Facile synthesis of hierarchical MoS2 microspheres composed of few-layered nanosheets and their lithium storage properties
Shujiang
Ding
ab,
Dongyang
Zhang
a,
Jun Song
Chen
bc and
Xiong Wen (David)
Lou
*bc
aDepartment of Applied Chemistry, School of Sciences, Xi'an Jiaotong University, Xi'an, P.R. China 710049. E-mail: dingsj@mail.xjtu.edu.cn
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, Singapore 637457. E-mail: xwlou@ntu.edu.sg
cEnergy Research Institute @ NTU, Nanyang Technological University, 50 Nanyang Drive, Singapore 637553
First published on 24th November 2011
Abstract
In this work, we demonstrate an interesting polystyrene microsphere-assisted synthesis of hierarchical MoS2 spheres composed of ultrathin nanosheets. The as-prepared sample exhibits promising lithium storage properties with improved cyclic capacity retention and rate capability.
As a typical two-dimensional (2D) nanostructure with a large lateral size and a small thickness, nanosheets (NSs) have attracted increasing research interest due to their high surface area, controlled exposed crystal facets,1–7 diverse compositions such as TiO2,1–12SnO29,13–17 and SnO18,19 and wide application potentials in many fields such as lithium-ion batteries (LIBs),1,8–11,13,14,20 gas sensing,21glucose sensing12 and catalysis.3,5,7,16
MoS2 has a similar layered structure with graphite, in which molybdenum atoms are sandwiched between two layers of sulfur atoms. MoS2 has been found attractive in many applications, such as lubricants,22catalysts,23 and transistors.24 This layered structure is also beneficial for the intercalation of guest ions, such as Li+ ions25–30 and Mg2+ ions.31,32 As a result, many nanostructures of MoS2 such as nanoflakes, nanotubes and nanoflowers have been reported as electrode materials for LIBs.29,33,34 Although these MoS2 nanostructures exhibit high capacities up to 1000 mA h g−1, their cycling stability cannot meet the requirement for practical use in LIBs. Some methods have been proposed to improve the cycling performance of MoS2 by integrating MoS2 with different carbonaceous materials, like amorphous carbon, graphene25,26 and carbon nanotubes (CNTs).22,35
Herein, we report a facile synthesis of uniform hierarchically structured MoS2 microspheres composed of NSs (designated as MoS2-NS microspheres thereafter), in which polystyrene (PS) microspheres are added to facilitate the assembly of MoS2 NSs to form uniform microspheres. After an annealing treatment in inert atmosphere, the PS microspheres are decomposed and uniform MoS2-NS microspheres are obtained. The high surface area resulting from the assembly of nanosheets should help store more lithium ions and the large void space between nanosheets will buffer the volume variation accompanying the charge–discharge process, thus leading to improved cycling stability. As expected, in comparison to MoS2 flakes, these MoS2-NS microspheres manifest improved lithium storage properties with better cycling performance and higher capacity.
Fig. 1 shows the morphology of the as-prepared products. From the scanning electron microscopy (SEM; Fig. 1A) image, it is very obvious that the product is composed of PS microspheres and MoS2-NS microspheres with sizes of ∼1.5 μm and ∼1 μm, respectively. After being treated in the atmosphere of H2/N2 mixture at 800 °C, PS microspheres are decomposed and the MoS2-NS microspheres are preserved. Fig. 1B shows the morphology of the MoS2-NS microspheres. It can be seen that these MoS2-NS microspheres are composed of sheet-like subunits and the size is slightly reduced to 700–900 nm. With a closer examination with transmission electron microscopy (TEM) (Fig. 1C), the nanosheets with a lateral size of around 500 nm aggregate to form relatively loose microspheres. The high resolution TEM (HRTEM) image (Fig. 1D) shows that the interlayer distance is about 0.62 nm, which is assigned to the (002) plane of MoS2. The nanosheets are composed of 3–5 layers of MoS2 molecular sheets.
 | ||
| Fig. 1 (A) Scanning electron microscopy image (SEM) of the as-synthesized material; (B) SEM and (C) transmission electron microscopy (TEM) images of the MoS2-NS microspheres; (D) a high-resolution TEM image of several MoS2 NSs; the inset shows a HRTEM image of a single MoS2 nanosheet. | ||
The crystallinity of the samples is measured by X-ray diffraction (XRD), and the XRD pattern of MoS2-NS microspheres is shown in Fig. 2A. It is clear that the diffraction peaks at 2θ = 14, 33 and 59° can be assigned to (002), (100) and (110) planes of MoS2 (JCPDS 37-1492) respectively. The surface structure of the MoS2-NS microspheres is further studied by the gas sorption measurement. From the N2 adsorption–desorption isotherm (Fig. 2B), a distinct hysteresis loop is identified at a relative pressure of 0.1–0.9, indicating the presence of a mesoporous structure, which leads to a Brunauer–Emmett–Teller (BET) surface area of 36 m2 g−1. The pore size distribution is calculated by the Barrett–Joyner–Halenda (BJH) method. The plots (inset of Fig. 2B) show that most pores are in the mesoporous range with a peak centered at 3–10 nm. These pores are formed from the stacking of MoS2 nanosheets.
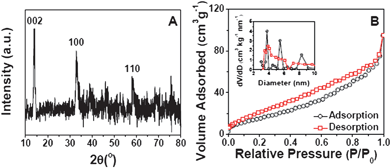 | ||
| Fig. 2 (A) XRD pattern of MoS2-NS microspheres; (B) N2 adsorption–desorption of MoS2-NS microspheres; the inset shows the pore size distributions calculated from both the adsorption and desorption branches. | ||
We have studied the effect of PS on the morphologies of the products. The morphology of the MoS2 sample prepared by the same hydrothermal route without adding PS is shown in Fig. 3. As can be seen, the MoS2 product obtained without adding PS is composed of micrometre-sized flakes, which are formed by dense stacking of MoS2 nanosheets. By comparing the morphologies of MoS2-NS microspheres and MoS2 flakes, it is evident that the PS microspheres have played a key role on the formation of MoS2-NS microspheres. The exact mechanism for the function of PS microspheres is unclear at this moment. It is hypothesized that in the presence of PS microspheres in the solution, nanosheets with smaller thickness and lateral size are formed easily initiated by the functional groups on the PS surface. At the same time, the PS microspheres may act as physical confinement to effectively prevent the further growth of MoS2 NSs into thick flakes and facilitate the assembly of MoS2 NSs into microspheres.

Next, we investigate the electrochemical properties of MoS2-NS microspheres as an anode material for LIBs. Fig. 4A shows the representative cyclic voltammograms (CVs) of MoS2-NS microspheres. As can be seen, the CV behavior is generally consistent with those reported previously.25–27,35 In the first cathodic sweep, the peak at 0.74 V is attributed to the intercalation of lithium ions into the MoS2 lattice which transforms the triangular prism (coordination of Mo by six S atoms) into an octahedral structure.25,36 This peak disappears in the second cathodic sweep because only nearly amorphous MoS2 is reformed after the charge process (lithium extraction) in the first cycle. The other peak at 0.1 V is attributed to the complete reduction process:25–27
| MoS2 + 4Li ↔ Mo + 2Li2S | (1) |
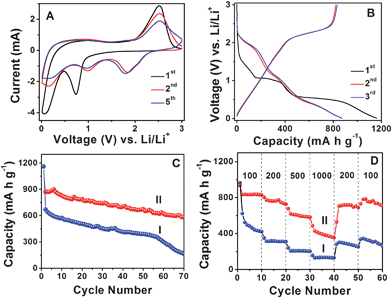 | ||
| Fig. 4 (A) Representative CVs at a scan rate of 0.5 mV s−1 for the first, second, and fifth cycles of MoS2-NS microspheres. (B) Charge–discharge voltage profiles at a current density of 100 mA g−1 of MoS2-NS microspheres. (C) Comparative cycling performance of MoS2 flakes (I) and MoS2-NS microspheres (II) at a current density of 100 mA g−1. (D) Cycling performance of MoS2 flakes (I) and MoS2-NS microspheres (II) at different current densities indicated (mA g−1). | ||
The initial discharge and charge capacities of MoS2-NS microspheres are found to be 1160 and 791 mA h g−1. Such a high initial lithium storage capacity might be associated with the unique hierarchical structure of the stacked MoS2 nanosheets. The irreversible capacity loss of around 31.8% may be mainly attributed to the irreversible processes, such as formation of solid–electrolyte interface (SEI) layer and trapping of some lithium in the lattice. The discharge and charge capacities in the second cycle are 873 and 822 mA h g−1, respectively, giving rise to a Coulombic efficiency of 94.2% and this value further increases to 95.4% in the third cycle. Fig. 4C shows the cycling performance at a current density of 100 mA g−1. After 50 discharge–charge cycles, the MoS2-NS microspheres can still deliver a capacity of 672 mA h g−1, while the MoS2 flakes only deliver 390 mA h g−1. After 70 cycles, the capacities of MoS2-NS microspheres and MoS2 flakes further decrease to 585 mA h g−1 and 163 mA h g−1, respectively. Furthermore, the MoS2-NS microspheres also exhibit better performance compared to the nanocomposite composed of several layered MoS2 nanosheets on CNTs backbone, whose capacity is about 350 mA h g−1 after 50 cycles.37Fig. 4D further displays the comparative cycling performance of MoS2-NS microspheres and MoS2 flakes at various current densities. At current densities of 200, 500 and 1000 mA g−1, the capacities of MoS2-NS microspheres are 726, 581 and 353 mA h g−1, respectively as compared to 311, 207 and 134 mA h g−1 only for pure MoS2 flakes. It is thus evident that the MoS2-NS microspheres exhibit much improved lithium storage performance compared to the pure MoS2 flakes. The enhanced lithium storage properties might be attributed to the unique hierarchical structure of MoS2-NS microspheres. Specifically, the large surface area offered by the ultrathin NSs endows the MoS2-NS microspheres with high specific capacity as a result of increased reactive sites and interface between the active material and electrolyte. Moreover, the presence of sufficient void space between the organized MoS2 nanosheets not only allows fast lithium ion diffusion but also effectively buffers the mechanical stress and volume variation accompanying the lithium inserting/de-inserting process. Furthermore, the flexible nature of such ultrathin MoS2 NSs enhances the robustness of the electrode structure.
In summary, we have prepared hierarchical spheres composed of ultrathin MoS2 nanosheetsvia a facile PS microsphere-assisted hydrothermal method. It is found that the presence of PS microspheres is critical for the formation of such interesting hierarchical spheres. In the absence of PS microspheres, only MoS2 flakes are formed. Both MoS2 hierarchical microspheres and flakes have been evaluated as anode materials for lithium-ion batteries. The results show that these unique MoS2-NS microspheres exhibit greatly enhanced lithium storage properties compared to the MoS2 flakes.
Experimental section
Materials preparation
In a typical synthesis of MoS2-NS microspheres, 50 mg of PS microspheres were dispersed into 40 mL of de-ionized water by ultrasonication for 5 minutes. 0.25 g sodium molybdate (Na2MoO4·2H2O, Sigma-Aldrich) was then added to the above solution. After stirring for 5 minutes, 0.5 g of thiourea was added. After stirring for another 5 minutes, the reaction solution was then transferred to a 60 mL Teflon-lined stainless steel autoclave and kept in an electric oven at 200 °C for 24 h. The autoclave was then left to cool down to room temperature in the oven. The black precipitate was collected by centrifugation, washed thoroughly with ethanol, and dried at 80 °C for 12 h. The as-prepared MoS2-NS microspheres were further treated at 800 °C in the atmosphere of 5% H2 balanced by N2 for 2 h with a heating rate of 1 °C min−1 to obtain the highly crystalline MoS2-NS microspheres. The preparation process of MoS2 flakes is similar to the one for MoS2-NS microspheres, except the addition of PS.Materials characterization
The product morphology was examined using a field-emission scanning electron microscope (FESEM; JEOL, JSM-6700F, 5 kV) and a transmission electron microscope (TEM; JEOL, JEM-2100F, 200 kV). Crystallographic information of the samples was collected using powder X-ray diffraction (XRD; Bruker, D8 Advance X-ray diffractometer, Cu Kα radiation, λ = 1.5406 Å). The nitrogen sorption measurement was performed using a Quantachrome Instrument (Autosorb AS-6B).Electrochemical measurements
The electrochemical measurements were carried out using two-electrode Swagelok-type cells (X2 Labwares, Singapore) with pure lithium metal as both the counter and the reference electrodes at room temperature. The working electrode consisted of the active material, a conductive agent (carbon black, Super-P-Li), and a polymer binder (poly(vinylidene difluoride), PVDF, Aldrich) in a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10 weight ratio. The electrolyte used was 1.0 M LiPF6 in a 50:50 (w/w) mixture of ethylene carbonate and diethyl carbonate. Cell assembly was carried out in an Ar-filled glovebox with concentrations of moisture and oxygen below 1.0 ppm. Cyclic voltammetry (0.005–3.0 V, 0.5 mV s−1) was performed using an electrochemical workstation (CHI 660C). The charge/discharge tests were performed using a NEWARE battery tester at different current densities with a voltage window of 0.01–3.0 V.
:20:10 weight ratio. The electrolyte used was 1.0 M LiPF6 in a 50:50 (w/w) mixture of ethylene carbonate and diethyl carbonate. Cell assembly was carried out in an Ar-filled glovebox with concentrations of moisture and oxygen below 1.0 ppm. Cyclic voltammetry (0.005–3.0 V, 0.5 mV s−1) was performed using an electrochemical workstation (CHI 660C). The charge/discharge tests were performed using a NEWARE battery tester at different current densities with a voltage window of 0.01–3.0 V.
Notes and references
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Y. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124–6130 CrossRef CAS.
- Y. Q. Dai, C. M. Cobley, J. Zeng, Y. M. Sun and Y. N. Xia, Nano Lett., 2009, 9, 2455–2459 CrossRef CAS.
- X. G. Han, Q. Kuang, M. S. Jin, Z. X. Xie and L. S. Zheng, J. Am. Chem. Soc., 2009, 131, 3152 CrossRef CAS.
- G. Liu, H. G. Yang, X. W. Wang, L. N. Cheng, J. Pan, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2009, 131, 12868–12869 CrossRef CAS.
- S. W. Liu, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS.
- D. Q. Zhang, G. S. Li, X. F. Yang and J. C. Yu, Chem. Commun., 2009, 4381–4383 RSC.
- J. S. Chen and X. W. Lou, Electrochem. Commun., 2009, 11, 2332–2335 CrossRef CAS.
- S. J. Ding, J. S. Chen and X. W. Lou, Adv. Funct. Mater., 2011, 21, 4120–4125 CrossRef CAS.
- S. J. Ding, J. S. Chen, D. Y. Luan, F. Y. C. Boey, S. Madhavi and X. W. Lou, Chem. Commun., 2011, 47, 5780–5782 RSC.
- S. J. Ding, J. S. Chen, Z. Y. Wang, Y. L. Cheah, S. Madhavi, X. Hu and X. W. Lou, J. Mater. Chem., 2011, 21, 1677–1680 RSC.
- P. Si, S. J. Ding, J. Yu, X. W. Lou and D. H. Kim, ACS Nano, 2011, 5, 7617–7626 CrossRef CAS.
- S. J. Ding and X. W. Lou, Nanoscale, 2011, 3, 3586–3588 RSC.
- S. J. Ding, D. Y. Luan, F. Y. C. Boey, J. S. Chen and X. W. Lou, Chem. Commun., 2011, 47, 7155–7157 RSC.
- C. Wang, Y. Zhou, M. Y. Ge, X. B. Xu, Z. L. Zhang and J. Z. Jiang, J. Am. Chem. Soc., 2010, 132, 46–47 CrossRef CAS.
- W. W. Wang, Y. J. Zhu and L. X. Yang, Adv. Funct. Mater., 2007, 17, 59–64 CrossRef CAS.
- X. M. Yin, C. C. Li, M. Zhang, Q. Y. Hao, S. Liu, L. B. Chen and T. H. Wang, J. Phys. Chem. C, 2010, 114, 8084–8088 CAS.
- B. Kumar, D. H. Lee, S. H. Kim, B. Yang, S. Maeng and S. W. Kim, J. Phys. Chem. C, 2010, 114, 11050–11055 CAS.
- K. Sakaushi, Y. Oaki, H. Uchiyama, E. Hosono, H. S. Zhou and H. Imai, Small, 2010, 6, 776–781 CrossRef CAS.
- J. H. Chae, K. C. Ng and G. Z. Chen, Proc. Inst. Mech. Eng., Part A, 2010, 224, 479–503 CrossRef.
- J. H. Lee, Sens. Actuators, B, 2009, 140, 319–336 CrossRef.
- X. F. Zhang, B. Luster, A. Church, C. Muratore, A. A. Voevodin, P. Kohli, S. Aouadi and S. Talapatra, ACS Appl. Mater. Interfaces, 2009, 1, 735–739 CAS.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS.
- K. Chang and W. X. Chen, Chem. Commun., 2011, 47, 4252–4254 RSC.
- K. Chang, W. X. Chen, L. Ma, H. Li, H. Li, F. H. Huang, Z. D. Xu, Q. B. Zhang and J. Y. Lee, J. Mater. Chem., 2011, 21, 6251–6257 RSC.
- G. D. Du, Z. P. Guo, S. Q. Wang, R. Zeng, Z. X. Chen and H. K. Liu, Chem. Commun., 2010, 46, 1106–1108 RSC.
- C. Q. Feng, J. Ma, H. Li, R. Zeng, Z. P. Guo and H. K. Liu, Mater. Res. Bull., 2009, 44, 1811–1815 CrossRef CAS.
- J. Xiao, D. W. Choi, L. Cosimbescu, P. Koech, J. Liu and J. P. Lemmon, Chem. Mater., 2010, 22, 4522–4524 CrossRef CAS.
- X. L. Li and Y. D. Li, J. Phys. Chem. B, 2004, 108, 13893–13900 CrossRef CAS.
- Y. L. Liang, R. J. Feng, S. Q. Yang, H. Ma, J. Liang and J. Chen, Adv. Mater., 2011, 23, 640–643 CrossRef CAS.
- H. Li, W. J. Li, L. Ma, W. X. Chen and J. M. Wang, J. Alloys Compd., 2009, 471, 442–447 CrossRef CAS.
- R. Dominko, D. Arcon, A. Mrzel, A. Zorko, P. Cevc, P. Venturini, M. Gaberscek, M. Remskar and D. Mihailovic, Adv. Mater., 2002, 14, 1531–1534 CrossRef CAS.
- S. J. Ding, J. S. Chen and X. W. Lou, Chem.–Eur. J., 2011, 17, 13142–13145 CrossRef CAS.
- Y. Miki, D. Nakazato, H. Ikuta, T. Uchida and M. Wakihara, J. Power Sources, 1995, 54, 508–510 CrossRef CAS.
- Q. Wang and J. H. Li, J. Phys. Chem. C, 2007, 111, 1675–1682 CAS.
| This journal is © The Royal Society of Chemistry 2012 |