Functionalization of glass substrates: mechanistic insights into the surface reaction of trialkoxysilanes†
Clifford A.
Schlecht
and
Joshua A.
Maurer
*
Department of Chemistry and Center for Materials Innovation, Washington University, One Brookings Drive, St. Louis, MO 63130, USA. E-mail: maurer@wustl.edu; Fax: +1 (314) 935-4481; Tel: +1 (314) 935-4695
First published on 5th October 2011
Abstract
Modification of substrates by trialkoxysilane reagents is the most common method employed for surface functionalization. Classically, the mechanism for surface reactivity in trialkoxysilanes is described as proceeding through a trisilanol intermediate. Here, we present data that contradict this mechanism and propose an alternative mechanism, in which trialkoxysilanes react directly with surface hydroxyl groupsvia an addition–elimination type mechanism.
Modification of glass substrates by trialkoxysilane reagents is the most common method employed for the functionalization of glass surfaces. These functionalized surfaces play increasingly important roles in the development of new industrial applications and research tools for a broad array of areas, ranging from microelectronics to gene chips and biosensors.1,2,3,4 Moreover, alkyl trialkoxysilanes are preferred over alkyl trichlorosilanes for these applications, since the monomers are less reactive and, thus, can incorporate a broader array of functional groups.
The classical mechanism for surface reactivity of trichlorosilanes and trialkoxysilanes involves hydrolysis of the silane reagent to the trisilanol followed by surface reaction and crosslinking (Scheme 1).5,6,7,8 In some cases it has been proposed that the hydrolysis reaction occurs in bulk solution, while in other cases water at the substrate–solvent interface is postulated to hydrolyze a pre-organized monolayer. Despite significant evidence for this mechanism in the case of trichlorosilanes, we believe that hydrolysis of trialkoxysilanes to trisilanols prior to surface reaction is unlikely. Here we present evidence that suggests that a trisilanol intermediate is not involved in the surface reaction of trialkoxysilanes under standard deposition conditions. Furthermore, we suggest an alternative mechanism involving direct reaction of trialkoxysilanes with surface hydroxyl groups. The precedent for our proposed mechanism comes from solution reactions, such as alkoxysilane transesterification and the similarity of silane-containing reactions to phosphorus coupling agents.9 To examine surface reactivity, we use self-assembled monolayers (SAMs) as a probe to elucidate reactivity at the solid–liquid interface, which is difficult to study using classical techniques.
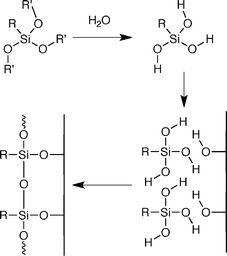 | ||
| Scheme 1 Classical mechanism for the reaction of trichlorosilane and trialkoxysilane molecules with glass substrates. | ||
Surface modification of glass substrates with trialkoxysilanes commonly involves three steps: coating the substrate with the silane dissolved in an organic solvent, annealing the substrate, and washing the substrate to remove unreacted silane. In the case of long-chain trialkoxysilanes, the extent of surface coverage can then be inferred from infrared analysis of the symmetric and asymmetric methylene stretching frequencies. As the silane packing density increases, the resulting monolayer becomes more ordered, which gives rise to shifts in the symmetric and asymmetric methylene stretching frequencies to lower wavenumbers and a corresponding narrowing of peak widths.10,11,12 Furthermore, for trialkoxysilanes, it has been demonstrated that silane surface coverage can be increased by repeating the coating, annealing, and washing steps.
In this study, we examine surfaces prepared from two related long-chain trialkoxysilanes; eicosyltrimethoxysilane and eicosyltriisopropoxysilane (Fig. 1). These silanes contain identical long alkyl chains, but differ substantially in the size of the silane group, with the triisopropoxy group being much larger than the trimethoxy group. Synthetic details for the preparation of these compounds are provided in the ESI.† Despite significant differences in silane group size, based on the reaction mechanism shown in Scheme 1, we would expect these silanes to produce surfaces with similar packing densities.
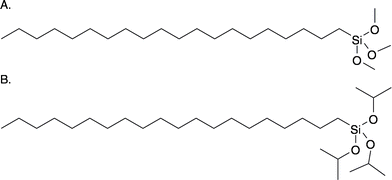 | ||
| Fig. 1 Structure of eicosyltrimethoxysilane (A) and eicosyltriisopropoxysilane (B). | ||
Glass substrates were coated with the silane monomers dissolved in dry tetrahydrofuran, annealed at 150 °C for 15 min, washed with organic solvents to remove unbound material, and analyzed by transmission infrared spectroscopy. This cycle was then repeated multiple times to increase the silane surface density. Fig. 2 shows the positions of the asymmetric methylene stretches for samples as a function of the number of silane coatings and additional data are provided in the ESI.†
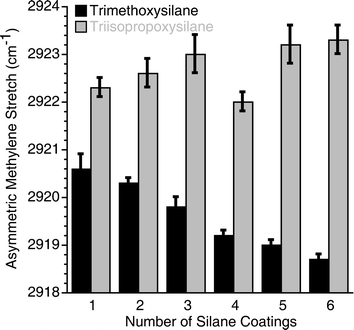 | ||
| Fig. 2 Asymmetric methylene stretching frequencies for surfaces coated with eicosylsilane reagents as a function of coating number. | ||
For the long chain trimethoxysilane, the position of the symmetric and asymmetric methylene stretching frequency shifts to lower wavenumbers as the number of monomer coatings increases, which indicates a denser packing of silane groups on the surface. Whereas, for the long chain triisopropoxysilane, no significant change in the stretching frequency is observed, indicating that additional triisopropoxysilane molecules do not react with the surface after the initial monomer coating reaction. These results do not agree with the classical mechanism of trialkoxysilane reactivity, since this mechanism would involve the formation of an identical intermediate for both the triisopropoxysilane and trimethoxysilane monomers (Scheme 1). Instead these results show that the size of the silane moiety affects surface density, suggesting a direct reaction between surface hydroxyl groups and the silane moiety. To further explore this relationship, we prepared surfaces with a single coating of one silane reagent and back-filled with additional coatings of the other silane reagent. If the initial coating was carried out with the trimethoxysilane and subsequent coatings were carried out with the triisopropoxysilane, no change in ordering was observed upon subsequent coatings. If the initial coating was carried out with the triisopropoxysilane and subsequent coatings were carried out with the trimethoxysilane, then the subsequent coatings resulted in increased surface coverage. The asymmetric methylene stretching frequency shifted from 2922.9 ± 0.2 cm−1, after an initial coating with triisopropoxysilane, to 2920.9 ± 0.2 cm−1 with four subsequent coatings with trimethoxysilane. However, despite a significant increase in surface coverage, these changes are much smaller than those observed for coatings with trimethoxysilane monomers alone (Fig. 2). This suggests that the size of the monomer head group not only influences both the initial surface density, but also affects the “holes” on the substrate that remain after coating. In the case of trimethoxysilane, methanol is lost upon reaction with the surface, which most likely occurs during the annealing step, and the resulting “holes” within the surface are too small to be filled by reactive triisopropoxysilane moieties. While if the initial reaction is carried out with triisopropoxysilane, isopropanol is lost upon surface reaction and the resulting “holes” can accommodate the trimethoxysilane monomer. These observations argue against the formation of a trisilanol intermediate, as proposed in Scheme 1, since this intermediate would lead to identical monolayer structures for the trimethoxysilane and triisopropoxysilane monomers. Furthermore, the rates of trialkoxysilane hydrolysis in solution, as measured by 1H, 13C and 29Si NMR, are extremely slow under neutral conditions, are not very alkoxide dependent, and, in most cases, spontaneous hydrolysis is indetectable.13,14 To fit all of these observations, we propose that trialkoxysilane monomers react directly with glass surfaces, through an addition–elimination type mechanism, with the central silicon atom forming a pentavalent intermediate as shown in Scheme 2. Pentavalent silicon intermediates are well established in main group chemistry15,16 and the proposed reaction mechanism accounts for our striking observations of how silane group size affects surface density.
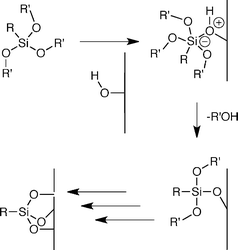 | ||
| Scheme 2 Proposed addition–elimination mechanism for the reaction of trialkoxysilanes with glass substrates. | ||
In summary, this study provides further insight into the mechanism by which trialkoxysilanes react with glass substrates under standard solution deposition conditions. Based on our results, we propose a mechanism that involves direct reaction of a trialkoxysilane with the substrate via an addition–elimination type mechanism. Moreover, the proposed mechanism indicates that the functional group density for silanized surfaces can be controlled by simply altering the size of the trialkoxysilane moiety and provides a starting point for tuning the reactivity of trialkoxysilane monomers.
Acknowledgements
This work was supported in part by the Monsanto-Washington University Plant Biology Research Program and in part by the National Institute of Mental Health (1R01MH085495). Mass Spectral analysis was provided by the Washington University Mass Spectrometry Resource Center, which is partially supported by NIH P41RR00954.References
- S. K. Arya, A. K. Prusty, S. P. Singh, P. R. Solanki, M. K. Pandey, M. Datta and B. D. Malhotra, Anal. Biochem., 2007, 363, 210 CrossRef CAS.
- J. W. F. Robertson, D. J. Tiani and J. E. Pemberton, Langmuir, 2007, 23, 4651 CrossRef CAS.
- S. Petersen, J. M. Alonso, A. Specht, P. Duodu, M. Goeldner and A. del Campo, Angew. Chem., Int. Ed., 2008, 47, 3192 CrossRef CAS.
- X. L. Gao, E. Gulari and X. C. Zhou, Biopolymers, 2004, 73, 579 CrossRef CAS.
- P. Silberzan, L. Leger, D. Ausserre and J. J. Benattar, Langmuir, 1991, 7, 1647 CrossRef CAS.
- J. Sagiv, J. Am. Chem. Soc., 1980, 102, 92 CrossRef CAS.
- S. R. Carino, H. Tostmann, R. S. Underhill, J. Logan, G. Weerasekera, J. Culp, M. Davidson and R. S. Duran, J. Am. Chem. Soc., 2001, 123, 767 CrossRef CAS.
- B. Y. Chow, D. W. Mosley and J. M. Jacobson, Langmuir, 2005, 21, 4782 CrossRef CAS.
- M. B. Smith and J. MarchMarch's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley Interscience, Hoboken, NJ, 2007 Search PubMed.
- A. Y. Fadeev and T. J. McCarthy, Langmuir, 2000, 16, 7268 CrossRef CAS.
- H. Byrd, J. K. Pike and D. R. Talham, Chem. Mater., 1993, 5, 709 CrossRef CAS.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559 CrossRef CAS.
- M.-C. B. Salon, P.-A. Bayle, M. Abdelmouleh, S. Boufi and M. N. Belgacem, Colloids Surf., A, 2008, 312, 83–91 CrossRef.
- F. D. Osterholtz and E. R. Pohl, J. Adhes. Sci. Technol., 1992, 6, 127–149 CrossRef CAS.
- A. R. Bassindall and P. G. Taylor, in The Chemistry of Organic Silicon Compounds, ed. S. Patai and Z. Rappoport, John Wiley & Sons, New York, 1989, pp. 839–892 Search PubMed.
- C. Eaborn, R. Eidenschink and D. R. M. Walton, J. Chem. Soc., Chem. Commun., 1975, 388b RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and infrared spectra in tabular form. See DOI: 10.1039/c1ra00421b/ |
| This journal is © The Royal Society of Chemistry 2011 |