
Anion-induced chiral assembly: construction of Ag(I) coordination polymers for photocatalytic degradation of organic dyes†
Chao
Huang
 *,
Jin-Xia
Liang
and
Bi-Xue
Zhu
*
*,
Jin-Xia
Liang
and
Bi-Xue
Zhu
*
Key Laboratory of Macrocyclic and Supramolecular Chemistry of Guizhou Province, Guizhou University, Guiyang 550025, China. E-mail: chuang1@gzu.edu.cn; bxzhu@gzu.edu.cn
First published on 24th October 2024
Abstract
A racemic bis(pyridyl) ligand (L) was synthesized and characterized following our previous work. Subsequently, a series of Ag(I) coordination polymers (CPs) were synthesized from this ligand with silver(I) salts containing nine different anions and further characterized by single-crystal X-ray diffraction analysis. The crystal structures of CPs 1–9 show that the anions play a vital role in the chiral assembly process of L with different silver(I) salts, resulting in homochiral or heterochiral complexes. CPs 1–3 exist as 2D homochiral conglomerates formed by spontaneous chiral resolution. CPs 4–8 exist as 1D heterochiral tube-like chains, and CP 9 adopts a 1D heterochiral zig-zag chain structure. Furthermore, CPs 2, 4, and 9 were chosen as representatives to investigate their photocatalytic performances for the degradation of organic dyes. The results showed that CP 4 is much more active than CPs 2 and 9 for the degradation of both rhodamine B (RhB) and methylene blue (MB) under UV light irradiation, and it is thus inferred that CPs 4–8 are suitable as potential photocatalysts for the degradation of RhB and MB in water. This work has revealed that the chiral assembly structures induced by anions lead to differences in their photocatalytic performances.
Introduction
Chirality is considered as a basic characteristic in nature and plays an important role in many biological and chemical processes.1–3 Metal complexes derived from chiral ligands are widely used in chiral reagents, enantioselective sensors, chiral luminescent materials, and chiral liquid crystals.4–7 Coordination reactions of metal ions with racemic ligands can result in two types of complexes, namely, heterochiral complexes through a ligand self-discrimination process or homochiral complexes through a ligand self-recognition process,8–10 which facilitates the construction of frameworks with novel topologies and interesting functions.11Crystal engineering through self-assembly techniques offers a robust approach for the design and construction of supramolecular structures with distinctive structural characteristics and adjustable properties.12–16 Over the past few decades, there has been growing interest in metal cages, clusters, macrocycles, and helical polymers due to their ability to combine the attributes of metal ions and organic ligands, resulting in assemblies with diverse applications.17–21 Supramolecular chemistry based on Ag(I) complexes is rapidly progressing with many advances in the control of framework structures and topologies. The versatile coordination number of the Ag(I) ion allows the design of different geometrical configurations and dimensionalities, ranging from 0D, 1D, 2D to 3D networks.22–27 In reactions of Ag(I) with organic ligands, the structure of the assembled product depends not only on the nature of the ligand and anion, but also on the stoichiometry, reaction solvent, concentration, and crystallization conditions.28–30 Furthermore, the properties of the obtained Ag(I) complexes are also affected by weak non-covalent interactions of Ag⋯X, Ag⋯Ag, and Ag⋯π contacts, which play important roles in the self-assembly process.31–33 As a result, grasping the key factors that influence self-assembly reactions is important for the construction of novel Ag(I)-based complexes with designed architectures and functional properties.
On the other hand, dyes are widely used in many industries, such as textiles, food processing, plastics, cosmetics, printing, and dyeing.34,35 Compared with other organic pollutants, these aromatic compounds are highly chemically stable and difficult to degrade, and can cause serious environmental pollution.36,37 For example, even a small amount of methylene blue (MB) may induce adverse effects, including hemolytic anemia in infants, fever, hypotension, and bluish discoloration of the skin.38–40 Among the various methods for dye removal, the photocatalytic technique for decomposition of dyes to less lethal components may be a potential and beneficial method.41–43 This approach is economical and works efficiently under normal environmental conditions. To date, due to the semiconducting properties, a number of Ag(I)-based complexes have been comprehensively studied as photocatalysts for the decomposition of organic dyes.44–46
In our previous work, we synthesized a racemic bis(pyridyl) ligand (L) and demonstrated its homochiral or heterochiral assembly with Hg(II), Cd(II), and Ag(I) ions, respectively (Scheme 1).47 Thereafter, three new racemic ligands with similar structures to L were successively designed and synthesized. We have reported the synthesis, characterization, and structural diversity of a series of complexes derived from these ligands with Zn(II), Cd(II), and Hg(II) ions, which showed that the coordination of racemic ligands with different metal salts resulted in various supramolecular structures with 0D, 1D, 2D, or 3D frameworks.48–50 Furthermore, the influencing factors for the chiral assembly modes of racemic ligands were discussed based on the different conformations of the obtained complexes. In the present work, we continued to focus on the chiral assembly processes of racemic L with a series of Ag(I) salts containing different counter anions. The anion-induced chiral assemblies were further analyzed and summarized in depth according to the diverse structures of nine Ag(I)-based coordination polymers (CPs). Moreover, the photocatalytic degradation behavior of these CPs toward organic dyes was investigated in detail based on their semiconductive properties.
 | ||
| Scheme 1 The mirror structure of racemic ligand L. | ||
Experimental section
Materials and general methods
All reagents and solvents were commercially available and used without further purification. Ligand L was synthesized and characterized according to our previous work.47 CHN elemental analyses were carried out using a PerkinElmer 240C elemental analyzer. IR spectra were recorded using a Bruker VERTEX 70 instrument. Single-crystal X-ray diffraction data were obtained using a Bruker D8 Venture diffractometer. Circular dichroism (CD) spectra were measured using an applied photophysics Chirascan spectrophotometer. Powder X-ray diffraction (PXRD) patterns were collected using a Bruker D8 Advance diffractometer. Thermogravimetric analyses (TGAs) were carried out using a NETZSCH TG 209 thermal analyzer. Solid-state UV-vis diffuse-reflectance spectra and UV-vis absorption spectra were measured using Shimadzu UV-3600 and Shimadzu UV-2700 spectrophotometers, respectively. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific K-Alpha instrument.Synthesis of CPs 1–9
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N).
N).
N).
N).
N).
N).
N).
N).
N).
N).
N).
N).
N).
N).
N).
N).
X-ray crystallography
X-ray diffraction data for CPs 2–9 were collected using a Bruker D8 Venture diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at room temperature. The crystal structures were solved by direct methods of SHELXT and refined by the full matrix least-squares technique on F2 using the OLEX2 program.51,52 The disordered lattice water molecules in CPs 4, 5, and 6 were removed using the PLATON/SQUEEZE routine, implemented in the OLEX2 program. Crystal data and refinement parameters of CPs 2–9 are summarized in Table 1. Selected bond lengths and bond angles are summarized in Table S1 (ESI†).| Compound | 2a | 2b | 3a | 3b | 4 |
|---|---|---|---|---|---|
| Empirical formula | C40H33AgBF4N11O2 | C40H33AgBF4N11O2 | C40H33AgClN11O6 | C40H33AgClN11O6 | C40H33AgF6N11O2Sb |
| Formula weight | 894.45 | 894.45 | 907.09 | 907.09 | 1043.39 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | triclinic |
| Space group | C2 | C2 | C2 | C2 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/(Å) | 14.812(2) | 14.7993(7) | 14.892(9) | 14.859(2) | 12.3419(7) |
| b/(Å) | 18.154(3) | 18.1492(10) | 18.146(10) | 18.158(3) | 12.4304(8) |
| c/(Å) | 8.4652(11) | 8.4593(4) | 8.470(5) | 8.4544(11) | 15.0532(10) |
| α/(deg) | 90 | 90 | 90 | 90 | 72.737(2) |
| β/(deg) | 120.558(4) | 120.5260(10) | 120.357(17) | 120.335(4) | 78.353(2) |
| γ/(deg) | 90 | 90 | 90 | 90 | 87.717(2) |
| V/(Å3) | 1960.2(5) | 1957.21(17) | 1975(2) | 1968.7(5) | 2159.4(2) |
| Z | 2 | 2 | 2 | 2 | 2 |
| D c/(g cm−3) | 1.515 | 1.518 | 1.525 | 1.530 | 1.605 |
| θ range/(deg) | 2.24–26.00 | 2.24–26.00 | 2.66–26.00 | 3.18–25.00 | 2.36–26.00 |
| Absorption coefficient/mm−1 | 0.585 | 0.586 | 0.641 | 0.643 | 1.153 |
| F(000) | 908 | 908 | 924 | 924 | 1036 |
| Reflections collected | 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 495 495 |
27688 |
22453 |
21236 |
54766 |
| Independent reflections | 3873 | 3844 | 3882 | 3450 | 8486 |
| Observed reflections (I > 2σ(I)) | 3532 | 3798 | 3678 | 3348 | 6645 |
| Number of parameters | 270 | 271 | 270 | 270 | 551 |
| Goodness-of-fit on F2 | 1.026 | 1.030 | 1.050 | 1.074 | 1.072 |
| R int | 0.0848 | 0.0694 | 0.0556 | 0.0638 | 0.0919 |
| Final R indices (I >2σ(I)) | R 1 = 0.0281, wR2 = 0.0611 | R 1 = 0.0256, wR2 = 0.0644 | R 1 = 0.0261, wR2 = 0.0597 | R 1 = 0.0285, wR2 = 0.0713 | R 1 = 0.0545, wR2 = 0.1569 |
| R indices (all data) | R 1 = 0.0341, wR2 = 0.0635 | R 1 = 0.0260, wR2 = 0.0647 | R 1 = 0.0291, wR2 = 0.0611 | R 1 = 0.0298, wR2 = 0.0722 | R 1 = 0.0694, wR2 = 0.1668 |
| Largest peak/hole (e Å−3) | 0.215/−0.324 | 0.281/−0.312 | 0.193/−0.302 | 0.407/−0.334 | 1.410/−0.879 |
| Compound | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|
| Empirical formula | C41H33AgF3N11O5S | C42H33AgF3N11O4 | C45H41AgN10O7S | C47H40AgN11O6 | C28H23AgN6O3 |
| Formula weight | 956.71 | 920.66 | 973.81 | 962.77 | 599.39 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Monoclinic |
| Space group |
P |
P |
P |
P |
P2/c |
| a/(Å) | 12.265(3) | 12.347(7) | 12.6679(15) | 12.545(4) | 14.435(7) |
| b/(Å) | 12.471(3) | 12.411(7) | 12.8540(14) | 13.113(4) | 7.367(3) |
| c/(Å) | 14.971(4) | 14.893(9) | 14.4865(18) | 14.901(4) | 24.627(12) |
| α/(deg) | 76.925(8) | 73.604(16) | 109.170(3) | 104.696(10) | 90 |
| β/(deg) | 73.214(7) | 77.732(17) | 100.546(4) | 108.283(9) | 96.652(16) |
| γ/(deg) | 88.047(7) | 86.394(18) | 92.703(4) | 95.127(10) | 90 |
| V/(Å3) | 2134.3(9) | 2139(2) | 2175.8(4) | 2212.9(11) | 2601(2) |
| Z | 2 | 2 | 2 | 2 | 4 |
| D c/(g cm−3) | 1.489 | 1.429 | 1.486 | 1.445 | 1.531 |
| θ range/(deg) | 2.36–26.00 | 2.35–25.00 | 2.22–28.31 | 2.39–26.00 | 2.31–25.00 |
| Absorption coefficient/mm−1 | 0.592 | 0.539 | 0.575 | 0.518 | 0.817 |
| F (000) | 972 | 936 | 1000 | 988 | 1216 |
| Reflections collected | 48510 |
27918 |
36643 |
49629 |
33602 |
| Independent reflections | 8348 | 7505 | 10715 |
8693 | 4590 |
| Observed reflections (I > 2σ(I)) | 6387 | 5070 | 5961 | 6286 | 2746 |
| Number of parameters | 554 | 551 | 584 | 579 | 346 |
| Goodness-of-fit on F2 | 1.086 | 1.079 | 1.018 | 1.149 | 1.117 |
| R int | 0.0954 | 0.0840 | 0.1032 | 0.0981 | 0.1405 |
| Final R indices (I > 2σ(I)) | R 1 = 0.0673, wR2 = 0.1979 | R 1 = 0.0642, wR2 = 0.1754 | R 1 = 0.0530, wR2 = 0.1041 | R 1 = 0.0493, wR2 = 0.1240 | R 1 = 0.0917, wR2 = 0.2416 |
| R indices (all data) | R 1 = 0.0860, wR2 = 0.2137 | R 1 = 0.0997, wR2 = 0.1926 | R 1 = 0.1232, wR2 = 0.1290 | R 1 = 0.0777, wR2 = 0.1350 | R 1 = 0.1530, wR2 = 0.2693 |
| Largest peak/hole (e Å−3) | 1.849/−1.248 | 0.699/−0.705 | 0.515/−0.718 | 0.974/−0.685 | 3.099/−3.601 |
Photocatalytic degradation measurements
The photocatalytic degradation behavior of CPs 2, 4, and 9 was investigated in aqueous solution. Rhodamine B (RhB) and MB were selected as two model dye contaminants. In practice, powder samples (50 mg) of the synthesized CPs were added to the dye aqueous solutions (150 mL, 10 mg L−1) for the photocatalytic degradation. Subsequently, the mixed solution was stirred under dark conditions for 30 min for the purpose of bringing the CPs and dyes into adsorption–desorption equilibrium. Then 1 mL of 30% H2O2 was added to the solution, and the solution was stirred constantly under a 200 W high pressure Hg lamp. In order to assess the degradation behavior, 5 mL of clear solution was taken out at intervals of 15 min over the course of a 120-min period and subjected to UV-vis spectral measurements. The control experiments were completed under the same conditions without any catalyst.Results and discussion
Crystal structures of CPs 1–3
Solvent evaporation from dilute solutions of racemic L in acetonitrile containing Ag(I) nitrate, Ag(I) tetrafluoroborate, and Ag(I) perchlorate afforded CPs 1–3 as yellow crystals, respectively. Suitable crystals were randomly selected and structurally characterized by single-crystal X-ray diffraction analysis. The results showed that spontaneous chiral resolution had occurred during crystallization of the complexes. Since CPs 1–3 are isostructural except for the variations in the counter anions, and the corresponding CPs a and b are enantiomers, only 2a is discussed below as a representative example.CP 2a crystallizes in a monoclinic unit cell with the chiral space group C2. The asymmetric unit of 2a comprises one independent LR, half of an Ag(I) ion, half of a free tetrafluoroborate anion, and half of a lattice acetonitrile molecule. In CP 2a, each Ag(I) center is four-coordinated by four pyridyl nitrogen atoms from four LR ligands, lying in a distorted tetrahedral geometry with τ4 = 0.79 (Fig. 1a).53 The Ag–N bond lengths are in the range of 2.267(3)–2.423(3) Å, and the bond angles around the Ag(I) centers are comparable to those in reported tetrahedral Ag(I) complexes.54,55 The extended connection of the ligands with Ag(I) ions leads to a 2D homochiral cationic coordination network parallel to the ab plane, in which the anions are dispersed within the voids to neutralize the overall charge (Fig. 1b). In addition, the 2D sheets stack into a 3D supramolecular framework through weak C–H⋯O hydrogen bonds, and the top view of this framework shows 1D M-helical channels (Fig. 1c). The enantiomeric nature of CP 2b can be simply represented by the mirror structure; thus, corresponding 1D P-helical channels are formed in exactly the same coordination framework (Fig. 1d).
 | ||
| Fig. 1 (a) The coordination environments around the Ag atoms in 2a. Symmetry codes: a = 1 − x, y, −1 − z; b = −1/2 + x, −1/2 + y, –1 + z; and c = 3/2 − x, −1/2 + y, −z. (b) 2D sheet structure viewed along the ab plane (the acetonitrile molecules are omitted for clarity). (c) 1D M-helical channels formed in the 3D framework of 2a (LR enantiomers are colored in red). (d) 1D P-helical channels formed in the 3D framework of 2b (LS enantiomers are colored in blue). | ||
Crystal structures of CPs 4–8
With SbF6−, CF3SO3−, CF3CO2−, tosylate, and salicylate as the counter anions, yellow crystals of CPs 4–8 were obtained with drastically different coordination configurations compared to 1–3. Since CPs 4–8 show very similar assembly modes, only the structure of 4 is discussed herein as a representative example. CP 4 crystallizes in a triclinic unit cell with the space group P. The asymmetric unit comprises two ligands, an Ag(I) ion, a free hexafluoroantimonate anion, a lattice acetonitrile molecule, and a lattice water molecule. Each Ag(I) center is four-coordinated by four pyridyl nitrogen atoms from four adjacent ligands in a distorted tetrahedral geometry with τ4 = 0.77 (Fig. 2a). The Ag–N bond lengths are in the range of 2.268(4)–2.436(4) Å, and the bond angles around the Ag(I) centers vary from 96.43(15)° to 135.90(14)°. In contrast to structures 1–3, two antiparallel ligands connect with two Ag(I) ions to form a 26-membered metallomacrocycle, which is further bridged by adjacent ligands in the μ2-mode to yield a 1D tube-like chain. The hexafluoroantimonate anions are located in the 1D tube to neutralize the overall charge (Fig. 2b). To clearly demonstrate the chiral assembly mode in CP 4, ligands LR and LS are colored in red and blue, respectively, as shown in Fig. 2c. It is of much interest to observe that each Ag2L2 metallomacrocycle comprises two ligands with opposite chirality, and that this extended coordination results in a heterochiral chain constructed through a chiral self-discrimination process. The 1D chains are associated through weak hydrogen bonds to generate a 3D network within 1D channels formed along the a axis (Fig. 2d).
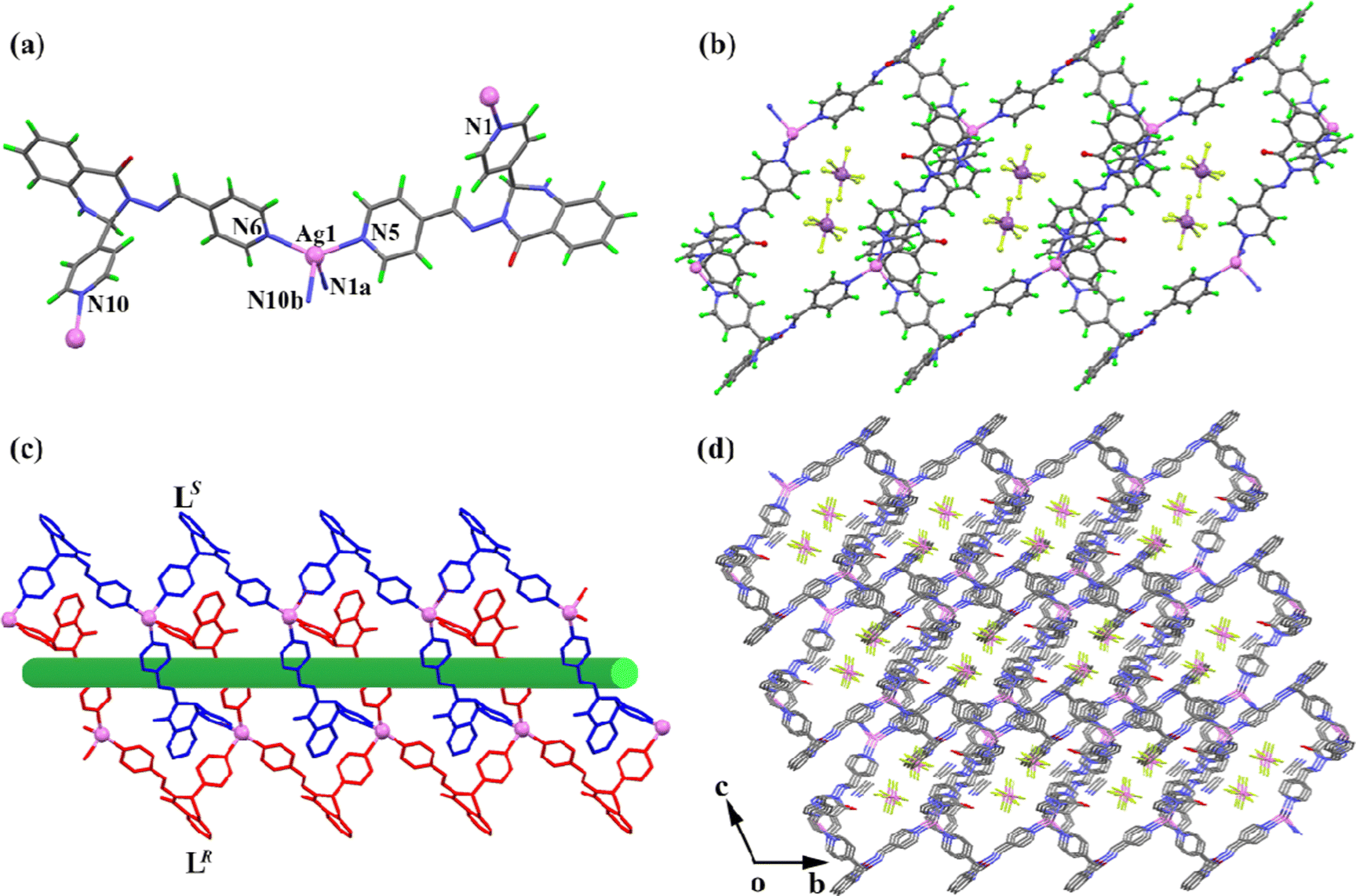 | ||
| Fig. 2 (a) The coordination environments around the Ag atoms in 4. Symmetry codes: a = x, 1 + y, z; b = 2 − x, 2 − y, 1 − z. (b) 1D tube-like chain structure of 4 (the solvent molecules are omitted for clarity). (c) The chiral assembly mode in 4. (d) 1D channels viewed along the a axis. | ||
Crystal structure of CP 9
The coordination reaction of racemic L with Ag(I) benzoate in acetonitrile solution afforded CP 9 as yellow crystals. CP 9 crystallizes in a monoclinic unit cell with the space group P2/c. The asymmetric unit is comprised of a ligand L, an Ag(I) ion, a coordinated benzoate anion, and a lattice acetonitrile molecule. There are two types of Ag(I) ions with different coordination modes in the complex (Fig. 3a). Each Ag1 center is four-coordinated by two pyridyl nitrogen atoms and two carboxyl oxygen atoms from adjacent benzoate anions, lying in a distorted tetrahedral geometry with τ4 = 0.84, while each Ag2 center is two-coordinated by two pyridyl nitrogen atoms from two adjacent ligands in a linear geometry. The distance between the adjacent Ag(I) centers is 12.557 Å, and the bond lengths and bond angles around these Ag(I) centers are consistent with those in other Ag(I) complexes.56,57 The dihedral angle between the terminal pyridyl rings bridged by the Ag1 center is 36.34°, and the two pyridyl rings bridged by the Ag2 center are parallel to each other. This extended coordination of the ligands with Ag(I) ions results in a 1D zig-zag chain polymer (Fig. 3b). It is worth noting that each four-coordinated Ag(I) ion is connected with two ligands of the same chirality, while each two-coordinated Ag(I) ion is connected with two ligands of opposite chirality, thus resulting in a heterochiral structure. Furthermore, π⋯π stacking interactions with a centroid-to-centroid distance of 3.771 Å are formed between the adjacent 1D chains to stabilize the 2D packing structure (Fig. 3c), and the 2D structure self-assembles through N–H⋯O hydrogen bonds to generate a 3D supramolecular network (Fig. 3d). | ||
| Fig. 3 (a) Coordination environments around the Ag(I) atoms in 9. Symmetry codes: a = 1 − x, y, 3/2 − z; b = 2 − x, −1 − y, 1 − z. (b) 1D zig-zag structure and the chiral assembly mode in 9 (the solvent molecules are omitted for clarity). (c) 2D packing structure formed by π⋯π stacking interactions of 1D zig-zag chains. (d) 3D supramolecular framework formed by N–H⋯O hydrogen bonds. | ||
Determining factors for the chiral assembly
In this work, nine AgX salts (X = NO3−, BF4−, ClO4−, SbF6−, CF3SO3−, CF3CO2−, tosylate, salicylate, and benzoate) were selected for the synthesis of CPs 1–9 under similar conditions. The simplified structures of CPs 1–9 and the chiral assembly modes of racemic L are summarized in Fig. 4. It is noted that all of the CPs adopt 1D or 2D structures, which can be classified into three kinds of topologies according to the different anions employed. CPs 1–3 have 2D sheet structures, whereas CPs 4–8 have 1D tube-like structures. In these CPs, all of the Ag(I) centers are four-coordinated by the terminal pyridyl rings of L, and the noncoordinated anions are dispersed within the voids to maintain charge balance. With benzoate as the anion, a 1D zig-zag structure was obtained, in which the benzoate anion acts as a monodentate ligand in coordinating with an Ag(I) ion. By comparing the chiral assembly modes of racemic L in these CPs, it is found that CPs 1–3 exist as homochiral polymers formed by spontaneous chiral resolution, while CPs 4–9 exist as heterochiral polymers, which indicate their chiral self-recognition and self-discriminating processes, respectively. In summary, the assembly of racemic L with Ag(I) salts conforms to the following three characteristics. (i) The flexible coordination number of the Ag(I) ion results in two different coordination modes in CP 9. (ii) The assembly of racemic L with Ag(I) ions leads to homochiral polymers in the presence of anions of small size (NO3−, BF4−, and ClO4−), while the assembly leads to heterochiral polymers in the presence of other larger sizes of anions, implying that the size of the anions easily affects the final coordination structures. (iii) The coordination ability of benzoate is superior to those of the other anions, making the structure of CP 9 obviously different from the other CPs. These characteristics show that the coordination configuration of metal ions, as well as the size and coordination ability of anions, exert a synergistic influence on the chiral assembly of CPs.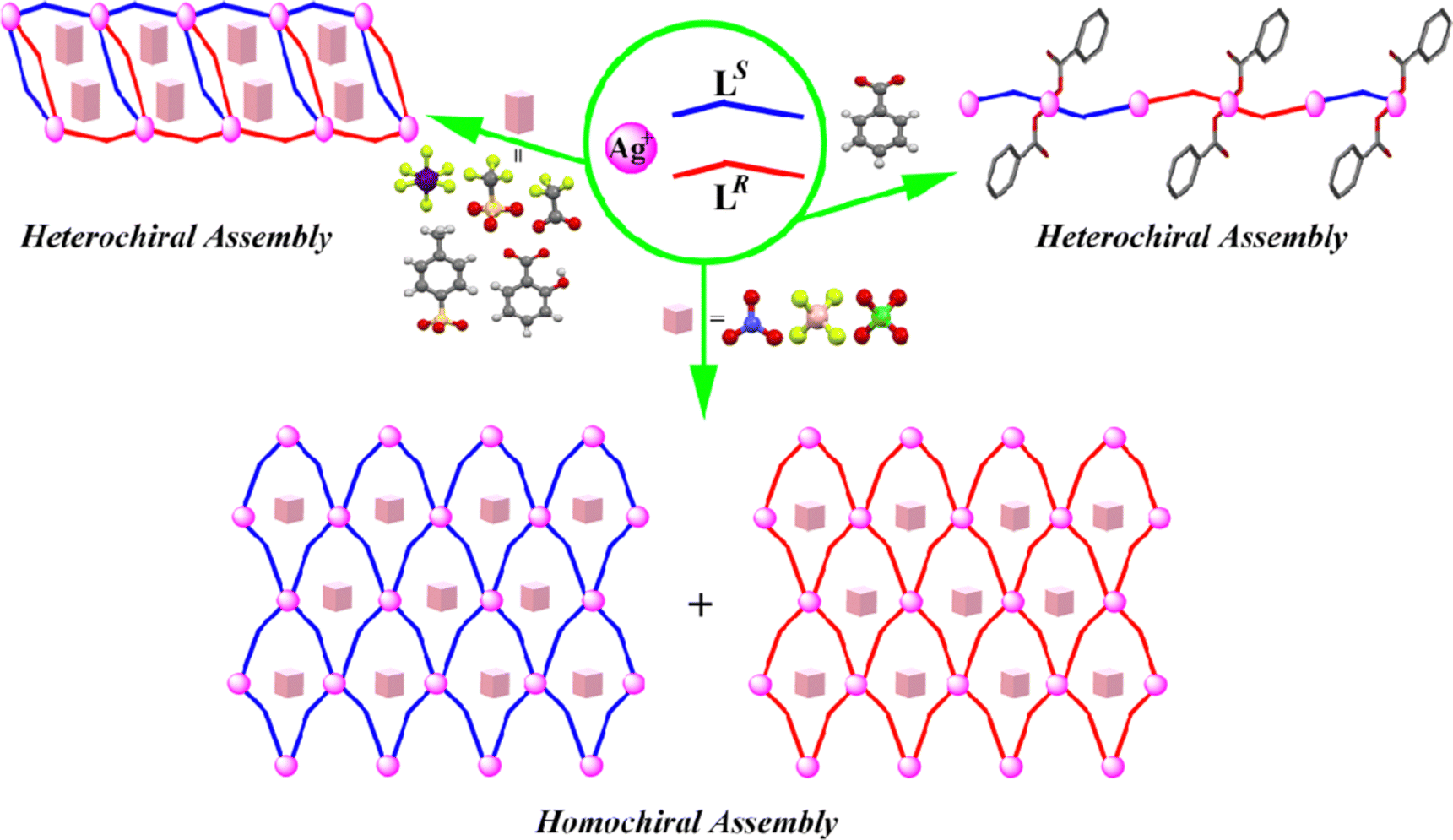 | ||
| Fig. 4 Structural differences of chiral assembly in CPs 1–9 in the presence of different anions (the ligands and the anions have been simplified for clarity). | ||
CD, PXRD and TGA characterization
The CD spectra of CPs 1–3 were recorded in acetonitrile solution (Fig. S11, ESI†). The CD spectrum of 1a exhibits negative Cotton effects at 240 and 281 nm, and exhibits positive Cotton effects at 266 nm and 360 nm, while 1b shows opposite signals at the same positions to form a mirror image, implying that 1a and 1b are enantiomers. The CD spectra of 2 and 3 are similar to those of 1, as might be expected in view of the high similarity of the self-assembled structures. PXRD patterns of the synthesized samples in the solid state were obtained and analyzed at room temperature (Fig. S12, ESI†). The experimental data match well with the simulated data, indicating the bulk phase purity of the CPs. Also, the TGA of these CPs were performed under a nitrogen atmosphere to assess their thermal stabilities (Fig. S13 and S14, ESI†). The CPs show slight weight losses with an increase in temperature, corresponding to the release of the lattice acetonitrile and/or water molecules. Upon further heating, the organic skeleton structures show the onset of collapse. The starting temperatures for the decomposition of the CPs conform to the following order: 127 °C (9) < 164 °C (8) < 172 °C (6) < 188 °C (3) < 198 °C (1) < 200 °C (2) < 202 °C (5) < 204 °C (7) < 212 °C (4).Optical properties
The solid-state UV-vis diffuse-reflectance spectra of CPs 1–9 were recorded at room temperature. All of the CPs exhibit strong absorptions in the UV region (Fig. S15, ESI†), implying that they may be sensitive to UV light and show potential photocatalytic activity. The band-gap energies (Eg) of CPs 1–9 were measured by the Kubelka–Munk method based on their UV-vis diffuse-reflectance spectra (Fig. S16, ESI†). The Eg values are estimated to lie in the range of 1.678–1.695 eV for CPs 1–9, indicating their semiconductive behavior and hence their potential for use as photocatalysts.58Photocatalytic properties
Considering that CPs 1–9 can be divided into three kinds of self-assembled structures, CPs 2 (representing CPs 1–3), 4 (representing CPs 4–8), and 9 with relatively low Eg values were selected to evaluate their catalytic degradation performances toward RhB and MB, which are regarded as two model organic dyes in wastewater. CPs 2, 4, and 9 are all insoluble in water, and the PXRD patterns of pristine CPs 2, 4, and 9 are consistent with those after the treatments of different aqueous solutions with different pH values (3–12) or temperatures (25–75 °C) for 3 h (Fig. S17, ESI†), indicating their good structural stability. Therefore, they were tested as catalysts in powder form for the degradation reactions in aqueous solution. In the presence of H2O2 (1 mL), the degradation reactions of RhB and MB were monitored by recording UV-vis spectra at intervals of 15 min, following the characteristic absorptions at 554 and 664 nm, respectively. The photocatalytic degradation activities of RhB by CPs 2, 4, and 9 are shown in Fig. 5a–c, and the concentration ratios (C/C0, where C0 represents the initial RhB concentration of the solution and C represents the RhB concentration of the solution at time t) in the absence or presence of the catalyst versus irradiation time (t) are shown in Fig. 5d. | ||
| Fig. 5 UV-vis absorption spectra of RhB (10 mg L−1) in the presence of CPs 2 (a), 4 (b), and 9 (c). (d) Plots of the concentration ratios of RhB (C/C0) versus irradiation time in the presence of the CPs and without any catalyst during the decomposition reaction under UV irradiation. | ||
In the absence of a catalyst, the addition of H2O2 leads only to a slight decrease in the characteristic absorption, even after 120 min. Thus, there is no significant dye degradation without a catalyst. In contrast, the degradation of RhB under UV light irradiation leads to remarkable absorption changes upon addition of CP 4, and the degradation efficiency [R = (C0 − C)/C0] of RhB reaches 94.6% within 120 min. The UV-vis absorption spectra indicate a degradation efficiency of 49.7% in the presence of CP 2, while the degradation efficiency is only 24.5% in the presence of CP 9 after 120 min, indicating that CP 4 exhibits much higher photocatalytic activity toward RhB than CP 2 or 9. Likewise, the photocatalytic activities of CPs 2, 4, and 9 for the degradation of MB were also examined (Fig. 6a–c). Approximately 86.1% of MB is decomposed in the presence of CP 4 within 120 min, while only 46.5% and 15.0% of degradation efficiencies are observed in the presence of CPs 2 and 9, respectively (Fig. 6d). Overall, the degradation experiments imply that CP 4 exhibits good photocatalytic activity toward both RhB and MB under UV light irradiation, whereas CP 9 shows relatively poor degradation performances toward these two dyes.
 | ||
| Fig. 6 UV-vis absorption spectra of MB (10 mg L−1) in the presence of CPs 2 (a), 4 (b), and 9 (c). (d) Plots of the concentration ratios of MB (C/C0) versus irradiation time in the presence of the CPs and without any catalyst during the decomposition reaction under UV irradiation. | ||
Furthermore, kinetic studies were carried out to analyze the degradation of RhB and MB in the presence of CP 4. The obtained kinetic data were fitted to a pseudo-first-order kinetic model (Fig. S18, ESI†). Linear fitting of plots ofln(C0/C) versus irradiation time (t) by the pseudo-first-order equation shows the applicability of this model, with the corresponding square of correlation coefficient (R2) values of 0.9814 (RhB) and 0.9948 (MB), respectively. The rate constants (k) are determined from the slopes of the pseudo-first-order plots as 0.0245 min−1 for RhB and 0.0166 min−1 for MB. Compared to the previously reported values,42,59 CP 4 shows a relatively good photocatalytic degradation performance toward these two dyes.
According to the above experimental observations and previously reported literature by Chen and Paola,60,61 the photocatalytic mechanism of Ag(I) CPs is assumed to rely on a hydroxyl radical (˙OH)-mediated route (Fig. S19, ESI†). H2O2 is an effective and active oxidizing substance. The photocatalytic reaction proceeds when the Ag(I) CPs are irradiated with UV light. When the photon energy is greater than the semiconductor forbidden bandwidth, the electrons (e−) in the valence band (VB) are excited to the conduction band (CB), leaving the same amount of holes (h+) in the VB, which leads to the formation of e−CB–h+VB pairs.62,63 Subsequent interactions of h+ with H2O2 or H2O generate extremely active •OH. Simultaneously, the reaction of e− with O2 derived from the decomposition of H2O2 generates a superoxide radical (˙O2−), which is ultimately transformed into ˙OH.64,65 The resulting ˙OH radical, which acts as a strong oxidizing agent, can effectively oxidize most organic dyes into CO2, H2O, and other small molecules.
To verify the above photocatalytic mechanism of the synthesized Ag(I) CPs toward the dyes, we selected the reaction system containing CP 4 and RhB by introducing tert-butyl alcohol (TBA). It is known that TBA is an effective quenching agent and could be used to capture the resulting ˙OH radicals in the reaction system. Upon the addition of 0.2 mL of excess TBA into the solution containing CP 4, H2O2, and RhB, the degradation of RhB was evidently hindered as the TBA brought about the capture of ˙OH radicals, with only a degradation efficiency of 9.3% under UV light irradiation for 2 h (Fig. S20, ESI†), indicating the ˙OH-mediated mechanism.
We also tried to collect the final degradation products by using a previous method to further confirm the mechanism.66 The solution after the degradation reaction was extracted using chloroform, and the organic phase was characterized by gas chromatography–mass spectrometry (GC/MS). No new organic species were detected, which implied that RhB was completely decomposed in the solution. The proposed CO2 evolved from the reaction was collected by hanging a small bottle of saturated Ba(OH)2 solution inside the reaction tube containing CP 4 and RhB. During UV light irradiation, a white precipitate of BaCO3 was observed, which was collected, dried and weighed. The yield of CO2 based on the degraded RhB was about 94% under UV light irradiation for 2 h. These results also confirm the degradation of RhB and the ˙OH-mediated mechanism described above.
It is known that the metal ion, the coordination environment, and the assembly mode are key factors influencing the photocatalytic activity of the synthesized CPs. In order to rationalize the different photocatalytic performances of CPs 2, 4, and 9, we carried out DFT calculations using the Gaussian 16 program67 to determine the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) in each case (Fig. 7). In the calculations, the hybrid method B3LYP was employed, along with an effective core potential basis set such as SDD for the Ag atom and 6-31++G* for all other atoms. The .cif files of the crystal structures were used as starting geometries for the optimization processes. The HOMO–LUMO (H–L) energy gaps of these CPs are listed in Table 2, which increase as follows: 9 (0.94 eV) < 2 (1.08 eV) < 4 (3.47 eV). The corresponding wavelengths  are calculated to be 4 (357 nm) < 2 (1148 nm) < 9 (1319 nm). Under 365 nm UV irradiation, a more suitable H–L gap results in much easier electronic excitation, facilitating the separation of h+ and e−. In view of this, the photocatalytic activity order 4 > 2 > 9 is in good agreement with the sequence of the H–L energy gaps. It is further inferred that CPs 5–8, which show similar structures to CP 4, may also exhibit good photocatalytic activity toward these two dyes under the same conditions.
are calculated to be 4 (357 nm) < 2 (1148 nm) < 9 (1319 nm). Under 365 nm UV irradiation, a more suitable H–L gap results in much easier electronic excitation, facilitating the separation of h+ and e−. In view of this, the photocatalytic activity order 4 > 2 > 9 is in good agreement with the sequence of the H–L energy gaps. It is further inferred that CPs 5–8, which show similar structures to CP 4, may also exhibit good photocatalytic activity toward these two dyes under the same conditions.
 | ||
| Fig. 7 HOMO and LUMO Frontier molecular orbitals of CPs 2, 4, and 9 calculated with DFT at the B3LYP level. | ||
| CP | HOMO (H) energy (eV) | LUMO (L) energy (eV) | H–L energy gap (eV) |
|
|---|---|---|---|---|
| 2 | −4.87 | −3.79 | 1.08 | 1148 |
| 4 | −6.54 | −3.07 | 3.47 | 357 |
| 9 | −3.77 | −2.83 | 0.94 | 1319 |

Additionally, to check the reusability of CP 4 as a catalyst for the photocatalytic degradation of RhB and MB, recycling experiments were performed (Fig. 8). After the dye degradation experiment, the catalyst was collected by centrifugation, washed with distilled water, and dried at 60 °C for 5 h. It was then reused in a new experiment. The results show that the degradation efficiency of recycled CP 4 toward RhB within 2 h decreases from 94.6% to 85.0% after 10 cycles, and the degradation efficiency toward MB decreases from 86.1% to 77.1%. The slow decline in the degradation efficiencies may be due to poisoning of the catalyst by the oxidation products. Moreover, the experimental PXRD patterns of CP 4 after the degradation experiment match well with the original patterns, and the characteristic elemental peaks of C, O, N, and Ag in the XPS wide spectra (Fig. S21, ESI†) of CP 4 after the photocatalytic degradation reaction are consistent with those before the reaction, indicating the good structural stability of CP 4. The recycling experiments suggest that CP 4 maintains its photocatalytic activity and structural integrity during repeated photocatalytic cycles, which is important for its potential use as a photocatalyst.
 | ||
| Fig. 8 Recycling catalytic performances of CP 4 for the degradation of RhB and MB under UV irradiation. | ||
Conclusions
In summary, nine CPs were successfully constructed from a racemic ligand with different Ag(I) salts. In the presence of anions of small sizes, CPs 1–3 were obtained as 2D homochiral sheets, whereas in the presence of anions of larger sizes, CPs 4–8 were obtained as 1D heterochiral tube-like chains. In these CPs, all of the anions are noncoordinated and are dispersed in the voids to maintain charge balance. In the presence of an anion with strong coordination ability, CP 9 was obtained as a 1D heterochiral zig-zag chain, in which the benzoate acts as a monodentate ligand. The results provide sufficient evidence that the coordination configuration of the metal ions, as well as the size and coordination ability of the anions, influenced the chiral assembly of racemic ligands with metal ions. In addition, based on the structural differences and the band-gap energies of these Ag(I) CPs, the photocatalytic degradation performances of CPs 2, 4, and 9 toward RhB and MB were further studied. The results revealed that the catalytic activities for the degradation of both dyes are in the order 4 > 2 > 9. In combination with the photocatalytic performance, the structural stability, and the relatively good catalytic recycling performance of CP 4, it is thus inferred that CPs 4–8 may be suitable for the photocatalytic degradation of RhB and MB in aqueous solution based on their structural similarity, which is of potential significance in relation to environmental protection. Moreover, the study of the chiral resolution of CPs 1–3 induced by chiral reagents is in progress.Data availability
The data supporting this article have been included as part of the ESI.† CCDC 2361654–2361663 contains the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22461009) and the Guizhou Provincial Basic Research Program (Natural Science) in China (ZK[2024]080).References
- Y. Machida, Y. Tanaka, Y. Masuda, A. Kimura and T. Kawasaki, Chem. Sci., 2023, 14, 4480 RSC.
- X. Yang, X. Jin, L. Zhou, P. Duan, Y. Fan and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202115600 CrossRef CAS PubMed.
- J. Karst, N. H. Cho, H. Kim, H.-E. Lee, K. T. Nam, H. Giessen and M. Hentschel, ACS Nano, 2019, 13, 8659 CrossRef CAS PubMed.
- N. del Giudice, G. Voegeli, J.-M. Strub, B. Heinrich and L. Douce, Inorg. Chem., 2024, 63, 6103 CrossRef CAS PubMed.
- J. Liu, M. Morikawa, H. Lei, K. Ishiba and N. Kimizuka, Langmuir, 2016, 32, 10597 CrossRef CAS PubMed.
- X. He, Q. Zhang, W. Wang, L. Lin, X. Liu and X. Feng, Org. Lett., 2011, 13, 804 CrossRef CAS.
- P. S. Steinlandt, L. Zhang and E. Meggers, Chem. Rev., 2023, 123, 4764 CrossRef CAS PubMed.
- C. S. Arribas, O. F. Wendt, A. P. Sundin, C.-J. Carling, R. Wang, R. P. Lemieux and K. Wärnmark, Chem. Commun., 2010, 46, 4381 RSC.
- X. Shang, I. Song, J. H. Lee, W. Choi, H. Ohtsu, G. Y. Jung, J. Ahn, M. Han, J. Y. Koo, M. Kawano, S. K. Kwak and J. H. Oh, ACS Appl. Mater. Interfaces, 2019, 11, 20174 CrossRef CAS.
- J. Dupont, B. Hartwig, K. L. Barbu-Debus, V. Lepere, R. Guillot, M. A. Suhm and A. Zehnacker, Phys. Chem. Chem. Phys., 2024, 26, 10610 RSC.
- X. Huang, F. Chen, H. Sun, L. Yang, Q. Yang, Z. Zhang, Y. Yang, Q. Ren and Z. Bao, J. Am. Chem. Soc., 2024, 146, 617 CrossRef CAS.
- Z. Zhou, C. E. Hauke, B. Song, X. Li, P. J. Stang and T. R. Cook, J. Am. Chem. Soc., 2019, 141, 3717 CrossRef CAS.
- X. Yang, T.-C. Cheng and A. J. Morris, J. Mater. Chem. C, 2024, 12, 4562 RSC.
- R. Adam, M. Mon, R. Greco, L. H. G. Kalinke, A. Vidal-Moya, A. Fernandez, R. E. P. Winpenny, A. Doménech-Carbó, A. Leyva-Pérez, D. Armentano, E. Pardo and J. Ferrando-Soria, J. Am. Chem. Soc., 2019, 141, 10350 CrossRef CAS.
- S. Li, B. Wang, G. Liu, X. Li, C. Sun, Z. Zhang and X. Wang, Inorg. Chem. Front., 2024, 11, 1561 RSC.
- X.-Y. Ren, F.-Y. Chen, C.-H. Zhang, Z.-G. Liang, X.-Y. Yu, S.-D. Han and G.-M. Wang, Chem. – Eur. J., 2024, e202402581 CrossRef CAS.
- Q. Zhang, Y. Wang, P. Braunstein and J.-P. Lang, Chem. Soc. Rev., 2024, 53, 5227 RSC.
- Y. Wang, Q. Zhang, Q. Liu, B. F. Abrahams and J.-P. Lang, Angew. Chem., Int. Ed., 2024, 63, e202409472 CrossRef CAS PubMed.
- X.-Q. Ma, H.-P. Xiao, Y. Chen, Q.-S. Lai, X.-X. Li and S.-T. Zheng, Coord. Chem. Rev., 2024, 510, 215818 CrossRef CAS.
- W.-H. Zhang, Z.-G. Ren and J.-P. Lang, Chem. Soc. Rev., 2016, 45, 4995 RSC.
- T.-Y. Gu, M. Dai, D. J. Young, Z.-G. Ren and J.-P. Lang, Inorg. Chem., 2017, 56, 4668 CrossRef.
- H. Song, N. Wang, X. Shi, H. Meng, Y. Han, J. Wu, J. Xu, Y. Xu, T. Sun and X. Zhang, Appl. Organomet. Chem., 2020, 34, e5972 CrossRef CAS.
- J. Zhang, W. Gao, Y. Lv, C. Fei, Z. Wu and H. Wu, J. Mol. Struct., 2024, 1311, 138448 CrossRef CAS.
- S. Kumar, S. Liu, B. Mohan, M. Zhang, Z. Tao, Z. Wan, H. You, F. Sun, M. Li and P. Ren, Inorg. Chem., 2021, 60, 7070 CrossRef CAS PubMed.
- L. Mistry, O. El-Zubir, T. Pope, P. G. Waddell, N. Wright, W. A. Hofer, B. R. Horrocks and A. Houlton, Cryst. Growth Des., 2021, 21, 4398 CrossRef CAS.
- Á. García-Romero, J. M. Martín-Álvarez, D. Miguel, D. S. Wright, C. M. Álvarez and R. García-Rodríguez, Inorg. Chem., 2021, 19206 CrossRef.
- J.-Y. Cheng, P. Wang, J.-P. Ma, Q.-K. Liu and Y.-B. Dong, Chem. Commun., 2014, 50, 13672 RSC.
- C.-P. Li, J.-Y. Ai, H. He, M.-Z. Li and M. Du, Cryst. Growth Des., 2019, 19, 2235 CrossRef CAS.
- M. I. Rogovoy, T. S. Frolova, D. G. Samsonenko, A. S. Berezin, I. Y. Bagryanskaya, N. A. Nedolya, O. A. Tarasova, V. P. Fedin and A. V. Artem'ev, Eur. J. Inorg. Chem., 2020, 1635 CrossRef CAS.
- R. Das, V. Parihar and C. M. Nagaraja, Inorg. Chem. Front., 2022, 9, 2583 RSC.
- M. Garai, K. Maji, V. V. Chernyshev and K. Biradha, Cryst. Growth Des., 2016, 16, 550 CrossRef CAS.
- C. Huang, H.-Q. Tian, R.-F. Li, Y. Xiong, T. Jiang, D.-M. Chen and B.-X. Zhu, Inorg. Chem., 2022, 61, 19512 CrossRef CAS PubMed.
- J. Moussa, L. M. Chamoreau, M. P. Gullo, A. D. Esposti, A. Barbieri and H. Amouri, Dalton Trans., 2016, 45, 2906 RSC.
- V. Selvaraj, T. S. Karthika, C. Mansiya and M. Alagar, J. Mol. Struct., 2021, 1224, 129195 CrossRef CAS.
- T. Islam, M. R. Repon, T. Islam, Z. Sarwar and M. M. Rahman, Environ. Sci. Pollut. Res., 2023, 30, 9207 CrossRef CAS PubMed.
- S. E. H. Etaiw, T. A. Fayed, D. M. A. El-Aziz and H. M. Khatab, Appl. Organometal. Chem., 2020, 34, e5301 CrossRef CAS.
- X.-F. Qi, F. Zhang, Z.-P. Chen, X. Chen, M.-C. Jia, H.-F. Ji and Z.-F. Shi, J. Mater. Chem. C, 2023, 11, 3715 RSC.
- V. Shukla, M. Ahmad, R. L. LaDuca and K. A. Siddiqui, J. Mol. Struct., 2023, 1294, 136371 CrossRef CAS.
- C. Shi, Z.-H. Nie, L. Zhao, L. Lu, F. Cheng, X. Chen, G. Tan, Q.-Q. Liu, J. Wang, R. Chauhan and A. Kumar, Polyhedron, 2021, 207, 115362 CrossRef CAS.
- Y.-Y. Yang, L.-X. Zhou, H.-L. Zhu, W.-Y. Li and Y.-Q. Zheng, Polyhedron, 2018, 148, 161 CrossRef CAS.
- H. Li, Y. Wang, Y. He, Z. Xu, X. Zhao and Y. Han, New J. Chem., 2017, 41, 1046 RSC.
- J.-Z. Gu, Y. Cai, M. Wen, Z.-F. Shi and A. M. Kirillov, Dalton Trans., 2018, 47, 14327 RSC.
- L. Nandi, S. Barman, A. Das, P. Brandão, E. Zangrando, A. Basu and S. Dalai, J. Mol. Struct., 2023, 1293, 136291 CrossRef CAS.
- C.-Y. Liu, L.-Y. Xu, Z.-G. Ren, H.-F. Wang and J.-P. Lang, Cryst. Growth Des., 2017, 17, 4826 CrossRef CAS.
- H. Yuan, P. Shang, J. Yang, Q. Huang, L. Song and X.-F. Jiang, Inorg. Chem., 2023, 62, 2652 CrossRef CAS.
- C.-F. Liu, C.-Y. Liu, Z.-G. Ren and J.-P. Lang, Eur. J. Inorg. Chem., 2019, 1816 CrossRef CAS.
- C. Huang, Y.-J. Liu, Y.-T. Chen, D.-M. Chen and B.-X. Zhu, Eur. J. Inorg. Chem., 2020, 704 CrossRef CAS.
- Y.-J. Liu, Y.-T. Chen, M.-Z. Chen, X.-J. Mo, C. Huang, D.-M. Chen and B.-X. Zhu, Inorg. Chim. Acta, 2020, 510, 119702 CrossRef CAS.
- Y.-Q. Lin, X.-M. Tian, B.-X. Zhu, D.-M. Chen and C. Huang, Inorg. Chem., 2023, 62, 12099 CrossRef CAS.
- Y.-Q. Lin, X.-M. Tian, Y. Xiong, C. Huang, D.-M. Chen and B.-X. Zhu, Inorg. Chem., 2023, 62, 19887 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Cryst., 2009, 42, 339 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- G. Durá, M. C. Carrión, F. A. Jalón, A. M. Rodríguez and B. R. Manzano, Cryst. Growth Des., 2013, 13, 3275 CrossRef.
- L.-L. Han, X.-Y. Zhang, J.-S. Chen, Z.-H. Li, D.-F. Sun, X.-P. Wang and D. Sun, Cryst. Growth Des., 2014, 14, 2230 CrossRef CAS.
- Z.-L. Chu, H.-B. Zhu, D.-H. Hu, W. Huang and S.-H. Gou, Cryst. Growth Des., 2008, 8, 1599 CrossRef CAS.
- T. H. Noh, Y. J. Choi, Y. K. Ryu, Y.-A. Lee and O.-S. Jung, CrystEngComm, 2009, 11, 2371 RSC.
- S. Mandal, S. P. Nanavati, D. J. Willock and R. Ananthakrishnan, J. Phys. Chem. C, 2019, 123, 23940 CrossRef CAS.
- W. Hu, D. Liu, A. Singh, S. W. Gosavi, R. Chahuan, H. Sakiyama and M. Muddassir, J. Mol. Struct., 2022, 1248, 131510 CrossRef CAS.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206 RSC.
- A. D. Paola, E. García-López, G. Marcì and L. Palmisano, J. Hazard. Mater., 2012, 211–212, 3 CrossRef.
- N. K. Shee, H. J. Jo and H.-J. Kim, Inorg. Chem. Front., 2022, 9, 1270 RSC.
- F. Wang, F.-L. Li, M.-M. Xu, H. Yu, J.-G. Zhang, H.-T. Xia and J.-P. Lang, J. Mater. Chem. A, 2015, 3, 5908 RSC.
- Z.-G. Wang, H.-Y. Yin, T.-H. Liu, Z.-Q. Wu, L.-H. Chen, J. Fei and Q. Li, J. Mol. Struct., 2024, 1295, 136651 CrossRef CAS.
- J.-F. Wang, S.-Y. Liu, C.-Y. Liu, Z.-G. Ren and J.-P. Lang, Dalton Trans., 2016, 45, 9294 RSC.
- X.-Y. Wu, H.-X. Qi, J.-J. Ning, J.-F. Wang, Z.-G. Ren and J.-P. Lang, Appl. Catal., B, 2015, 168–169, 98 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Za-krzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2361654–2361663. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc03372h |
| This journal is © The Royal Society of Chemistry 2025 |