TEMPO-oxidized cellulose nanofibers
Akira
Isogai
*,
Tsuguyuki
Saito
and
Hayaka
Fukuzumi
Department of Biomaterial Sciences, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo, 113-8657, Japan. E-mail: aisogai@mail.ecc.u-tokyo.ac.jp
First published on 19th October 2010
Abstract
Native wood celluloses can be converted to individual nanofibers 3–4 nm wide that are at least several microns in length, i.e. with aspect ratios >100, by TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl radical)-mediated oxidation and successive mild disintegration in water. Preparation methods and fundamental characteristics of TEMPO-oxidized cellulose nanofibers (TOCN) are reviewed in this paper. Significant amounts of C6 carboxylate groups are selectively formed on each cellulose microfibril surface by TEMPO-mediated oxidation without any changes to the original crystallinity (∼74%) or crystal width of wood celluloses. Electrostatic repulsion and/or osmotic effects working between anionically-charged cellulose microfibrils, the ζ-potentials of which are approximately −75 mV in water, cause the formation of completely individualized TOCN dispersed in water by gentle mechanical disintegration treatment of TEMPO-oxidized wood cellulose fibers. Self-standing TOCN films are transparent and flexible, with high tensile strengths of 200–300 MPa and elastic moduli of 6–7 GPa. Moreover, TOCN-coated poly(lactic acid) films have extremely low oxygen permeability. The new cellulose-based nanofibers formed by size reduction process of native cellulose fibers by TEMPO-mediated oxidation have potential application as environmentally friendly and new bio-based nanomaterials in high-tech fields.
 Akira Isogai | Akira Isogai received his PhD in 1985 for work on reactions of cellulose in non-aqueous and homogeneous media from The University of Tokyo. As a post-doc, he joined The Institute of Paper Chemistry, Appleton, USA, working with Prof. Rajai H. Atalla. In 1986, he returned to The University of Tokyo as an Assistant Professor, and started polysaccharide and paper chemistry. He spent 1989 at Forest Products Lab, USDA, Madison, USA as a visiting scientist. He was promoted to Associate and full Professors in 1994 and 2003, respectively. Since 1996 he has worked on TEMPO-mediated oxidation of cellulose and other polysaccharides as the regioselective and effective chemical modification. |
 Tsuguyuki Saito | Tsuguyuki Saito received his PhD from The University of Tokyo in 2008 under the supervision of Prof. Akira Isogai for TEMPO-mediated oxidation of native cellulose. Whilst a graduate student he spent 2005 with Dr Michel R. Vignon in the CERMAV-CNRS, France as a Marie-Curie fellow. He received President and Dean Awards for his sophisticated PhD thesis from The University of Tokyo in 2008. After JSPS postdoctoral study with Prof. Takashi Kato in the Graduate School of Engineering, The University of Tokyo, he again joined Prof. Isogai’s group as an Assistant Professor in 2009. His research interests now involve construction of biomimetic materials using bio-based nanofibers by hierarchical self-assembly. |
 Hayaka Fukuzumi | Hayaka Fukuzumi received her Bachelor and Master degrees in Biomaterial Sciences from The University of Tokyo in 2007 and 2009, respectively. She is currently pursuing a PhD at the Graduate School of Agricultural and Life Sciences, The University of Tokyo, under the supervision of Prof. Isogai. She received Dean Awards for her Bachelor and Master theses from the University of Tokyo. In 2009 she received a Research Fellowship for Young Scientists from the Japan Society for the Promotion of Science (JSPS). Her research is focused on establishing clear relationships between the nano-sized structures in the film and gas-barriers of cellulose and chitin nanofiber films. |
1 Introduction
Nanofibers are defined as nano-sized fibers <100 nm wide or micron-sized fibers with nano-dimension cross sectional structures. Nanofibers have extremely large specific surface areas, the properties of which are sometimes significantly different from those of the bulk materials; therefore, the research and development of nanofibers has been extensively promoted by both academia and industry in the nanotechnology field. Potential applications of nanofibers include efficient catalysts, electro-optical films, nanofiber-reinforced composites, microelectronics, gas-barrier films, cosmetics, flame-resistant materials and other high-tech and high-performance materials.1–4 Recent environmental issues and the creation of recycle-based and sustainable societies has driven the fundamental research and application of bio-nanofibers in this decade.Cellulose is one of the crystalline structural polysaccharides, that accumulates as the most abundant biopolymer present primarily in wood biomass. The architecture of the unique hierarchical structures of cellulose, i.e. linear glucan chains → crystalline cellulose microfibrils 3–4 nm wide consisting of 30–40 cellulose chains → bundles of microfibrils → cell walls → fibers → plant tissue → trees or other plants, comprises hemicellulose and lignin to reinforce the plant living bodies (Fig. 1). Humans have long history of using chemically and mechanically modified celluloses as well as the original cellulose fibers in various fields from commodities such as paper and textile to high-tech materials such as flat panel components in liquid crystal displays, hollow fibers for artificial kidney dialysis, components of medicines, and food additives.
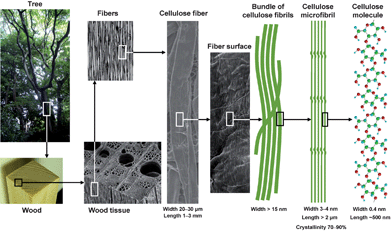 | ||
| Fig. 1 Hierarchical structure of wood biomass and the characteristics of cellulose microfibrils. | ||
Partially or significantly fibrillated cellulose fibers have been used as beaten pulps in papermaking and micro-fibrillated celluloses (MFC), respectively. MFC has been produced from wood pulp/water slurries at the industrial level by repeated high-pressure homogenization treatment,5,6 and has been used as a filter aid and thickener. Generally, high energy consumption is unavoidable for nanofibrillation of wood and other plant celluloses by partial cleavage of numerous numbers of inter-fibrillar hydrogen bonds, and complete individualization of wood cellulose fibers to 3–4 nm wide fibril elements (Fig. 1) without damages has not yet been achieved. Mechanical fibrillation of wood cellulose/water slurries by grinder treatment at the laboratory level has been found to be more effective in both terms of nanofibrillation and energy consumption.79 Wood cellulose fibers for papermaking have been converted to 20–100 nm in wide nanofibers by treatment with a grinder.7–9 Other than wood cellulose fibers, agricultural waste and never-dried wood holocelluloses were studied as resources for the nanofibrillation of cellulose.10–14
Chemistry-assisted nano-fibrillations of various materials, such as wood, cotton, ramie, and bacterial and tunicate celluloses, have been extensively studied at the laboratory level. In most cases, anionically charged functional groups are introduced on the cellulose microfibril surfaces to form strong electrostatic repulsion between cellulose microfibrils in water. A traditionally significant and representative method to introduce charged groups onto cellulose microfibril surfaces is acid hydrolysis with 64% H2SO4 at 45 °C for 1–4 h.15–19 Successive mechanical disintegration of the acid hydrolyzed residues in water using, for example, an ultrasonic homogenizer, provides cellulose nanocrystals. When wood celluloses are subjected to acid hydrolysis followed by sonication, cellulose nanocrystals or nanowhiskers 5–10 nm wide and 50–200 nm long, a part of which form spindle-like bundles, are obtained. However, weight recovery ratios are low at only 30–50%.16Cellulase-pretreatment or partial carboxymethylation of wood celluloses enabled a reduction in energy consumption for nanofibrillation processes in water using refiner and high-pressure homogenizer treatments.20–22 Graft-polymerization of acrylonitrile onto wood celluloses and successive mechanical treatment seems to cause dispersion of partially anionic groups-grafted nanocellulose in water.23MFC, enzymatically or chemically modified MFCs, and acid-hydrolyzed cellulose nanocrystals thus obtained have been studied primarily for applications as fillers in nanocomposites and electro-optical films, and related reviews have been recently published.24,25
Plant cellulose microfibrils can be regarded as bio-based nanofibers with uniform widths, high crystallinity and high aspect ratios originally present in the plant cell walls. However, it has been impossible to isolate completely individualized cellulose microfibrils from plant cellulose fibers without serious damage or yield losses, because numerous hydrogen bonds are present between cellulose microfibrils in cellulose fibers (Fig. 1).
We have developed a new method to prepare completely individualized cellulose nanofibers 3–4 nm wide and at least a few microns long from wood cellulose fibers by 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO)-mediated oxidation under moderate aqueous conditions.26–29 In this paper, fundamental characteristics and applications of TEMPO-oxidized cellulose nanofibers (TOCN) are reviewed with some recently obtained data.
2 TEMPO-mediated oxidation of celluloses
TEMPO and its analogues are water-soluble, commercially-available and stable nitroxyl radicals, most of which have a negative result to the Ames test. Catalytic oxidation using TEMPO has opened a new field of efficient and selective conversion chemistry of alcoholic hydroxyl groups to aldehydes, ketones and carboxyl groups under mild conditions. Many related studies have been extensively carried out in the last two decades, and have been reviewed in detail.30,31 Particularly, de Nooy et al. first applied TEMPO-mediated oxidation to water-soluble polysaccharides such as starch, amylodextrin and pullulan for regioselective conversion of C6 primary hydroxyls to carboxylate groups.32 In this system, catalytic amounts of TEMPO and NaBr were dissolved in polysaccharide solutions at pH 10–11, and oxidation was started by the addition of NaClO solution as a primary oxidant. The efficient conversion of primary hydroxyl groups to carboxylates viaaldehydes is hypothesized to proceed according to the mechanism shown in Fig. 2.33,34 Various TEMPO-mediated oxidation reactions of mono-, oligo- and polysaccharides for regioselective conversion of primary hydroxyls to carboxylate groups have been reviewed elsewhere.35,36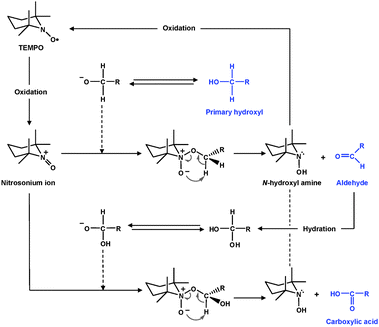 | ||
| Fig. 2 TEMPO-mediated oxidation of primary hydroxyls to carboxyl groupsviaaldehydes.32 | ||
According to the scheme shown in Fig. 3, the C6 primary hydroxyls of cellulose are expected to be oxidized to C6 carboxylate groups by TEMPO/NaBr/NaClO oxidation in water at pH 10–11. The oxidation process can be monitored from the pattern of aqueous NaOH consumption, which is continuously added to the reaction mixture to maintain the pH at 10 during the oxidation. When TEMPO/NaBr/NaClO oxidation was applied to native celluloses such as cotton linters, bleached kraft pulps and bacterial cellulose, even under harsh oxidation conditions or for extended reaction times, almost no or only small amounts of water-soluble products were obtained.37 On the other hand, slurries of regenerated and mercerized celluloses, and ball-milled and liquid NH3-treated native celluloses became clear solutions as the oxidation proceeded; water-soluble oxidized products were obtained in these cases.37–39 The oxidized products had almost homogeneous chemical structures of sodium (1 → 4)-β-D-polyglucuronate or Na salt of cellouronic acid (CUA) consisting of D-glucuronosyl units alone. Hence, the C6 primary hydroxyls of celluloses can be entirely and selectively converted to C6 sodium carboxylate groups by TEMPO-mediated oxidation.37–39 It was confirmed that C6-carboxylate groups are formed by not only the oxidized TEMPO but also NaBrO and/or NaClO present in the TEMPO/NaBr/NaClO system at pH 10 (Fig. 3).40
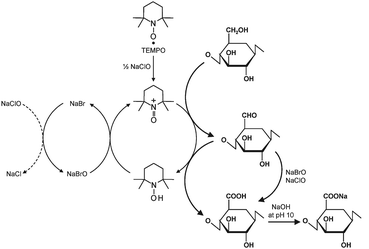 | ||
| Fig. 3 Regioselective oxidation of C6 primary hydroxyls of cellulose to C6 carboxylate groups by TEMPO/NaBr/NaClO oxidation in water at pH 10–11. | ||
However, weight-average degrees of polymerization (DPw) for CUA prepared by TEMPO/NaBr/NaClO oxidation at pH 10 ranged from 40 to 80, and were far lower than those of the original celluloses (DPw 380–1200) used as starting materials.38,41 Two routes for the depolymerization of CUA are plausible. One route is β-elimination due to C6 aldehyde groups formed as intermediate structures (Fig. 3) under alkaline conditions,42–44 and another is cleavage of the (1 → 4)-β-glycoside bonds of CUA by hydroxyl and/or other active radical species formed in situ as side reactions during TEMPO-mediated oxidation.45
TEMPO/NaClO/NaClO2 oxidation systems in aqueous–organic solvents have been proposed as an alternative, where catalytic amounts of TEMPO and NaClO are combined with NaClO2 as the primary oxidant.46 This TEMPO/NaClO/NaClO2 oxidation system (Fig. 4) was applied to a commercial regenerated cellulose with a viscosity average DP (DPv) of 680 suspended in a buffer at pH 4.8–6.8. CUAs with DPw values higher than 490 were quantitatively obtained as water-soluble products in this case.47 Almost pure CUA was obtained by the TEMPO/NaClO/NaClO2 oxidation at pH 4.8, according to the nuclear magnetic resonance (NMR) spectrum (Fig. 5).47CUA is significantly resistant to acid hydrolysis, but susceptible to β-elimination under alkaline conditions. These properties are opposite to those of the original celluloses, which are susceptible to acid hydrolysis, but are relatively stable under alkaline conditions.48
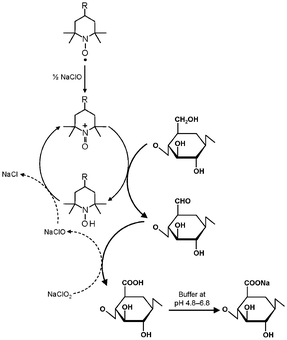 | ||
| Fig. 4 Selective oxidation of C6 primary hydroxyls of cellulose to carboxylate groups by TEMPO/NaClO/NaClO2 oxidation in water at pH 4.8–6.8. | ||
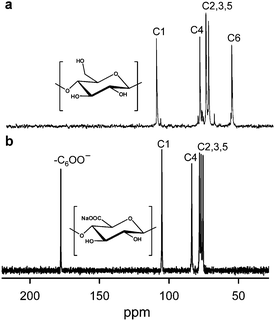 | ||
| Fig. 5 13C NMR spectra of (a) cellulose oligomers dissolved in DMSO-d6 and (b) CUA with DPw 490 dissolved in D2O. CUA was prepared by TEMPO/NaClO/NaClO2 oxidation in water at pH 4.8.47Reproduction of the lower NMR spectrum from ref. 47 with permission from Elsevier (© Elsevier 2009). | ||
When a thin CUA film was formed on a poly(ethylene terephthalate) (PET) film by a coating method, the oxygen permeability at 30 °C and 70% relative humidity decreased from 15000 mL m−2 day−1kPa−1 for the base PET film to 100–300 mL m−2 day−1kPa−1 (Table 1).49 Thus, oxygen barrier properties appear on the thin CUA layer-containing PET films.
| O2 permeability/mL m−2 day−1kPa−1 | |
|---|---|
| PET film (Blank) | 15000 |
| PET film coated with: | |
| CUA-COONa | 240 |
| CUA-COOH | 120 |
| Sodium alginate | 4190 |
| Pectin | 1520 |
| Carboxymethyl cellulose Na salt | 10000 |
| Hydroxyethyl cellulose | 1430 |
| Chitosan | 11200 |
| Water-soluble starch | 3000 |
| Poly(vinylidene chloride) | 200–1000 |
| Ethylene vinyl alcohol copolymer | 100 |
| Poly(vinyl alcohol) | <100 |
| Nylon | 4000 |
Various TEMPO-mediated oxidation systems and conditions have been applied to cellulose, chitin, curdlan and other mono- and polysaccharides in terms of regioselectivity at the C6 primary hydroxyls, oxidation efficiency, suppression of depolymerization or side reactions, and utilization of non-chlorine primary oxidants.50–71
3 Biodegradation of TEMPO-oxidized cellulose
Even though CUA is a new sodium polyglucuronate artificially prepared from cellulose by TEMPO-mediated oxidation, depolymerization of CUA occurs by commercial crude cellulase treatment. Because pure endo- or exo-type cellulases cannot depolymerize CUA at all, some enzymes present as contaminants in the commercial cellulases may participate in the depolymerization of CUA.72,73 Moreover, CUA is easily metabolized to CO2 and water by microorganisms present in natural soils, and both endo- and exo-type CUA lyases (CUL-I and CUL-II, respectively) have been isolated and purified from a bacterial strain Brevundimonas sp. SH203 present in nature (Fig. 6).74,75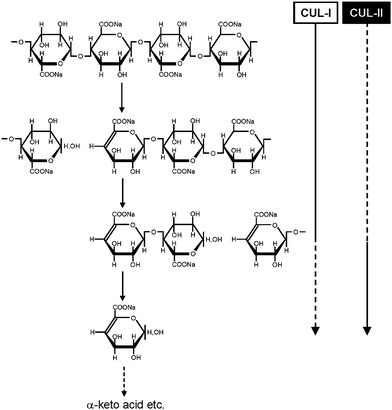 | ||
| Fig. 6 Schematic degradation system of CUA by endo- and exo-type CUA lyases (CUL-I and CUL-II, respectively).74,75Reproduction of illustration from ref. 75 with permission from Springer (© Springer 2008). | ||
The cDNA encoding CUA lyase produced by Trichoderma reesei, a filamentous fungus, was cloned and the recombinant enzyme was heterologously expressed in Pichia pastoris. The recombinant TrichodermaCUA lyase (TrCUL-I) degrades CUA endolytically by β-elimination.76 The three-dimensional structure of TrCUL-I was defined from its crystals (Fig. 7), showing that a calcium ion is bound to the site far from the cleft and appears to contribute to the stability.77
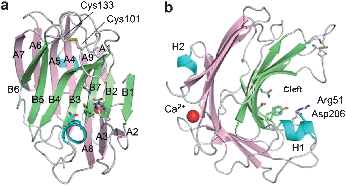 | ||
| Fig. 7 Overall structure of TrCUL-I. (a) Front and (b) side view of the ribbon representation.77Reproduction of images from ref. 77 with permission from Elsevier (© Elsevier 2009). | ||
CUAs are both biodegradable and metabolizable, and have relatively low-molecular weights, so they have potential application as new biodegradable polymeric builders in detergents to capture calcium ions in laundry water.78
4 TEMPO-mediated oxidation of native celluloses
When TEMPO/NaBr/NaClO oxidation at pH 10 and room temperature is applied to native celluloses with the cellulose I crystal structure, such as cotton linters, bleached kraft and sulfite pulps (wood celluloses), softwood thermomechanical pulp, bacterial cellulose and ramie fiber, the original fibrous morphologies are unchanged even after oxidation with sufficient amounts of reagents. However, significant amounts of sodium carboxylate groups and small amounts of aldehyde groups are formed in the products by oxidation of a part of C6 primary hydroxyls.26–29,79–90 The carboxyl content of the TEMPO-oxidized celluloses has been determined by the electric conductivity titration method.80,84Aldehyde groups are selectively converted to carboxyl groups by NaClO2 treatment in water at room temperature and pH 4–5 for 1 day, and carboxyl groups additionally formed by NaClO2 oxidation are regarded as aldehyde groups in the TEMPO-oxidized celluloses.80,83,86,91A bleached softwood kraft pulp for papermaking, one of the wood celluloses that contains approximately 90% cellulose and 10% hemicelluloses, was oxidized using the TEMPO/NaBr system in water at pH 10 with various amounts of NaClO as the primary oxidant (Fig. 8). The sodium carboxylate content increased from 0.01 to 1.70 mmol g−1, i.e. a 170-fold increase, by the oxidation with NaClO of 10 mmol g−1-pulp, while small amounts of aldehyde groups were present in the oxidized celluloses.
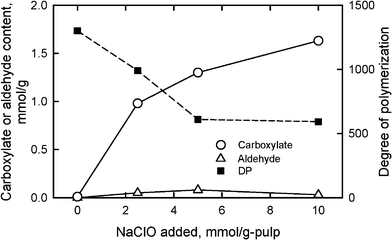 | ||
| Fig. 8 Relationships between the amount of NaClO added in the TEMPO/NaBr/NaClO oxidation of bleached softwood kraft pulp at pH 10 and room temperature, and either carboxylate and aldehyde contents or degree of polymerization of the oxidized pulps. | ||
Because the TEMPO-oxidized celluloses containing both glucosyl and glucuronosyl units are totally soluble in 0.5 M copper ethylenediamine hydroxide (cuen) solution, DPv values can be roughly estimated by calculation from the intrinsic viscosities using the conventional Mark–Houwink–Sakurada equation.92 If small amounts of C6 aldehyde groups are present in the TEMPO-oxidized celluloses, the viscosity method would provide lower DPv values than the accurate values by β-elimination, which takes place at the C6 aldehyde groups in the alkaline cuen solution. Thus, additional oxidation with NaClO2 in water at pH 4–5 for selective conversion of C6 aldehydes to C6 carboxyl groups is required before the viscosity measurement to obtain more accurate DPv values. Fig. 8 shows that the DPv values decreased from 1200 to approximately 600 by TEMPO-mediated oxidation with NaClO of 5–10 mmol g−1 pulp. Thus, significant depolymerization is inevitable during the TEMPO/NaBr/NaClO oxidation of native celluloses in water at pH 10.
Moreover, trace amounts of C2 or C3 ketones are formed as side reactions, depending on oxidation conditions and reactivity of TEMPO derivatives used. The ketone and aldehyde groups formed in the TEMPO-oxidized celluloses cause brown discoloration by heating, though the amounts are quite small. Relationships between oxidation conditions and side reactions have been studied for mono- or disaccharides,36,51–53 but not yet for cellulose. These issues are now under investigation.
Oxidation using another system, TEMPO/NaClO/NaClO2, in water at pH 6.8 and 60 °C suppressed the depolymerization of the oxidized celluloses to some extent (Fig. 9).29,93DPv values of more than 1000 were obtained from the oxidized celluloses from the same bleached softwood kraft pulp in Fig. 8. Even though no aldehyde groups were present, carboxylate contents were lower than 1 mmol g−1; the efficiency for the formation of carboxylate groups by this oxidation system is somewhat lower than for TEMPO/NaBr/NaClO oxidation at pH 10.
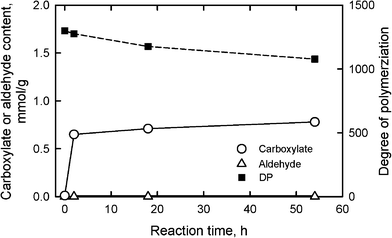 | ||
| Fig. 9 Relationships between the reaction time of bleached softwood kraft pulp under TEMPO/NaClO/NaClO2 oxidation at pH 6.8 and 60 °C, and either carboxylate and aldehyde contents or degree of polymerization of the oxidized pulps. | ||
Fig. 8 and 9 show that significant amounts of carboxylate groups were formed in the TEMPO-oxidized wood celluloses. Nevertheless, the original crystal structure of cellulose I, the crystallinity indices and the crystal sizes were unchanged after the oxidation (Fig. 10).27,29,80 These results indicate that the carboxylate groups formed by TEMPO-mediated oxidation are selectively present on cellulose microfibril surfaces without any internal cellulose crystallites. Solid-state 13C NMR spectra of TEMPO-oxidized celluloses also support the hypothesis that only C6 primary hydroxyls exposed on the original cellulose microfibrils are converted to carboxylate groups by oxidation.29,94
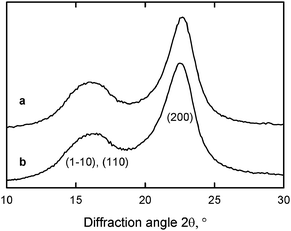 | ||
| Fig. 10 X-ray diffraction patterns of (a) the original wood cellulose with carboxylate content of 0.01 mmol g−1 and (b) TEMPO-oxidized wood cellulose with carboxylate content of 1.70 mmol g−1.29Reproduction of this figure from ref. 29 with permission from American Chemical Society (© American Chemical Society 2009). | ||
When various native celluloses having different microfibril widths were treated with an excess amount of NaClO in the TEMPO-mediated oxidation, the maximum contents of C6-oxidized groups (both carboxylate and aldehyde groups) that were formed varied depending on the cellulose I crystal widths. The relationship between the microfibril width of various native celluloses and either calculated content of C6 primary hydroxyls exposed on the microfibril surfaces or the experimentally-obtained maximum content of C6-oxidized groups are plotted in Fig. 11.95 The dashed line is calculated from the crystal widths of cellulose I determined from X-ray diffraction patterns.
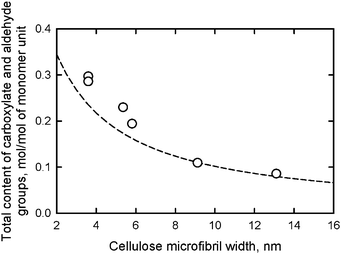 | ||
| Fig. 11 Relationships between the microfibril width of various native celluloses and the total content of carboxylate and aldehyde groups formed in the corresponding TEMPO-oxidized native celluloses. The dashed line shows the relationship between microfibril width and the content of C6 primary hydroxyls exposed on the surfaces of cellulose I crystallites.95Reproduction of the figure from ref. 95 with permission from American Chemical Society (© American Chemical Society 2010). | ||
The results in Fig. 11 reveal that the calculated amounts of surface C6 primary hydroxyls were in good agreement with the total contents of C6-oxidized groups experimentally determined; smaller microfibril widths of the native cellulose resulted in more oxidized groups being formed on the microfibril surfaces by TEMPO-mediated oxidation. Thus, TEMPO-mediated oxidation selectively converts C6 primary hydroxyls exposed on the surfaces of crystalline cellulose microfibrils to sodium carboxylate groups. Every one of two glucosyl units in the cellulose chains on the microfibril surface are converted to C6 carboxylate groups by oxidation (Fig. 12).27,29,80,94,95
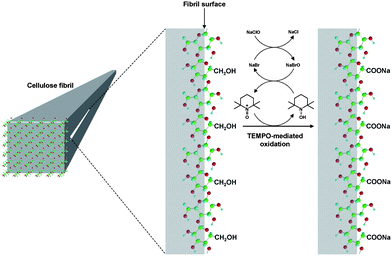 | ||
| Fig. 12 Schematic model for the oxidation of C6 primary hydroxyls on native cellulose microfibril surfaces to C6 carboxylate groups using the TEMPO/NaBr/NaClO system.95Reproduction of illustration from ref. 95 with permission from American Chemical Society (© American Chemical Society 2010). | ||
Thus, TEMPO-mediated oxidation is a type of regioselective surface modification of crystalline native cellulose microfibrils, and allows the formation of anionic carboxylate groups with quite high densities on the microfibril surfaces. Moreover, when the TEMPO-oxidized native celluloses with maximum carboxylate contents were extracted with aqueous NaOH, regularly alternating copolysaccharides consisting of glucosyl and glucuronosyl units were obtained as water-soluble fractions by surface-peeling. This result also justified the surface structures of TEMPO-oxidized native celluloses shown in Fig. 12.96
In the case of the TEMPO/NaBr/NaClO oxidation of lignin-containing samples such as softwood thermomechanical pulp (TMP), lignin and hemicelluloses in TMP were removed as water-soluble degraded fractions as NaClO consumption proceeded.89,90 TEMPO-oxidized cellulose was then obtained from TMP as a water-insoluble fraction by filtration of the oxidation mixture. Even though additional amounts of NaClO and extended oxidation times were required for complete TMP oxidation, compared with the oxidation of wood celluloses such as bleached kraft pulps, TEMPO-oxidized native celluloses with carboxylate contents of 1–1.5 mmol g−1 were obtained as water-insoluble products, the weight recovery ratios of which were approximately 40%.90
Significant amounts of sodium carboxylate groups are present in the TEMPO-oxidized native celluloses, which is different from the original native celluloses, so that the hydrophilic nature, thermal degradation behavior and other properties of TEMPO-oxidized celluloses can be controlled to some extent by various metal-ion exchange, methyl esterification and other chemical modification of the carboxylate groups.81,97,98
5 Preparation of TEMPO-oxidized cellulose nanofibers dispersed in water
When the carboxylate contents formed from the C6 primary hydroxyl groups of bleached hardwood and softwood kraft pulps were more than approximately 0.8 mmol g−1, the oxidized cellulose/water slurries were mostly converted to transparent and highly viscous dispersions by mechanical disintegration treatment using a household blender, double-cylinder-type homogenizer or ultrasonic homogenizer for 2–10 min.26,27,29,90,95Transmission electron microscopy (TEM) observation revealed that the dispersions consisted of mostly individualized TOCN of 3–4 nm in width and a few microns long (Fig. 13a–c). This was the first time that wood cellulose fibers 20–40 μm in width were fined down to the smallest elements next to cellulose molecules, consisting of approximately 30–40 cellulose chains, i.e. individual cellulose nanofibers with high aspect (length/width) ratios of more than 100 and in high yields (>90%), were found by TEMPO-mediated oxidation followed by mild mechanical disintegration of the oxidized wood celluloses in water. | ||
| Fig. 13 TEM images (a–c) of a dried dispersion of TEMPO-oxidized hardwood cellulose with carboxylate and aldehyde contents of 0.8 and 0.0 mmol g−1, respectively, observed by diffraction-contrast method in the bright-field mode using a low-dose exposure. The insets show a typical dispersion (d) and (e) that observed between cross polarizes.29Reproduction of image a from ref. 29 with permission from American Chemical Society (© American Chemical Society 2009). | ||
The nanofiber widths are almost constant, irrespective of wood species examined so far, whereas the lengths are widely distributed and varied depending on the oxidation and disintegration conditions. Even though no intrinsic differences were found between never-dried and once-dried wood pulps for the preparation of nano-dispersions, never-dried wood pulps were converted to transparent dispersions within shorter times and in higher yields when the amount of NaClO added was reduced to 3 mmol g−1 pulp or lower.27 The presence of birefringence (Fig. 13e) is usually taken as an indication of individual nanofiber dispersion.99 Other native celluloses such as cotton linters, ramie, and bacterial, tunicate and algal celluloses were also converted to individual nanofibers by TEMPO-mediated oxidation, while the optimum oxidation conditions to provide nanofibers in high yields varied between the original celluloses used as the starting materials.26,95
When the amount of NaClO added to the TEMPO/NaBr/NaClO oxidation of a bleached hardwood kraft pulp at pH 10 varied from 2.5 and 5 mmol g−1-pulp, the carboxylate content formed was correspondingly varied from 1.0 to 1.5 mmol g−1. The transmittance of the aqueous dispersions prepared under the same moderate mechanical disintegration conditions varied from translucent to highly transparent (Fig. 14). Hence, carboxylate content is the key factor influencing the degree of nanofiber conversion of TEMPO-oxidized celluloses. The UV-vis transmittance spectrum in Fig. 14a shows that the transmittance is wavelength-dependent and related to the nanofiber widths.100–102
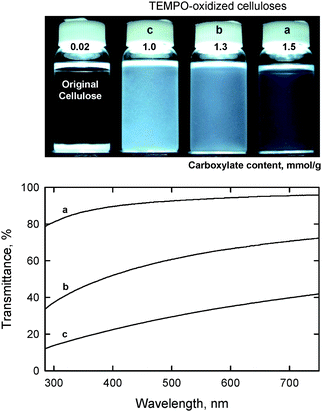 | ||
| Fig. 14 Dispersion states and UV-vis transmittance of TEMPO oxidized hardwood celluloses with different carboxylate contents.27Reproduction of both photograph and figure from ref. 29 with permission from American Chemical Society (© American Chemical Society 2007). | ||
Unfibrillated and partly fibrillated fractions, if present, can be removed from the dispersions at approximately 0.1% consistency by centrifugation at 12,000 gravity for 20 min. The degrees of individualization or yields of individual TOCNs are thus estimated from the weight ratios of the precipitates separated from dispersions by centrifugation. In the case of TEMPO/NaBr/NaClO oxidation at pH 10, yields of individual hardwood TOCN were slightly lower than those for softwood TOCN with the same carboxylate contents of approximately 1 mmol g−1. Relatively hydrophobic xylan, which has no C6 primary hydroxyls and may be present to some extent in the TEMPO-oxidized hardwood celluloses, would probably lead to the lower nanofiber yields. When carboxylate content was more than 1.5 mmol g−1, the nanofiber yields for both hardwood and softwood TEMPO-oxidized celluloses prepared using the TEMPO/NaBr/NaClO system at pH 10 were more than 95%.
Fig. 15 shows TEM images of hardwood TOCN with carboxylate contents of 1.0 and 1.5 mmol g−1. The former cellulose nanofibers clearly formed aggregates, resulting in low-light transmittance of the dispersion. In contrast, the TEMPO-oxidized cellulose with a carboxylate content of 1.5 mmol g−1 was mostly converted to individual nanofibers with almost uniform widths of 3–4 nm. Nanofiber widths that are smaller than the wavelength of visible light cause the dispersion to be almost transparent. The large amounts of anionically charged sodium carboxylate groups present in high densities on the TOCN surfaces may cause the formation of such sufficiently individualized and long cellulose nanofibers by electrostatic repulsion and/or osmotic effects in water.27
 | ||
| Fig. 15 TEM images of dried dispersions of TEMPO-oxidized hardwood celluloses with carboxylate contents of 1.0 and 1.5 mmol g−1, observed by negative staining with uranyl acetate. The insets show the corresponding aqueous dispersions.27Reproduction of images from ref. 27 with permission from American Chemical Society (© American Chemical Society 2007). | ||
Fig. 16 shows ζ-potentials and carboxylate contents of the various TEMPO-oxidized native celluloses presented in Fig. 11. Although the oxidized celluloses had clearly different carboxylate contents that were dependent on the crystalline microfibril widths of the native celluloses, all the TOCNs had similar ζ-potentials of approximately −75 mV. This indicates that all the TEMPO-oxidized celluloses have almost the same density of carboxylate groups on the surfaces of crystalline cellulose microfibrils, because the ζ-potentials are directly related to the surface density of dissociated carboxyl groups. The average density of C6 primary hydroxyls exposed on the crystallite surfaces was calculated to be approximately 1.7 groups nm−2 from the surface structures of cellulose I crystallites.103
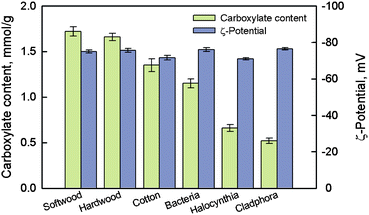 | ||
| Fig. 16 Carboxylate contents of various TEMPO-oxidized celluloses and ζ-potentials of the individualized TOCNs dispersed in water.95Reproduction of figure from ref. 95 with permission from American Chemical Society (© American Chemical Society 2010). | ||
The nanofiber yield, light-transmittance and viscosity of the TEMPO-oxidized cellulose/water dispersions were dependent on the apparatus and conditions used during mechanical treatment. In small-scale experiments at the laboratory level, one of the following apparatus, or a combination of two or three apparatus, can be used for mechanical disintegration of TEMPO-oxidized celluloses in water: magnetic stirrer, household mixer, double-cylinder-type homogenizer and ultrasonic homogenizer. For any of these disintegration methods, nanofiber yields of more than 95% were achieved for TEMPO-oxidized celluloses having carboxylate contents greater than a necessary level, although the time required for disintegration to achieve the maximum nanofiber yield was significantly different.
The TOCN/water dispersions had similar shear-thinning behavior, irrespective of the mechanical disintegration methods used. However, the viscosities of the obtained dispersions were different and dependent on the disintegration methods, and were generally in the order, magnetic stirring > household mixer treatment > ultrasonic homogenizer treatment. Probably, the ultrasonic homogenizer treatment efficiently converts TEMPO-oxidized celluloses to individual nanofibers in the shortest disintegration time. Mechanical disintegration times for magnetic stirring, household mixer and ultrasonic homogenizer treatments to reach nanofiber yields of more than 95% were, for example, ca. 10 days, 4 min and 2 min, respectively. TOCN/water dispersions prepared by ultrasonic homogenizer treatment generally had the highest nanofiber yields, although the viscosities were relatively low. The rheological properties of TOCN/water dispersions have been studied in detail.27,104
Once such transparent dispersions of TOCNs in water were obtained, the transparencies were maintained for more than 6 months at approximately 4 °C in a refrigerator. However, because TOCNs are biodegradable, the viscosities of dispersions without small drops of ethanol or the addition of a suitable fungicide may decrease to some extent with increasing storage time. Transparent TOCN/water dispersions sometimes turn to mould within several days when exposed to air at room temperature.
There are two advantageous key points to produce TOCNs at the industrial level for future utilization as new bio-based nanofibers in high-tech fields. One is that the original fibrous morphologies of wood celluloses such as bleached kraft and sulfite pulps are maintained at the micron level without serious swelling in water before and after TEMPO-mediated oxidation, even though significant amounts of hydrophilic sodium carboxylate groups are formed in the TEMPO-oxidized wood celluloses (Fig. 17).27 Thus, after TEMPO-mediated oxidation, the oxidized wood cellulose fibers can be thoroughly washed with water by simple suction filtration, and then squeezed to high solid consistencies for transportation to a mill, where mechanical disintegration of the TEMPO-oxidized cellulose in water is available on site to produce nanofiber/water dispersions. If partial dispersion to individual nanofibers takes place during the TEMPO-mediated oxidation, it is difficult and costly to separate the nanofibers from the chemicals and other contaminants present in the aqueous oxidation mixtures. Moreover, the transportation of aqueous TOCN dispersion with quite low solid consistencies would be costly and energy-consumptive.
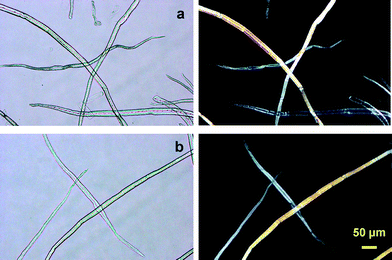 | ||
| Fig. 17 Optical micrographs of the (a) original and (b) TEMPO-oxidized hardwood celluloses with carboxylate contents of 0.05 and 1.5 mmol g−1, respectively. Images were taken with or without cross polarizers.27Reproduction of images from ref. 27 with permission from American Chemical Society (© American Chemical Society 2007). | ||
The other advantageous point is that inexpensive wood celluloses such as bleached kraft and sulfite pulps are the most suitable resources for the efficient production of TOCNs. These pulps have been produced on a large scale from wood chips at the industrial level using environmentally friendly, effective and lower energy consumptive pulping and bleaching processes. These pulps generally contain 85–95% cellulose and correspondingly 15–5% disordered hemicelluloses, which are present in the spaces between cellulose microfibrils. During the TEMPO-mediated oxidation of wood celluloses, the oxidized TEMPO molecules or nitrosonium ions should penetrate into the spaces between cellulose microfibrils and oxidize every one of two glucosyl units in the cellulose chains present on the microfibril surfaces. The hemicelluloses present between cellulose microfibrils form sponge-like layers, into which the oxidized TEMPO molecules can easily penetrate during oxidation (Fig. 18).
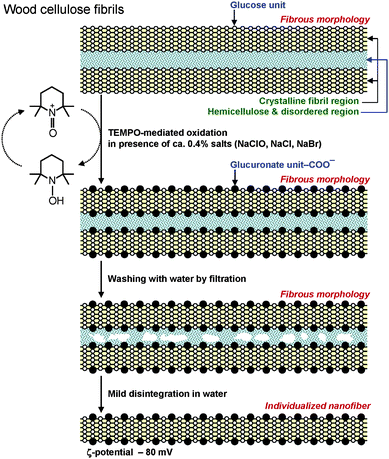 | ||
| Fig. 18 Schematic model of sponge-like hemicelluloses present in the spaces between cellulose microfibrils, and their role in the TEMPO-mediated oxidation of wood celluloses. | ||
A representative TEMPO/NaBr/NaClO oxidation process performed at pH 10 for the preparation of TEMPO-oxidized wood celluloses is illustrated in Fig. 19. The expensive TEMPO used as the catalyst can be recycled and reused by desalination of the washing effluent. Thus, it is possible to significantly reduce the production cost of TEMPO-oxidized celluloses. The reuse of once-used TEMPO in washing effluents was studied to establish a TEMPO-recycling system.105
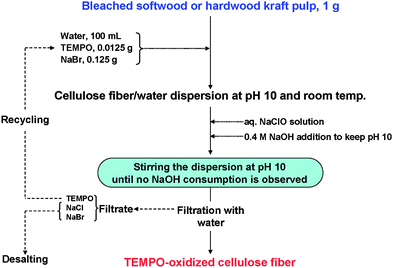 | ||
| Fig. 19 Representative process to prepare TEMPO-oxidized wood celluloses by the TEMPO/NaBr/NaClO system in water at pH 10. | ||
Fig. 20 shows the relationships between carboxylate content and nanofiber yield of TEMPO-oxidized hardwood celluloses prepared using TEMPO and its analogues by TEMPO/NaBr/NaClO oxidation in water at pH 10. From the catalyst compounds examined to date, TEMPO, 4-acetamido-TEMPO and 4-methoxy-TEMPO provide high and similar carboxylate contents and nanofiber yields.106 The stability of the TEMPO analogues to NaClO is one of the factors that influenced the results shown in Fig. 20,107 while the redox potentials of the TEMPO analogues seem to have no correlation to the results.
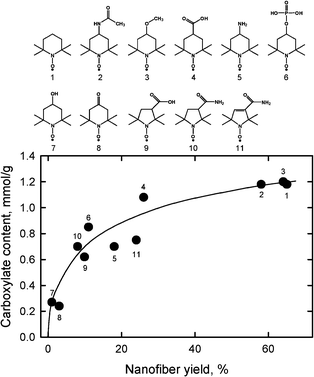 | ||
| Fig. 20 Relationship between carboxylate content and nanofiber yield of TEMPO-oxidized hardwood celluloses prepared using TEMPO and its analogues by TEMPO/NaBr/NaClO oxidation in water at pH 10.106Reproduction of figure from ref. 106 with permission from Elsevier (© Elsevier 2010). | ||
1) TEMPO/NaBr/NaClO oxidation in water at pH 10 and room temperature and 2) TEMPO/NaClO/NaClO2 oxidation in water at pH 6.8 and 60 °C yield mostly individualized wood TOCN/water dispersions by mild mechanical disintegration treatment of the oxidized celluloses in water. TEMPO-electromediated oxidation without the use of any chlorine-containing chemicals is also applicable to the preparation of individualized TOCNs, although longer oxidation times are required.40 Other TEMPO-mediated oxidation systems, such as TEMPO/peroxidase/H2O2 and TEMPO/laccase/O2 systems,37,65 are ineffective for the oxidation of wood celluloses, and the resulting carboxylate contents in the oxidized products were lower than 0.3 mmol g−1, even after oxidation for more than 4 days.
Commercially available MFC has been produced from wood cellulose/water slurries by repeated high-pressure homogenizer treatments.5,6Cellulose nanocrystals (CNC) or cellulose nanowhiskers (CNW) have been prepared from wood and other native celluloses by acid hydrolysis using, for example, 64% H2SO4, and successive mechanical disintegration of the hydrolyzed residues in water.15–19 Small amounts of sulfate ester groups (<0.3 mmol g−1) are formed on the CNC surfaces, so that electrostatic repulsion formed between CNC whiskers leads to partial nano-dispersion in water. An optical micrograph of MFC, and typical TEM images of wood TOCN27 and CNC16 are depicted in Fig. 21.
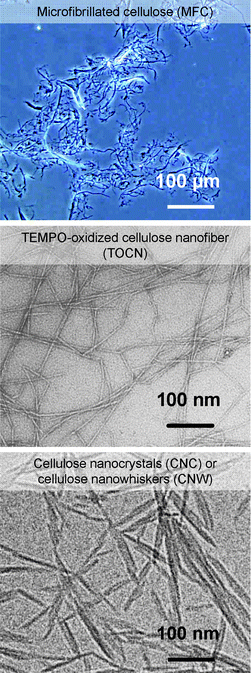 | ||
| Fig. 21 Optical micrograph of MFC, and TEM images of wood TOCN27and CNC.16Reproduction of the TOCN image from ref. 27 with permission from American Chemical Society (© American Chemical Society); reproduction of the CNC image from ref. 16 with permission from Springer (© Elsevier 1998). | ||
Significant amounts of clearly unfibrillated bundles and fibers are still present in MFC. CNC whiskers have spindle-like morphologies due to the partial removal of cellulose molecules by acid hydrolysis. The TEM image of CNC shows that CNC whiskers form some bundles by incomplete individualization of cellulose microfibrils, which is probably due to an insufficient sulfate ester content of less than 0.3 mmol g−1.16 Differences in the preparation method, yield and other properties and factors of MFC, TOCN and CNC (or CNW)16 are summarized in Table 2. The high aspect ratios, uniform widths and complete nano-dispersion of individual TOCNs are different from the others, which clearly indicates the advantages of TOCN for applications as new bio-based nanofibers in high-tech fields.
| TOCN | MFC | CNC or CNW | |
|---|---|---|---|
| Preparation method | TEMPO-mediated oxidation of wood cellulose, and mild disintegration of the oxidized cellulose in water | Repeated high-pressure homogenizer treatment of wood cellulose/water slurry | Acid hydrolysis of wood cellulose with 64% H2SO4, and disintegration of the residues in water |
| Yield | >90% | ∼100% | <50% |
| Morphology | Uniform width of 3–4 nm, <2–3 μm in length | Uneven width of 10–2000 nm forming bundles | Uneven width of 5–10 nm, <300 nm in length, spindle-like whiskers forming partial bundles |
| Energy consumption in nano-conversion | <7 MJ kg−1 | 700–1400 MJ kg−1 | <7 MJ kg−1 |
| Potential applications | High gas-barrier films for packaging & display, fine separation filters, health care materials, nanofibers for composites | Filter aid nanofiber for composites, thickeners | Nanofiller for composites |
6 TEMPO-oxidized cellulose nanofiber films and composites
Self-standing films with sufficient light transparency and flexibility were prepared from softwood and hardwood TOCN/water dispersions. UV-vis transmittance in Fig. 22 show that the film prepared from hardwood cellulose had lower transmittance, probably due to the presence of a small amount of hydrophobic xylan in the film.28 The refractive index of the TOCN film was 1.545 ± 0.002 at room temperature, and this value is consistent with those previously reported for cellulose.108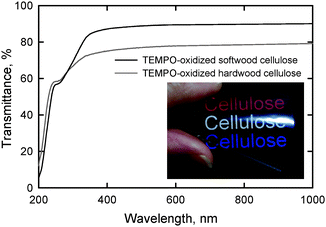 | ||
| Fig. 22 UV-vis transmittance of nanofiber films prepared from TEMPO-oxidized softwood and hardwood celluloses. The photograph shows the light transmittance behavior of the nanofiber film prepared from TEMPO-oxidized softwood cellulose.28Reproduction of image and figure from ref. 28 with permission from American Chemical Society (© American Chemical Society 2009). | ||
The densities and moisture contents of TOCN films conditioned at 23 °C and 50% relative humidity were approximately 1.45–1.47 g cm−3 and 13–15%, respectively. The tensile strengths and elastic moduli of the films were 200–300 MPa and 6–7 GPa, respectively,28 which were higher than those of cellophane films (Fig. 23). Both the tensile strength and elastic modulus of a poly(vinyl alcohol) (PVA) film were remarkably improved by 20% TOCN addition. However, similar results would be expected by the addition of only a few % TOCN if each TOCN is present individually in the TOCN/PVA composite film.1 Therefore, distribution control of individualized TOCN to obtain more homogeneous distribution in a composite is important to achieve the effect of high aspect ratio TOCNs on the mechanical properties.
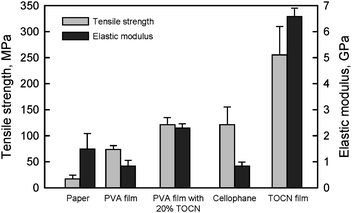 | ||
| Fig. 23 Tensile strength and elastic modulus of TOCN film, 20% TOCN/PVA composite film and others. | ||
Moreover, because sodium carboxylate groups abundantly present on the TOCN surfaces cannot form hydrogen bonds, it may be possible to further improve the mechanical properties of TOCN films by converting the sodium carboxylate structures to free carboxylate groups by, for example, gaseous acid treatment. Self-standing films prepared from TEMPO-oxidized wood celluloses by TEMPO/NaClO/NaClO2 oxidation in water at pH 6.8 had higher tensile strength than those prepared from TEMPO/NaBr/NaClO oxidation in water at pH 10, probably because the former had higher DP values.29 However, the elastic moduli of the films were similar, despite the different TEMPO-mediated oxidation methods employed. TOCN/ethylcellulose and TOCN/phenol resin composites have been studied to improve mechanical and thermal properties of the matrix materials.109,110
The highly hydrophilic nature of TOCN films leads in turn to low contact resistance to water. The contact angle of a water droplet placed on a TOCN film decreased from 47° over time as a result of partial penetration of water into the film. Alkyl ketene dimer (AKD) is a typical hydrophobizing chemical used in papermaking processes. TOCN film treated with a 0.05% AKD dispersion had a water-contact angle of 94°, and this value was maintained for 10 s (Fig. 24). Therefore, hydrophilic TOCN films can be hydrophobized using a simple soaking method with a cationic AKD dispersion. The abundant carboxylate groups in the TOCN film are likely to behave as adsorption sites for cationic AKD-dispersed particles.28
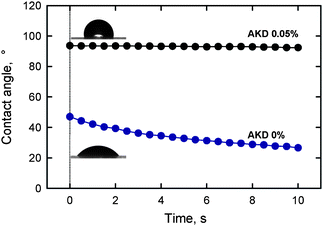 | ||
| Fig. 24 Change in the contact angle over time of a water droplet on a nanofiber film prepared from TEMPO-oxidized softwood cellulose before and after treatment with a 0.05% alkyl ketene dimer dispersion.28Reproduction of images and figure from ref. 28 with permission from American Chemical Society (© American Chemical Society 2009). | ||
Thermal degradation of both the TEMPO-oxidized softwood cellulose and TOCN films started to occur at approximately 200 °C in a nitrogen atmosphere, while for the original cellulose, degradation began at 300 °C. Thus, the formation of sodium carboxylate groups from the C6 primary hydroxyls of cellulose microfibril surfaces by TEMPO-mediated oxidation leads to a significant decrease in the thermal degradation point (Fig. 25).28,60,98,111 The thermal expansivity of TOCN films was measured in a nitrogen atmosphere after heating the film at 120 °C for 10 min to remove residual moisture. The coefficient of thermal expansion (CTE) of the TOCN film was approximately 2.7 ppm K−1, which is much lower than that of glass (ca. 9 ppm K−1). The extremely low CTE is caused by the high crystallinity of the nanofibers comprising the TOCN film.28
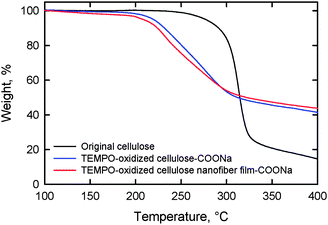 | ||
| Fig. 25 Thermogravimetric curves of the original cellulose, TEMPO-oxidized cellulose and TOCN film.98Reproduction of figure from ref. 98 with permission from Elsevier (© Elsevier 2010). | ||
Atomic force microscopy (AFM) observation of TOCN films showed that the film surface consists of randomly assembled nanofibers (Fig. 26).28 The nanofiber widths were estimated to be 3–4 nm from the height of the AFM images, which is in good agreement with TEM measurements.27,29 The elastic modulus of single TEMPO-oxidized tunicate cellulose nanofibers was measured by three-point bending test using an AFM cantilever to be approximately 145 GPa.112
 | ||
| Fig. 26 AFM image of the surface of a nanofiber film prepared from TEMPO-oxidized softwood cellulose.28Reproduction of image from ref. 28 with permission from American Chemical Society (© American Chemical Society 2009). | ||
The oxygen permeability of a poly(lactic acid) (PLA) film surface-coated with TOCN was examined under dry conditions. The original PLA film had an oxygen permeability of approximately 7.4 mL m−2 day−1kPa−1. Surprisingly, the oxygen permeability was decreased to <0.001 mL m−2 day−1kPa−1 (Fig. 27) by the thin TOCN layer.28 The oxygen permeability of the TOCN-coated PLA film was close to that for typical synthetic polymer films such as poly(vinylidene chloride) and poly(ethylene)-poly(vinyl alcohol) copolymer that have high oxygen barrier functionality. The refractive index of PLA (1.48–1.50)113 was close to that of TOCN (1.55). Therefore, transparent and highly oxygen-impermeable films can be obtained by coating of TOCN dispersion on PLA films, which would make wholly biomass-sourced film packaging available for foods and medicines. The interaction or adhesion strength between PLA and TOCN layers at the interfaces in the TOCN-coated PLA films were improved by hydrophilic modification of the PLA surfaces by graft-polymerization treatment.114 The oxygen barrier properties of chemically modified MFC films has also studied by other researchers.115
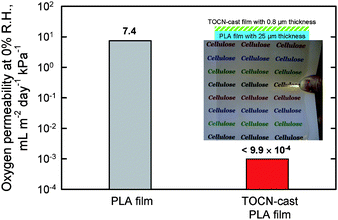 | ||
| Fig. 27 Light transparency and flexibility of poly(lactic acid) (PLA) film coated with softwood TOCN film, and oxygen permeability values of the base PLA and TOCN/PLA films. | ||
The degree of nanofibrillation or swelling behavior of TEMPO-oxidized wood celluloses can be controlled by the disintegration conditions in water and addition of coagulants; therefore, TOCN-containing paper-like sheets can be prepared by conventional papermaking processes without any serious drainage problems. These TOCN sheets have potential applications as ion-exchange materials for water and high-performance air-filters116,117 The adsorption behavior of cationic compounds on TEMPO-oxidized cellulose fibers possessing anionic charges has also been reported.118
7 Concluding remarks
TOCN are categorized as new bio-based nano-materials prepared from abundantly available wood biomass by the position-selective catalytic oxidation of C6 primary hydroxyls exposed on crystalline cellulose microfibril surfaces under moderate aqueous conditions similar to enzymatic or biological reactions. TOCNs thus obtained have unique and characteristic properties in terms of uniform and ultrafine widths of 3–4 nm, high crystallinity and high aspect ratios. TEMPO-mediated oxidation of wood celluloses has a high possibility of becoming a breakthrough nano-technology to bridge biomass/forest refining and high-tech/cutting-edge fields, and to contributing to the establishment of sustainable societies and environmentally friendly systems based on renewable resources to partly replace fossil resources. We have started a Nanotech Challenge Project entitled, “Development of environmentally-friendly and high-performance packaging materials using TOCN”, which was supported by the New Energy and Industrial Technology Development Organization (NEDO) from 2007.119 The main research & development subjects of the present project focus on more cost-effective and environmentally friendly oxidation systems, safety-issues of the products and processes, further improvement of gas-barrier, water-resistant, thermal and mechanical properties of TOCN films and TOCN-containing composites, practical technologies for multilayer coating process of TOCN.Acknowledgements
This research has been continuously supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) from 1996 to 2013. The TOCN Nanotech Challenge Project has been supported by NEDO from 2007 to 2012.References
- D. R. Paul and L. M. Robeson, Polymer, 2008, 49, 3187–3204 CrossRef CAS.
- R. Rusli and S. J. Eichhorn, Appl. Phys. Lett., 2008, 93, 0331111-1–3.
- A. Šturcová, G. R. Davies and S. J. Eichhorn, Biomacromolecules, 2005, 6, 1055–1061 CrossRef CAS.
- H. M. C. De Azeredo, Food Res. Int., 2009, 42, 1240–1253 CrossRef CAS.
- F. W. Herrick, R. L. Casebier, J. K. Hamilton and K. R. Sandberg, J. Appl. Polym. Sci., Appl. Polym. Symp., 1983, 37, 797–813 CAS.
- A. F. Turbak, F. W. Snyder and K. R. Sandberg, J. Appl. Polym. Sci., Appl. Polym. Symp., 1983, 37, 815–827 CAS.
- T. Taniguchi and K. Okamura, Polym. Int., 1998, 47, 291–294 CrossRef CAS.
- S. Iwamoto, A. N. Nakagaito, H. Yano and M. Nogi, Appl. Phys. A: Mater. Sci. Process., 2005, 81, 1109–1112 CrossRef CAS.
- S. Iwamoto, A. N. Nakagaito and H. Yano, Appl. Phys. A: Mater. Sci. Process., 2007, 89, 461–466 CrossRef CAS.
- E. Dinand, H. Chanzy and M. R. Vignon, Cellulose, 1996, 3, 183–188 CrossRef CAS.
- E. Dinand, H. Chanzy and M. R. Vignon, Food Hydrocolloids, 1999, 13, 275–283 CrossRef CAS.
- E.-L. Hult, T. Iversen and J. Sugiyama, Cellulose, 2003, 10, 103–110 CrossRef CAS.
- K. Abe, S. Iwamoto and H. Yano, Biomacromolecules, 2007, 8, 3276–3278 CrossRef CAS.
- S. Iwamoto, K. Abe and H. Yano, Biomacromolecules, 2008, 9, 1022–1026 CrossRef CAS.
- R. H. Marchessault, F. F. Morehead and N. M. Walter, Nature, 1959, 184, 632–633 CrossRef CAS.
- X. M. Dong, J.-F. Revol and D. G. Gray, Cellulose, 1998, 5, 19–32 CrossRef CAS.
- S. Beck-Candanedo, M. Roman and D. G. Gray, Biomacromolecules, 2005, 6, 1048–1054 CrossRef CAS.
- O. van den Berg, J. R. Capadona and C. Weder, Biomacromolecules, 2007, 8, 1353–1357 CrossRef CAS.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J.-L. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2008, 9, 57–65 CrossRef CAS.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolainen, J. Laine, P. T. Larsson, O. Ikkala and T. Lindström, Biomacromolecules, 2007, 8, 1934–1941 CrossRef CAS.
- M. Henriksson, G. Henriksson, L. A. Berglund and T. Lindström, Eur. Polym. J., 2007, 43, 3434–3441 CrossRef CAS.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795 CrossRef.
- P. Lepoutre, S. H. Hui and A. A. Robertson, J. Macromol. Sci., Part A: Pure Appl. Chem., 1976, A10, 681–693 Search PubMed.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- Y. Habibi, L. A Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691 CrossRef CAS.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS.
- H. Fukuzumi, T. Saito, Y. Kumamoto, T. Iwata and A. Isogai, Biomacromolecules, 2009, 10, 162–165 CrossRef CAS.
- T. Saito, M. Hirota, N. Tamura, S. Kimura, H. Fukuzumi, L. Heux and A. Isogai, Biomacromolecules, 2009, 10, 1992–1996 CrossRef CAS.
- W. Adam, C. R. Saha-Möller and P. A. Ganeshpure, Chem. Rev., 2001, 101, 3499–3547 CrossRef CAS.
- A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 10, 1153–1174 CrossRef.
- A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Carbohydr. Res., 1995, 269, 89–98 CrossRef CAS.
- W. F. Bailey, J. M. Bobbitt and K. B. Wiberg, J. Org. Chem., 2007, 72, 4504–4509 CrossRef CAS.
- S. Goldstein and A. Samuni, J. Phys. Chem. A, 2007, 111, 1066–1072 CrossRef CAS.
- A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Tetrahedron, 1995, 51, 8023–8032 CrossRef CAS.
- P. L. Bragd, H. van Bekkum and A. C. Besemer, Top. Catal., 2004, 27, 49–66 CrossRef CAS.
- A. Isogai and Y. Kato, Cellulose, 1998, 4, 153–164 CrossRef.
- T. Isogai, M. Yanagisawa and A. Isogai, Cellulose, 2009, 16, 117–127 CrossRef CAS.
- D. da Silva Perez, S. Montanari and M. R. Vignon, Biomacromolecules, 2003, 4, 1417–1425 CrossRef CAS.
- T. Isogai, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1593–1599 CrossRef CAS.
- I. Shibata, M. Yanagisawa, T. Saito and A. Isogai, Cellulose, 2006, 13, 73–80 CrossRef CAS.
- A. E. J. de Nooy, A. C. Besemer, H. van Bekkum, J. A. P. P. van Dijk and J. A. M. Smit, Macromolecules, 1996, 29, 6541–6547 CrossRef CAS.
- A. Potthast, T. Rosenau and P. Kosma, Adv. Polym. Sci., 2006, 205, 1–48 CAS.
- A. Potthast, S. Schiehser, T. Rosenau and M. Kostic, Holzforschung, 2009, 63, 12–17 CrossRef CAS.
- I. Shibata and A. Isogai, Cellulose, 2003, 10, 151–158 CrossRef CAS.
- M. Zhao, J. Li, E. Mano, Z. Song, D. M. Tschaen, E. J. J. Grabowski and P. J. J. Reider, J. Org. Chem., 1999, 64, 2564–2566 CrossRef CAS.
- M. Hirota, N. Tamura, T. Saito and A. Isogai, Carbohydr. Polym., 2009, 78, 330–335 CrossRef CAS.
- S. Fujisawa, T. Isogai and A. Isogai, Cellulose, 2010, 17, 607–605 CrossRef CAS.
- Y. Kato, J. Kaminaga, R. Matsuo and A. Isogai, J. Polym. Environ., 2005, 13, 261–266 CrossRef CAS.
- C. Fraschini and M. R. Vignon, Carbohydr. Res., 2000, 328, 585–589 CrossRef CAS.
- P. L. Bragd, A. C. Besemer and H. van Bekkum, Carbohydr. Res., 2000, 328, 355–363 CrossRef CAS.
- P. L. Bragd, A. C. Besemer and H. van Bekkum, J. Mol. Catal. A: Chem., 2001, 170, 35–42 CrossRef CAS.
- M. Ibert, F. Marsais, N. Merbouh and C. Brückner, Carbohydr. Res., 2002, 337, 1059–1063 CrossRef CAS.
- P. L. Bragd, A. C. Besemer and H. van Bekkum, Carbohydr. Polym., 2002, 49, 397–406 CrossRef CAS.
- I. Shibata and A. Isogai, Cellulose, 2003, 10, 335–341 CrossRef CAS.
- Y. Kato, R. Matsuo and A. Isogai, Carbohydr. Polym., 2003, 51, 69–75 CrossRef CAS.
- R. N. Desai and L. F. Blackwell, Synlett., 2003, 13, 1981–1984.
- A. Zanka, Chem. Pharm. Bull., 2003, 51, 888–889 CrossRef CAS.
- Y. Kato, J. Kaminaga, R. Matsuo and A. Isogai, Carbohydr. Polym., 2004, 58, 421–426 CrossRef CAS.
- S. Gomez-Bujedo, E. Fleury and M. R. Vignon, Biomacromolecules, 2004, 5, 565–571 CrossRef CAS.
- B. Sun, C. Gu, J. Ma and B. Liang, Cellulose, 2005, 12, 59–66 CrossRef CAS.
- D. Liaigre, T. Breton and E. M. Belgsir, Electrochem. Commun., 2005, 7, 312–316 CrossRef CAS.
- M. Lei, R.-J. Hu and Y.-G. Wang, Tetrahedron, 2006, 62, 8928–8932 CrossRef CAS.
- M. Shibuya, M. Tomizawa, I. Suzuki and Y. Iwabuchi, J. Am. Chem. Soc., 2006, 128, 8412–8413 CrossRef CAS.
- I. W. C. E. Arends, Y.-X. Li and R. A. Sheldon, Biocatal. Biotransform., 2006, 24, 443–448 CrossRef CAS.
- M. Barbier, T. Breton, K. Servat, E. Grand, B. Kokoh and J. Kovensky, J. Carbohydr. Chem., 2006, 25, 253–266 CrossRef CAS.
- X. Wang, R. Liu, Y. Jin and X. Liang, Chem.–Eur. J., 2008, 14, 2679–2685 CrossRef CAS.
- N. Tamura, M. Wada and A. Isogai, Carbohydr. Polym., 2009, 77, 300–305 CrossRef CAS.
- M. Hirota, N. Tamura, T. Saito and A. Isogai, Cellulose, 2009, 16, 841–851 CrossRef CAS.
- M. Hirota, N. Tamura, T. Saito and A. Isogai, Cellulose, 2010, 17, 279–288 CrossRef CAS.
- N. Tamura, M. Hirota, T. Saito and A. Isogai, Carbohydr. Polym., 2010, 81, 592–598 CrossRef CAS.
- Y. Kato, N. Habu, J. Yamaguchi, Y. Kobayashi, I. Shibata, A. Isogai and M. Samejima, Cellulose, 2002, 9, 75–81 CrossRef CAS.
- C. Delattre, P. Michaud, R. Elboutachfaiti, B. Courtois and J. Courtois, Cellulose, 2006, 13, 63–71 CrossRef CAS.
- N. Konno, N. Habu, I. Maeda, N. Azuma and A. Isogai, Carbohydr. Polym., 2006, 64, 589–596 CrossRef CAS.
- N. Konno, A. Isogai, N. Habu and N. Iihashi, Cellulose, 2008, 15, 453–463 CrossRef CAS.
- N. Konno, K. Igarashi, N. Habu, M. Samejima and A. Isogai, Appl. Environ. Microbiol., 2009, 75, 101–107 CrossRef CAS.
- N. Konno, T. Ishida, K. Igarashi, S. Fushinobu, M. Samejima, N. Habu and A. Isogai, FEBS Lett., 2009, 538, 1323–1326 CrossRef.
- T. Yoshimura, M. Okutsu and M. Hiroshima, WO Patent 2009/122953, 2009.
- T. Kitaoka, A. Isogai and F. Onabe, Nord. Pulp Pap. Res. J., 1999, 14, 274–279.
- T. Saito and A. Isogai, Biomacromolecules, 2004, 5, 1983–1989 CrossRef CAS.
- T. Saito and A. Isogai, Carbohydr. Polym., 2005, 61, 183–190 CrossRef CAS.
- T. Saito, I. Shibata, A. Isogai, N. Suguri and N. Sumikawa, Carbohydr. Polym., 2005, 61, 414–419 CrossRef CAS.
- T. Saito and A. Isogai, TAPPI J., 2005, 4(3), 3–8 Search PubMed.
- S. Montanari, M. Roumani, L. Heux and M. R. Vignon, Macromolecules, 2005, 38, 1665–1671 CrossRef CAS.
- T. Saito, Y. Okita, T. T. Nge, J. Sugiyama and A. Isogai, Carbohydr. Polym., 2006, 65, 435–450 CrossRef CAS.
- T. Saito and A. Isogai, Colloids Surf., A, 2006, 289, 219–225 CrossRef CAS.
- T. Saito and A. Isogai, Ind. Eng. Chem. Res., 2007, 46, 773–780 CrossRef CAS.
- Z. Dang, J. Zhang and A. J. Ragauskas, Carbohydr. Polym., 2007, 70, 310–317 CrossRef CAS.
- L. Mao, K. Law, D. Claude and B. Francois, Ind. Eng. Chem. Res., 2008, 47, 2809–2812.
- Y. Okita, T. Saito and A. Isogai, Holzforschung, 2009, 63, 529–535 CrossRef CAS.
- E. J. Parks and R. L. Hebert, TAPPI J., 1972, 55(10), 1510–1514 Search PubMed.
- M. Marx, Makromol. Chem., 1955, 16, 157–176 CrossRef CAS.
- T. Saito, M. Hirota, N. Tamura and A. Isogai, J. Wood Sci., 2010, 56, 227–232 CrossRef CAS.
- T. Saito, I. Shibata, A. Isogai, N. Suguri and N. Sumikawa, Carbohydr. Polym., 2005, 61, 414–419 CrossRef CAS.
- Y. Okita, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1696–1700 CrossRef CAS.
- M. Hirota, K. Furihata, T. Saito, T. Kawada and A. Isogai, Angew. Chem. Int. Ed., 2010, 49, 7670–7672 CrossRef.
- S. Zhang, B. Sun, W. Wang, M. Zhu and J. Chen, J. Macromol. Sci. A, 2006, 43, 1895–1906 CrossRef CAS.
- H. Fukuzumi, T. Saito, Y. Okita and A. Isogai, Polym. Degrad. Stab., 2010, 95, 1502–1508 CrossRef CAS.
- M. M. De Souza Lima and R. Borsali, Macromol. Rapid Commun., 2004, 25, 771–787 CrossRef.
- M. E. Carr Jr, L. I. Shen and J. Hermans, Biopolymers, 1977, 16, 1–15 CrossRef CAS.
- M. E. Carr Jr and J. Hermans, Macromolecules, 1978, 11, 46–50 CrossRef CAS.
- R. R. Hatgan and J. Hermans, J. Biol. Chem., 1979, 254, 11272–11281.
- A. A. Baker, W. Helbert and J. Sugiyama, J. Struct. Biol., 1997, 119, 129–138 CrossRef CAS.
- E. Lasseuguette, D. Roux and Y. Nishiyama, Cellulose, 2008, 15, 425–433 CrossRef CAS.
- L. Mao, P. Ma, K. Law, C. Daneault and F. Brouillette, Ind. Eng. Chem. Res., 2010, 49, 113–116 CrossRef CAS.
- S. Iwamoto, W. Kai, T. Isogai, T. Saito, A. Isogai and T. Iwata, Polym. Degrad. Stab., 2010, 95, 1394–1498 CrossRef CAS.
- I. Pater, M. Opietnik, S. Böhmdorfer, M. Becker, A. Potthast, T. Saito, A. Isogai and T. Rosenau, Holzforschung, 2010, 64, 549–554 CrossRef.
- M. Nogi and H. Yano, Appl. Phys. Lett., 2009, 94, 233117 CrossRef.
- R. K. Johnson, A. Zink-Sharp, S. H. Renneckar and W. G. Glasser, Cellulose, 2009, 16, 227–238 CrossRef CAS.
- Z. Li, S. Renneckar and J. R. Barone, Cellulose, 2010, 17, 57–68 CrossRef CAS.
- E. Jakab, E. Meszaros and J. Borsa, J. Anal. Appl. Pyrolysis, 2010, 87, 117–123 CrossRef CAS.
- S. Iwamoto, W. Kai, T. Iwata and A. Isogai, Biomacromolecules, 2009, 10, 2571–2576 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS.
- W. Kai, S. Iwamoto, K. Akamatsu, S. Nakao, A. Isogai and T. Iwata, Polym. Degrad. Stab., 2010, 95, 1004–1010 CrossRef CAS.
- K. Syverud and P. Stenius, Cellulose, 2009, 16, 75–85 CrossRef CAS.
- M. Ishizuka, Y. Kazumori, T. Enomae and A. Isogai, Japan TAPPI J., 2010, 64, 437–477 Search PubMed.
- M. Ishizuka, Y. Kazumori, T. Enomae and A. Isogai, Japan TAPPI J., 2010, 64, 889–902 Search PubMed.
- S. Alila, F. Aloulou, D. Beneventi and S. Boufi, Langmuir, 2007, 23, 3723–3731 CrossRef CAS.
- N. E. D. O. Website ( 2009); http://app3.infoc.nedo.go.jp/informations/koubo/press/EF/nedopress.2009-02-10.5277370827/, http://app3.infoc.nedo.go.jp/informations/koubo/kaiken/BE/nedopressorder.2009-02-10.0007481708/cellulose.pdf, http://www.tech.nedo.go.jp/PDF/100012422.pdf.
| This journal is © The Royal Society of Chemistry 2011 |