Preparation of graphene by a low-temperature thermal reduction at atmosphere pressure
Wufeng
Chen
and
Lifeng
Yan
*
Hefei National Laboratory for Physical Sciences at the Microscale and CAS Key Laboratory of Soft Matter Chemistry, Department of Chemical Physics, University of Science and Technology of China, Hefei 230026, P. R. China
First published on 16th January 2010
Abstract
It is still a challenge to prepare graphene from graphene oxide (GO) under a mild thermal reduction. Here we report that GO can even be reduced at a low temperature (below 150 °C) at atmospheric pressure in a mixture of N,N-dimethylacetamide (DMAc) and water (10 : 3, v/v) under the protection of nitrogen gas. FT-IR, XRD, XPS, Raman, and AFM studies confirmed the thermal reduction of GO and the formation of graphene. The as-prepared graphene can be fully dispersed in pure DMAc to form an organic suspension, and the suspension is stable for at least two months at room temperature.
Introduction
As a member of the carbon family, graphene has attracted much attention recently for its dazzling electronic and mechanical properties.1,2 Recently, a lot of efforts have been made to find easy methods of preparing graphene in a high yield. Several typical methods have been developed and reviewed,3 such as chemical vapor deposition (CVD),4 micromechanical exfoliation of graphite,5 epitaxial growth on an electrically insulating surface,6 solvothermal synthesis,7 and the reduction of GO.8 Generally, the reduction of GO is carried out by chemical methods, using different reductants such as hydrazine,8 dimethylhydrazine,9 hydroquinone10 or NaBH4,11 and usually the reductants are hazardous. Recently, it was reported that GO can be reduced under alkaline conditions12 or with thermal methods.13 The thermal method is believed to be a green method in which no hazardous reductants are used but this process requires a rapid heating (>2000 °C min−1) up to 1050 °C14 in an oven under argon gas or up to 800 °C under hydrogen gas.15 It is still difficult to thermally reduce GO under mild conditions.It is believed that the thermal reduction of GO was accompanied by the elimination of epoxy and carboxyl groups. In a typical thermogravimetry (TGA) experiment, it is usually found that the dry GO powder can be decomposed near 210 °C with a mass loss caused by releasing oxygen from GO.16 The process gives a typical oxidation of the GO surface, and forms O2, CO, CO2 and H2O. Chua et al.17 recently showed direct evidence of oxygen elimination from GO functionalized with octadecylamine (ODA) by STM studies at 150–200 °C. Nethravathi and Rajamathi18 found that chemically modified graphene sheets can be produced by a solvothermal reduction of GO suspension in water, or reducing solvents such as butanol, at temperatures of 120–200 °C. However, they found that high pressure is an important factor in the conversion of GO to graphene—no apparent reduction occurs at atmosphere pressure. The high pressure requirement makes it difficult to use a continuous process for graphene preparation.
Here, an effort to prepare graphene from GO by a low temperature thermal reduction at atmospheric pressure was carried out.18N,N-dimethylacetamide (DMAc) with a boiling point of 165 °C was utilized as the solvent, and the as-prepared GO was dispersed into a mixture of DMAc/H2O (10 : 3, v/v) to give a suspension. Then the suspension was heated at low temperatures (below 150 °C) and at atmospheric pressure for various lengths of time, and the conversion of GO was studied by means of FT-IR, Raman, XRD, XPS, TG and AFM studies.
Experimental
Materials
Graphite powder, natural, briquetting grade, ∼100 mesh, 99.9995% (metals basis) was purchased from Alfa Aesar. Analytical grade DMAc, NaNO3, and KMnO4 and 98% H2SO4, 30% H2O2 aqueous solution were purchased from Shanghai Chemical Reagents Company, and were used directly without further purification. Ultra-pure water (18 MΩ) was produced by a Millipore System (Millipore Q, USA).Preparation of graphene oxide (GO)
GO was prepared from natural graphite by the well-known Hummers method.19,20 GO powder was obtained after freeze-drying the suspension.Thermal reduction of GO
First, 20 mg of the as-prepared GO were dispersed into 6 mL water under mild ultrasound for 2 min. Then 20 mL of DMAc was added under mild ultrasound. Finally, a transparent yellow–brown suspension of GO was obtained. After that, nitrogen gas was bubbled through the suspension for 30 min to remove the dissolved oxygen gas, and it was heated in an oil-bath at 100 °C, 125 °C or 150 °C for 1 h, and 150 °C for 5 h, under the protection of nitrogen gas. After the reaction, the suspensions were filtered through micropore filters and the resulting cakes were washed by anhydrous ethanol three times. Furthermore, each cake was re-dispersed into 15 mL water, and freeze-dried. At the end we obtained reduced GO as a black powder.Colloid of graphene in DMAc
1.5 mg of the above black powder of reduced GO were added into 10 mL of pure DMAc under mild ultrasound. A colloid of graphene in DMAc with a concentration of 0.15 mg mL−1 was obtained.Characteristics of the samples
Fourier transform infrared (FT-IR) spectra of the samples were recorded by a Bruker vector-2 spectrophotometer (Germany). The test specimens were prepared by the KBr-disk method. Wide-angle X-ray diffraction (XRD) analyses were carried out by employing an X-ray diffractometer (D/MAX-1200, Rigaku Denki Co. Ltd., Japan). The X-ray diffraction patterns with Cu Kα radiation (λ = 1.5406 nm) at 40 kV and 100 mA were recorded in the range of 2θ = 5–70°. The thermal properties of the samples were characterized by a thermogravimeter (TGA, DTA-50, Shimazu, Japan), and all the measurements were carried out under nitrogen gas over a temperature range of 30–700 °C with a ramp rate of 5 °C min−1. A commercial atomic force microscope (Nanoscope IIIa; Digital Instruments, Santa Barbra, CA), equipped with a J scanner was used to measure the morphology of the sample. A Si3N4 tip (Nanoprobes, Digital Instruments Inc.) was used in the contact mode. The scan rates were in between 1.0 and 2.4 Hz. XPS spectra were recorded by an Escalab MK II photoelectron spectrometer (VG Scientific Ltd., United Kingdom). Raman spectra were taken by a RAMAMLOG 6 (Spex, USA) with a 50× objective lens and 514.5 nm laser excitation.Results and discussion
First, GO was prepared by a typical Hummer method, and it can be fully dispersed in water to obtain a yellow–brown aqueous suspension as shown in Fig. 1a. However, when the aqueous suspension was heated at 100 °C for 1 h under the protection of nitrogen gas its color changed to brown–black (Fig. 1b), indicating a partial reduction of GO. However, the extent of the reduction is not high even when heated at 150 °C for 5 h. It was found that the as-prepared GO could also be dispersed in a mixture of DMAc/H2O (10 : 3, Fig. 1c). After the suspension was heated at 100 °C for 1 h under the protection of nitrogen gas the color of the suspension also went black (Fig. 1d), and the extent of the reduction is higher than that in aqueous suspension. Fig. 1e shows the image of the organic suspension after it was heated at 125 °C for 1 h, and clearly the suspension became very black. Similarly when the suspension was heated at 150 °C for 5 h, it results in a deep black suspension as shown in Fig. 1f, indicating a high extent of reduction of GO. | ||
| Fig. 1 Photographs of GO suspension with a concentration of 0.77 mg mL−1 under different conditions: (a) in water, (b) after heating the aqueous suspension at 100 °C for 1 h, (c) in DMAc/H2O (10 : 3), (d) after heating the organic suspension at 100 °C for 1 h, (e) at 125 °C for 1 h, and (f) 150 °C for 5 h. | ||
Fig. 2 shows the FT-IR spectra of GO (curve 1), the suspensions of GO in DMAc/H2O after they were heated at 100 °C for 1 h (curve 2), 125 °C for 1 h (curve 3), 150 °C for 1 h (curve 4), 150 °C for 5 h (curve 5) and a graphene chemically reduced by hydrazine (curve 6) as produced using the reported method.7 For GO, the characteristic peaks appear for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1735 cm−1), aromatic CC (1622 cm−1), carboxy C–O (1414 cm−1), epoxy C–O (1228 cm−1), and C–O (1116 cm−1).21 After the thermal treatment, the peaks for oxygen functional groups were reduced significantly, and the peak at 1414 cm−1 for carboxy C–O was nearly entirely removed by the thermal treatment at 150 °C for 5 h. The peaks at 1735 cm−1 for thermal treatment samples were also decreased. Especially for the thermal treatment at 150 °C for 5 h, the curve is very similar to that of the chemically reduced graphene, indicating the high efficiency of reduction by heating in the mixture. The peak at 1573 cm−1 still exists and is attributed to the aromatic CC group.
O (1735 cm−1), aromatic CC (1622 cm−1), carboxy C–O (1414 cm−1), epoxy C–O (1228 cm−1), and C–O (1116 cm−1).21 After the thermal treatment, the peaks for oxygen functional groups were reduced significantly, and the peak at 1414 cm−1 for carboxy C–O was nearly entirely removed by the thermal treatment at 150 °C for 5 h. The peaks at 1735 cm−1 for thermal treatment samples were also decreased. Especially for the thermal treatment at 150 °C for 5 h, the curve is very similar to that of the chemically reduced graphene, indicating the high efficiency of reduction by heating in the mixture. The peak at 1573 cm−1 still exists and is attributed to the aromatic CC group.
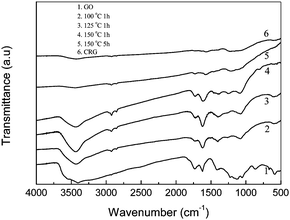 | ||
| Fig. 2 FT-IR spectra of GO, and the products obtained by thermal reduction of the GO suspension in DMAc/H2O with a concentration of 0.77 mg mL−1 at 100 °C for 1 h, 125 °C for 1 h, 150 °C for 1 h and 150 °C for 5 h. FT-IR spectrum of the chemical reduced graphene from GO was also showed here. | ||
The reduction of GO to graphene should result in structural change, especially the distance between the layers. Fig. 3 shows the X-ray diffraction patterns for the natural graphite, GO and the product after heating at 150 °C for 5 h. For natural graphite, the peak at 26.48° of 2θ corresponds to the (002) diffraction line with the interlayer spacing along the c-axis of 0.34 nm.10 However, for GO the peak disappeared and there appears a wide diffraction peak at 21.78°, corresponding to a d-spacing of 0.41 nm. After the thermal treatment, the fact that all the peaks disappeared indicates the efficient exfoliation of the multiplayer during the thermal reduction of GO.
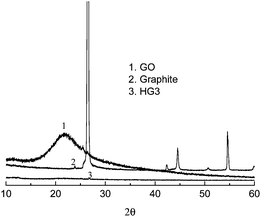 | ||
| Fig. 3 XRD pattern of natural graphite, GO, and the products obtained by the thermal reduction of GO at 150 °C for 5 h (HG 3). | ||
XPS measurements could provide the direct evidence of the reduction of GO during the thermal treatment. Fig. 4 shows the XPS patterns of GO and the thermal reduced graphene at 150 °C for 5 h. For GO, two bands at 284.7 eV and 286.9 eV are observed which could be attributed to the graphitic CC species and C–O species respectively. Also the fit of the curve gives a small peak at 287.9 eV, which is attributed to the CO species.22 However, after thermal treatment, the peaks at 286.9 eV and 287.9 eV decrease significantly, and a little peak appears at 285.6 eV. The C/O ratio changes from 2.09 to 4.70, indicating the efficient deoxygenation of GO and the formation of graphene. The degree of the thermal reduction is similar to the typical chemical reduction process.
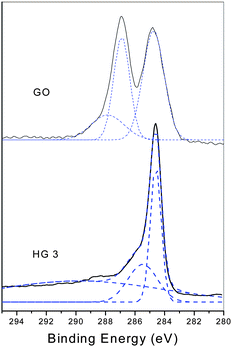 | ||
| Fig. 4 High-resolution C1s XPS spectra of GO and the thermal reduced GO at 150 °C for 5 h. | ||
Fig. 5 shows the Raman spectra of GO and the thermally reduced graphene. Both spectra show the existence of the D band and G band. For GO, the G band locates at 1600 cm−1, while for the reduced graphene the G band moved to 1586 cm−1, which equals the value of pristine graphite (1586 cm−1), indicating the reduction of GO during the thermal treatment.22 At the same time there exists the D band for the two spectra, which locates at 1350 cm−1 and 1347 cm−1 for GO and the thermally reduced sample respectively, and which corresponds to the defect of the sample and the size of the in-plane sp2 domain.18,23
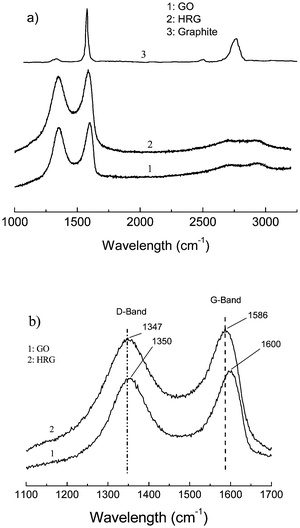 | ||
| Fig. 5 (a) Raman spectra of the natural graphite, GO and the thermal reduced graphene (HG 3). (b) Enlarged view of the same spectra showing the shift in the G-band. | ||
Fig. 6 shows the typical AFM height images of GO and the thermally reduced GO at 150 °C for 5 h. For GO, the typical thickness is about 0.67 nm, while for the reduced GO the thickness is about 0.82 nm.22 The results reveal that the as-prepared graphene is in a single layer, and the obtained graphene is of the order of micrometres in size.
 | ||
| Fig. 6 AFM height images of GO (a) and the thermally reduced GO (c). (d) is a close-up of (c), and (b) and (e) are the section line analyses as indicated. | ||
TGA measurement provides further proof of the reduction of GO under thermal treatment. Fig. 7 shows the TG curves for the natural graphite, GO and the thermally reduced GO at 150 °C for 1 h and 5 h. For GO, three steps appeared for mass loss on increasing temperature. The main mass loss is about 10% at around 100 °C, which can be ascribed to the removal of the adsorbed water. The mass loss at around 200 °C is about 30%, and this was attributed to the decomposition of labile oxygen functional groups.24 For the product prepared after thermal treatment at 150 °C for 1 h, only 10% of the mass was lost at around 200 °C, indicating the decreasing number of labile oxygen functional groups. However, for the product prepared by the thermal treatment at 150 °C for 5 h, there is no mass loss at around 200 °C and most of the mass loss occurs at about 500 °C, indicating the near-complete reduction of GO.25 Here the reduction of GO to graphene is also believed to be by oxygen elimination due to the thermal effect, for DMAc is a good solvent for graphene, and it is suggested that the quick dispersion of the as-formed graphene by thermal reduction in the solution may be a dynamic driving mechanism for efficient reduction, and the role of DMAc will be studied next.
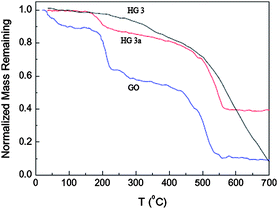 | ||
| Fig. 7 Normalized remaining mass of GO and the thermally reduced GO at 150 °C for 1 h (HG 3a) and 5 h (HG 3) as a function of temperature. | ||
The paper of the thermally reduced product was prepared by a simple filtration of relative suspensions. Fig. 8 shows the photo of the obtained HG 3 paper, and its color of light black. The thickness is about 10 μm. The electrical conductivity of the papers was measured by the Van der Pauw method, and it was found that the conductivity of HG 3 paper is 230 S m−1 while it is 0.015 S m−1 for GO paper. It reveals that the conductivity of papers increases about 104 times after the thermal reduction.
 | ||
| Fig. 8 Photo of the thermally reduced graphene paper. | ||
The as-prepared graphene can be fully dispersed into pure DMAc, and a stable graphene suspension in organic solvent was obtained (Fig. 9a). The suspension is very stable, and it has no phase separation even after more than two months at room temperature (Fig. 9b). It provides a different way of preparing new derivatives or novel materials of graphene in this organic solvent in future.
 | ||
| Fig. 9 Photographs of the suspension of the as-prepared garphene in pure DMAc (0.15 mg mL−1) for 5 min (a) and two months (b). | ||
Conclusions
In summary, a very simple method was developed to prepare graphene from GO by heating its suspension in DMAc/H2O below 150 °C at atmospheric pressure, and the reduced degree depends on the heating temperature and time. After the product was dried, a black graphene powder was obtained in high yield. The as-prepared graphene can be fully dispersed in pure DMAc to form a stable suspension with a concentration of 1.0 mg ml−1. The suspension is useful for the further modification of graphene and the preparation of novel materials.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 20874095), the National Key Technology R&D Program (No. 2006BAF02A09). Prof. Jinlong Yang is appreciated for his valuable suggestions.References
- D. Li and R. B. Kaner, Science, 2008, 320, 1170–1171 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- F. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS.
- X. Lu, M. Yu, H. Huang and R. S. Ruoff, Nanotechnology, 1999, 10, 269–272 CrossRef CAS.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1196 CrossRef CAS.
- M. Choucair, P. Thordarson and J. A. Stride, Nat. Nanotechnol., 2009, 4, 30–33 CrossRef CAS.
- S. Stankovich, D. A. Dikin and et al, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS.
- G. Wang, J. Yang, J. Oark, X. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192–8195 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- G. Williams, B. Serger and P. V. Kamat, ACS Nano, 2008, 2, 1487–1491 CrossRef CAS.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schnlepp, A. A. Abdalam, J. Liu and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- H. C. Schniepp, J. Li, M. J. McAllister, H. Sai, D. H. Adamson, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- L. L. Chua, S. Wang, P. J. Chia, L. Chen, L. H. Zhao, W. Chen, A. T. S. Wee and P. K. H. Ho, J. Chem. Phys., 2008, 129, 114702 CrossRef.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS.
- W. Hummers and R. Offema, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- S. Park, J. An, J. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS.
- D. Li, M. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2009, 25, 5957–5968 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prudhomme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS.
- S. Park, J. An, R. D. Piner, I. Jung, D. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS.
- W. Chen, L. Yan and P. R. Bangal, Carbon, 2010 DOI:10.1016/j.carbon.2009.11.037.
| This journal is © The Royal Society of Chemistry 2010 |