A comparative study of the SET-LRP of oligo(ethylene oxide) methyl ether acrylate in DMSO and in H2O
Nga H.
Nguyen
a,
Jakov
Kulis
ab,
Hao-Jan
Sun
a,
Zhongfan
Jia
ab,
Bart
van Beusekom
ac,
Martin E.
Levere
a,
Daniela A.
Wilson
ac,
Michael J.
Monteiro
ab and
Virgil
Percec
*a
aRoy & Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: percec@sas.upenn.edu
bAustralian Institute for Bioengineering and Nanotechnology, University of Queensland, Brisbane QLD 4072, Australia
cRadboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands
First published on 8th October 2012
Abstract
A comparative analysis of the SET-LRP of oligo(ethylene oxide) methyl ether acrylate (OEOMEA) in DMSO and in H2O at 25 °C is reported. Both the catalysis with activated Cu(0) wire/Me6-TREN and with mimics of “nascent” Cu(0) nanoparticles/Me6-TREN resulted in a higher rate of polymerization in water than in DMSO. This result is consistent with the acceleration expected for SET-LRP by a more polar reaction solvent, and with the difference between the equilibrium constants of disproportionation of CuBr in DMSO (Kd = 1.4–4.4) and in water (Kd = 106 to 107), both much higher in the presence of Me6-TREN. The inefficient access of the Cu(0) catalyst to the hydrophobic reactive centers of the monomer and initiator assembled in micellar structures explains the induction time observed in the SET-LRP of OEOMEA in water. This induction period is longer for Cu(0) wire. The use of “nascent” Cu(0) nanoparticles prepared by the disproportionation of CuBr in DMSO, in combination with 5 mol% CuBr2, led to an extremely efficient SET-LRP of OEOMEA in water. This SET-LRP in water is fast and follows first order kinetics to complete monomer conversion with linear dependence of experimental Mn on conversion, and narrow molecular weight distribution. Under the polymerization conditions investigated in both water and DMSO, no reduction in the absorbance of CuBr2/Me6-TREN was observed by online UV-vis spectroscopy. This excludes the formation of CuBr by reduction of CuBr2 by Cu(0) during the SET-LRP in DMSO and in water.
Introduction
Hydrophilic polymers are of considerable interest for a wide range of applications in the biomedical field.1,2 For example, poly(hydroxyethyl methacrylate) (poly(HEMA) and poly(hydroxyethyl acrylate) (poly(HEA) have pioneered the field of contact lenses and enzyme-immobilization.2 Thermal-responsive polymers such as poly(N-isopropylacrylamide) (poly(NIPAM) with a lower critical solution temperature (LCST) in aqueous media close to physiological temperature3 and polymers derived from poly(ethylene oxide) (PEO) with an LCST that can be engineered by varying their molecular weight, composition and topology are also extensively investigated for applications in biomedical research.1,2Living polymerization discovered by Szwarc in 1956![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 provides access to the construction of well-defined polymers with perfect and near-perfect chain-end functionality, complex macromolecular topology, and architecture.5–7 Metal-catalyzed living radical polymerization (LRP)5 has been utilized for the precise synthesis of polymers, including hydrophilic polymers in aqueous media. LRP of HEA8 and protected HEMA9 were first reported using CuX-catalyzed atom transfer living radical polymerization (ATRP) in bulk and in water using 2,2′-bipyridine (bpy) as a ligand in combination with an alkyl halide initiator at elevated temperature. It was later reported that unprotected HEMA could be successfully polymerized in methanol and water–methanol mixtures using CuX/bpy as a catalyst, and a water-soluble initiator derived from PEO.10 CuCl or CuBr/bpy-catalyzed ATRP of oligo(ethylene oxide) methyl ether methacrylate (OEOMA) performed in an aqueous environment was first described by the same research group.11,12 Later, it was shown that the pyridylmethanimine ligand (N-(n-propyl)-2-pyridylmethanimine) can be used to replace bpy in order to provide the successful polymerization of OEOMA in water at ambient temperature.13 Recently, well-defined poly(OEOMA) was prepared via Activator Generated by Electron Transfer (AGET ATRP)14 and Activator Regenerated by Electron Transfer (ARGET ATRP),15 both by slow feeding of ascorbic acid to reduce CuBr2/tris[(2-pyridyl)-methyl]amine (TMPA) to CuBr. In all cases, CuX/L was considered the active catalyst. However, CuX is known to be unstable in water, with quantitative disproportionation to Cu(0) and CuX2 (Kd = 0.89 × 106 to 5.8 × 107) even in the absence of a ligand that favors its disproportionation such as tris[2-(dimethylamino)ethyl]amine (Me6-TREN).16 With the exception of the CuBr/N-(n-propyl)-2-pyridylmethanimine complex that was demonstrated experimentally to be stable towards disproportionation in water at room temperature,13 the stability of the CuX complex with bpy or TMPA ligands in water was estimated based on the stability constants βI and βII of CuX and CuX2 species (βI = [CuIL]/[Cu1+][L] and βII = [CuIIL]/[Cu2+][L]).17 Therefore, whether or not CuX/L is the activator under the previously mentioned polymerization conditions remains uncertain.
4 provides access to the construction of well-defined polymers with perfect and near-perfect chain-end functionality, complex macromolecular topology, and architecture.5–7 Metal-catalyzed living radical polymerization (LRP)5 has been utilized for the precise synthesis of polymers, including hydrophilic polymers in aqueous media. LRP of HEA8 and protected HEMA9 were first reported using CuX-catalyzed atom transfer living radical polymerization (ATRP) in bulk and in water using 2,2′-bipyridine (bpy) as a ligand in combination with an alkyl halide initiator at elevated temperature. It was later reported that unprotected HEMA could be successfully polymerized in methanol and water–methanol mixtures using CuX/bpy as a catalyst, and a water-soluble initiator derived from PEO.10 CuCl or CuBr/bpy-catalyzed ATRP of oligo(ethylene oxide) methyl ether methacrylate (OEOMA) performed in an aqueous environment was first described by the same research group.11,12 Later, it was shown that the pyridylmethanimine ligand (N-(n-propyl)-2-pyridylmethanimine) can be used to replace bpy in order to provide the successful polymerization of OEOMA in water at ambient temperature.13 Recently, well-defined poly(OEOMA) was prepared via Activator Generated by Electron Transfer (AGET ATRP)14 and Activator Regenerated by Electron Transfer (ARGET ATRP),15 both by slow feeding of ascorbic acid to reduce CuBr2/tris[(2-pyridyl)-methyl]amine (TMPA) to CuBr. In all cases, CuX/L was considered the active catalyst. However, CuX is known to be unstable in water, with quantitative disproportionation to Cu(0) and CuX2 (Kd = 0.89 × 106 to 5.8 × 107) even in the absence of a ligand that favors its disproportionation such as tris[2-(dimethylamino)ethyl]amine (Me6-TREN).16 With the exception of the CuBr/N-(n-propyl)-2-pyridylmethanimine complex that was demonstrated experimentally to be stable towards disproportionation in water at room temperature,13 the stability of the CuX complex with bpy or TMPA ligands in water was estimated based on the stability constants βI and βII of CuX and CuX2 species (βI = [CuIL]/[Cu1+][L] and βII = [CuIIL]/[Cu2+][L]).17 Therefore, whether or not CuX/L is the activator under the previously mentioned polymerization conditions remains uncertain.
Cu(0)-catalyzed single-electron transfer living radical polymerization (SET-LRP) was first reported in 2002.18,19 The key features of SET-LRP include activation of the alkyl halide initiator or dormant polymer chain-end via heterogeneous SET from the electron-donor Cu(0), and the disproportionation of the in situ generated CuX/L to regenerate the Cu(0) activator and CuX2 deactivator.7,18,19 The latter allows for accumulation of the CuX2/L deactivator without the need for the bimolecular termination required to generate the persistent radical effect20 that is demanded by metal-catalyzed LRP such as ATRP.6 As a result, SET-LRP has been shown to produce polymers with perfect and near perfect retention of chain-end functionality even at complete monomer conversion.21 Thus, critical to the success of SET-LRP is the appropriate selection of a polar solvent and ligand that would stabilize CuX2 over CuX species. For example, while the disproportionation of CuX in the absence of a ligand is quantitative in H2O,16Kdis in DMSO is significantly lower (1.4–4.4).22 However, the extent of disproportionation is significantly enhanced in the presence of CuX2-stabilizing N-ligands such as Me6-TREN or tris(2-aminoethyl)amine (TREN). Kdis of the CuBr/Me6-TREN complex can be 1–3 orders of magnitude higher than that of CuBr alone in DMSO.7,23
The nature of the solvent in the Cu(0)-mediated SET-LRP plays an important role. Previous reports demonstrated that addition of a small amount of water (5–10%) in an organic solvent greatly accelerated the SET-LRP of methyl acrylate (MA) and at the same time improved the control of molecular weight distribution.24 Both the increase in rate and improved control have been attributed to the enhanced disproportionation of CuX in aqueous media as well as to the ability of the polar solvent mixtures to stabilize the polar transition state between the Cu(0) electron donor and the alkyl halide electron acceptor.7,24 The positive effect of water in other metal-catalyzed LRPs was also observed in several other laboratories.13,25 Unfortunately, due to the limited solubility of poly(methyl acrylate) (PMA) in aqueous media, a direct comparison of the kinetic behavior of the SET-LRP of MA in DMSO and in H2O was not possible.
In this report, the comparative SET-LRP of oligo(ethylene oxide) methyl ether acrylate (OEOMEA) in DMSO and in water was analyzed. OEOMEA was selected due to its solubility both in DMSO and in water, as well as in THF to facilitate the analysis of molecular weight and molecular weight distribution by gel permeation chromatography (GPC). Mechanistically, the analysis of the SET-LRP in water would provide further insights into the role of solvent polarity and the extent of disproportionation of CuX. Technologically, water is an environmentally benign solvent that provides a green solution to the synthesis of functional hydrophilic polymers by SET-LRP on both a laboratory and industrial scale.
Results and discussion
Cu(0) wire-catalyzed SET-LRP of OEOMEA in DMSO
Scheme 1 outlines the synthesis of poly(OEOMEA) by Cu(0) wire/Me6-TREN mediated polymerization of OEOMEA in DMSO. Two initiators commonly used for the homopolymerization of hydrophilic monomers in aqueous media were examined for this polymerization.12,13 The oligo(ethylene oxide)-derived macroinitiator (1) was synthesized by reacting a monomethoxy-capped oligo(ethylene oxide) with 2-bromoisobutyryl bromide according to Wang and Armes (Scheme 2a).12 1,2-Dihydroxypropane-3-oxy-(2-bromo-2-methylpropionyl) (2) was prepared in two steps by esterification of solketal alcohol with 2-bromoisobutyryl bromide, followed by removal of the acetonide protecting group (Scheme 2b).13 The polymerization was carried out under the following conditions: [OEOMEA]0/[initiator]0/[Me6-TREN]0 = 50/1/0.1, OEOMEA = 1 g, DMSO or H2O = 0.5 mL with hydrazine-activated Cu(0) wire as a catalyst (4.5 cm of 20 gauge wire, diameter = 0.0812 cm) (1.16 cm2 surface area (SA)). The results are summarized in Table 1.
| No. | I | Cu(0) length/SA (cm cm−2) | Solvent | CuBr2 (mol%) | k appp (min−1) | Time (min) | Conv. (%) | M (th) | M n (GPC) | M w/Mn |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 4.5 (1.16) | DMSO | 0 | 0.034 | 180 | 98 | 23910 |
20900 |
1.29 |
| 2 | 1 | 1.0 (0.27) | DMSO | 0 | 0.016 | 310 | 97 | 23780 |
16000 |
1.26 |
| 3 | 2 | 4.5 (1.16) | DMSO | 0 | 0.034 | 150 | 97 | 23520 |
17200 |
1.26 |
| 4 | 2 | 1.0 (0.27) | DMSO | 0 | 0.015 | 1280 | 99 | 23750 |
19800 |
1.30 |
| 5 | 1 | 4.5 (1.16) | H2O | 0 | 0.050 | 260 | 95 | 23260 |
19200 |
1.57 |
| 6 | 1 | 1.0 (0.27) | H2O | 10 | 0.050 | 300 | 97 | 23780 |
15100 |
1.28 |
| 7 | 1 | 1.0 (0.27) | H2O | 20 | 0.052 | 115 | 94 | 23060 |
15000 |
1.19 |
| 8 | 2 | 4.5 (1.16) | H2O | 0 | 0.039 | 150 | 97 | 23520 |
17200 |
1.26 |
| 9 | 2 | 1.0 (0.27) | H2O | 0 | 0.042 | 380 | 99 | 23980 |
18400 |
1.25 |
The kinetics of the SET-LRP of OEOMEA in DMSO catalyzed by Cu(0) wire/Me6-TREN and initiated with initiators 1 and 2 are shown in Fig. 1a and c. Both initiators provide a living polymerization with first order kinetics in the concentration of monomer and propagating radicals. It is important to note that a tertiary alkyl halide initiator is preferred over the secondary alkyl halide for the polymerization of OEOMEA, as poor control over the molecular weight distribution resulted from the initiation with a secondary bromide initiator.26 This is in agreement with a publication reporting that a tertiary alkyl bromide is the true electronic mimic for polyacrylates.26
![Conversion and ln([M]0/[M]) vs. time kinetic plots (a and c); and experimental Mn and Mw/Mnvs. theoretical Mth (b and d); in SET-LRP of OEOMEA in DMSO initiated with initiator 1 (a and b); and initiator 2 (c and d). Reaction conditions: OEOMEA = 1 g, DMSO = 0.5 mL, [OEOMEA]0/[initiator]0/[Me6-TREN]0 = 50/1/0.1, hydrazine activated Cu(0) wire of 4.5 cm of 20 gauge wire (diameter = 0.0812 cm, surface area = 1.16 cm2).](/image/article/2013/PY/c2py20782f/c2py20782f-f1.gif) | ||
| Fig. 1 Conversion and ln([M]0/[M]) vs. time kinetic plots (a and c); and experimental Mn and Mw/Mnvs. theoretical Mth (b and d); in SET-LRP of OEOMEA in DMSO initiated with initiator 1 (a and b); and initiator 2 (c and d). Reaction conditions: OEOMEA = 1 g, DMSO = 0.5 mL, [OEOMEA]0/[initiator]0/[Me6-TREN]0 = 50/1/0.1, hydrazine activated Cu(0) wire of 4.5 cm of 20 gauge wire (diameter = 0.0812 cm, surface area = 1.16 cm2). | ||
Fig. 1b and d show a linear dependence of the experimental molecular weight, Mn, with conversion and narrow molecular weight distribution (Mw/Mn) at all monomer conversions in the Cu(0) wire-catalyzed SET-LRP of OEOMEA in DMSO. When the length of the Cu(0) wire catalyst was decreased from 4.5 cm to 1 cm, corresponding to a reduction in surface area from 1.16 cm2 to 0.27 cm2, the kappp of the SET-LRP of OEOMEA decreased from 0.034 min−1 to 0.016 min−1 (Table 1, entries 2 and 4). This result is in agreement with a previous publication confirming a surface mediated activation of SET-LRP of OEOMEA during catalysis with activated Cu(0) wire.27
Cu(0) wire-catalyzed SET-LRP of OEOMEA in H2O at 25 °C
The kinetics of the SET-LRP of OEOMEA in H2O catalyzed by Cu(0) wire/Me6-TREN and initiated with initiators 1 and 3 are shown in Fig. 2. Two important observations must be noted. First, these polymerizations exhibited a very long induction time of 3 h. Second, a very small amount of gel-like precipitate (about 1%) formed during the induction period and throughout the polymerization. This precipitate could not be dissolved in any good solvent for poly(OEOMEA) such as THF, suggesting that cross-linking occurred during this polymerization in water. This behavior was not observed in the SET-LRP of OEOMEA in DMSO under identical conditions. However, despite the induction period and the formation of the precipitate, the polymerization of OEOMEA in water proceeded with linear kinetics to near complete monomer conversion. A linear evolution of experimental Mn with the theoretical values and relatively narrow molecular weight distribution are shown in Fig. 2 and Table 1, entries 5 and 8. Compared to the polymerizations in DMSO under identical conditions, the SET-LRP of OEOMEA in water was much faster. Specifically, the polymerization of OEOMEA initiated with initiator 1 catalyzed by activated Cu(0) wire (1 cm of 20 gauge wire) in water is 3 times faster than the polymerization in DMSO (Table 1, compare entries 2 and 5). The greater kappp in water than in DMSO is determined by the higher solvent polarity, as well as a higher disproportionation constant of CuBr in water than in DMSO.24![Conversion and ln([M]0/[M]) vs. time kinetic plots (a and c); and experimental Mn and Mw/Mnvs. theoretical Mth (b and d); in SET-LRP of OEOMEA in H2O initiated with initiator 1 (a and b); and initiator 2 (c and d). Reaction conditions: OEOMEA = 1 g, H2O = 0.5 mL, [OEOMEA]0/[initiator]0/[Me6-TREN]0 = 50/1/0.1, hydrazine-activated Cu(0) wire 4.5 cm of 20 gauge wire (diameter = 0.0812 cm, surface area = 1.16 cm2).](/image/article/2013/PY/c2py20782f/c2py20782f-f2.gif) | ||
| Fig. 2 Conversion and ln([M]0/[M]) vs. time kinetic plots (a and c); and experimental Mn and Mw/Mnvs. theoretical Mth (b and d); in SET-LRP of OEOMEA in H2O initiated with initiator 1 (a and b); and initiator 2 (c and d). Reaction conditions: OEOMEA = 1 g, H2O = 0.5 mL, [OEOMEA]0/[initiator]0/[Me6-TREN]0 = 50/1/0.1, hydrazine-activated Cu(0) wire 4.5 cm of 20 gauge wire (diameter = 0.0812 cm, surface area = 1.16 cm2). | ||
Armes laboratory previously observed the formation of insoluble precipitate within 2–3 min during the polymerization of HEMA in water, and suggested cross-linking reactions as a result of the extremely fast initiation by the very reactive catalyst in aqueous media.10 In an attempt to eliminate the formation of the gel-like precipitate, the Cu(0) wire length was decreased from 4.5 cm to 1 cm, corresponding to a decrease from 1.16 cm2 to 0.27 cm2 in surface area, in the polymerization of OEOMEA initiated with 1 and 2 (Table 1, entries 6 and 9). However, despite the reduction of the surface area of the Cu(0) catalyst, the formation of the precipitate during the initial induction period was still observed. Interestingly, the kappp of the SET-LRP of OEOMEA did not change by decreasing the surface area of the Cu(0) wire catalyst (Table 1, entries 6 and 9). This result is in contrast with the previous observation that a slower kappp was obtained by lowering the active surface area of the Cu(0) catalyst27,28 and suggests that under these reaction conditions the polymerization was saturated in the Cu(0) catalyst in water. To eliminate the cross-linking side-reaction, the CuBr2 deactivator was added during the Cu(0) wire-catalyzed SET-LRP of OEOMEA in water (Table 1, entry 7). It was observed that the addition of 20 mol% CuBr2 with respect to initiator concentration successfully suppressed the formation of the gel-like precipitate, while at the same time shortened the induction period from 3 h to 1.5 h. In addition, despite the introduction of a significant amount of CuBr2 (20 mol%), the rate of the polymerization (kappp = 0.052 min−1) is still comparable to the kappp value obtained from the polymerization performed at the same surface area with a lower CuBr2 concentration (kappp = 0.050 min−1) and that at higher surface area Cu(0) wire in the absence of CuBr2 (kappp = 0.050 min−1) (Table 1, entries 5–7). This result supports the saturation with the Cu(0) wire catalyst in the SET-LRP of OEOMEA in H2O.
A recent publication reported the extremely fast Cu(0) wire/Me6-TREN catalyzed SET-LRP of HEA in water with no induction period in the polymerization.29 This is in contrast to the long induction time under the polymerization conditions reported. In addition, the polymerization of OEOMEA in DMSO during catalysis with Cu(0) wire did not show any induction time. Thus, the behavior of each individual component, OEOMEA monomer and initiators in water, was investigated. All molecules were dispersed in water by directly dissolving the molecules in water at various concentrations, or by injection of 50 μL of THF solutions of the monomer and initiators into ultrapure water (1 mL, final concentration = 5, 10 or 20 mg mL−1) followed by 5 s of vortex mixing.30 The resulting dispersions were analyzed by Dynamic Light Scattering (DLS) and cryogenic Transmission Electron Microscopy (cryo-TEM). The DLS results are summarized in Table 2.
| Compound | Concentration (mg mL−1) | Direct dissolution | Injection method | ||
|---|---|---|---|---|---|
| Z-Average (nm) | PDI | Z-Average (nm) | PDI | ||
| OEOMEA | 5 | 156.0 | 0.265 | 75.96 | 0.199 |
| OEOMEA | 10 | 169.1 | 0.274 | 105.8 | 0.183 |
| OEOMEA | 20 | 189.5 | 0.207 | 77.8 | 0.234 |
| 1 | 5 | 221.1 | 0.232 | 126.1 | 0.099 |
| 1 | 10 | 226.1 | 0.211 | 140.7 | 0.080 |
| 1 | 20 | 220.3 | 0.200 | 157.2 | 0.103 |
| 2 | 5 | 200.2 | 0.433 | 99.3 | 0.116 |
| 2 | 10 | 233.8 | 0.345 | 114.8 | 0.126 |
| 2 | 20 | 291.3 (not stable) | 0.389 | 125.5 | 0.081 |
In all cases, DLS indicated the self-assembly of the amphiphilic OEOMEA and initiators in water, into micellar aggregate structures with narrower PDI when the self-assembly was generated by the injection method of their THF solutions in water. The self-assembly of OEOMEA and initiators, 1 and 2, into micellar aggregates in water is an unexpected result and, to the best of our knowledge, has not been reported previously in the literature. Both OEOMEA with 7/8 EO repeating units and the two initiators investigated in this study have always been regarded as water-soluble molecules.10,12–15 However, the amphiphilic property of the PEO-derived macromonomers and their corresponding polymers are not without precedent. Methoxy-capped poly(ethylene glycol) methacrylate or styrene with 25–45 EO repeating units self-assembled in micellar structures, in which it was proposed that the PEO chains fully extend to allow a compact arrangement of the hydrophobic polymerization groups.31a Likewise, poly(OEOMA) (OEOMEA Mn = 475 and 1100) self-assembles into micellar structures, as demonstrated by DLS and TEM studies.32 It was reported that the tendency for micellar structure formation depends on the length of the PEO side chains.32
Fig. 3 illustrates the cryo-TEM images of the spherical self-assembled structures derived from OEOMEA and initiators 1 and 2 at 20 mg mL−1. Fig. 3 suggests that the more hydrophobic core, conjugated alkene in OEOMEA or tertiary alkyl bromide, is shielded by the hydrophilic tail, oligo(ethylene oxide) chains in OEOMEA and initiator 1, or the 1,3-dihydroxy moiety in initiator 2. This raises the question of whether the cause of the long induction period in the Cu(0) wire-catalyzed SET-LRP of OEOMEA in water is due to the inability of the Cu(0) wire catalyst to physically access the hydrophobic reactive sites (acryloyl or bromine chain-end) from the monomer, initiator or the dormant polymer chains. It was previously observed that the conventional free radical polymerization of PEO-macromonomers in water was significantly accelerated compared to their polymerization in benzene as a solvent. This was attributed to the ability of the amphiphilic monomers to form micelles with the polymerizable units in the core and the hydrophilic chains in the shell. This aggregation locally concentrates the hydrophobic polymerizing groups, allowing a rapid polymerization when conventional radical initiators are used.31
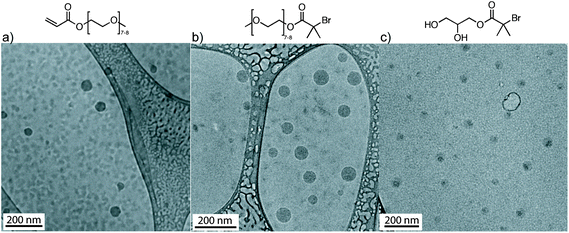 | ||
| Fig. 3 Cryo-TEM of OEOMEA, and initiators 1 and 2 in H2O at 20 mg mL−1. The dispersions were prepared by the injection of a THF solution of OEOMEA and of initiators 1 and 2 in water. | ||
SET-LRP of OEOMEA in H2O catalyzed by Cu(0) powder prepared by disproportionation of CuBr/Me6-TREN
To address the physical barrier between the Cu(0) catalyst with the reactive center of the monomer and initiators, the extremely reactive Cu(0) powder prepared from the disproportionation of CuBr/Me6-TREN33 was investigated as the catalyst for SET-LRP of OEOMEA in water.Fig. 4 shows the transmission electron micrographs of the Cu(0) particles prepared by the disproportionation of CuBr/Me6-TREN in DMSO before (Fig. 4a and b) and after filtration of the disproportionation mixture with a 0.45 μm nylon filter (Fig. 4c and d). These micrographs show the presence of very small Cu(0) particles (<20 nm) formed during the disproportionation of CuBr/Me6-TREN in DMSO, and their more dense agglomerates isolated on the filter after filtration. The larger Cu(0) nanoparticles were isolated after filtration, which is due to nucleation and growth and/or agglomeration and loss of the much smaller Cu(0) colloidal particles during the filtration process with a 0.45 μm filter. Fig. 4c and d show that the particles have rough surfaces with a relatively uniform size distribution of about 500 nm. Different batches of Cu(0) particles prepared using the same procedure show similar sizes and surface roughness.
![TEM micrographs of “nascent” Cu(0) particles formed during the disproportionation of CuBr/Me6-TREN in DMSO (a and b) and obtained by filtration with a 0.45 μm nylon filter (c and d). [CuBr] = 50 mM, [Me6-TREN] = 25 mM, DMSO = 10 mL.](/image/article/2013/PY/c2py20782f/c2py20782f-f4.gif) | ||
| Fig. 4 TEM micrographs of “nascent” Cu(0) particles formed during the disproportionation of CuBr/Me6-TREN in DMSO (a and b) and obtained by filtration with a 0.45 μm nylon filter (c and d). [CuBr] = 50 mM, [Me6-TREN] = 25 mM, DMSO = 10 mL. | ||
SET-LRP of OEOMEA was then performed in water using Cu(0) particles prepared from disproportionation of CuBr/Me6-TREN in H2O at 25 °C under the following conditions: [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0 = 50/1/0.1/0.1, OEOMEA = 1 g, H2O = 0.5 mL. Gel formation was observed within 5 min of addition of 10 mol% Cu(0) particles (with respect to initiator concentration). It should be noted that unlike the Cu(0) wire-catalyzed polymerization in water, in which only a small fraction of gel was formed, in the Cu(0) nanoparticles-catalyzed SET-LRP in water the entire reaction mixture was converted into a gel-like material. The isolated gel was not soluble in any solvent, and therefore the monomer conversion could not be determined. Its formation suggests the extremely rapid initiation catalyzed by Cu(0) particles formed from disproportionation of CuBr that resulted in rapid chain-transfer to the OE side chain. A decrease in the Cu(0) concentration to 2.5 mol% with respect to initiator concentration still resulted in gel formation, with the extremely high molecular weight polymer and broad molecular weight distribution. Nevertheless, no induction period was observed during the use of the “nascent” Cu(0) powder catalyst, suggesting a more difficult accessibility problem by the Cu(0) wire catalyst to the reactive center of OEOMEA and the initiators in the Cu(0) wire-catalyzed SET-LRP in water.
To effectively suppress the gel formation, mediated by the chain transfer process, 5 mol% CuBr2 was introduced at the beginning of the polymerization. Fig. 5 shows the kinetics of the SET-LRP of OEOMEA initiated with initiator 1 during catalysis with “nascent” Cu(0) particles in water in the presence of 5 mol% externally added CuBr2 at 25 °C. Despite the small negligible induction time (about 3–5 min) required to access the inner parts of the micellar aggregates by the nanopowder catalyst, the polymerization is extremely fast, reaching complete monomer conversion within 20 min with no gel formation. The polymerization follows first order kinetics, linear dependence of theoretical molecular weight with conversion and narrow molecular weight distribution (Mw/Mn = 1.3 at all conversions) (Fig. 5a and b). Fig. 6a shows the gel permeation chromatograms (GPC) of poly(OEOMEA) prepared by SET-LRP in H2O, initiated with 1 and catalyzed with the mimics of nascent Cu(0) colloidal particles, at different conversions. The polymer molecular weight distribution decreases from 1.3 at 61% conversion to 1.31 at 93% conversion, and the chromatograms remain symmetrical. At near complete monomer conversion, a slight shoulder at the higher molecular weight region was observed, indicative of a very small extent of bimolecular termination. Similarly, the SET-LRP of OEOMEA initiated with initiator 2 exhibits the typical behavior of a living polymerization, achieving complete monomer conversion within 25 min. At 100% monomer conversion, the final polymer has a Mw/Mn of 1.25 (Fig. 5c and d, 6b).
![Conversion and ln([M]0/[M]) vs. time kinetic plots (a and c); and experimental Mn and Mw/Mnvs. theoretical Mth (b and d); in SET-LRP of OEOMEA in H2O initiated with initiator 1 (a and b); and initiator 2 (c and d). Reaction conditions: OEOMEA = 1 g, H2O = 0.5 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, “nascent” Cu(0) particle prepared by the disproportionation of CuBr/Me6-TREN in DMSO. Data presented in symbols with different colors are from duplicated experiments.](/image/article/2013/PY/c2py20782f/c2py20782f-f5.gif) | ||
| Fig. 5 Conversion and ln([M]0/[M]) vs. time kinetic plots (a and c); and experimental Mn and Mw/Mnvs. theoretical Mth (b and d); in SET-LRP of OEOMEA in H2O initiated with initiator 1 (a and b); and initiator 2 (c and d). Reaction conditions: OEOMEA = 1 g, H2O = 0.5 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, “nascent” Cu(0) particle prepared by the disproportionation of CuBr/Me6-TREN in DMSO. Data presented in symbols with different colors are from duplicated experiments. | ||
![Gel permeation chromatograms (GPC) for SET-LRP of OEOMEA in H2O initiated with initiator 1 (a); and initiator 2 (b). Reaction conditions: OEOMEA = 1 g, H2O = 0.5 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, “nascent” Cu(0) particle prepared by the disproportionation of CuBr/Me6-TREN in DMSO.](/image/article/2013/PY/c2py20782f/c2py20782f-f6.gif) | ||
| Fig. 6 Gel permeation chromatograms (GPC) for SET-LRP of OEOMEA in H2O initiated with initiator 1 (a); and initiator 2 (b). Reaction conditions: OEOMEA = 1 g, H2O = 0.5 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, “nascent” Cu(0) particle prepared by the disproportionation of CuBr/Me6-TREN in DMSO. | ||
Dependence of the rate of SET-LRP of OEOMEA on the concentration of water
Fig. 7 demonstrates the relationship between the kappp of the SET-LRP of OEOMEA and the amount of H2O used in the polymerization. In these sets of experiments, the amount of monomer, initiator (1 or 2) and ligand was kept constant while the amount of H2O was changed. As the amount of water was increased from 0.1 to 0.75 mL, the apparent rate constant of the polymerization increased from 0.147 min−1 to 0.35 min−1 for initiator 1, and from 0.139 min−1 to 0.30 min−1 for initiator 2. Upon further addition of water, from 0.75 to 1.1 mL, the kappp of the SET-LRP of OEOMEA in water reaches a plateau at 0.35 min−1 for initiator 1, and at 0.30 min−1 for initiator 2 (Fig. 7 and Table 3). The rate enhancement at higher H2O content demonstrates the catalytic effect of H2O and is in agreement with a previous study on the external rate order of DMSO on SET-LRP of MA.19 At much higher water content, the kappp decreased, as a result of the opposing effect between the enhanced polarity of the aqueous reaction media and the decreased monomer and catalyst concentrations (Fig. 7, Table 3, entry 9).![The dependence of kappp on the volume of H2O in SET-LRP of OEOMEA in H2O initiated with 1 and 2. Polymerization conditions: OEOMEA = 1 g, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, “nascent” Cu(0) particles prepared by the disproportionation of CuBr/Me6-TREN in DMSO were used as a catalyst.](/image/article/2013/PY/c2py20782f/c2py20782f-f7.gif) | ||
| Fig. 7 The dependence of kappp on the volume of H2O in SET-LRP of OEOMEA in H2O initiated with 1 and 2. Polymerization conditions: OEOMEA = 1 g, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, “nascent” Cu(0) particles prepared by the disproportionation of CuBr/Me6-TREN in DMSO were used as a catalyst. | ||
| No. | Solvent | Initiator | V H2O (mL) | k appp (min−1) | Time (min) | Conv. (%) | M (th) | M n (GPC) | M w/Mn |
|---|---|---|---|---|---|---|---|---|---|
| 1 | H2O | 1 | 0.1 | 0.147 | 23 | 91 | 22180 |
15200 |
1.25 |
| 2 | H2O | 1 | 0.2 | 0.215 | 17 | 95 | 23060 |
15900 |
1.26 |
| 3 | H2O | 1 | 0.3 | 0.257 | 16 | 96 | 23400 |
17600 |
1.25 |
| 4 | H2O | 1 | 0.4 | 0.280 | 19 | 98 | 23780 |
15200 |
1.24 |
| 5 | H2O | 1 | 0.5 | 0.346 | 22 | 100 | 24250 |
18000 |
1.31 |
| 6 | H2O | 1 | 0.75 | 0.345 | 13 | 97 | 23540 |
17000 |
1.36 |
| 7 | H2O | 1 | 0.9 | 0.344 | 23 | 100 | 24250 |
17600 |
1.49 |
| 8 | H2O | 1 | 1.1 | 0.335 | 16 | 98 | 23780 |
29700 |
1.51 |
| 9 | H2O | 1 | 2 | 0.283 | 20 | 99 | 24010 |
17400 |
1.50 |
| 10 | DMSO | 1 | 0.75 | 0.067 | 40 | 95 | 23310 |
19800 |
1.26 |
| 11 | H2O | 2 | 0.1 | 0.139 | 30 | 96 | 23600 |
20100 |
1.26 |
| 12 | H2O | 2 | 0.1 | 0.139 | 30 | 96 | 23600 |
20000 |
1.26 |
| 13 | H2O | 2 | 0.3 | 0.187 | 27 | 99 | 23040 |
17000 |
1.28 |
| 14 | H2O | 2 | 0.4 | 0.235 | 20 | 99 | 23750 |
21700 |
1.25 |
| 15 | H2O | 2 | 0.5 | 0.296 | 20 | 100 | 24250 |
20300 |
1.29 |
| 16 | H2O | 2 | 0.75 | 0.297 | 25 | 100 | 24250 |
21600 |
1.35 |
| 17 | H2O | 2 | 0.9 | 0.304 | 19 | 99 | 23750 |
23300 |
1.63 |
| 18 | DMSO | 2 | 0.75 | 0.063 | 28 | 83 | 19950 |
14300 |
1.22 |
Table 3 shows that the Mw/Mn values of poly(OEOMEA) prepared by SET-LRP in water increased from 1.25 to 1.5–1.6 as the amount of water increased (entries 1–9 and 11–17). One explanation could be the dissociation of the Br anion from the CuBr2/Me6-TREN deactivator complex in a highly aqueous medium.14,15,34 This would result in less efficient deactivation, and therefore, less effective control of the molecular weight distribution. In a polymerization with a lower water content, it may suggest that relatively hydrophobic particles formed by the OEOMEA monomer or initiator will reduce the extent of ligand substitution of the CuBr2/Me6-TREN complex by the water molecule, leading to good control over the polymer molecular weight and distribution.
Comparative analysis of the kinetics of SET-LRP of OEOMEA in DMSO and in H2O
Fig. 8 compares the kinetics of the polymerization performed in DMSO and in water. As expected, the SET-LRP of OEOMEA proceeded much faster in water under similar polymerization conditions. This is in agreement with previous results from this and other laboratories. The kappp values in the SET-LRP of OEOMEA initiated with initiators 1 and 2 are almost 5 times higher than those obtained from the polymerization in DMSO (Table 3, entries 6, 10, 16 and 18). This can be attributed to the higher polarity of water that accelerate reactions with polar transition states, and the much higher disproportionation constant of CuBr in water.7,24![ln([M]0/[M]) vs. time kinetic plots (a and c); and experimental Mn and Mw/Mnvs. theoretical Mth (b and d); in SET-LRP of OEOMEA in H2O (red) and in DMSO (green) initiated with initiator 1 (a and b); and initiator 2 (c and d). Reaction conditions: OEOMEA = 1 g, H2O or DMSO = 0.75 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, the “nascent” Cu(0) particle prepared from the disproportionation of CuBr/Me6-TREN in DMSO. Data in red symbols were obtained in water while the data in green symbols were obtained in DMSO.](/image/article/2013/PY/c2py20782f/c2py20782f-f8.gif) | ||
| Fig. 8 ln([M]0/[M]) vs. time kinetic plots (a and c); and experimental Mn and Mw/Mnvs. theoretical Mth (b and d); in SET-LRP of OEOMEA in H2O (red) and in DMSO (green) initiated with initiator 1 (a and b); and initiator 2 (c and d). Reaction conditions: OEOMEA = 1 g, H2O or DMSO = 0.75 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, the “nascent” Cu(0) particle prepared from the disproportionation of CuBr/Me6-TREN in DMSO. Data in red symbols were obtained in water while the data in green symbols were obtained in DMSO. | ||
The GPC chromatograms of poly(OEOMEA) obtained at 97% conversion from the SET-LRP of OEOMEA in water, initiated with 1 and 2 and catalyzed with Cu(0) particles generated by disproportionation are shown in Fig. 9. In both cases, the chromatograms are symmetrical, and narrow molecular weight distribution (Mw/Mn = 1.36) was demonstrated.
![Gel permeation chromatograms (GPC) for SET-LRP of OEOMEA in H2O initiated with initiator 1 (a); and initiator 2 (b). Reaction conditions: OEOMEA = 1 g, H2O = 0.75 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, “nascent” Cu(0) particle prepared by the disproportionation of CuBr/Me6-TREN in DMSO.](/image/article/2013/PY/c2py20782f/c2py20782f-f9.gif) | ||
| Fig. 9 Gel permeation chromatograms (GPC) for SET-LRP of OEOMEA in H2O initiated with initiator 1 (a); and initiator 2 (b). Reaction conditions: OEOMEA = 1 g, H2O = 0.75 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, “nascent” Cu(0) particle prepared by the disproportionation of CuBr/Me6-TREN in DMSO. | ||
UV-vis monitoring of the SET-LRP of OEOMEA in H2O and in DMSO
It has been suggested that the active catalyst in the Cu(0)-mediated polymerization is CuX formed from reduction of CuX2 by Cu(0), and that the polymerization follows an ARGET or SARA-ATRP mechanism.35 This was concluded from the observation that CuX2 was reduced using an excess of Cu(0) powder or wire and Me6-TREN in MeCN, DMF and DMSO in the absence of a polymerization process.36 By contrast, when the Cu(0)-catalyzed SET-LRP of MA in a mixture of methanol and toluene was monitored by a photodiode array, CuX2 absorbance was shown to increase with conversion in the presence of a polymerization process.37 This result suggested that there is no reduction of CuX2 by Cu(0) in the presence of activation during the polymerization, thereby eliminating the propensity of CuX formed via reduction of CuX2 as the active catalyst. In a separate manuscript,38 SET-LRP polymerizations were performed in an UV-vis cell and the absorbance of CuX2 produced was monitored as a function of conversion during the polymerization. This methodology aimed to separate Cu(0)-catalyzed SET-LRP, in which no reduction of CuX2 should be observed, from the CuX-mediated polymerization.The SET-LRP of OEOMEA catalyzed with “nascent” Cu(0)/Me6-TREN initiated with initiator 1 in water in the presence of externally added CuBr2 was performed in a cuvette cell and the absorbance of CuBr2 was monitored by UV-vis spectroscopy as a function of time (Fig. 10a). The overlaid kinetics of the polymerization, plotted in different colors, was shown in filled symbols. The absorbance 865 nm, corresponding to CuBr2/Me6-TREN in H2O from a control experiment, relative to the baseline value at 548 nm was shown as green empty symbols. Fig. 10a shows that the absorbance at 865 nm increased continuously, relative to the baseline value (548 nm) with time, indicating that there was no measurable reduction of CuBr2 to CuBr by Cu(0) during the polymerization. The absorbance vs. time data from four reactions overlay showed that the kinetics of the formation of CuBr2 is experimentally reproducible.
![Evolution of conversion and UV-absorbance for SET-LRP of PEGMEA in H2O (a and b); and in DMSO (b and d); initiated with 1 (a and b); and 2 (c and d). Reaction conditions: OEOMEA = 1 g, H2O or DMSO = 0.75 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, the “nascent” Cu(0) particle prepared from the disproportionation of CuBr/Me6-TREN in DMSO was used as a catalyst. Data presented in symbols with different colors and in symbols with different shapes are from duplicated or triplicated experiments.](/image/article/2013/PY/c2py20782f/c2py20782f-f10.gif) | ||
| Fig. 10 Evolution of conversion and UV-absorbance for SET-LRP of PEGMEA in H2O (a and b); and in DMSO (b and d); initiated with 1 (a and b); and 2 (c and d). Reaction conditions: OEOMEA = 1 g, H2O or DMSO = 0.75 mL, [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05, the “nascent” Cu(0) particle prepared from the disproportionation of CuBr/Me6-TREN in DMSO was used as a catalyst. Data presented in symbols with different colors and in symbols with different shapes are from duplicated or triplicated experiments. | ||
The same trends were observed for the SET-LRP of OEOMEA initiated with initiator 1 in the presence of 5 mol% of CuBr2 in DMSO (Fig. 10b) and the polymerizations initiated with initiator 2 in H2O and in DMSO under otherwise similar conditions (Fig. 10c and d). In all cases, no decrease in the absorbance of CuBr2 was observed at any stage of the polymerization. These experimental observations support a reaction mechanism that produces CuBr2via disproportionation rather than by the persistent radical effect generated by bimolecular termination.21,22
Conclusions
This report demonstrates the synthesis of poly(OEOMEA) via SET-LRP initiated with a PEO-derived macroinitiator (1) and 1,2-dihydroxypropane-3-oxy-(2-bromo-2-methylpropionyl) initiator (2) in the polar solvents DMSO and H2O at 25 °C. When hydrazine-activated Cu(0) wire/Me6-TREN was used as the catalyst, the SET-LRP in DMSO provided a living polymerization, producing polymers with narrow molecular weight distribution. When the polymerization was performed in water under otherwise identical conditions, a long induction time was observed with formation of a gel precipitate. The latter was suppressed with the introduction of 20 mol% CuBr2 with respect to the initiator concentration. The presence of the induction time in the Cu(0) wire-catalyzed SET-LRP of OEOMEA in water was attributed to the physical barrier between the active Cu(0) wire catalyst with the hydrophobic reactive center of the OEOMEA monomer and initiators that are self-assembled in water. DLS and cryo-TEM studies demonstrated the formation of the self-assembled structures of the monomer and initiators in water, in which the hydrophobic moieties of the monomers and initiators are shielded and therefore prevent access of the Cu(0) wire catalyst. This hypothesis was confirmed by the successful polymerization of OEOMEA in water using “nascent” Cu(0) nanoparticles prepared from the disproportionation of CuBr/Me6-TREN in DMSO, which showed a negligible induction period and the absence of gel formation. The use of 5 mol% of CuBr2 with respect to the initiator concentration was necessary to suppress cross-linking reaction that otherwise occurred at the start of the polymerization. The SET-LRP of OEOMEA performed in water using “nascent” Cu(0) particles in the presence of 5 mol% CuBr2 is extremely fast, reaching complete monomer conversion within 25 min, and provides polymers with relatively narrow molecular weight distribution. Under the same reaction conditions, the SET-LRP of OEOMEA in water is much faster than that in DMSO, which is consistent with the increase in reaction polarity and the enhanced disproportionation of CuBr in water. When a UV-vis spectrophotometer was utilized to monitor the SET-LRP of OEOMEA performed in water and in DMSO using “nascent” Cu(0) particles in the presence of 5 mol% CuBr2, no reduction in the absorbance of CuBr2 by Cu(0) was observed at any stage of the polymerization and even after the polymerization reached complete conversion. This result excludes the possibility of CuBr formed from the reduction of CuBr2 by Cu(0) as the active catalyst during the polymerization of OEOMEA in water using Cu(0)/Me6-TREN as a catalyst.Experimental
Materials
Oligo(ethylene oxide) methyl ether acrylate (average Mn 480) (Aldrich) was passed through two short columns of basic Al2O3 prior to use in order to remove the radical inhibitor. Copper (0) wire (20 gauge wire (0.812 mm diameter), Fischer) was activated with hydrazine hydrate (100%, Acros) (hydrazine 64%) prior to use.39 Dimethyl sulfoxide (DMSO) (Fisher, Certified ACS, 99.9) was used as received. Hexamethylated tris(2-aminoethyl)amine (Me6-TREN) was synthesized as described in the literature.40Techniques
500 MHz 1H NMR spectra were recorded on a Bruker DRX500 NMR instrument at 23 °C in D2O or CDCl3 with tetramethylsilane (Me4Si) as an internal standard. Gel Permeation Chromatography (GPC) analyses of the polymer samples were done on a Perkin-Elmer Series 10 high-performance liquid chromatograph, equipped with an LC-100 column oven (30 °C), a Nelson Analytical 900 Series integration data station, a Perkin-Elmer 785 UV-vis detector (254 nm), a Varian star 4090 refractive index (RI) detector, and three AM gel columns (500 Å, 5 μm; 1000 Å, 5 μm; and 104 Å, 5 μm). THF (Fisher, HPLC grade) was used as an eluent at a flow rate of 1 mL min−1. The number-average (Mn) and weight-average (Mw) molecular weights of poly(OEOMEA) samples were determined with poly(methyl methacrylate) (PMMA) standards purchased from American Polymer Standards. UV-vis spectra were recorded on a Shimadzu 1601 spectrometer with Shimadzu UV-Probe software. Polymerizations were performed in a rectangular glass Starna UV-vis cuvette cell (3.5 mL) with 1 cm path length and airtight screw cap fitting. A Teflon coated cylindrical stirring bar of 0.8 cm length and 0.4 cm diameter was used for the polymerization experiments performed in the cuvette cell. The glovebox was an Innovative Technology Inc. model operating under a nitrogen atmosphere, deoxygenated with a copper catalyst and with the moisture level ideally maintained below 25 ppm. Dynamic light scattering (DLS) measurements were performed with a Malvern Instruments particle sizer (Zetasizer® Nano S, Malvern Instruments, UK). Transmission electron microscopy (TEM) and cryogenic transmission electron microscopy (cryo-TEM) experiments were conducted with a JEOL 2010 TEM operating at an accelerating voltage of 200 kV. All images were recorded with a cooled Gatan 694 1K CCD camera. For cryo-TEM, the specimen temperature was maintained at −178 °C using a Gatan CT3500 cryo transfer holder.Synthesis of initiator 1 (ref. 12)
To a round bottom flask equipped with a dropping funnel were added polyethylene glycol monomethyl ether, 350 (5 g, 0.0143 mol), distilled triethylamine (1.67 g, 0.0165 mol), and distilled THF (10 mL). The mixture was cooled to 0 °C with an icebath. 2-Bromoisobutyryl bromide (3.65 g, 0.0162 mol) in THF (15 mL) (33% w/v) was added dropwise. Upon complete addition, the reaction solution was allowed to warm to 25 °C and stirred for another 4 h. The reaction mixture was filtered and the THF was removed on a rotary evaporator. The resulting yellow crude product was dissolved in water and extracted with diethyl ether. The organic layer was combined and dried over dry MgSO4. The product was obtained upon removal of the solvent followed by several washes with n-hexane (75%). δH (500 MHz, CDCl3, Me4Si) 4.32 (2H, dd, CH2), 3.80–3.47 (25H, m, CH2O), 3.38 (2H, s, OCH3), 1.95 (6H, s, CH3).Synthesis of 2,3-dihydroxypropyl 2-bromo-2-methylpropanoate (initiator 2)13
Initiator 2 was synthesized in two steps according to the literature.13 (2,2-Dimethyl-1,3-dioxolan-4-yl)methyl 2-bromo-2-methylpropanoate was prepared by the esterification of solketal with 2-bromoisobutyryl bromide. Solketal (5.315 g, 0.04 mol), distilled triethylamine (8.1 g, 0.08 mol), and distilled tetrahydrofuran (THF) (35 mL) were charged in a 100 mL round-bottom flask equipped with a magnetic stirrer and cooled to 0 °C with an icebath. 2-Bromo-2-methylpropionyl bromide (9.3 g, 0.0404 mol) in distilled THF (15 mL) was added dropwise to the reaction mixture under N2. The mixture was stirred for another 3.5 h while being brought to room temperature. THF was removed using the rotary evaporator and the residue was taken up in ether and filtered off. The organic layer was washed with 10% HCl followed by washes with brine and finally with saturated aqueous solution of NaHCO3. The crude product was isolated as a slightly yellowish oil (85%) and was used in the next step without further purification. δH (500 MHz, CDCl3, Me4Si) 4.35–4.28 (1H, m), 4.20 (2H, m), 4.06 (1H, dd), 3.80 (1H, dd), 1.91 (6H, s, CH3), 1.47–1.26 (6H, m, CH3).A mixture of 3 g of (2,2-dimethyl-1,3-dioxolan-4-yl)methyl 2-bromo-2-methylpropanoate (0.011 mol), 9 mL of glacial acetic acid, 24 mL of deionized water, and a catalytic amount of 4-methoxybenzene were stirred under N2 at 80 °C for 1 h. The solution was cooled to 25 °C prior to the addition of 30 mL of ethyl acetate. After extraction the aqueous layer was saturated with NaHCO3 by portion-wise addition of NaHCO3 powder. The aqueous layer was extracted three times with ethyl acetate. The crude product was obtained from the combined organic layers followed by removal of the solvent as a yellowish oil. The crude product was recrystallized from toluene (∼1 g in 25 mL) to yield a white crystal (77%). δH (500 MHz, CDCl3, Me4Si) 4.28 (2H, qd), 4.00 (1H, dd), 3.75 (1H, ddd), 3.66 (1H, dt), 2.51 (1H, d), 2.07 (1H, t), 1.96 (6H, s).
DLS measurements of the dispersion of the monomer and initiators in water
The standard procedure for the dispersion of monomer and initiator molecules in water is as follows: injection of 50 μL of THF solution containing 20 mg of monomer or initiator molecules into 1 mL of ultrapure water followed by 5 s of vortex mixing. DLS experiments were carried out using a Zetasizer Nano S from Malvern instruments equipped with 4 mW He–Ne laser 633 nm and avalanche photodiode positioned at 175° to the beam and temperature controlled cuvette holder. Instrument parameters were determined automatically along with measurement times. Experiments were performed in triplicate.Sample preparation for cryo-TEM
Samples for cryo-TEM were prepared with a Gatan Cp3 cryo plunger operating at room temperature and 70–90% relative humidity. A copper TEM grid (300 mesh pre-coated with lacey carbon film, Ted Pella) was treated with oxygen and hydrogen plasma (Gatan 950 Solarus plasma cleaner) to create a hydrophilic surface, and then 2 μL aqueous solution was placed on the grid held by non-magnetic tweezers. Excess solution was blotted away with filter paper mounted on the plunger to obtain a thin film of solution spanning the grid. A vitrificated water film was created by plunging the grid into a liquid ethane reservoir (∼90 K) placed in a workstation filled with liquid nitrogen. The grids with the vitrificated film were stored in liquid nitrogen until imaging.Sample preparation for TEM
Samples for TEM were prepared by placing 10–20 μL solution on a 300 mesh grid (SPI) pre-coated with continuous Formar resin and carbon film. The film was made hydrophilic by treating with oxygen and hydrogen plasma (Gatan 950 Solarus plasma cleaner) before use. The excess solution was blotted away with filter paper after about 30 minutes. The grid was then dried and stored in an oven at 55 °C overnight before imaging.Preparation of “nascent” Cu(0) particles obtained by the disproportionation of CuBr/Me6-TREN in DMSO33
A solution of Me6-TREN (57.6 mg, 0.25 mol, 25 mM) in DMSO (10 mL) was degassed using six freeze–pump–thaw cycles, and brought to the glovebox. In a glovebox, CuBr powder (72 mg, 0.50 mmol) was loaded into a 20 mL vial, and mixed with 10 mL degassed solution of Me6TREN/DMSO (0.25 mol, 25 mM). The disproportionation reaction mixtures were stirred for 1 h under N2 atmosphere, and then filtered through a 0.45 μm nylon membrane. The collected Cu(0) nanoparticles were washed with deoxygenated acetonitrile (Ar-purged) (10 mL) to remove residual CuBr and deoxygenated methanol (10 mL) to remove residual CuBr2/Me6-TREN. After the acetonitrile wash, the filtrate was colorless. Finally, the Cu(0) nanoparticles were rinsed with deoxygenated acetone (2 × 10 mL), and the volatile components were removed under high vacuum. The resulting Cu(0) nanoparticles are fine, brick-red colored powder, and were stored under vacuum or in a nitrogen-filled glovebox for future use.Typical procedure for SET-LRP of OEOMEA in a Schlenk tube
In a 25 mL Schlenk tube, the reagents were added in the following order under gentle stirring: initiator 1 (21 mg, 0.042 mmol), monomer (OEOMEA, 1 g, 2.1 mmol), ligand (Me6-TREN, 1.1 μL, 4.1 μmol) and solvent (DMSO or H2O) (0.5 mL). The solvent, particularly H2O, was added last, after addition of the monomer to prevent the formation of the self-assembly of the initiator in H2O. When the polymerization was performed in the presence of externally added CuBr2, a stock solution of CuBr2 in DMSO or H2O was prepared. The mixture was purged with argon for 20 min, and deoxygenated using six freeze–pump–thaw cycles with liquid N2. After the last deoxygenation cycle, Cu(0) nanoparticles (0.27 mg, 0.42 mmol) weighed on a small filter paper (1 × 1 cm), or hydrazine-activated Cu(0) wire was loaded into the reaction vessel under positive argon pressure, defining t = 0. The reaction vessel was placed in a water bath thermostated at 25 °C with stirring. The side arm of the flask was purged with argon before it was opened for sampling at the predetermined times with an airtight syringe. Samples taken during the polymerization were dissolved in CDCl3 (when using DMSO as a solvent) or D2O (when using H2O as a solvent) for the analysis of conversion by 1H NMR. After removing the solvents, the samples were used for analysis of molecular weight and molecular weight distribution by GPC.Procedure for the SET-LRP of OEOMEA in the absence of CuBr2 in a UV cuvette: [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0 = 50/1/0.1/0.1; OEOMEA = 1 g; solvent = 0.75 mL
Two reactions were performed in parallel using the same batch of reaction mixture and stopped at different times to rapidly obtain a complete set of data. A small Teflon coated magnetic stirring bar (0.8 cm length and 0.4 cm diameter from Fisher), Cu(0) nanoparticle (0.27 mg) were charged to an airtight screw cap cuvette (3.5 mL from Starna Cells) and placed into a moisture and oxygen-free glovebox. A deoxygenated mixture of initiator 1 (21 mg, 0.042 mmol), monomer (OEOMEA, 1 g, 2.1 mmol), ligand (Me6-TREN, 1.1 μL, 4.1 μmol) and solvent (DMSO or H2O) (0.75 mL) was brought into the glovebox. An aliquot of 1.75 g of the reaction mixture was transferred into the UV-vis cuvette, defining t = 0. The cuvette was sealed, removed from the glovebox and placed in a thermostatted water bath operating at 25 °C with a magnetic stirring bar. UV-vis spectra were periodically recorded. The UV-cuvette was opened to oxygen to stop the reaction after an appropriate time, and the conversion and molecular weight of the polymer were determined. The procedure was repeated several times, with the reaction time varied, to obtain a complete set of data.Procedure for the SET-LRP of OEOMEA in the presence of CuBr2 in a UV cuvette: [OEOMEA]0/[initiator]0/[Cu(0)]0/[Me6-TREN]0/[CuBr2]0 = 50/1/0.1/0.15/0.05; OEOMEA = 1 g; solvent = 0.75 mL
The UV-vis monitoring of the polymerizations in the presence of CuBr2 followed similar procedures as described above. A deoxygenated mixture of initiator 1 (21 mg, 0.042 mmol), monomer (OEOMEA, 1 g, 2.1 mmol), ligand (Me6-TREN, 1.7 μL, 6.3 μmol) and solvent (DMSO or H2O) (0.75 mL) containing CuBr2 (0.47 mg, 2.1 μmol) was brought into the glovebox. For reactions in the presence of CuBr2, the initial absorbance measured at t = 0 had to be consistent and within 10% uncertainty, representing slight variations in batch-to-batch production of the reaction mixture.Acknowledgements
Financial support by the National Science Foundation (DMR-1120901 and DMR-1066116) and P. Roy Vagelos at Penn are gratefully acknowledged. J.K. acknowledges funding from The University of Queensland Graduate School International Travel Award (GSITA). D.A.W. and B.v B. acknowledge funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-20012)/ERC-StG 307679 “StomaMotors” and the Radboud Honours Academy Award of the Faculty of Science.Notes and references
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- H. Kakwere and S. Perrier, Polym. Chem., 2011, 2, 270–288 RSC.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- (a) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3746 CrossRef CAS; (b) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS.
- S. Coca, C. B. Jasieczek, K. L. Beers and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1417–1424 CrossRef CAS.
- K. L. Beers, S. Boo, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1999, 32, 5772–5776 CrossRef CAS.
- K. L. Robinson, M. A. Khan, M. V. de Paz Banez, X. S. Wang and S. P. Armes, Macromolecules, 2001, 34, 3155–3158 CrossRef CAS.
- X. S. Wang, S. F. Lascelles, R. A. Jackson and S. P. Armes, Chem. Commun., 1999, 1817–1818 RSC.
- X. S. Wang and S. P. Armes, Macromolecules, 2000, 33, 6640–6647 CrossRef CAS.
- (a) S. Perrier, S. P. Armes, X. S. Wang, F. Malet and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1696–1707 CrossRef CAS; (b) S. Perrier and D. M. Haddleton, Macromol. Symp., 2002, 182, 261–272 CrossRef CAS.
- J. K. Oh, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 3161–3167 CrossRef CAS.
- A. Simakova, S. E. Averick, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 6371–6379 CrossRef CAS.
- (a) S. Ahrland and J. Rawsthorne, Acta Chem. Scand., 1970, 24, 157–172 CrossRef CAS; (b) L. Ciavatta, D. Ferri and R. Palombari, J. Inorg. Nucl. Chem., 1980, 42, 593–598 CrossRef CAS.
- (a) N. V. Tsarevsky, W. A. Braunecker and K. Matyjaszewski, J. Organomet. Chem., 2007, 692, 3212–3222 CrossRef CAS; (b) N. V. Tsarevsky and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5098–5112 CrossRef CAS.
- (a) V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS; (b) V. Percec, A. V. Popov, E. Ramirez-Castillo and O. Weichold, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3283–3299 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS.
- (a) H. Fischer, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1885–1901 CrossRef CAS; (b) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS.
- (a) N. H. Nguyen, M. E. Levere, J. Kulis, M. J. Monteiro and V. Percec, Macromolecules, 2012, 45, 4606–4622 CrossRef CAS; (b) N. H. Nguyen, M. E. Levere and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 860–873 CrossRef CAS; (c) F. Nyström, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. R. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5313–5321 CrossRef; (d) A. H. Soeriyadi, C. Boyer, F. Nystrom, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS; (e) C. Boyer, A. Derveaux, P. B. Zetterlund and M. R. Whittaker, Polym. Chem., 2012, 3, 117–123 RSC.
- S. Ahrland, P. Blauenstein, B. Tagesson and D. Tuhtar, Acta Chem. Scand., Ser. A, 1980, 34a, 265–272 CrossRef.
- B. M. Rosen, X. Jiang, C. J. Wilson, N. H. Nguyen, M. J. Monteiro and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5606–5628 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, X. Jiang, S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5577–5590 CrossRef CAS.
- G. Coullerez, A. Carlmark, E. Malmstrom and M. Jonsson, J. Phys. Chem. A, 2004, 108, 7129–7131 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1235–1247 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
- G. Lligadas, B. M. Rosen, C. A. Bell, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8365–8371 CrossRef CAS.
- E. Nicol, T. Derouineau, F. Puaud and A. Zaitsev, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3885–3894 CrossRef CAS.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS.
- (a) K. Ito, K. Tanaka, H. Tanaka, G. Imai, S. Kawaguchi and S. Itsuno, Macromolecules, 1991, 24, 2348–2354 CrossRef CAS; (b) V. Percec, C. H. Ahn and B. Barboiu, J. Am. Chem. Soc., 1997, 119, 12978–12979 CrossRef CAS; (c) V. Percec, C. H. Ahn, W. D. Cho, A. M. Jamieson, J. Kim, T. Leman, M. Schmidt, M. Gerle, M. Moller, S. A. Prokhorova, S. S. Sheiko, S. Z. D. Cheng, A. Zhang, G. Ungar and D. J. P. Yeardley, J. Am. Chem. Soc., 1998, 120, 8619–8631 CrossRef CAS; (d) V. Percec, C. H. Ahn, G. Ungar, D. J. P. Yeardley, M. Moller and S. S. Sheiko, Nature, 1998, 391, 161–164 CrossRef CAS.
- H. Hussain, K. Y. Mya and C. He, Langmuir, 2008, 24, 13279–13286 CrossRef CAS.
- X. Jiang, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 403–409 CrossRef CAS.
- (a) N. Bortolamei, A. A. Isse, A. J. D. Magenau, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 11391–11394 CrossRef CAS; (b) H. Nagatani, H. Tanida, I. Watanabe and T. Sagara, Anal. Sci., 2009, 25, 475–480 CrossRef CAS.
- (a) K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS; (b) Y. Kwak, A. J. D. Magenau and K. Matyjaszewski, Macromolecules, 2011, 44, 811–819 CrossRef CAS.
- (a) K. Matyjaszewski, N. V. Tsarevsky, W. A. Braunecker, H. Dong, J. Huang, W. Jakubowski, Y. Kwak, R. Nicolay, W. Tang and J. A. Yoon, Macromolecules, 2007, 40, 7795–7806 CrossRef CAS; (b) T. Guliashvili, P. V. Mendonça, A. C. Serra, A. V. Popov and J. F. J. Coelho, Chem.–Eur. J., 2012, 18, 4607–4612 CrossRef CAS.
- M. E. Levere, I. Willoughby, S. O'Donohue, P. M. Wright, A. J. Grice, C. Fidge, C. Remzi Becer and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1753–1763 CrossRef CAS.
- (a) M. E. Levere, N. H. Nguyen and V. Percec, Macromolecules, 2012 DOI:10.1021/ma301547n; (b) M. E. Levere, N. H. Nguyen, H.-J. Sun and V. Percec, Polym. Chem., 2012 10.1039/c2py20791e.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |