Iron-catalysed reduction of carbonyls and olefins
Bryden A. F.
Le Bailly
and
Stephen P.
Thomas
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS. E-mail: stephen.thomas@bristol.ac.uk
First published on 31st October 2011
Abstract
The transition metal-catalysed reduction of olefins and carbonyl groups are amongst the most used reactions in synthetic chemistry. Recent developments using iron catalysts have shown that iron species are able to equal the activity of the traditionally used transition metals in these reductions. This review will focus on the iron-catalysed hydrosilylation, hydrogenation and transfer hydrogenation of carbonyl groups and the hydrogenation of olefins.
 Bryden A. F. Le Bailly | Bryden A. F. Le Bailly was born near Cambridge in 1987. He received his MSci in Chemistry from the University of Bristol in 2011, where he developed the iron-catalysed reductive cross-coupling reaction under the guidance of Dr Stephen P. Thomas. For this he was awarded the Lilly prize at Bristol. As of September 2011 he is undertaking a PhD in the group of Professor Jonathan Clayden at the University of Manchester. |
 Stephen P. Thomas | Stephen P. Thomas is a Lecturer in Organic and Biological Chemistry at the University of Bristol. He received his PhD from Churchill College, Cambridge University in 2007 working with Dr Stuart Warren. He then undertook postdoctoral work with Professor Andreas Pfaltz at the University of Basel, Switzerland. His research interests are focussed on organometallic catalysis and mechanism, and the use on non-precious metals to replace and expand upon the reactivity of traditionally used 2nd and 3rd row transition-metals. |
Introduction
Selective carbonyl and olefin hydrogenations are important transformations for the synthesis of both fine and bulk chemicals, finding application for a wide range of substrates and functionalities.1,2 Transition-metal-catalysed reactions currently provide the optimal methods for the reduction of carbonyls and olefins in high yield with both chemo- and stereoselectivity, with high atom efficiency, reducing waste and energy consumption.3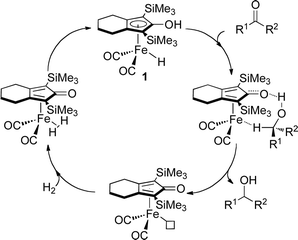 | ||
| Fig. 1 Proposed catalytic cycle (simplified) for the hydrogenation of ketones by 1.25 | ||
Recently there has been considerable interest in the replacement of established transition-metal catalysts with iron-based species within catalytic applications.4,5 Iron presents an intriguing alternative to the precious metals currently used as it is inexpensive, highly abundant, non-toxic and environmentally benign.6,7 Furthermore, residual metal traces allowed in active pharmaceutical ingredients are 100 times greater for iron than that of Pt, Ir, Pd, Ru or Rh.8 It is therefore somewhat surprising that until recently iron catalysts have not been used in hydrogenation reactions, particularly as iron-based systems have been used on vast-scale in the Harber–Bosch and Fischer–Tropsch processes.
Much attention has been given to tri- and tetradentate ligands bearing phosphorus and nitrogen donors such as porphyrin and diiminodiphosphine systems for iron-catalysed hydrogenations.9,10 These redox-active structures function to stabilise the iron centre11 as well as acting as a scaffold framework which can be modulated to give varying activity and selectivity.1,11 Practicalities including the ease of synthesis, tuneability and stability of such ligands are central to the efficacy of catalyst systems, and many successful approaches herein have focused on such issues to give previously unseen reactivity.
Carbonyls
Early examples of achiral hydrogenations of carbonyl substrates focused on the use of iron carbonyls but suffer from extreme conditions required to achieve sufficient reactivity.3 The reductive potential of iron was first observed when addition of stoichiometric Fe(CO)5 to a cobalt-catalysed hydroformylation process gave the hydrogenated alcohol products.12 The first catalytic approach was reported by Markó in 1983, using Fe(CO)5 at 150 °C and 100 bar H2 with triethylamine as both solvent and base to form the active catalyst [HFeCO4]−.13 It was found that activity increased with stronger basicity, a trend throughout iron carbonyl approaches.13,14,15In quick succession, Vancheesen reported the use of a similar hydridoiron carbonylate species as the active catalyst for transfer hydrogenation, using 1-phenylethanol or isopropanol as the hydride source and a phase transfer catalyst (Table 1).16 This system was based on well established Ru clusters17 and Abbayes' aryl nitrate reduction using a similar precursor.18 Although milder conditions were used, low turnover frequencies19 (TOFs) up to 13 h−1 were observed.
|
|
||
|---|---|---|
| R1 | R2 | Yield (%) |
| Ph | Me | 36 |
| 4-Cl-C6H4 | Me | 42 |
| Et | Me | 20 |
| – (CH2)5– | 78 | |
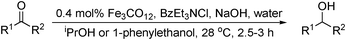
Hydrogenation using dihydrogen
The first proficient iron-catalysed hydrogenation of ketones using H2 was reported by Casey in 2007.20,21 At 3 atm hydrogen pressure and ambient temperature, excellent conversions were observed for the reduction of a variety of aromatic ketones (Table 2). Challenging substrates, such as benzophenone, took up to 72 h to achieve a moderate yield. The catalyst was found to be active for the reduction of ketones, aldehydes and imines, however α,β-unsaturated ketones gave both the unsaturated and saturated alcohol products. Casey's catalyst is believed to operate via an outer-sphere ionic mechanism,22,23 similar to Noyori's DAIPEN catalyst,24 where OH is generated inside a solvent cage and then coordinated to the iron centre. By using molecular traps, Casey reported a concerted hydride/proton transfer, and reactivation of the catalyst by heterolytic H2 cleavage (Fig. 1). Density functional theory (DFT) calculations identified the intermediate structures in the catalytic cycle and the previously asserted mechanism was supported by kinetic studies.25 The catalyst was also found to be active under transfer hydrogenation conditions.20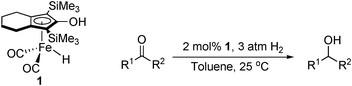
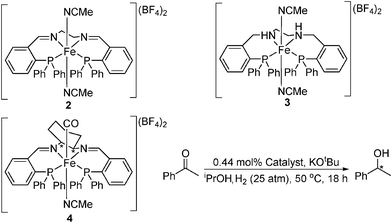
Morris reported the use of iron catalysts for the enantioselective reduction of acetophenone using dihydrogen in 2008 (Table 3).10a The dicationic complex 4 was found to be less active than the catalyst reported by Casey, with TOFs of approximately 5 h−1 at 50 °C observed (compared to 2 h−1 at 25 °C for 1).26 As with Casey's work, an outer-sphere mechanism was proposed for this transformation (Fig. 2), where heterolytic splitting of H2 gives an H-Fe-N-H structure that permits the outer-sphere transfer of proton and hydride.10d Similar activities were reported for both the imine and amine ligand catalysts (2 and 3). As with Ru catalysts of this kind,27 these species are most likely converted to a similar active catalyst via hydrogenation of the imine groups. DFT calculations gave similar activation barriers for both the Ru and Fe systems.10d
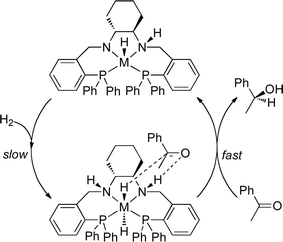 | ||
| Fig. 2 Proposed outer-sphere with ligand assistance (HOL) mechanism for the hydrogenation of acetophenone for Ru and possibly Fe.26 | ||
|
|
||
|---|---|---|
| Catalyst | Conversion (%) | ee (%) |
| 2 | 95 | — |
| 3 | 99 | — |
| 4 | 40 | 27 (S) |
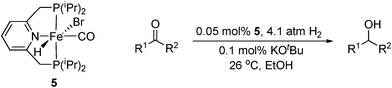
More recently, Milstein has used iron pincer complexes such as 5 to hydrogenate ketones under an atmosphere of dihydrogen.28 With very low (0.05 mol%) catalyst and base loadings, a range of ketones were reduced in up to 94% yield and with TOFs of up to 87 h−1 (Table 4). Some functional group tolerance has been illustrated but amine and nitrile substituents led to no product conversion. The reaction protocol was also applied to aldehydes and benzaldeyde was reduced in 36% yield. As with similar Ru complexes used for ester and amide reduction, it was proposed from NMR studies that the pyridine ring undergoes dearomatisation to form the active catalyst species.29
|
|
||
|---|---|---|
| R1 | R2 | Yield (%) |
| Ph | Me | 94 |
| 4-Cl-C6H4 | Me | 86 |
| Ph | CF3 | 54 |
| – (CH2)5– | 64 | |
| Ph | H | 36 |
Transfer Hydrogenation
Transfer hydrogenation is an alternative method for the synthesis of enantiopure alcohols from ketones.30 Because of the frequent need for high dihydrogen pressure and the dangers associated with use of a hazardous gas, transfer hydrogenation has received significant focus. Established Rh- and Ru-transfer hydrogenation catalysts currently give the highest yields, selectivities and activities and have TOFs for acetophenone of 100–4000 h−1.13a,31Based on the catalysts of the Markó and Vancheesen groups,13,16 Chen and coworkers reported an enantioselective iron hydrogenation catalyst from the complex [Et3NH][HFe3CO11] and enantiopure diaminodiphosphine ligands (Table 5).14 As proposed by Vancheesen, the Fe3 cluster is thought to remain intact throughout the reaction.16 The best results were observed for aryl-alkyl ketones, and ee was found to increase with increasing steric bulk of the carbonyl. A range of yields and ee's were obtained, including 1,1-diphenylacetone which gave 98% ee of the corresponding alcohol in only 18% yield. For acetophenone, the activity increased slightly but with decreased ee (TOF = 13 h−1 at 82 °C).
|
|
|||||
|---|---|---|---|---|---|
| R1 | R2 | T/°C | Yield (%) | Time (h) | ee (%) |
| Ph | Me | 45 | 92 | — | 61 (R) |
| Ph | Et | 45 | 87 | 4.5 | 72 (S) |
| 1-Tetralone | 82 | 30 | 7 | 45 (R) | |
| 6-Cl-C6H4 | Me | 82 | 5 | 11 | 16 (R) |
| CH(Ph)2 | Me | 65 | 18 | 21 | 98 (S) |
Beller and coworkers have introduced a series of iron catalysts generated in situ for transfer hydrogenation using a monodentate phosphine, a tridentate amine and an iron source.15 The resulting tuneability gives this system good potential for use in industrial applications, which is reinforced by the commercial availability of the materials used. The 2,2′:6′,2′′-terpyridine/PPh3/Fe3(CO)12 system reduced aryl-alkyl ketones in good yield (Table 6). and activity was found to be highly base-dependent, with NaOiPr giving the best results. Kinetic studies revealed a monohydride pathway, whereby formation of an Fe-monohydride species promotes H-transfer from donor to substrate.3,15
|
|
|||
|---|---|---|---|
| R1 | R2 | Yield (%) | |
| Cat1: | Ph | Me | 95 |
| Ph | Et | 81 | |
| Cy | Me | 93 | |
| 4-Cl-C6H4 | Me | >99 | |
| Ph | CH2Cl | 5 | |
| Cat2: | Ph | Me | 94 |
| Ph | C3H5 | 22 | |
| 4-Cl-C6H4 | Me | 95 | |
| Ph | Et | 87 | |
| t Bu | Me | 90 | |
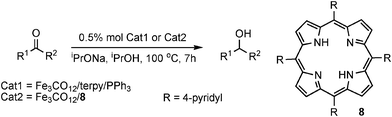
Beller has reported a biomimetic approach whereby iron-porphyrin catalysts were shown to also give excellent activity, with TOFs of up to 642 h−1 (Table 6).9a Aryl-alkyl and dialkyl ketones were reduced in 68–99% yield, though those bearing α-substituents gave variable results. Fe3(CO)12 was again found to be one of the most active iron sources, and interestingly there was no relationship found between the oxidation state of the iron source and catalytic activity, though a small amount of water was found to increase the TOF to 2500 h−1.9a Hemin was also tested and although lower activity was observed, precatalyst formation is not needed and it is naturally abundant, making this a worthwhile strategy.3 Both the porphyrin and the three-component constructs have been modified to reduce α-substituted alkoxyketones using FeCl2 as the iron source, showing the versatility and chemoselectivity of the method for a wide substrate scope.3,9b
Morris has achieved promising results for transfer hydrogenation10 using catalysts based on Fe- and Ru- ligand systems33,34,35 where hydride transfer occurs from the metal and proton transfer from the ligand via an outer-sphere mechanism. A number of aromatic ketones were reduced in 35–98% yield and 50–99% ee, with more challenging non-aryl substrates hydrogenated in up to 57% ee (Table 7).10c Very low catalysts loadings (0.025–0.17%) and ambient temperatures were used.10b Sterically demanding, prochiral substrates were reduced with generally higher enantioselectivities but with lower catalyst activities. There was some loss of activity after 30 min, possibly due to decomposition of the catalyst. Initial TOFs of up to 2600 h−1 were reported, which on increasing ketone concentration were increased to up to 4900 h−1, as high as the most active Ru systems for transfer hydrogenation of aromatic ketones.10c These systems represent some of the most active catalysts for carbonyl reduction to date, giving the corresponding alcohols in less than 1 h in most cases.
|
|
||||
|---|---|---|---|---|
| R1 | R2 | Conversion (%) | Time (h) | ee (%) |
| Ph | Me | 90 | 0.5 | 82 (S) |
| Ph | Et | 90 | 0.4 | 94 (S) |
| Ph | t Bu | 35 | 3.3 | 99 (S) |
| 4-Cl-C6H4 | Me | 96 | 0.3 | 80 (S) |
| 4-MeO-C6H4 | Me | 65 | 0.66 | 54 (S) |
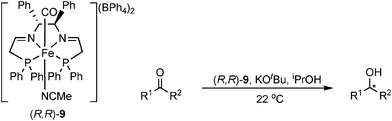
Morris' catalysts were also found to be active for an α,β-unsaturated ketone, selectively giving the unsaturated alcohol in 82% yield and 60% ee. The mechanism and active catalyst species are as yet unknown, but trends in reactivity were similar to Ru3P2N2 complexes36 involving a cluster-catalysed mechanism, and the selectivity towards C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O suggests an outer-sphere (HOL) mechanism.10c,37 The high activity of the catalysts is possibly due to the use of strong field ligands which keep the iron centre in a low spin-state, stabilising the catalyst.26,38 This is akin to the hydrogenase active site which contains CO and CN ligands.39
O suggests an outer-sphere (HOL) mechanism.10c,37 The high activity of the catalysts is possibly due to the use of strong field ligands which keep the iron centre in a low spin-state, stabilising the catalyst.26,38 This is akin to the hydrogenase active site which contains CO and CN ligands.39
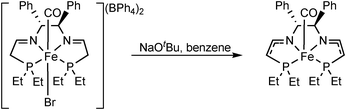
A series of complexes have been prepared by Morris via deprotonation α to the phosphine, to give a species that is active for the catalytic reduction of ketones in the absence of base (Fig. 3).10f It was shown the role of base in such reactions is only for this purpose and plays no further role in the catalytic process. Hence species such as these are likely intermediates in the catalytic cycle and were shown to give similar activities to the precatalyst 9 (in the absence of base).
A novel approach involving tetradentate iminophosphorane ligands for transfer hydrogenation of ketones has been reported by Buchard.40 With very low catalyst loading (0.1 mol%) the iron-iminophosphoranes tested gave conversions of 75–91% for acetophenone after 8 h, with all systems giving full conversion after 24 h.
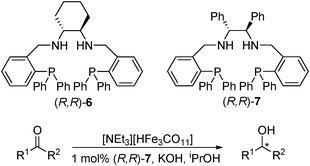
Recently, Beller has applied the method used by Chen (Table 5) for the asymmetric transfer hydrogenation of ketones, to reduce imine substrates with high enantioselectivities.41 With the iron hydridocarbonylate catalyst and (R,R)-6, a selection of aromatic N-(diphenylphosphinyl)-imines were hydrogenated in most cases with excellent yield and ee (Table 8). The scope of the reaction was also extended to heteroaromatic and dialkyl imines, the latter of which gave the amines with slightly lower yield and ee.
|
|
|||
|---|---|---|---|
| R1 | R2 | Yield (%) | ee (%) |
| Ph | Me | 87 | 96 (R) |
| 5-Me-C6H4 | Me | 95 | 97 (R) |
| naphthyl | Me | 98 | 98 (R) |
| 5-Cl-thiophenyl | Me | 78 | 95 (R) |
| Et | Me | 62 | 39 (R) |
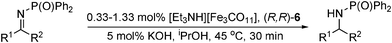
Hydrosilylation
Hydosilylation, followed by cleavage of the Si–O bond on aqueous work-up, offers an alternative method for the reduction of aldehydes and ketones to the corresponding alcohols. Nishiyama has developed a hydrosilylation-based reduction using low valence iron species generated from Fe(OAc)2 and bi- and tridentate nitrogen ligands such as N,N,N',N'-tetramethylethylenediamine (tmeda), to reduce ketones with good yield (Table 9).42 Ethers, esters and bromide groups were all tolerated under the reaction conditions. Chemoselectivity was also observed for benzalacetone, which was reduced to its corresponding unsaturated alcohol in 80% yield. It was later reported that with specific ketones, the Fe-tmeda system acted to catalyse dehydrogenative silylation of the ketone to form a silyl enol ether. This was overcome by the addition of sodium thiophene-2-carboxylate (STC) to give almost complete selectivity for the alcohol.43|
|
||
|---|---|---|
| R1 | R2 | Yield (%) |
| Ph | Me | 93 |
| 4-MeO-C6H4 | Me | 94 |
| 1-Tetralone | 62 | |
| (CH2)2Ph | Me | 88 |
| (CH)2Ph | Me | 80 |
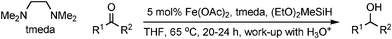
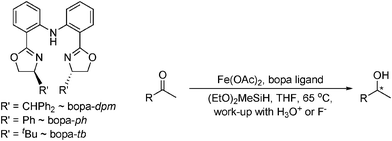
The first iron-catalysed asymmetric hydrosilylation of ketones was also reported by Nishiyama in 2007, using N,N,N-bis(oxazolinylphenyl)amine (bopa) ligands and (EtO)2MeSiH as the hydride source. More recently these bopa pincer ligands have been modified to reduce a range of aromatic ketones in excellent yield (88–99%) and moderate to high enantioselectivity (50–88% ee) with bopa-dpm (Table 10) in 24 h.44 All active catalysts were found to be highly air-sensitive and as yet only an [FeIIICl2-bopa-ip] species has been isolated, but is not an active reduction catalyst.44 Loadings as low as 2 mol% Fe(OAc)2 and 3 mol% bopa-dpm were used, though increased ee was observed when using a [Co-bopa-ph] system or [Ru-(phebox)] (bis(oxazolinyl)phenyl) catalyst.45
|
|
||
|---|---|---|
| R | Yield (%) | ee (%) |
| a 5 mol% bopa-tb ligand and Fe(OAc)2, for 48 h. b 2 mol% bopa-dpm ligand and Fe(OAc)2, for 24 h. c 5 mol% Co(OAc)2 and bopa-ph, for 24 h, all other conditions the same. | ||
| Pha | 75 | 79 (R) |
| Naphthyla | 59 | 65 (R) |
| Phb | 96 | 80 (R) |
| Naphthylb | 99 | 65 (R) |
| 2-MeO-C6H4b | 95 | 50 (R) |
| 4-MeO-C6H4b | 98 | 78 (R) |
| 4-Morpholino-C6H4b | 93 | 88 (R) |
| Naphthylc | 99 | 92 (R) |
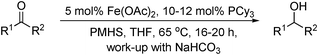
In addition to transfer hydrogenation, Beller has developed a hydrosilylation of aldehydes using electron-rich phosphorus ligands and hydrosilanes.46 The iron salts, ligand system (tricyclohexylphosphine) and stoichiometric reductant (polymethylhydrosiloxane, PMHS) used are all inexpensive and carry few hazards. Aromatic substrates were reduced in 72–99% yield, with heteroaromatic and aliphatic aldehydes reduced in 80–92% and 60–90% yield respectively (Table 11). Even aldehydes bearing groups susceptible to reduction such as –NO2, –CN, halides and carbon-carbon double bonds were reduced chemoselectively and trans-cinnamaldehyde was reduced to trans-cinnamyl alcohol in 82% yield. The scope of this simple system has since been extended to include the hydrosilylation of ketones in good to excellent yield, including benzophenone and dialkyl ketones which gave slightly lower yield (Table 11).47 Chemoselectivity was observed for α,β-unsaturated ketones to give allylic alcohols.
|
|
||
|---|---|---|
| R1 | R2 | Yield (%) |
| a PMHS = polymethylhydrosiloxane. | ||
| Ph | H | 93 |
| 4–F–C6H4 | H | 72 |
| 3-pyridyl | H | 92 |
| t Bu | H | 62 |
| (CH)2Ph | H | 82 |
| Ph | Ph | 80 |
| 4–CF3–C6H4 | Me | 96 |
| Cy | Me | 58 |
| (CH)2Ph | Me | 82 |
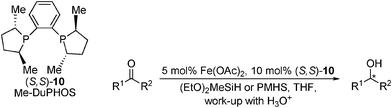
Based on the success of the aldehyde hydrosilylation, an asymmetric adaptation for ketones was reported.48 Instead of ligand modularity, the focus was on creating an efficient in situ catalyst from commercially available starting materials. Increasingly basic phosphorus ligands were found to give higher ee, and after extensive screening [Fe(OAc)2–Me–DuPHOS]49 was found to reduce acetophenone in 82% yield and 80% ee in just 1 h at 100 °C. Aryl-alkyl ketones, including substituted acetophenones used in the pharmaceutical industry,50 were reduced in moderate yield and ee (Table 12). The catalyst was also tested with a dialkyl and an α,β-unsaturated ketone, giving the respective alcohols in 45% ee and 79% ee, the former being higher than that obtained when using Ru- or Ti-catalysed hydrosilylations.51 As with the aldehyde hydrosilylation, ligand loadings of 10 mol% were used. Most recently the substrate scope of this hydrosilylation method has been broadened and acetylpyridines, α,β-unsaturated- and benzyl-alkyl ketones have all been reduced to the corresponding alcohols in good to excellent yield, but with low to moderate enantioselectivity (Table 12).47
|
|
|||||
|---|---|---|---|---|---|
| R1 | R2 | T/°C | Yield (%) | Time (h) | ee (%) |
| Ph | Me | 23 | 80 | 48 | 79 (R) |
| Ph | Me | 65 | 85 | 2 | 75 (R) |
| 2,4,6-Me-C6H2 | Me | 65 | >99 | 20 | 99 (R) |
| 6-Me-C6H4 | Me | 65 | 60 | 16 | 51 (R) |
| Cy | Me | 45 | 57 | 16–24 | 45 (R) |
| Cyclohexene | Me | 45 | 68 | 16–24 | 79 (R) |
| 2-Pyridyl | Me | 65 | 68 | 45 | 28 |
| CH2Ph | Me | 23 | 86 | 42 | 22 |
| 3,4,5-OMe-C6H2 | Me | 23 | 84 | 32 | 89 |
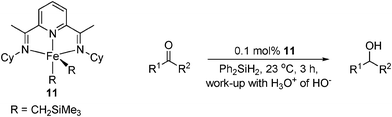
Chirik has developed a series of highly active bis(imino)pyridine (PDI) iron(0) bis(dinitrogen) [(iPrPDI)Fe(N2)2] complexes for the hydrogenation and hydrosilylation of alkenes,52 which have since been modified to catalyse the hydrosilylation of carbonyls.53 Using Ph2SiH2, several aromatic ketones and aldehydes were reduced in >99% conversion in under 3 h (Table 13). Cyclohexenones were hydrosilylated in 12 h to give the unsaturated alcohol as the sole product, however the reaction was less chemoselective for the reduction of non-cyclic enones.
|
|
||
|---|---|---|
| R1 | R2 | Conversion (%) |
| a After 12 h. | ||
| Ph | Me | >99 |
| 1-tetralone | 82 | |
| 4-CF3-C6H4 | Me | >99 |
| 3,5-CF3-C6H3 | Me | 68 |
| Cyclohexenone | >99a | |
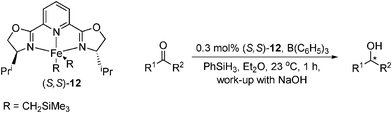
The Chirik group have more recently prepared enantiopure iron-bis(oxazoline) (box) and pyridine-box (pybox) complexes for the asymmetric hydrosilylation of ketones.54 Excellent conversions (TOFs of up to 330 h−1) and modest to good ee was obtained in most cases, though high asymmetric induction was observed for hindered aryl ketones and tetralone with activation by B(C6F5)3 (Table 14); mesityl ketone was reduced by catalyst 12 in 93% ee, but in only 16% conversion.
|
|
|||
|---|---|---|---|
| R1 | R2 | Conversion (%) | ee (%) |
| a With 1 mol% [(iPrBox)Fe(CH2SiMe3)2]. | |||
| Ph | R1 = Ph, R2 = Me | 99 | 54 (R) |
| 1-Tetralone | 99 | 41 (R) | |
| 4-OMe-C6H4 | R1 =, R2 = Me | 55 | 25 (R) |
| n-Bu | R1 =, R2 = Me | 99 | 11 (R) |
| 2,4,6-Me-C6H2 | R1 =, R2 = Mea | 16 | 93 (R) |
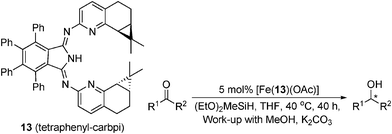
A novel approach to asymmetric catalytic hydrosilylation has been reported by Gade, where the stereocontrol of the [Fe-BPI] (bis(2-pyridylimino)isoindole) catalysts relies on the stereodirecting pyridyl wings and a protective “hedge” preventing backside attack on the metal (Table 15).55 The BPI ligands are readily prepared in a one-pot synthesis from enantiopure aminopyridines, terpenes and Fe(OAc)2. The carene-derived [Fe(tetraphenyl-carbpi)(OAc)] was found to be the most selective, reducing acetophenone in 86% ee. A range of aromatic ketones were reduced in up to 93% ee, and dialkyl ketones in 55–60% ee. These are some of the highest selectivities reported for iron-catalysed ketone reductions and are synthetically useful and importantly, comparable to non-Fe catalysts.29,56,57 Compared to Morris' catalysts, which show similar reactivity and selectivity, this catalyst requires higher loadings and increased reaction times of 16—40 h.
|
|
|||
|---|---|---|---|
| R1 | R2 | Yield (%) | ee (%) |
| Ph | Me | 85 | 86 (S) |
| 1-Tetralone | 72 | 80 (S) | |
| 4-Cl-C6H4 | Me | 79 | 85 (S) |
| 2,4,6-Me-C6H2 | Me | 83 | 93 (S) |
| t Bu | Me | 51 | 59 (S) |
Tilley has used the simple iron silylamide catalyst [Fe(N(SiMe3)2)2] and Ph2SiH2 to perform the hydrosilylation of a number of ketones and aldehydes with generally quantitative conversion (Table 16).58 A range of functionalities including CC, cyano- and cyclopropyl- were tolerated and TOFs of up to 2400 h−1 were observed. The hydrosilylation was also found to occur without solvent with no loss of conversion. NMR studies showed the formation of an NH(SiMe3)2, which is hypothesised to coordinate to the iron and initiate the hydrosilylation.
|
|
|||
|---|---|---|---|
| R1 | R2 | Time (h) | Conversion (%) |
| Ph | Me | 0.7 | >98 |
| Et | Et | 0.7 | >98 |
| CH2CH(CH2)2 | Me | 5.5 | 95 |
| 4-NMe2-C6H4 | H | 0.5 | >98 |
| 4-CN-C6H4 | Me | 17 | >98 |
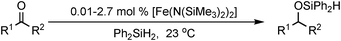
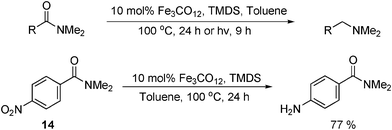
Following their work on the reduction of carboxamides to amines using Pt or Rh catalysts with PMHS,59 Nagashima and coworkers recently reported the same transformation with iron carbonyl catalysts.60 A number of carboxamides were reduced in good to excellent yield at either 100 °C for 24 h or by irradiation for 9 h at room temperature (Table 17). The catalyst system was also found to reduce nitroarenes at high temperature. Interestingly, when N,N-dimethyl-p-nitrobenzamide (14) was tested at 100 °C, Fe3(CO)12 selectively reduced the nitro- group and not the carbonyl. As with early iron carbonyl catalysts,4a,13,16,61 a limitation of this work is the large amount of energy required to produce the active iron carbonyl catalyst.
|
|
||
|---|---|---|
| R | High Temp. Yield (%) | Photoassisted Yield (%) |
| a TMDS = 1,1,3,3-tetramethyldisilazane. | ||
| (CH2)2Ph | 85 | 73 |
| Cy | 81 | 68 |
| 4-Br-C6H4 | 96 | 95 |
| 4-OMe-C6H4 | 82 | 93 |
An iron-catalysed reduction of amides has been reported by Beller, which has been proposed to proceed via a similar mechanism to the Ru-catalysed reaction by Nagashima.62 A selection of tertiary amides were reduced to their corresponding amines in good to excellent yield by the Fe3(CO)12 catalyst, including aromatic, aliphatic and heterocyclic substrates (Table 18). A secondary amide was also reduced in 99% yield using PhSiH3 as the hydrogen source.

Alkenes and alkynes
As with carbonyl substrates, early attempts using iron as a stoichiometric reductant were focused on iron carbonyls,61,63 including Noyori64 who used Fe(CO)5 to reduce α,β-unsaturated ketones to their corresponding saturated ketones in generally excellent yield. At high temperature, Fe(CO)5 is converted to the active catalyst species Fe(CO)3 with vacant sites for coordination.63,65 Wrighton found the activity of Fe(CO)5 increased when using constant irradiation under ambient conditions to reduce alkenes,66 though low alkane yields were obtained for a limited range of substrates. All of these Fe0 carbonyl systems require either high temperatures and pressures13,16,61,67 or continued photoirradiation66 for useful TOFs to be achieved.Biomimetic approaches have also been developed, such as Kano who, like Beller,9a used Fe-porphyrins and base (NaBH4) to catalyse the hydrogenation of styrene via a radical process, giving TOFs up to 81 h−1.68 Sakaki later used similar catalysts with α,β-unsaturated esters, and reported impressive TOFs of up to 4580 h−1.69 Labelling studies showed proton transfer from MeOH and hydride from NaBH4. Inoue has used Fe4S4 clusters to catalyse the hydrogenation of octene and stilbene after activation by PhLi.70
The general trend of using P and N ligands in carbonyl reduction is also seen with olefin substrates, such as the use of multidentate P ligands by Bianchini to hydrogenate alkynes.71 The reduction is proposed to proceed via an intramolecular acid/base reaction of an η2–H2 species and coordinated substrate (following alkyne insertion). The catalyst was found to be chemoselective towards alkynes due to a greater tendency to undergo insertion, rather than a difference in coordinating ability compared to alkenes. TOFs up to 7.9 h−1 were observed under ambient conditions, which increased up to 45.2 h−1 on heating to 63 °C (Table 19).
|
|
||
|---|---|---|
| R | T/°C | TOF (h−1) |
| Ph | 20 | 7.6 |
| Ph | 63 | 45.2 |
| n-C3H7 | 20 | 7.9 |
| CHCH(OMe) |
63 | 21.2 |
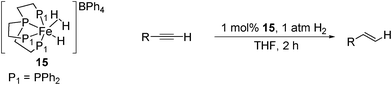
Bianchini later used the same catalyst to reduced α,β-unsaturated ketones72 using a cyclopentanol hydrogen donor. The products from this reaction were unpredicatble, though good selectivity for either the CC or CO was observed in most cases (Table 20). Strong-field tetradentate and hydride ligands were used to maintain the FeII d6 low-spin diamagnetic configuration.26 The mechanism of the reaction is proposed to proceed via an inner-sphere mechanism where the substrate coordinates cis to the hydride and therefore reduction of CC and CO can occur. As with the gaseous hydrogen system, the catalyst was found to be inactive for unfunctionalised alkenes under transfer hydrogenation conditions.
|
|
|||
|---|---|---|---|
| R1 | R2 | Time (h) | Major Product(s), Conversion (%) |
| Ph | Me | 7 | B, 95 |
| Ph | Ph | 7 | C, 30 |
| H | Et | 1 | C, 100 |
| Cyclohexenone | 5 | A, 44; B, 28 | |
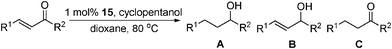
Peters has reported a series of [{PhB(CH2PiPr2)3}FeII-alkyl] catalyst precursors for the hydrogenation of alkenes using dihydrogen.73 Interestingly, Peters proposed an FeII/IV redox couple as the active species in the hydrogenations. A number of alkenes were reduced with TOFs up to 24 h−1 and catalyst loadings of 10 mol% (Table 21). 2-pentyne was also reduced to the fully saturated hydrocarbon in 6 h with a TOF of 1.6 h−1, though other alkynes remain problematic due to unwanted side reactions such as reductive dimerisation and polymerisation. At lower catalyst loadings (2.5 mol%) only three turnovers were observed as a result of catalyst decomposition.
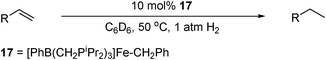
The Chirik group has made significant breakthroughs in the field of iron-catalysed olefin hydrogenation and hydrosilylation using a series of bis(imino)pyridine [(PDI)Fe0(N2)2] precatalysts, based on the polymerisation catalysts of Gibson and Brookhart.52,74,75,76,77,78,79 The L3Fe0 species' PDI ligand framework can delocalise charge over its conjugated π-system, making them ideal for redox processes.80 It is proposed that the ligand can accept up to three electrons from the metal centre, acting as an electron reservoir for its activation.74d The precatalyst [(iPrPDI)Fe(N2)2] 18, (Table 22) has been shown to be effective for the hydrogenation,52,74e hydrosilylation and [2π + 2π] cyclisation81 of olefins.
|
|
|||
|---|---|---|---|
| R1 | R2 | Time (min) | TOF (h−1) |
| H | C4H9 | 12 | 1814 |
| (+)-(R)-Limonene | 210 | 104 | |
| –(CH2)4– | 380 | 57 | |
| H | CH2NHCH3 | 60 | 320 |
| Ph | CO2Me | 5 | >240 |
| Ph | Me (trans) | 5 | >240 |
| diphenylacetylene | Successful but TOF not reported | ||
| Ph | COPh (trans) | Induced catalyst decomposition | |
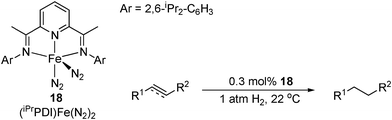
Using just 0.3 mol% catalyst loading, 18 was shown to have excellent activity for the hydrogenation of olefins, giving TOFs up to 1814 h−1, reducing 1-hexene in minutes at ambient temperature and 1 atm H2 (Table 22).52 The highest activities were seen for terminal alkenes, with internal alkenes requiring longer reaction times. The catalyst system can also be applied in neat alkene and reaches optimal activity over an extended reaction time. Some internal alkynes were also hydrogenated with 18. Excellent functional group tolerance was observed, including unprotected amines, ethers, esters and ketones.74e The catalyst was found to decompose with α,β-unsaturated ketones, though α,β-unsaturated esters were reduced to saturated esters with TOFs up to 240 h−1. Activities up to 364 h−1 for olefin hydrosilylation were also reported with PhSiH3 to give the anti-Markovnikov product, albeit with longer reaction times.52
Recently dimeric iron-nitrogen complexes have been prepared by reduction of the corresponding [(PDI)FeBr2] complex with sodium naphthalenide (Scheme 1).76 As with the dinitrogen monomers, the dimeric compounds are intermediate spin FeII complexes with dianionic PDI ligands following a 2e− reduction.74c,82 Complex 19 was found be significantly more active in the catalytic hydrogenation of ethyl-3-methylbut-2-enoate, giving complete conversion in just 1.5 h, compared to 18 which gave a maximum 60% conversion in 72 h.74e
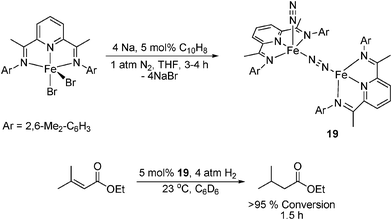 | ||
| Scheme 1 Preparation of the catalyst precursor 19 and reduction of ethyl-3-methylbut-2-enoate.76 | ||
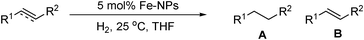
De Vries has reported a non-ligand approach to catalytic hydrogenation of both alkenes and alkynes using Fe-nanoparticles (NPs).83 Fe-NPs prepared by the method of Bedford,5n gave quantitative conversion for cis-1,2- and 1,1-disubstitued alkenes after 15 min. 1,2-trans-disubstituted and cyclic cis-alkenes were reduced at a lower rate (Table 23). On increasing the temperature to 80 °C and at dihydrogen pressures of 30 bar, a TOF of 118 h−1 was achieved for 1-octyne. The Fe-NPs were found to be inactive for the hydrogenation of tri- and tetra-substituted alkenes, though several alkynes were also successfully reduced to alkanes in 15 h. For alkynes the presence of OH groups was found to retard the rate of reduction.
|
|
||||
|---|---|---|---|---|
| Substrate | Conversion (%) | Time (h) | H2 pressure (bar) | A:B |
| a cyclooctene 100% conversion at 100 °C. | ||||
| Norbornene | 100 | 0.5 | 5 | — |
| trans-2-Hexene | 22 | 15 | 5 | — |
| cis-2-Hexene | 100 | 15 | 5 | — |
| Cyclooctene | 11a | 15 | 5 | — |
| 1-Hexyne | 100 | 15 | 20 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 :0 |
| 1-Hexyne | 100 | 0.5 | 10 | 0.6:1 |
| 3-Hexyn-1-ol | 100 | 15 | 20 | 1:0 |
| 2-Methyl-3-butyl-1-ol | 56 | 15 | 20 | 1:2 |
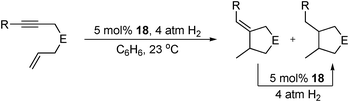
The precatalyst 18 has also been applied to the reductive cyclisation of 1,6- enynes and diynes.75 At ambient temperatures, a range of enynes and diynes were cyclised in moderate to excellent yield with TOFs up to 20 h−1 (Table 24). Good functional group tolerance was observed, including malonate and ester substituted enynes. It is proposed that a FeII oxidation-state is maintained throughout the catalytic cycle, as electron transfer for the reductive C–C bond formation proceeds via oxidation of the ligand and not the metal centre.76 The substrate scope and activity observed are comparable to existing Rh catalysts.84
|
|
|||||
|---|---|---|---|---|---|
| R | E | Time (min) | Yield (%) | TOF (h−1) | cis/trans |
| H | NtBu | 180 | 68 | 6.7 | >99:1 |
| H | NTs | 60 | 79 | 20 | 75:25 |
| Me | O | 180 | 62 | 6.7 | — |
| SiMe3 | NTs | 540 | 82 | 2.2 | — |
More recently, Enthaler has reported the reduction of alkynes to alkenes using an iron-catalysed hydrosilylation.85 Using 5 mol% Fe2(CO)9, 10 mol% PBu3 and (EtO)3SiH as the reductant, a series of alkynes were reduced to the corresponding (Z)-alkenes with good selectivity (Table 25). The reaction was shown to be efficient for the reduction of diaryl-, aryl-alkyl- and ester substituted alkynes.
|
|
|||
|---|---|---|---|
| R1 | R2 | Conversion (%) | (Z) : (E) |
| Ph | H | >99 | — |
| Ph | Ph | 68 | >99 : <1 |
| Ph | 4-F-Ph | 48 | 78:22 |
| Ph | n-Bu | 44 | 99:1 |
| Ph | 2-Py | 55 | 36:64 |
| Ph | CO2Et | 83 | 19:81 |
| CO2Me | CO2Me | >99 | 43:57 |
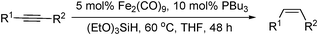
Conclusions
The use of iron catalysts for the catalytic reduction of carbonyls and olefins has experienced a renaissance in recent years. Although there are few examples of ketone hydrogenation using gaseous hydrogen, a number of transfer hydrogenations and hydrosilylations have produced promising activities, yields and enantioselectivities. The highest TOFs reported for an iron catalyst by Morris (4900 h−1) are as good as that of the well established Noyori transfer catalyst (4600 h−1),31a which are achieved at much higher catalyst loadings and at 80 °C. Although many of the iron systems reported have activity equal to or higher than Noyori's catalyst under the standard reaction conditions (TOF = 227 h−1), the ee's reported to date are generally lower (Table 26).86|
|
||||
|---|---|---|---|---|
| Precatalysta | Yield (%) | Catalyst Loading (mol%) | Time (h) | ee (%) |
| a See relevant scheme for full precatalyst and reaction conditions. b Conversion. | ||||
| 1 20 | 83 | 2 | 20 | — |
| [NEt3][HFe3CO11], (R,R)-714 | 92 | 1 | — | 61 (R) |
| Fe3CO12/terpy/PPh315 | 95 | 0.5 | 7 | — |
| Fe3CO12/porphyrin 89 | 94 | 0.5 | 7 | — |
| 3 10a | 99 | 0.44 | 18 | — |
| (R,R)-910c | 90b | 0.14 | 0.5 | 82 (S) |
| Fe(OAc)2,(S,S)-1048 | 80 | 5 | 48 | 79 (R) |
| 11 53 | >99b | 0.1 | 3 | — |
| (S,S)-1254 | >99b | 0.3 | 1 | 54 (R) |
| [Fe(13)(OAc)]55 | 85 | 5 | 40 | 86 (S) |
| Fe(OAc)2, tmeda42 | 93 | 5 | 20-24 | — |
| Fe(OAc)2, bopa-dpm44 | 96 | 5 | 48 | 80 (R) |
| [Fe(N(SiMe3)2)2]58 | >98b | 2.7 | 0.7 | — |
| Noyori transfer catalyst85 | >99 | 0.5 | 20 | 98 (S) |

A key component of the most efficient catalysts seems to be the use of modular ancillary ligand systems, already seen in established hydrogenation processes.11,56,57,87 The importance of modifying catalysts to specific substrates and reactions is seen in the work of Chirik, Gade and others. A vast range of active iron complexes can be easily prepared from a number of readily available, inexpensive precursors such as Fe(OAc)2. The economic and toxicity benefits of iron systems make them good candidates for use in industrial and pharmacological syntheses.3 However, there has been an emphasis on the economics of iron catalysts and their inexpense has been slightly overstated. For enantioselective reactions the cost of the ligand system is typically highest, for example the enantiopure Me–DuPHOS ligand48 is four times that of rhodium metal.88 Iron reduces this cost, but there is still a limited availability of varied metal precursors (particularly anhydrous substrates). Costs can be most significantly driven down by the use of widely available reductants and iron precursors, modular ligands and by using in situ or short-step catalyst syntheses.
Much fewer examples of iron-catalysed olefin hydrogenation have been reported, though the catalysts introduced by Chirik, as well as those from de Vries and Bianchini have delivered promising reactivity. The range of iron catalysts developed by Chirik are highly air sensitive, in fact the reaction mixtures are exposed to air on completion to aid the removal of the catalyst. In comparison, the widely used Pfaltz87,89 and Crabtree90 Ir-catalysts for olefin hydrogenation are relatively air-stable and easy to handle. Although the most progress has been made with PDI–Fe–dinitrogen systems, the range of substrates is still limited and tri- and tetra- substituted olefin hydrogenation is yet to be reported with iron.91 Thus as yet there have been no asymmetric olefin hydrogenations catalysed by iron species, though the high activity and low catalyst loadings of Chirik's precatalysts are promising (Table 27).
|
|
|||
|---|---|---|---|
| Precatalyst | Substrate | Conversion (%) | Time (h) |
| 17 73 | Ethylene | >95 | 0.4 |
| 1-Hexene | >95 | 2.2 | |
| 18 52,74e | 1-Hexene | >98 | 0.2 |
| (+)-(R)-Limonene | >98 | 3.5 | |
| trans-β-Methylstyrene | >98 | 3 | |
| R = Me, R' = CO2Et | >99 | 0.08 | |
| R= (Me)2, R' = CO2Et | 50, 24 h | 24 | |
| 19 76 | Ethyl-3-methylbut-2-enoate | >95, 1.5 h | 1.5 |
| Fe-NPs83 | trans-Stilbene | >99 | 15 |
| 1-Octene | >99 | 15 | |
| Cyclohexene | >99 | 15 | |
| Norbornene | >99 | 0.5 | |

Much of the work into the iron-catalysed homogenous reduction of carbonyls and olefins is still in its infancy, and although substrate scope is broadening it is still somewhat limited, particularly for olefins. Where there has been mechanistic and structural insight into the iron catalysts and their precursors, as with Morris and Chirik, rapid development and comparable activities to established catalysts have been achieved. A bench stable and selective iron hydrogenation catalyst with broad-scope is still needed for iron catalysis to compete with the established precious metals, though the economic and most importantly, toxicity benefits iron species present on a large-scale mean it has the potential to replace existing catalysts for industrial hydrogenation.
References
- H. U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103 CrossRef CAS.
- P. N. Rylander, Hydrogenation Methods, Academic Press, New York, 1990 Search PubMed.
- S. Enthaler, K. Junge and M. Bellerin: Iron Catalysis in Organic Chemistry: Reactions and Applications (Ed.: B. Plietker), Wiley-VCH, Weinheim, 2008 and references therein Search PubMed.
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS and references therein; (b) S. K. Ritter, Chem. Eng. News, 2008, 86, 53 CrossRef; (c) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS; (d) K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC; (e) For examples of iron-catalysed reduction reactions not involving carbonyls or olefins see S. Enthaler, ChemCatChem, 2011, 3, 666 Search PubMed; (f) S. Enthaler, ChemCatChem, 2010, 2, 1411 Search PubMed; (g) W. M. Czaplik, S. Grupe, M. Mayer and A. Jacobi von Wangelin, Chem. Commun., 2010, 46, 6350 RSC; (h) K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769 RSC; (i) H. U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210 Search PubMed.
- (a) For reviews see: E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 Search PubMed; (b) W. M. Czaplik, M. Mayer, J. Cvengros and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS; (c) A. Leitner, in: Iron Catalysis in Organic Chemistry: Reactions and Applications, B. Pleitker, Ed., Wiley-VCH, Weinheim, Germany, 2008, 147–176 Search PubMed; (d) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS; (e) B. M. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS; (f) For selected recent examples see A. K. Steib, T. Thaler, K. Komeyama, P. Mayer and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 3303 Search PubMed; (g) W. M. Czaplik, M. Mayer and A. Jacobi von Wangelin, ChemCatChem, 2011, 3, 135 Search PubMed; (h) M. Mayer, W. M. Czaplik and A. Jacobi von Wangelin, Adv. Synth. Catal., 2010, 352, 2147 CrossRef CAS; (i) R. B. Bedford, M. Huwe and M. C. Wilkinson, Chem. Commun., 2009, 600 RSC; (j) W. M. Czaplik, M. Mayer and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2009, 48, 607 CrossRef CAS; (k) G. Cahiez, V. Habiak, C. Duplais and A. Moyeux, Angew. Chem., Int. Ed., 2007, 46, 4364 CrossRef CAS; (l) G. Cahiez, C. Duplais and A. Moyeux, Org. Lett., 2007, 9, 3253 CrossRef CAS; (m) A. Fürstner, H. Krause and C. W. Lehmann, Angew. Chem., Int. Ed., 2006, 45, 440 CrossRef; (n) R. Bedford, M.. Betham, D. Bruce, S. Davis, R. Frost and M. Hird, Chem. Commun., 2006, 1398 RSC.
- V. W. Goldschmidt, Geochemistry, Oxford Press, London, 1958, 647 Search PubMed.
- Doc. Ref. EMEA/CHMP/SWP/4446/2000.
- F. Naud, F. Spindler, C. J. Rueggeberg, A. T. Schmidt and H. U. Blaser, Org. Process Res. Dev., 2007, 11, 519 Search PubMed.
- (a) S. Enthaler, G. Erre, M. K. Tse, K. Junge and M. Beller, Tetrahedron Lett., 2006, 47, 8095 CrossRef CAS; (b) S. Enthaler, B. Spilker, G. Erre, K. Junge, M. K. Tse and M. Beller, Tetrahedron, 2008, 64, 3867 CrossRef CAS.
- (a) C. Sui-Seng, F. Freutel, A. J. Lough and R. H. Morris, Angew. Chem., Int. Ed., 2008, 47, 940 CrossRef CAS; (b) N. Meyer, A. J. Lough and R. H. Morris, Chem.–Eur. J., 2009, 15, 5605 CrossRef CAS; (c) A. Mikhailine, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2009, 131, 1394 CrossRef CAS; (d) C. Sui-Seng, F. N. Haque, A. Hadzovic, A. Pütz, V. Reuss, N. Meyer, A. J. Lough, M. Zimmer-De luliis and R. H. Morris, Inorg. Chem., 2009, 48, 735 CrossRef CAS; (e) P. O. Lagaditis, A. Mikhailine, A. J. Lough and R. H. Morris, Inorg. Chem., 2010, 49, 1094 CrossRef CAS; (f) P. O. Lagaditis, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2011, 133, 9662 CrossRef CAS.
- J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832 CrossRef CAS.
- B. Fell, J. Shanschool and F. Asinger, J. Organomet. Chem., 1971, 33, 69 CrossRef CAS.
- (a) L. Markó, M. A. Radhi and I. Ötvös, J. Organomet. Chem., 1981, 218, 369 CrossRef CAS; (b) L. Markó and J. Palágyi, Transition Met. Chem., 1983, 8, 207 CrossRef CAS.
- J. S. Chen, L. L. Chen, Y. Ying, G. Chen, W. Y. Shen, Z. R. Dong, Y. Y. Li and J. X. Gao, Acta. Chim. Sin. (Huaxue Xuebao), 2004, 62, 1745 Search PubMed.
- S. Enthaler, B. Hagemann, G. Erre, K. Junge and M. Beller, Chem.–Asian J., 2006, 1, 598 CrossRef.
- (a) K. Jothimony and S. Vancheesen, J. Mol. Catal., 1989, 52, 301 CrossRef CAS; (b) K. Jothimony, S. Vancheesen and J. C. Kuriacose, J. Mol. Catal., 1985, 32, 11 CrossRef CAS.
- (a) S. Bhaduri and K. Sharma, J. Chem. Soc., Chem. Commun., 1988, 173 RSC; (b) Y. Blum and Y. Shvo, J. Organomet. Chem., 1984, 263, 93 CrossRef CAS.
- H. Des Abbayes and H. Alper, J. Am. Chem. Soc., 1977, 99, 98 CrossRef CAS.
- Turnover frequency = mol product/mol catalyst (per hour).
- C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816 CrossRef CAS.
- (a) H. J. Knölker, E. Baum, H. Goesmann and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 2064 CrossRef CAS; (b) Y. Blum, D. Czarkie, Y. Rahamim and Y. Shvo, Organometallics, 1985, 4, 1459 CrossRef CAS.
- C. P. Casey and H. Guan, J. Am. Chem. Soc., 2009, 131, 2499 CrossRef CAS.
- R. M. Bullock, Angew. Chem., Int. Ed., 2007, 46, 7360 CrossRef CAS.
- (a) R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS; (b) M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466 CrossRef CAS; (c) K. J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285 CrossRef CAS.
- H. Zhang, D. Chen, Y. Zhang, G. Zhang and J. Liu, Dalton Trans., 2010, 39, 1972 RSC.
- R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC.
- T. Li, R. Churland, A. J. Lough, K. Abdur-Rashid and R. H. Morris, Organometallics, 2004, 23, 6239 CrossRef CAS.
- R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120 CrossRef CAS.
- W. Baratta, K. Siega and P. Rigo, Chem.–Eur. J., 2007, 13, 7479 CrossRef CAS.
- S. Gaillard and J. Renaud, ChemSusChem, 2008, 1, 505 CrossRef CAS.
- (a) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS; (b) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300 CrossRef.
- As reported these ligands are (S,S) but have been drawn (R,R) in both the manuscript and experimental details.
- J. X. Gao, H. L. Wan, W. K. Wong, M. C. Tse and W. T. Wong, Polyhedron, 1996, 15, 1241 CrossRef CAS.
- J. X. Gao, T. Ikariya and R. Noyori, Organometallics, 1996, 15, 1087 CrossRef CAS.
- (a) V. Rautenstrauch, X. Hoang-Cong R. Churland, K. Abdur-Rashid and R. H. Morris, Chem.–Eur. J., 2003, 9, 4954 CrossRef CAS; (b) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201 CrossRef CAS.
- H. Zhang, C. B. Yang, Y. Y. Li, Z. R. Donga, J. X. Gao, H. Nakamura, K. Murata and T. Ikariya, Chem. Commun., 2003, 142 RSC.
- HOL - Hydride transfer to the substrate in an outer-sphere mechanism with ancillary ligand assistance.
- (a) P. I. Amrhein, S. D. Drouin, C. E. Forde, A. J. Lough and R. H. Morris, Chem. Commun., 1996, 1665 RSC; (b) C. E. Forde, S. E. Landau and R. H. Morris, J. Chem. Soc., Dalton Trans., 1997, 1663 RSC.
- G. J. Kubas, Chem. Rev., 2007, 107, 4152 CrossRef CAS.
- A. Buchard, H. Heuclin, A. Auffrant, X. F. Le Goff and P. Le Floch, Dalton Trans., 2009, 1659 RSC.
- S. Zhou, S. Fleischer, K. Junge, S. Das, D. Addis and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8121 CrossRef CAS.
- H. Nishiyama and A. Furuta, Chem. Commun., 2007, 760 RSC.
- A. Furuta and H. Nishiyama, Tetrahedron Lett., 2008, 49, 110 CrossRef CAS.
- T. Inagaki, L. T. Phong, A. Furuta, J. Ito and H. Nishiyama, Chem.–Eur. J., 2010, 16, 3090 CrossRef CAS.
- J. Ito, S. Ujiie and H. Nishiyama, Chem. Commun., 2008, 1923 RSC.
- N. S. Shaikh, K. Junge and M. Beller, Org. Lett., 2007, 9, 5429 CrossRef CAS.
- D. Addis, N. Shaikh, S. Zhou, S. Das, K. Junge and M. Beller, Chem.–Asian J., 2010, 5, 1687 CrossRef CAS.
- N. S. Shaikh, S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 2497 CrossRef CAS.
- (-)-1,2-bis[(2R,5R)-2,5-dimethylphospholano]benzene) = Me-DuPHOS.
- J. Wu, J. X. Ji and A. S. C. Chan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3570 CrossRef CAS.
- (a) Y. Nishibashi, I. Takei, S. Uemara and M. Hidai, Organometallics, 1998 Search PubMed; (b) Ti using, J. Yun and S. L Buchwald, J. Am. Chem. Soc., 1999, 121, 5640 CrossRef CAS.
- S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef.
- A. M. Tondreau, E. Lobkovsky and P. J. Chirik, Org. Lett., 2008, 10, 2789 CrossRef CAS.
- A. M. Tondreau, J. M. Darmon, B. M. Wile, S. K. Floyd, E. Lobkovsky and P. J. Chirik, Organometallics, 2009, 28, 3928 CrossRef CAS.
- B. K. Langlotz, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2008, 47, 4670 CrossRef CAS.
- J. M. Brown, T. Ohkuma and R. Noyori, in: Comprehensive Asymmetric Catalysis (Ed.: E. N. Jacobsen, A. Pfaltz and H. Yamamoto), Springer, Berlin, 1999 Search PubMed.
- H. Nishiyama, in: Comprehensive Asymmetric Catalysis (Ed.: E. N. Jacobsen, A. Pfaltz and H. : Yamamoto), Springer, Berlin, 1999 Search PubMed.
- J. Yang and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 10186 CrossRef CAS.
- (a) K. Matsubara, T. Iura, T. Maki and H. Nagashima, J. Org. Chem., 2002, 67, 4985 CrossRef CAS; (b) Y. Motoyama, C. Itonega, T. Ishida, M. Takasaki and H. Nagashima, Org. Synth., 2005, 82, 188 CAS; (c) Y. Motoyama, K. Mitsui, T. Ishida and H. Nagashima, J. Am. Chem. Soc., 2005, 127, 13150 CrossRef CAS; (d) S. Hanada, T. Ishida, Y. Motoyama and H. Nagashima, J. Org. Chem., 2007, 72, 7551 CrossRef CAS; (e) H. Sakasuma, Y. Motoyama and H. Nagashima, Chem. Commun., 2007, 4916 RSC.
- Y. Sunada, H. Kawakami, T. Imaoka, Y. Motoyama and H. Nagashima, Angew. Chem., Int. Ed., 2009, 48, 9511 CrossRef CAS.
- (a) E. N. Frenkel, E. A. Emken, H. M. Peters, V. L. Davidson and R. O. Butterfield, J. Org. Chem., 1964, 29, 3292 CrossRef CAS; (b) E. N. Frenkel, E. A. Emken and V. L. Davidson, J. Org. Chem., 1965, 30, 2739 CrossRef CAS.
- S. Zhou, K. Junge, S. Das and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9507 CrossRef CAS.
- M. A. Schroeder and M. S. Wrighton, J. Organomet. Chem., 1977, 128, 345 CrossRef CAS.
- R. Noyori, I. Umeda and T. Ishigami, J. Org. Chem., 1972, 37, 1542 CrossRef CAS.
- (a) C. U. Pittman Jr., W. D. Honnick, M. S. Wrighton, R. D. Sanner and R. G. Austin, Fundam. Res. Homogenous Catal., 1979, 3, 603 Search PubMed; (b) C. L. Randolph and M. S. Wrighton, J. Am. Chem. Soc., 1986, 108, 3366 CrossRef CAS.
- M. A. Schroeder and M. S. Wrighton, J. Am. Chem. Soc., 1976, 98, 551 CrossRef CAS.
- M. Cais and N. J. Maoz, J. Chem. Soc., 1971, 11, 1811 Search PubMed.
- K. Kano, M. Takeuchi, S. Hashimoto and Z. I. Yoshida, J. Chem. Soc., Chem. Commun., 1991, 1728 RSC.
- (a) S. Sakaki, T. Sagura, T. Arai, T. Kojima, T. Ogata and K. Ohkubo, J. Mol. Catal., 1992, 75, L33 CrossRef CAS; (b) S. Sakaki, T. I. Kojima and T. Arai, J. Chem. Soc., Dalton Trans., 1994, 7 RSC.
- (a) H. Inoue and M. Sato, J. Chem. Soc., Chem. Commun., 1983, 983 RSC; (b) H. Inoue and M. Suzuki, J. Chem. Soc., Chem. Commun., 1980, 817 RSC.
- C. Bianchini, A. Meli, M. Peruzzini, P. Frediani, C. Bohanna, M. A. Esteruelas and L. A. Oro, Organometallics, 1992, 11, 138 CrossRef CAS.
- C. Bianchini, E. Farnetti, M. Graziano, M. Peruzzini and A. Polo, Organometallics, 1993, 12, 3753 CrossRef CAS.
- E. J. Daida and J. C. Peters, Inorg. Chem., 2004, 43, 7474 CrossRef CAS.
- (a) S. C. Bart, E. J. Hawrelak, A. K. Schmisseur, E. Lobkovsky and P. J. Chirik, Organometallics, 2004, 23, 237 CrossRef CAS; (b) S. C. Bart, E. J. Hawrelak, E. Lobkovsky and P. J. Chirik, Organometallics, 2005, 24, 5518 CrossRef CAS; (c) R. J. Trovitch, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2006, 45, 7252 CrossRef CAS; (d) S. C. Bart, K. Chlopek, E. Bill, M. W. Bouwkamp, E. Lobkovsky, F. Neese, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13901 CrossRef CAS; (e) R. J. Trovitch, E. Lobkovsky, E. Bill and P. J. Chirik, Organometallics, 2008, 27, 1470 CrossRef CAS.
- K. T. Sylvester and P. J. Chirik, J. Am. Chem. Soc., 2009, 131, 8772 CrossRef CAS.
- S. K. Russell, J. M. Darmon, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2010, 49, 2782 CrossRef CAS.
- (a) G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 849 RSC; (b) G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728 CrossRef CAS.
- (a) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049 CrossRef CAS; (b) L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS; (c) B. L. Small and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 7143 CrossRef CAS.
- C. Bianchini, G. Giambastini, I. G. Rios, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391 CrossRef CAS.
- H. Sugiyama, I. Korobkov, S. Gambarotta, A. Möller and P. H. M. Budzelaar, Inorg. Chem., 2004, 43, 5771 CrossRef CAS.
- M. W. Bouwkamp, A. C. Bowman, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13340 CrossRef CAS.
- A. C. Bowman, C. Milsmann, C. C. H. Atienza, E. Lobkovsky, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 1676 CrossRef CAS.
- P. Phua, L. LeFort, J. A. F. Boogers, M. Tristany and J. G. de Vries, Chem. Commun., 2009, 3747 RSC.
- H. Y. Jang and M. J. Krische, Acc. Chem. Res., 2004, 37, 653 CrossRef CAS.
- S. Enthaler, M. Haberberger and E. Irran, Chem.–Asian J., 2011, 6, 1613 CrossRef CAS.
- A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521 CrossRef CAS.
- S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1407.
- All prices were taken from www.sigmaaldrich.com.
- S. Bell, B. Wüstenberg, S. Kaiser, F. Menges, T. Netscher and A. Pfaltz, Science, 2006, 311, 642 CrossRef CAS.
- (a) R. Crabtree, Acc. Chem. Res., 1979, 12, 331 CrossRef CAS; (b) R. H. Crabtree and M. W. Davis, J. Org. Chem., 1986, 51, 2655 CrossRef CAS.
- The reduction of a tri-substituted functionalised alkene has been reported by Chirik, with 50% conversion after 24 h.
| This journal is © The Royal Society of Chemistry 2011 |