan, C. Réthoré and N. Avarvari, Inorg. Chim. Acta, 2007, 360, 233 CrossRef CAS.
- (a) W. Liu, J. Xiong, Y. Wang, X.-H. Zhou, R. Wang, J.-L. Zuo and X.-Z. You, Organometallics, 2009, 28, 755 CrossRef CAS; (b) M. Nihei, N. Takahashi, H. Nishikawa and H. Oshio, Dalton Trans., 2011, 40, 2154 RSC; (c) G.-N. Li, Y. Liao, T. Jin and Y.-Z. Li, Inorg. Chem. Commun., 2013, 35, 27 CrossRef.
- (a) T. Murata, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui, M. Maesato, H. Yamochi, G. Saito and K. Nakasuji, Angew. Chem., Int. Ed., 2004, 43, 6343 CrossRef CAS PubMed; (b) J. Wu, N. Dupont, S.-X. Liu, A. Neels, A. Hauser and S. Decurtins, Chem. - Asian J., 2009, 4, 392 CrossRef CAS PubMed; (c) T. Murata, Y. Yamamoto, Y. Yakiyama, K. Nakasuji and Y. Morita, Bull. Chem. Soc. Jpn., 2013, 86, 927 CrossRef CAS.
- F. Riobé and N. Avarvari, Chem. Commun., 2009, 3753 RSC.
- (a) J. D. Crowley, P. H. Bandeen and L. R. Hanton, Polyhedron, 2010, 29, 70 CrossRef CAS; (b) P. M. Guha, H. Phan, J. S. Kinyon, W. S. Brotherton, K. Sreenath, J. T. Simmons, Z. Wang, R. J. Clark, N. S. Dalal, M. Shatruk and L. Zhu, Inorg. Chem., 2012, 51, 3465 CrossRef CAS PubMed; (c) L. Jiang, Z. Wang, S.-Q. Bai and T. S. A. Hor, CrystEngComm, 2013, 15, 10451 RSC; (d) J. P. Byrne, J. A. Kitchen, O. Kotova, V. Leigh, A. P. Bell, J. J. Boland, M. Albrecht and T. Gunnlaugsson, Dalton Trans., 2014, 43, 196 RSC.
- F. Gerson, A. Lamprecht and M. Fourmigué, J. Chem. Soc., Perkin Trans. 2, 1996, 1409 RSC.
- C. Goze, S.-X. Liu, C. Leiggener, L. Sanguinet, E. Levillain, A. Hauser and S. Decurtins, Tetrahedron, 2008, 64, 1345 CrossRef CAS.
- G. M. Sheldrick, Programs for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 982420–982423. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob00148f |
| This journal is © The Royal Society of Chemistry 2014 |