
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NHC–Ni nanoclusters covalently ligated on carbon nanotubes: highly active electrocatalysts for the oxygen evolution reaction†‡
Amalia
Rapakousiou
 *a,
Savvas G.
Chalkidis
b,
Michail P.
Minadakis
a,
María Luisa
Ruiz-González
c,
Cristina
Navio
d,
Georgios C.
Vougioukalakis
*b and
Nikos
Tagmatarchis
*a
*a,
Savvas G.
Chalkidis
b,
Michail P.
Minadakis
a,
María Luisa
Ruiz-González
c,
Cristina
Navio
d,
Georgios C.
Vougioukalakis
*b and
Nikos
Tagmatarchis
*a
aTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, Athens 11635, Greece. E-mail: arapak@eie.gr; tagmatar@eie.gr
bLaboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Athens, 15771, Greece. E-mail: vougiouk@chem.uoa.gr
cDepartamento de Química Inorgánica, Universidad Complutense de Madrid, Madrid, Spain
dIMDEA Nanoscience, Ciudad Universitaria de Cantoblanco, C/Faraday, 928049 Madrid, Spain
First published on 28th April 2025
Abstract
Developing robust and efficient oxygen evolution reaction (OER) electrocatalysts is essential for advancing renewable energy technologies like water electrolysis. Here, we present an electrocatalyst comprising N-heterocyclic carbene (NHC)-ligated nickel nanoclusters (NiNCs) covalently anchored on multi-walled carbon nanotubes (MWCNTs), with only 3.9% Ni/C atomic content. The unique nanoarchitecture, featuring a conductive NHC–Ni(0) core and an electroactive NiOx shell, was prepared via a dual metal-reduction and transmetalation strategy, offering exceptional electrocatalytic stability and efficiency, whereas MWCNTs offer mechanical robustness and a conductive nanocarrier support contributing to the enhanced OER kinetics. The electrocatalyst demonstrates low overpotentials of 320 mV and 500 mV at 10 mA cm−2 and 200 mA cm−2, respectively, in 1 M KOH, with very high specific (jECSA = 133 mA cm−2) and intrinsic (jnNi = 2.8 × 109 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mA mol−1 cm2) activities, as well as exceptional turnover frequencies (TOFs), reaching up to 7.4 s−1 under industrially relevant conditions (200 mA cm−2). Electrochemical impedance spectroscopy reveals rapid charge transfer kinetics (Rct: 66 Ω to 9.7 Ω) and efficient interfacial processes, driven by high dispersion and ultrafine NiNC size (<1.6 nm). The synergistic interplay between the NHC–Ni(0) core, which stabilizes the electroactive shell structure, and the MWCNTs, which enhance electron transport and provide mechanical robustness, ensures durability under prolonged cycling, with minimal losses (<1.3% overpotential gain and <0.5% apparent loss of Ni active sites). These results establish MWCNTs-NHC-ligated NiNCs as a scalable and high-performing electrocatalyst, surpassing RuO2 and competitive NiFe-based materials, highlighting how precise nanoengineering through innovative covalent architecture can enable efficient and durable OER technologies in clean energy systems.
mA mol−1 cm2) activities, as well as exceptional turnover frequencies (TOFs), reaching up to 7.4 s−1 under industrially relevant conditions (200 mA cm−2). Electrochemical impedance spectroscopy reveals rapid charge transfer kinetics (Rct: 66 Ω to 9.7 Ω) and efficient interfacial processes, driven by high dispersion and ultrafine NiNC size (<1.6 nm). The synergistic interplay between the NHC–Ni(0) core, which stabilizes the electroactive shell structure, and the MWCNTs, which enhance electron transport and provide mechanical robustness, ensures durability under prolonged cycling, with minimal losses (<1.3% overpotential gain and <0.5% apparent loss of Ni active sites). These results establish MWCNTs-NHC-ligated NiNCs as a scalable and high-performing electrocatalyst, surpassing RuO2 and competitive NiFe-based materials, highlighting how precise nanoengineering through innovative covalent architecture can enable efficient and durable OER technologies in clean energy systems.
Introduction
The Oxygen Evolution Reaction (OER) stands as a fundamental anodic process in electrochemical water splitting, essential for renewable energy technologies like water electrolysis and fuel cells.1–5 Efficient electrocatalysts for the OER are crucial for advancing these technologies and enabling the production of clean and sustainable energy. Nickel-based materials, including nanoclusters and nanoparticles, have attracted considerable attention due to their abundance, cost-effectiveness, and promising electrocatalytic activity in the OER.6–10 However, enhancing the performance and stability of nickel-based electrocatalysts remains a significant challenge.11 Recent research is increasingly focusing on novel catalyst–support interactions to enhance the electrochemical performance of Ni-based nanomaterials.12–16 In this context, exploring N-Heterocyclic Carbenes (NHCs) ligated to metal nanomaterials as electrocatalysts for the OER presents an underexplored but intriguing avenue for innovation.17–19 Among others, NHCs have emerged as promising ligands for tailoring the surface chemistry of metal nanomaterials.20 Renowned for their strong metal-binding affinity and tunable electronic properties, NHCs offer the potential to modulate the catalytic activity and stability of nickel nanoclusters and nanoparticles.21–23 Despite these promising attributes, the application of nanoscaled NHC–Ni interactions in electrocatalysis remains largely unexplored. This study aims to bridge this gap by investigating nanostructured MWCNTs-NHC-functionalized nickel nanoclusters (NiNCs) as electrocatalysts for the OER. Our nanoengineering strategy demonstrates the potential of rational nanoscale design in overcoming intrinsic material limitations to advance sustainable energy technologies.In this work, we present a highly innovative electrocatalyst for the Oxygen Evolution Reaction (OER), featuring NHC-ligated NiNCs chemically bonded on oxidized Multi-Walled Carbon Nanotubes (oMWCNTs). The oMWCNTs, known for their inherent electrocatalytic properties, pose a significant challenge as a nanocarrier due to their already high baseline activity.24 The synergistic interaction between NHC ligation, NiNCs, and MWCNT nanocarriers, however, achieves remarkable stability and electrocatalytic performance in the OER, outperforming oMWCNTs and surpassing the benchmark RuO2 electrocatalyst.
Results and discussion
The implementation of NHC ligands to stabilize and enhance the catalytic activity of metal catalysts has been extensively explored, leading to the development of robust catalytic protocols.25–27 In this regard, the synthetic strategy starts with the synthesis of an NHC-precursor ligand containing an alkyne group.28 Next, a copper-catalyzed azide–alkyne cycloaddition (CuAAC) with 4-azidoaniline 4 yields NHC-precursor 5, containing a 4-triazolylaniline group (Scheme 1, pages S5–S7 and Fig. S1–S11‡). The formation of NHC–Cu(I) complex 6 is achieved using a weak base route as outlined by Nolan's group,29 with mesityl substituents providing steric protection and stability to the in situ generated NHC ligands and the resulting copper complex (Scheme 1, page S8 and Fig. S12–S14‡).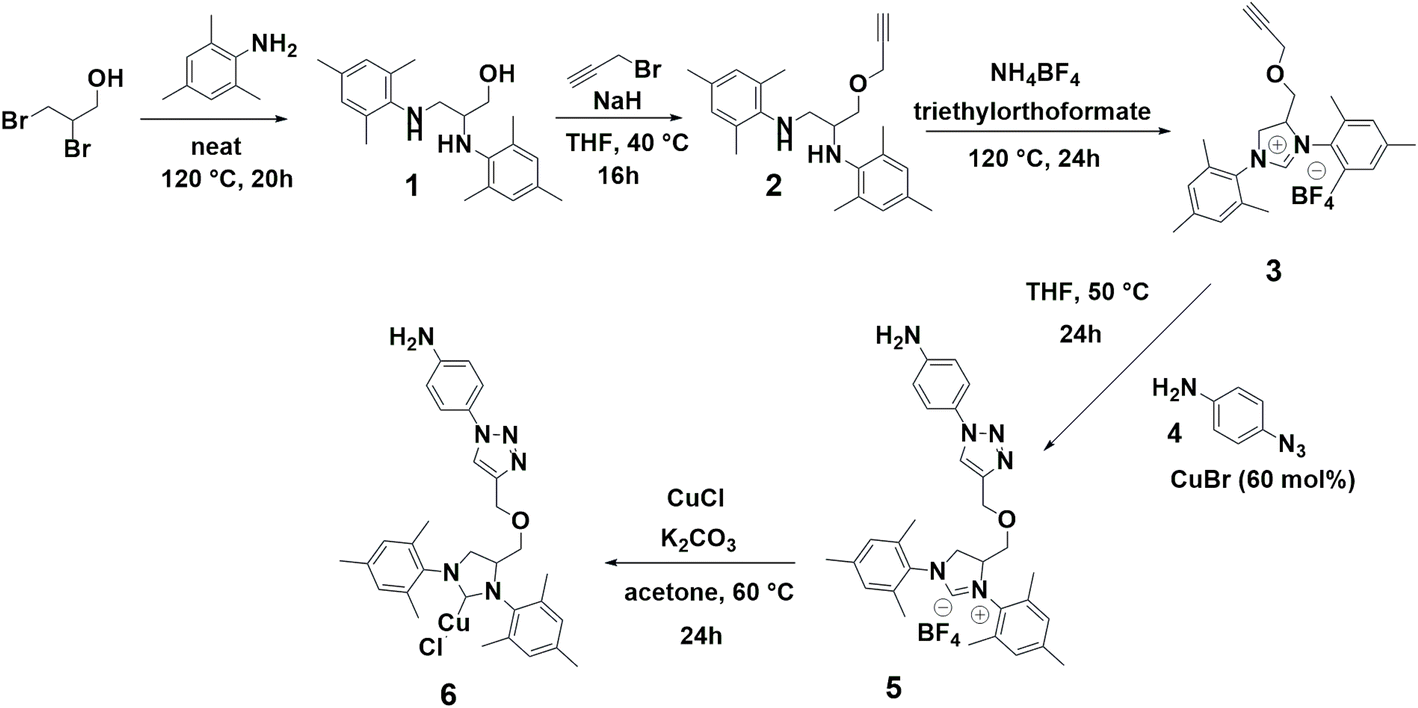 | ||
| Scheme 1 Synthesis of aniline functionalized NHC–Cu(I) complex 6. | ||
In parallel, pristine MWCNTs 7 were thoroughly purified to remove any residual metals (page S17 and Fig. S15–S17‡),30 before undergoing oxidation in a 3:1 v/v H2SO4:HNO3 mixture, resulting in oMWCNTs 8 bearing carboxylic acid groups (Scheme S1, page S19 and Fig. S18–S21‡).31 Nanomaterial 9 was then prepared via an amidation reaction between 8 and NHC–Cu(I) complex 6, (page S22 and Fig. S22–S26‡). The coordination of Ni(II) was achieved by reacting 9 with hexahydrate nickel chloride, facilitated by both triazolyl and amide coordinating functionalities.32,33In situ reduction of Ni(II), transmetallation of NHC–Cu(I) to NHC–Ni(0) and subsequent nucleation of NiNCs in a water/ethanol mixture at room temperature, using sodium borohydride (NaBH4) as the reducing agent, yielded the targeted electrocatalyst 10 (Scheme 2, page S26 and Fig. S27–S30‡).
 | ||
| Scheme 2 Preparation of electrocatalyst 10. | ||
Raman spectroscopy of MWCNTs 7 reveals an ID/IG ratio of 0.80 ± 0.01, with D and G bands at 1348 cm−1 and 1577 cm−1, respectively, indicating the graphitic structure (Fig. S15–S16 and Table S1‡).34,35 Upon oxidation to 8, the ID/IG ratio increases to 1.03 ± 0.01, accompanied by D and G band peaks blue-shifting to 1355 cm−1 and 1587 cm−1, confirming covalent surface functionalization (Fig. S18–S19 and Table S1‡).36,37 ATR-FTIR analysis shows a carbonyl stretch at 1702 cm−1, corresponding to –COOH groups (Fig. S20‡). TGA reveals a 6.5% weight loss at 600 °C, consistent with one –COOH group per 54 carbon atoms (Fig. S21‡).38
After amidation of 8, with complex 6, to form 9, the Raman ID/IG ratio remains unchanged, suggesting that functionalization occurs primarily on the ligand chain without altering the MWCNT framework. Notably, small shifts of the D (1353 cm−1) and G (1583 cm−1) bands reflect changes in the electronic properties of the carbon framework (Fig. S22–S23 and Table S1‡).39,40 ATR-FTIR shows an amide carbonyl stretching vibration at 1715 cm−1, confirming successful modification (Fig. S24‡), while TGA suggests a ligand-to-carbon ratio of 1:79, consistent with sole “on-ligand” modification (Fig. S25‡). XPS analysis confirms the targeted nanostructure, with the survey scan revealing characteristic peaks for C 1s, N 1s, O 1s, Cu 2p, and Cl 2p. High-resolution spectra of N 1s, C 1s, and Cu 2p validate the successful integration of complex 6 on oMWCNTs, highlighting the presence of triazole,41 and NHC moieties along with the Cu(I) oxidation state in 9 (Fig. S26‡).42
Following NHC-ligation with NiNCs in electrocatalyst 10, the ID/IG ratio as well as the G and D band positions remain stable, indicating a minimal impact on the MWCNT surface and the persistence of NHC–metal ligation, respectively (Fig. S27–S28 and Table S1‡). A 13 cm−1 downshift in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency found at 1702 cm−1 in the ATR-FTIR spectrum suggests an interaction between the carbonyl groups of 8 and NiNCs, supporting the NHC–NiNC ligation on oMWCNTs (8) (Fig. S29–S30‡).
O stretching frequency found at 1702 cm−1 in the ATR-FTIR spectrum suggests an interaction between the carbonyl groups of 8 and NiNCs, supporting the NHC–NiNC ligation on oMWCNTs (8) (Fig. S29–S30‡).
The XPS survey scan spectra confirm the engineered nanostructure of 10 (Fig. S31‡) detecting all relevant elements: C 1s, N 1s, O 1s and Ni 2p. Importantly, the absence of Cu 2p signals validates the efficiency of the transmetalation method in generating NHC–NiNCs (Fig. S31‡). Additionally, the atomic percentage of Ni/C, determined from the normalized areas of Ni and C peaks, is calculated to be only 3.9%. High-resolution N 1s spectra revealed three distinct components at binding energies (BE) of 399.2 eV, 400.2 eV and 401.8 eV (Fig. 1a and Table S2‡) in a 1:3:1 ratio. The lowest BE at 399.2 eV is attributed to the N–C–Ni(0) atom of the N-heterocyclic carbene, appearing at 1.0 eV lower than the analogous N–C–Cu(I) in 9.42 This shift reflects the electron-richer carbene ligand species bonded to Ni(0) atoms in the NiNCs. Narrow C 1s scans further corroborate the successful preparation, with characteristic components fitted and assigned (Fig. 1b and Table S2‡). Notably, the normalized areas show a 1:1 ratio between the N–C–Ni at 282.8 eV,43 and the MWCNTs-COONH- component at 289.1 eV, directly aligning with electrocatalyst 10's rational design. Ni 2p spectra (Fig. 1c) revealed a distinct component, at 852.7 eV, confirming the presence of Ni (0), alongside the predominant component at 856.8 eV, attributed to oxidized Nin+.44 Although this binding energy falls within the reported range of both Ni(II) and Ni(III), the observed shake-up satellites strongly support the assignment to Ni(II).45 The selective stabilization of Ni(0) by NHC ligands highlights their strong metal-binding affinity and electronic tuning properties, which confer electronic conductivity and structural integrity to electrocatalyst 10.21–23 The predominance of oxidized Nin+ is consistent with the very reactive Ni(0) nano species due to the very small size and clean surface state rendering Ni(0) highly prone to oxidation under atmospheric and aqueous conditions.46 This dual functionality—where the NHC-stabilized Ni(0) core ensures stability and provides a conductive support, while the oxidized NiOx shell acts as the electroactive surface for the OER—underscores the synergistic balance achieved in the engineered nanostructure of electrocatalyst 10.
 | ||
| Fig. 1 (a) XPS narrow scans of N 1s, (b) C 1s, (c) Ni 2p and (d) TEM image of 10, (e) HAADF-STEM image of 10 and (f) HRTEM-atomic resolved image of NiNCs of 10 with the corresponding lattice fringes (inset: FFT). | ||
Low magnification TEM images show the presence of MWCNTs (Fig. 1 d). The High-Resolution Transmission Electron Microscopy (HRTEM) study allows identifying the presence of crystalline nanoclusters on the surface of MWCNTs as well as graphitic and amorphous particles containing small crystalline nanoclusters in agglomerates surrounding MWCNTs' surface. The Energy-Dispersive X-ray Spectroscopy (EDS) analysis indicates the presence of Ni in both the MWCNT surface and the surface's surrounding agglomerates. Moreover, a Scanning Transmission Electron Microscopy (STEM) study has been performed. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images reflect the presence of more intense areas both in the surface and surrounding agglomerates suggesting—according to the higher atomic number of Ni against C—the presence of Ni (Fig. 1e, S32 and S33‡). This is confirmed with the simultaneous acquisition of EDS spectra in each scanning position (Fig. S32 and S33‡). EDS maps confirm the presence of Ni in the brighter areas and the existence of NiNCs. The interatomic distances of 2.08 Å measured in the NiNCs and the corresponding FFTs (Fig. 1f) are compatible with the fcc NiO unit cell along the [001] zone axis. The identification of NiO lattice fringes, a well-known Ni(II) phase, provides additional structural confirmation that the oxidized species observed by XPS corresponds to Ni(II) rather than Ni(III). The ensemble of these structural and compositional data allows validating the efficient decoration of NiNCs on MWCNTs. Given the exceptionally small size (<1.6 nm) and high surface area of the NiNCs, the oxidation easily occurs on the surface shell, while the NHC ligation stabilizes effectively the Ni(0) core, in accordance with the XPS findings.47–49
The electrocatalytic performance of electrocatalyst 10 was evaluated for the OER in aqueous 0.1 M KOH electrolyte solution at room temperature and compared against oMWCNTs 8, its MWCNTs-Cu(I)NHC precursor 9, and the benchmark RuO2. Linear sweep voltammetry (LSV) polarization curves were recorded within the potential range of 0.8–2.0 V vs. RHE, with iR compensation applied and normalized to the geometric electrode surface area (A = 0.196 cm2). Upon immersion of electrode 10 into 0.1 M KOH electrolyte, surface hydration of Ni(II) species to Ni(OH)2 occurs spontaneously on the NiNC surface, consistent with previous reports.49 During operation, the NiNCs in 10 undergo surface oxidation, leading to the formation of highly electroactive NiOOH species, as evidenced by the oxidation peak associated with the Ni(II)/Ni(III) transition (Ni(OH)2/NiOOH conversion), occurring just prior to the OER onset (Fig. 2a).50 Over the course of 1000 CV cycles, 10 undergoes progressive surface restructuring, with the Ni(II)/Ni(III) oxidation peak at 1.43 mV—with a full width half maximum (FWHM = 0.079), characteristic of highly uniform redox active species in nanoclusters—shifting to an even narrower peak at 1.39 mV (Fig. 2a) with a further 8% decrease in FWHM (Fig. S34‡). This shift reflects stabilization into a dynamic equilibrium between Ni(OH)2 and NiOOH, eventually resulting in a stable Ni(II)/Ni(III) oxidation potential.51,52
 | ||
| Fig. 2 (a) LSV polarization curves obtained at 1600 rpm rotation speed and 5 mV s−1 scan rate in 0.1 M KOH for pristine 10 and 10 after activation, (b) tabulated nNi (mol) of 10 before (pristine 10) and after activation, (c) LSV polarization curves obtained at 1600 rpm rotation speed and 5 mV s−1 scan rate in 0.1 M KOH of 10, 9, 8 and RuO2 before and after 104 successive LSV cycles, (d) Tafel slopes of 10, 8 and RuO2 before and after 104 successive LSV cycles, (e) tabulated comparative Tafel values of 10 (pristine 10, 10 and 10 after 104 cycles), 8 (8 and 8 after 104 cycles) and RuO2 (RuO2 and RuO2 after 104 cycles) and (f) tabulated comparative j0ECSA values of 10, 8 and RuO2. | ||
Linear sweep voltammetry (LSV) measurements recorded before and after electrode activation revealed that electrode 10 exhibited an overpotential of 370 mV (η10) to achieve a current density of 10 mA cm−2, which decreased to 350 mV following activation (Fig. 2a). Furthermore, at a higher current density of 80 mA cm−2, the electrode displayed an overpotential of 670 mV (Fig. S35‡), positioning 10 among the highly efficient electrocatalysts for the OER.5 To quantify the electroactive Ni sites (nNi, mol) available on electrode 10 and evaluate the impact of the activation process on surface restructuring and performance stabilization, the electroactive Ni surface species (nNi) participating in the OER catalytic process were determined both before and after activation (Fig. S34‡).53 By integrating the anodic oxidation peak prior to the OER onset, the electroactive Ni was calculated to be 6.05 × 10−8 mol before activation. Following the activation process, the electroactive Ni content increased significantly to 7.05 × 10−8 mol, representing a 16.5% enhancement in the available active sites in the optimized surface structure (Fig. 2b). Indeed, electrocatalyst 10 outperforms the highly efficient electrocatalyst 8 by 100 mV and slightly exceeds the benchmark electrocatalyst RuO2 by 30 mV (Table 1). Concretely, 8 exhibits efficient electrocatalytic activity with an overpotential η10 of 450 mV consistent with prior reports.24 However, after functionalization with the Cu(I)–carbene complex 6, nanomaterial 9 completely loses its electroactivity towards the OER. In contrast, the incorporation of NHC-ligated NiNCs in 10 not only restored but further enhanced its electrocatalytic activity by 100 mV (Fig. 2c). This stark difference in OER performance highlights the role of the metal center and surface functionality. In nanomaterial 8, surface –COOH groups facilitate proton-coupled electron transfer (PCET), enabling efficient OER activity. However, in 9, these are replaced by electronically inert –CONH linkages, while NHC–Cu(I) complexes further passivate the surface. Cu(I), with its d10 closed-shell configuration, remains redox-inactive under OER conditions and does not form catalytically active oxyhydroxide species. In contrast, nanomaterial 10 contains NHC–Ni(0)NiOx nanoclusters that dynamically restructure under increasing anodic potentials to form high-valent Ni oxyhydroxide phases, well-known as OER-active phases. These differences explain why 10 outperforms greatly both 8 and 9 in the OER. Notably, consistent performance was observed across five independent synthetic batches of electrocatalyst 10, confirming both its synthetic reproducibility and operational robustness in the OER (Table S3‡).
| Material | E 10 vs. RHE* | E 10 vs. RHE (104 c) | η 10 | R ct | Tafel slopes (1c/104c) | j0ECSA | Specific activity jECSA |
|---|---|---|---|---|---|---|---|
| a Nanomaterial presents no electroactivity and does not reach a current density of 10 mA cm−2. | |||||||
| 8 | 1.68 V | 1.69 V | 450 mV | 133 Ω | 80/94 mV dec−1 | 1.52 mA cmECSA−2 | 1.5 mA cmECSA−2 |
| 9 | —a | —a | —a | 6711 Ω | — | — | — |
| 10 | 1.58 V | 1.58 V | 350 mV | 55 Ω | 62/62 mV dec−1 | 1.28 mA cmECSA−2 | 15.3 mA cmECSA−2 |
| RuO2 | 1.62 V | 1.63 V | 390 mV | 114 Ω | 95/112 mV dec−1 | 0.11 mA cmECSA−2 | 0.6 mA cmECSA−2 |
The stability of electrocatalyst 10 was then evaluated over 104 cycles and compared to the previously demonstrated efficient electrocatalysts 8 and RuO2 (Fig. 2c and Table 1). Electrocatalyst 10 maintains its electrocatalytic performance with no measurable η10 gain, while 8 and RuO2 show only minor gains of 10 mV (Fig. 2c, dotted lines). In addition, the retention of active nickel sites after 104 OER cycles—as calculated with only a slight 0.46% decrease—highlights the excellent electroactive surface stability and sustained OER activity of 10 promoted by the effective ligation of MWCNTs–NHC–Ni(0) (Fig. S36‡). Long-term potentiometry for over 5 hours at 10 mA cm−2 was also conducted, showing a minimal gain of 15 mV in overpotential, corresponding only to a 0.94% increase (Fig. S37‡).
Tafel slopes were determined for 8, 10, and RuO2 in the low-potential region immediately after the interference from the Ni(II)/Ni(III) oxidation peak of 10 to ensure accuracy (Fig. 2d and Table 1). Electrocatalyst 10 demonstrates the lowest Tafel slope of 62 mV dec−1, outperforming 8 and RuO2, which exhibit slopes of 80 mV dec−1 and 95 mV dec−1, respectively, indicating significantly faster OER kinetics for 10. Notably, after 104 OER cycles, the Tafel slopes of 8 and RuO2 increase by 17.5% and 17.9%, respectively, indicating a decline in kinetic efficiency over prolonged cycling. In contrast, electrocatalyst 10 maintains its initial Tafel slope, showcasing superior kinetic stability under extended operational conditions (Fig. 2d and e). To gain deeper mechanistic insights into electrocatalyst 10,54 Tafel slopes were compared before and after activation (Fig. 2e and S38‡). Initially, pristine 10 exhibited a Tafel slope of 97 mV dec−1, indicating a more kinetically hindered surface-controlled process, with a rate-determining step (RDS) involving surface restructuring and the initial oxidation of Ni(II) to active NiOOH species. After activation, the Tafel slope decreased significantly to 62 mV dec−1, reflecting a diffusion-assisted regime, where OH− ion adsorption and PCET steps dominate, with an RDS likely involving PCET on stabilized NiOOH sites.55,56 Finally, the unchanged slope after prolonged cycling further indicates the robustness of the activated surface and the minimized impact of mass transport limitations.
The exchange current density (j0) provides key insight into the intrinsic efficiency of electron transfer at the electrode/electrolyte interface, reflecting the inherent activity of electrocatalysts.57 It was obtained by Tafel extrapolation and was normalized by the geometric electrode surface area. Before activation, electrocatalyst 10 exhibits a high j0 of 1.87 mA cm−2, which improves to 1.91 mA cm−2 after activation and remains stable even after 104 OER cycles, underscoring its exceptional kinetic robustness (page S36‡). Electrocatalyst 8 demonstrates a slightly higher j0 of 2.27 mA cm−2, while RuO2 shows a slightly lower value of 1.89 mA cm−2. Despite the inherently high j0 of 10, we further analyzed its intrinsic activity by normalizing j0 to the electrochemically active surface area (ECSA) for a fair comparison across materials with differing characteristics. The ECSA of 8, 9, 10 and RuO2 was calculated from double-layer capacitance (Cdl) measurements in non-faradaic regions of CVs at different scan rates (Fig. S39–S42 and Table S4‡).58,59 When normalized by ECSA, the j0ECSA of 10 is 1.28 mA cmECSA−2, only 0.24 mA cm−2 lower than that of 8 but surpassing the benchmark RuO2 by an impressive 12-fold (Fig. 2f and Table 1), highlighting the very efficient intrinsic kinetic activity of electrocatalyst 10.
To delve further into the performance of electrocatalyst 10, the specific activity per unit surface area (jECSA) and intrinsic activity per Ni electroactive site (jnNi) were evaluated across various overpotentials. Electrocatalyst 10 exhibits a remarkable specific activity, starting at 1.3 mA cmECSA−2 at an overpotential of 300 mV and reaching 15.3 mA cmECSA−2 at 380 mV, significantly outperforming both 8 (10-fold) and RuO2 (25-fold) (Fig. 3a and Table 1 and S43‡). This exceptional enhancement highlights the efficient utilization of electroactive Ni sites in 10, attributed to its rationally engineered nanostructure, which enables a high catalytic turnover per unit surface area. Furthermore, electrocatalyst 10 demonstrates an outstanding intrinsic activity (Fig. S44‡),60 increasing from 2.83 × 107 mA mol−1·cm−2 at 300 mV to 1.65 × 108 mA mol−1·cm−2 at 360 mV (Fig. 3b). This significant increase reflects the enhanced accessibility and reactivity of the NiIII active centers within the nanostructure of 10, facilitated by the synergistic interplay between the MWCNT-supported NHC-stabilized Ni(0) core providing a conductive support to the electroactive oxidized Nin+ shell species. This unique nanoarchitecture facilitates fast electron transfer kinetics and maximizes the utilization of every electroactive Ni site, positioning 10 as a highly efficient and intrinsically active OER electrocatalyst, significantly outperforming benchmark catalysts.61
 | ||
| Fig. 3 (a) Tabulated values of specific activity jECSA of 10, 8 and RuO2 across various overpotentials, (b) tabulated values of the intrinsic activity jnNi of 10 across various overpotentials, (c) TOF as a function of η for electrocatalyst 10, (d) logarithmic TOF (log TOF) as a function of η for electrocatalyst 10, (e) Nyquist plots of 10, 9, 8 and RuO2 (inset: applied Randles circuit fit) and (f) Rct and CPE.Y0 across various overpotentials for 10. RDS changes are marked with stars. | ||
The turnover frequency (TOF), a critical parameter reflecting the catalytic activity per active site, was also calculated for electrocatalyst 10 across various overpotentials (Fig. 3c).52,62 At 360 mV, corresponding to jgeo@10 mA cm−2, electrocatalyst 10 demonstrates a very high TOF of 0.46 s−1 per mole of Ni, underscoring the high activity and accessibility of its electroactive Ni sites (page S42‡) and highlighting the efficiency of its rationally engineered nanostructure. Furthermore, the linearity of the log TOF vs. η plot (Fig. 3d) and its slope of 62 mV dec−1 further validate the OER kinetics of 10, consistent with the Tafel slope, underlining the efficient, uniform activation of the electroactive Ni sites, further supporting PCET as the RDS.
Electrochemical impedance spectroscopy (EIS) was employed to investigate the charge transfer kinetics and capacitive properties of electrocatalyst 10,63 benchmark RuO2, and comparative starting materials 8 and 9 (Fig. 3e). The Nyquist plots modeled with a Randles circuit containing a constant-phase element (CPE) reveal significant differences in charge transfer resistances (Rct) among the samples (Table S5‡). Electrocatalyst 8 exhibits a moderate Rct of 133 Ω, while upon functionalization with the NHC–Cu(I) complex in 9, Rct skyrockets to thousands of Ω, underscoring the detrimental impact of this intermediate on charge transport. However, the formation of NHC–NiNCs in electrocatalyst 10 restores and dramatically enhances charge transfer efficiency, resulting in an Rct value 2.5 times lower than that of 8 and nearly half that of RuO2. The overall decrease in Rct reflects improved charge transport and enhanced electrical conductivity in 10.64 Additionally, Fig. 3f insets illustrate the detailed evaluation of Rct and capacitive properties at 0.1 M KOH, at various η. The bar chart (top inset, Fig. 3f) confirms a progressive decrease in Rct with increasing overpotential for electrocatalyst 10, demonstrating efficient electron transfer during the OER process. The relationship between η and Rct was analyzed across the kinetic region of the OER, corresponding to current densities up to 7 mA cm−2 (Fig. S45‡). Two distinct transitions are observed in the Nyquist-derived Rct values: a sharp decrease at approximately 2.5 mA cm−2 and an even steeper drop at 7 mA cm−2. These transitions likely correspond to changes in the RDS of the OER mechanism.59 At lower current densities, the initial Rct decrease suggests the onset of faster PCET kinetics, possibly associated with Ni(II)/Ni(III) oxidation. The second, more pronounced decline in Rct at 7 mA cm−2 likely reflects the transition to the oxygen evolution step, where surface oxygen species become the dominant intermediates. These observations underscore the dynamic restructuring of the electroactive surface of electrocatalyst 10, highlighting its adaptive behavior in achieving efficient charge transfer across different kinetic regimes of the OER process.13,65
After the comprehensive electrochemical evaluation in 0.1 M KOH, the study of 10 was extended to approach conditions closer to practical applications. The LSV polarization curve of 10 was recorded up to 200 mA cm−2 in the more concentrated alkaline environment, 1 M KOH (Fig. 4a), giving outstanding overpotentials η10 of 320 mV, η100 of 420 mV, and η200 of 500 mV. The improved ionic conductivity in 1 M KOH likely mitigates diffusion-limiting factors, enhancing charge transport and ensuring more efficient catalytic activity at higher current densities.66,67 This is also reflected by the smaller Tafel slope of 51 mV dec−1, suggesting that in 1 M KOH, the RDS involves the oxidation of adsorbed hydroxide species (OH*) to oxygen intermediates (O*), a step commonly associated with high-performance OER catalysts (Fig. 4b).68 The j0 and j0ECSA values are 1.86 mA cm−2 and 1.24 mA cm−2, respectively, whereas the jECSA at η350 is 17 mA cmECSA−2 reaching 133 mA cmECSA−2 at η500 (Fig. 4c and S46‡) and the intrinsic activity jnNi at η350 is 3.6 × 108 mA mol−1 cm2 with a jnNi = 2.8 x 109 mA mol−1 cm2 at η500 (Fig. 4d and S47‡).
 | ||
| Fig. 4 (a) LSV polarization curve obtained at 1600 rpm rotation speed and 5 mV s−1 scan rate in 1 M KOH for 10 reaching up to 200 mA cm−2, (b) Tafel slope of 10 in 1 M KOH, (c) tabulated values of the specific activity of 10 across various overpotentials in 1 M KOH, (d) tabulated values of the intrinsic activity of 10 across various overpotentials in 1 M KOH, (e) TOF plot of 10 across 350–500 mV overpotentials in 1 M KOH and (f) Nyquist plots of 10 (dots) with applied Randles circuit fit (lines) obtained at various j (mA cm−2) in 1 M KOH. | ||
The TOF values further reinforce these metrics, with 10 achieving a TOF of 7.4 s−1 at 500 mV (Fig. 4e). This substantial metric emphasizes how the increased hydroxide ion availability and improved charge transport in 1 M KOH enables significantly faster reaction kinetics and higher TOF values. Furthermore, catalyst 10 demonstrates highly rapid charge transfer kinetics at higher η, with a significant decrease in Rct from 66Ω at η1.25 to 9.7Ω at η20 (Table S6‡). The Cdl decreases consistently from 51.0μF to 35.6μF, due to increased surface coverage by reaction intermediates (OH*, O*) and thinning of the double layer, reflecting efficient interfacial processes. The substantially smaller solution resistance (Rs) in 1 M KOH (∼7.5 Ω) confirms the elimination of diffusion limitations, under these conditions. The Tafel slope derived from log(1/Rct), 54 mV dec−1 (Fig. S48‡), aligns closely with the polarization-derived Tafel slope, 51 mV dec−1, pointing to similar kinetic behavior, in which surface-controlled charge transfer processes dominate without significant contributions from diffusion complexities. Such alignment strengthens the hypothesis that the RDS involves the formation or oxidation of adsorbed intermediates (OH* or O*).
Finally, the durability was evaluated in 1 M KOH, at 10 mA cm−2 for ∼3 hours, demonstrating a minimal gain of 20 mV in overpotential, corresponding to only a 1.3% increase (Fig. S49‡). Interestingly, we recorded a post-test LSV (Fig. S50‡), which showed the near disappearance of the characteristic Ni(II)/Ni(III) redox peak, with only a faint shoulder visible at higher potentials and partially merged with the OER onset. This suggests progressive oxidation of surface Ni species into higher-valent Ni3+/Ni4+ states within the NiOx shell under prolonged anodic polarization. This phenomenon is consistent with prior studies, where extended electrochemical operation induced phase transitions and suppression of redox features due to the stabilization of higher oxidation states and restructuring of the active surface, while preserving electrocatalytic activity.69 Importantly, in electrocatalyst 10, this transformation correlates with only a minor performance loss (14 mV gain at 10 mA cm−2), confirming the robustness and adaptability of the NiOx catalytic surface during extended operation. Additionally, after 104 cycles in 1 M KOH, 10 maintained its performance with only a 15 mV overpotential gain and negligible change in Ni active sites (<0.1%), further confirming its durability (Fig. S51‡).
Conclusion
In conclusion, NHC-ligated NiNCs anchored covalently on MWCNTs were prepared via a dual metal-reduction and transmetalation strategy, creating a precisely engineered robust nanoarchitecture. Leveraging the unique tuning properties of NHC–M(0) ligation, the NHC–Ni(0) core provides exceptional stabilization and conductivity, supporting the formation of an electroactive NiOx shell, as confirmed by XPS and HRTEM studies. This synergistic design achieves efficient mechanistic progression through all OER steps. In 0.1 M KOH, the catalyst exhibits a low overpotential of 350 mV at 10 mA cm−2, with activation enhancing performance through the dynamic formation of a stabilized NiOOH shell. The catalyst outperforms the RuO2 benchmark in overpotential, j0, Tafel slopes, charge transfer resistance and specific activity, highlighting the efficient utilization of electroactive Ni sites. Under industrially relevant conditions (1 M KOH, 200 mA cm−2), electrocatalyst 10 achieves very low overpotentials (η10 = 320 mV, η200 = 500 mV), exceptional TOF values up to 7.4 s−1, and remarkable specific (jECSA = 133mA cm−2) and intrinsic (jnNi = 2.8 × 109 mA mol−1 cm2) activities, effectively overcoming diffusion limitations observed in less concentrated electrolytes. EIS analysis revealed highly efficient charge transfer kinetics, with Rct decreasing from 66 Ω to 9.7 Ω and Cdl declining consistently due to increased surface coverage by intermediates. The alignment of Tafel slopes derived from polarization (51 mV dec−1) and impedance (54 mV dec−1) confirms a surface-controlled charge transfer mechanism, with the RDS involving intermediate OH*/O* formation or oxidation. Durability tests confirm the catalyst's robustness, with minimal overpotential gains (0.9% and 1.3%) and <0.5% apparent loss of Ni active sites after 10000 cycles. The ultrafine size (<1.6 nm) and high dispersion of NiNCs expose nearly all electroactive Ni sites in the NiOx shell, maximizing catalytic efficiency. The NHC–Ni(0) core ensures structural stability and conductivity, while MWCNTs enhance electron transport and provide robust mechanical support, enabling efficient charge transport kinetics and enhanced durability under harsher electrochemical conditions. These findings position NHC-ligated NiNCs on MWCNTs as a scalable, durable, and highly efficient electrocatalyst for sustainable OER applications, surpassing state-of-the-art Ni-based and RuO2 benchmarks (Table S7‡).
Data availability
All data supporting the findings of this study are included in the manuscript and its ESI.‡ Additionally, the raw datasets are available from the corresponding authors, upon reasonable request.Author contributions
Rapakousiou Amalia: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing–original draft, writing–review and editing. Savvas G. Chalkidis: data curation, formal analysis, investigation, validation, writing–review and editing. Michail P. Minadakis: data curation, formal analysis, investigation, validation, writing–review and editing. María Luisa Ruiz-González: HRTEM/EDS/HAADF-STEM: data curation, formal analysis, investigation, resources, validation, writing–review and editing. Cristina Navio: XPS: data curation, formal analysis, investigation, resources, validation, visualization, writing–review and editing. Georgios C. Vougioukalakis: conceptualization, resources, supervision, validation, writing–review and editing. Nikos Tagmatarchis: conceptualization, resources, supervision, validation, writing–review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research project was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “2nd Call for H.F.R.I. Research Projects to support Post-Doctoral Researchers” granted to A.R. (NANOElectroCAT, project Number 913). A. R. acknowledges the support of ELECMI-CNME, Spain, for granting distinguished free access to ELECMI facilities and also thanks Dr A. Kagkoura for useful scientific discussions.References
- H. Wang, K. H. L. Zhang, J. P. Hofmann, V. A. de la Peña O'Shea and F. E. Oropeza, J. Mater. Chem. A, 2021, 9, 19465–19488 RSC.
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- S. R. Edea and Z. Luo, J. Mater. Chem. A, 2021, 9, 20131–20163 RSC.
- A. Vazhayil, L. Vazhayal, J. Thomas, S. Ashok C and N. Thomas, Appl. Surf. Sci. Adv., 2021, 6, 100184 CrossRef.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- K. S. Joya, L. Sinatra, L. G. AbdulHalim, C. P. Joshi, M. N. Hedhili, O. M. Bakr and I. Hussain, Nanoscale, 2016, 8, 9695–9703 RSC.
- X. Yan, L. Tian and X. Chen, J. Power Sources, 2015, 300, 336–343 CrossRef CAS.
- F.-T. Tsai, Y.-T. Deng, C.-W. Pao, J.-L. Chen, J.-F. Lee, K.-T. Lai and W.-F. Liaw, J. Mater. Chem. A, 2020, 8, 9939–9950 RSC.
- S. Funaki, T. Kawawaki, T. Okada, K. Takemae, S. Hossain, Y. Niihori, T. Naito, M. Takagi, T. Shimazaki, S. Kikkawa, S. Yamazoe, M. Tachikawa and Y. Negishi, Nanoscale, 2023, 15, 5201–5208 RSC.
- Y.-C. Zhang, C. Han, J. Gao, L. Pan, J. Wu, X.-D. Zhu and J.-J. Zou, ACS Catal., 2021, 11, 12485–12509 CrossRef CAS.
- L. Yang, Q.-X. Du, J. Cui, H.-W. Yang and H. Qian, Inorg. Chem., 2024, 63, 1954–1961 CrossRef.
- M. Kim, H. Han, K. Lee, S. Kang, S.-H. Lee, S. H. Lee, H. J, J. H. Ryu, C.-Y. Chung, K. M. Kim and S. Mhin, J. Mater. Chem. A, 2024, 12, 17596–17606 RSC.
- P. Chakthranont, J. Kibsgaard, A. Gallo, J. Park, M. Mitani, D. Sokaras, T. Kroll, R. Sinclair, M. B. Mogensen and T. F. Jaramillo, ACS Catal., 2017, 7, 5399–5409 CrossRef CAS.
- M. González-Ingelmo, M. López García, F. E. Oropeza, P. Álvarez, C. Blanco, R. Santamaría and V. G. Rocha, J. Mater. Chem. A, 2023, 11, 24248–24260 RSC.
- H. Lei, Z. Wang, F. Yang, X. Huang, J. Liu, Y. Liang, J. Xie, M. S. Javed, X. Lu, S. Tan and W. Mai, Nano Energy, 2020, 68, 104293 CrossRef CAS.
- D. M. Morales, M. A. Kazakova, S. Dieckhöfer, A. G. Selyutin, G. V. Golubtsov, W. Schuhmann and J. Masa, Adv. Funct. Mater., 2020, 30, 1–12 CrossRef.
- G. Chen, H. Xiang, Y. Guo, J. Huang, W. Chen, Z. Chen, T. Li and K. K. Ostrikov, Carbon Energy, 2024, 6, e522 CrossRef CAS.
- H. Shen, G. Tian, Z. Xu, L. Wang, Q. Wu, Y. Zhang, B. K. Teo and N. Zheng, Coord. Chem. Rev., 2022, 458, 214425 CrossRef CAS.
- M. Ghosh and S. Khan, ACS Catal., 2023, 13, 9313–9325 CrossRef CAS.
- A. Rapakousiou, M. P. Minadakis, S. G. Chalkidis, M. L. Ruiz-González, C. Navio, G. C. Vougioukalakis and N. Tagmatarchis, ACS Appl. Mater. Interfaces, 2025, 17(19), 28138–28150 CrossRef CAS.
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem, 2021, 5, 711–725 CrossRef CAS.
- J.-F. Soulé, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2013, 135, 10602–10605 CrossRef.
- D. Bouzouita, J. M. Asensio, V. Pfeifer, A. Palazzolo, P. Lecante, G. Pieters, S. Feuillastre, S. Tricard and B. Chaudret, Nanoscale, 2020, 12, 15736–15742 Search PubMed.
- X. Lu, W.-L. Yim, B. H. R. Suryanto and C. Zhao, J. Am. Chem. Soc., 2015, 137, 2901–2907 CrossRef CAS PubMed.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS.
- S. G. Chalkidis and G. C. Vougioukalakis, Eur. J. Org. Chem., 2023, 26, e202301095 CrossRef CAS.
- N. V. Tzouras, L. P. Zorba, E. Kaplanai, N. Tsoureas, D. J. Nelson, S. P. Nolan and G. C. Vougioukalakis, ACS Catal., 2023, 13, 8845–8860 CrossRef CAS.
- C. Deraedt, M. D'Halluin, S. Lesturgez, L. Salmon, G. Goglio, J. Ruiz and D. Astruc, Eur. J. Inorg. Chem., 2015, 2015, 1345–1350 CrossRef CAS.
- O. Santoro, A. Collado, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2013, 49, 10483 RSC.
- J.-M. Yuan, X.-H. Chen, X.-H. Chen, Z.-F. Fan, X.-G. Yang and Z.-H. Chen, Carbon, 2008, 46, 1266–1269 CrossRef CAS.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- D. Astruc, L. Liang, A. Rapakousiou and J. Ruiz, Acc. Chem. Res., 2012, 45, 630–640 CrossRef CAS.
- T. Bala, B. L. V. Prasad, M. Sastry, M. U. Kahaly and U. V. Waghmare, J. Phys. Chem. A, 2007, 111, 6183–6190 CrossRef CAS.
- L. Bokobza and J. Zhang, Express Polym. Lett., 2012, 6, 601–608 CrossRef CAS.
- A. Yoshida, Y. Kaburagi and Y. Hishiyama, Carbon, 2006, 44, 2333–2335 CrossRef CAS.
- S. L. H. Rebelo, A. Guedes, M. E. Szefczyk, A. M. Pereira, J. P. Araújo and C. Freire, Phys. Chem. Chem. Phys., 2016, 18, 12784–12796 RSC.
- S. Osswald, E. Flahaut, H. Ye and Y. Gogotsi, Chem. Phys. Lett., 2005, 402, 422–427 CrossRef CAS.
- D. D. Chronopoulos, C. G. Kokotos, N. Karousis, G. Kokotos and N. Tagmatarchis, Nanoscale, 2015, 7, 2750–2757 RSC.
- Z. Li, L. Deng, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2023, 135, 101089 CrossRef CAS.
- A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S. K. Saha, U. V. Waghmare, K. S. Novoselov, H. R. Krishnamurthy, A. K. Geim, A. C. Ferrari and A. K. Sood, Nat. Nanotechnol., 2008, 3, 210–215 CrossRef CAS PubMed.
- A. Rapakousiou, R. Sakamoto, R. Shiotsuki, R. Matsuoka, U. Nakajima, T. Pal, R. Shimada, A. Hossain, H. Masunaga, S. Horike, Y. Kitagawa, S. Sasaki, K. Kato, T. Ozawa, D. Astruc and H. Nishihara, Chem.–Eur. J., 2017, 23, 8443–8449 CrossRef CAS PubMed.
- R. Richstein, C. Eisen, L. Ge, M. Chalermnon, F. Mayer, B. K. Keppler, J. M. Chin and M. R. Reithofer, Chem. Commun., 2023, 59, 9738–9741 RSC.
- L. Sha, P. Gao, T. Wu and Y. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 40412–40419 CrossRef CAS.
- L. Zhi, W. Zhang, L. Dang, J. Sun, F. Shi, H. Xu, Z. Liu and Z. Lei, J. Power Sources, 2018, 387, 108–116 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. St. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- M. D. de los Bernardos, S. Pérez-Rodríguez, A. Gual, C. Claver and C. Godard, Chem. Commun., 2017, 53, 7894–7897 RSC.
- W. Su, F. Wu, L. Fang, J. Hu, L. Liu, T. Guan, X. Long, H. Luo and M. Zhou, J. Alloys Compd., 2019, 799, 15–25 CrossRef CAS.
- T. Yu, Y. Hou, P. Shi, Y. Yang, M. Chen, W. Zhou, Z. Jiang, X. Luo, H. Zhou and C. Yuan, Inorg. Chem., 2022, 61, 2360–2367 CrossRef CAS PubMed.
- B. Kim, A. Oh, M. K. Kabiraz, Y. Hong, J. Joo, H. Baik, S.-I. Choi and K. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 10115–10122 CrossRef CAS.
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS.
- N. Hales, T. J. Schmidt and E. Fabbri, Curr. Opin. Electrochem., 2023, 38, 101231 CrossRef CAS.
- S. Xie, Y. Yan, S. Lai, J. He, Z. Liu, B. Gao, M. Javanbakht, X. Peng and P. K. Chu, Appl. Surf. Sci., 2022, 605, 154743 CrossRef CAS.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef.
- L. Negahdar, F. Zeng, S. Palkovits, C. Broicher and R. Palkovits, ChemElectroChem, 2019, 6, 5588–5595 CrossRef CAS.
- G. Li, L. Anderson, Y. Chen, M. Pan and P.-Y. Abel Chuang, Sustain. Energy Fuels, 2018, 2, 237–251 RSC.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–426 CrossRef CAS.
- Q. Zhou, L. Liao, H. Zhou, D. Li, D. Tang and F. Yu, Mater. Today Phys., 2022, 26, 100727 CrossRef CAS.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2017, 29, 120–140 CrossRef CAS.
- Z. Wu, X. F. Lu, S. Zang and X. W. (David) Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS.
- F. Zheng, Z. Zhang, D. Xiang, P. Li, C. Du, Z. Zhuang, X. Li and W. Chen, J. Colloid Interface Sci., 2019, 555, 541–547 CrossRef CAS PubMed.
- A. C. Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3, 162–193 CrossRef CAS PubMed.
- R. Liu, G. Chen, Y. Guo, T. Li, J. Qiu, B. He and P. Tang, Chem. Eng. J., 2024, 500, 157098 CrossRef CAS.
- R. L. Doyle and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC.
- G.-F. Li, M. Divinagracia, M. F. Labata, J. D. Ocon and P.-Y. Abel Chuang, ACS Appl. Mater. Interfaces, 2019, 11, 33748–33758 CrossRef CAS.
- X. Li, Z. Zhang, X. Zhuang, Z. Jia and T. Wang, Chin. J. Chem., 2024, 42, 3533–3552 CrossRef CAS.
- Y. Hao, X. Cao, C. Lei, Z. Chen, X. Yang and M. Gong, Mater. Today Catal., 2023, 2, 100012 CrossRef.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
Footnotes |
| † Dedicated to Prof. Petra Rudolf on the occasion of her retirement, in profound appreciation of her outstanding contributions to science, her steadfast advocacy for women in research, and her enduring support and encouragement. |
| ‡ Electronic supplementary information (ESI) available: Methods and full experimental procedures, schemes and data, 1H, 13C, 19F and 2D NMR spectra, FTIR and ATR-FTIR spectra, Raman spectra and spectral maps, XPS spectra, TGA graphs, HAADF-STEM images and EDS maps, extensive electrochemical and electrocatalytic data, and comparative performance metrics. See DOI: https://doi.org/10.1039/d5ta00780a |
| This journal is © The Royal Society of Chemistry 2025 |