DOI:
10.1039/D5SC00623F
(Review Article)
Chem. Sci., 2025,
16, 5836-5848
Recent advances in catalytic asymmetric alkenyl C(sp2)–H bond functionalizations
Received
23rd January 2025
, Accepted 10th March 2025
First published on 10th March 2025
Abstract
Alkenes and their derivatives are widespread in numerous bioactive natural products and pharmaceutically relevant molecules. They are also synthetically versatile building blocks that have found broad applications in a plethora of organic transformations. The asymmetric alkenyl C(sp2)–H functionalization of readily available olefinic feedstocks allows the practical and straightforward synthesis of structurally diverse chiral compounds. As such, an ever-increasing number of robust and versatile strategies have been established to selectively functionalize the olefinic C(sp2)–H bonds in recent years. The current review provides a concise overview of these impressive achievements in the realm of asymmetric alkenyl C–H functionalization reactions, with a particular emphasis on substrate scopes, limitations, mechanistic studies, as well as their applications in the precise synthesis of diversely functionalized chiral molecules. Challenges and future opportunities regarding this area of research are also discussed. Through this review, we aim to inspire continuous efforts toward further development of more practical and broadly applicable strategies to advance this burgeoning field.
1. Introduction
Alkenes represent one of the most abundant and readily available chemical feedstocks, and are ubiquitous in countless agrochemicals, natural products, and biologically active compounds. Moreover, they can also serve as extraordinarily versatile synthons and precursors that have witnessed wide applications in a broad range of synthetic transformations. Accordingly, numerous conceptually different strategies have been developed for the expeditious synthesis of diversely functionalized alkenes and their derivatives.1 Among them, the direct alkenyl C–H functionalization of easily accessible alkenes has been recognized as one of the most straightforward tools to access functionalized alkenes in an atom- and step-economic manner.2–4 Continuous endeavors have been devoted to this emerging field, and a myriad of robust and powerful strategies regarding this fascinating research have been established, which significantly complements traditional methodologies for the facile synthesis of this important class of compounds.5–10
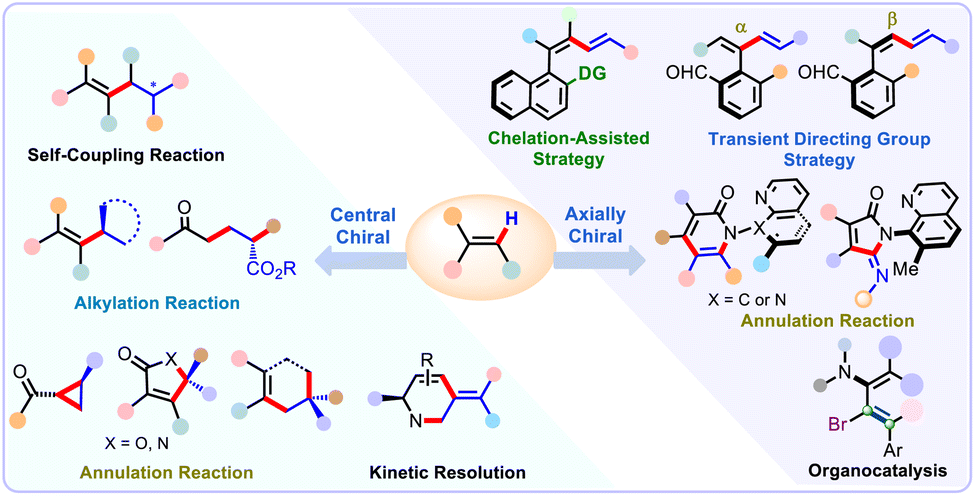
The asymmetric C–H functionalization has emerged rapidly as a transformative and versatile strategy to precisely fabricate diverse chiral architectures in a highly enantioselective manner.11–17 Despite splendid advances in alkenyl C(sp2)–H functionalization reactions, the development of an asymmetric version of these reactions is an attractive yet challenging task. The catalytic enantioselective functionalization of olefinic C–H bonds is drastically more difficult and has been relatively less explored than that of the aryl counterparts, mainly due to its strong π-coordinating ability of alkenes that may to some extent inhibit the reactivity and enantioselectivity of these reactions. Particularly, the side reactions such as E/Z isomerization, conjugate addition, cyclopropanation reaction as well as the competitive allylic C(sp3)–H bond functionalization also pose significant challenges to address. Moreover, these reactions seem more sensitive to steric hindrance in most cases, and densely substituted alkene substrates generally show remarkably low reactivity. To tackle these challenges and beyond, a large number of conceptually new and generally practical strategies have been established to elaborately modulate the reactivity and simultaneously enhance the stereoselectivity of these reactions, thereafter allowing the efficient enantioselective synthesis of a wealth of enantioenriched compounds (Scheme 1).
 |
| | Scheme 1 Asymmetric alkenyl C–H bond functionalizations. | |
The asymmetric alkenyl C–H functionalization reactions have made substantial strides in recent years. In this review, we attempt to highlight the latest advances and describe a concise overview of the enantioselective alkenyl C–H functionalization reactions. Special emphasis has been given to the substrate scopes, limitations, mechanistic investigations along with their applications in the streamlined assembly of multi-functionalized chiral molecules. The main challenges and future opportunities are also succinctly elucidated with the goal of expanding the application range of these reactions. Generally, there are two types of asymmetric C–H activation reactions: the enantioselective reactions based on C–H bond activation; and the enantioselective C–H activation reactions based on asymmetric C–H activation. The majority of the examples described in this review belong to the first category. For clarity, this review has been briefly categorized into two main sections: the first part covers the alkenyl C(sp2)–H functionalization reactions for the rapid synthesis of central chiral compounds; and the second part provides an overview of prominent methodologies established for the facile access to fabricate axially chiral compounds. We anticipate that this review will contribute to inspire continuous efforts and foster new breakthroughs for further development of more practical and innovative strategies to advance this fascinating field.
2. Alkenyl C(sp2)–H bond functionalization for the construction of central chiral compounds
As mentioned above, alkenes and their derivatives are widespread in a large variety of bioactive natural products and pharmaceutically relevant molecules. Remarkable advances have been achieved in the field of asymmetric alkenyl C(sp2)–H functionalization reactions. In this part of the review, we summarized the recent advances on these well-established enantioselective alkenyl C–H bond functionalization reaction of alkene feedstocks. Representative examples are discussed elaborately according to the different types of reactions, such as enantioselective self-coupling of alkenes, alkenyl C–H alkylation, alkenyl C–H functionalization/annulation sequence, as well as the intramolecular version of these reactions. Certain related mechanisms are also covered where appropriate.
2.1 Enantioselective self-coupling via alkenyl C–H functionalizations
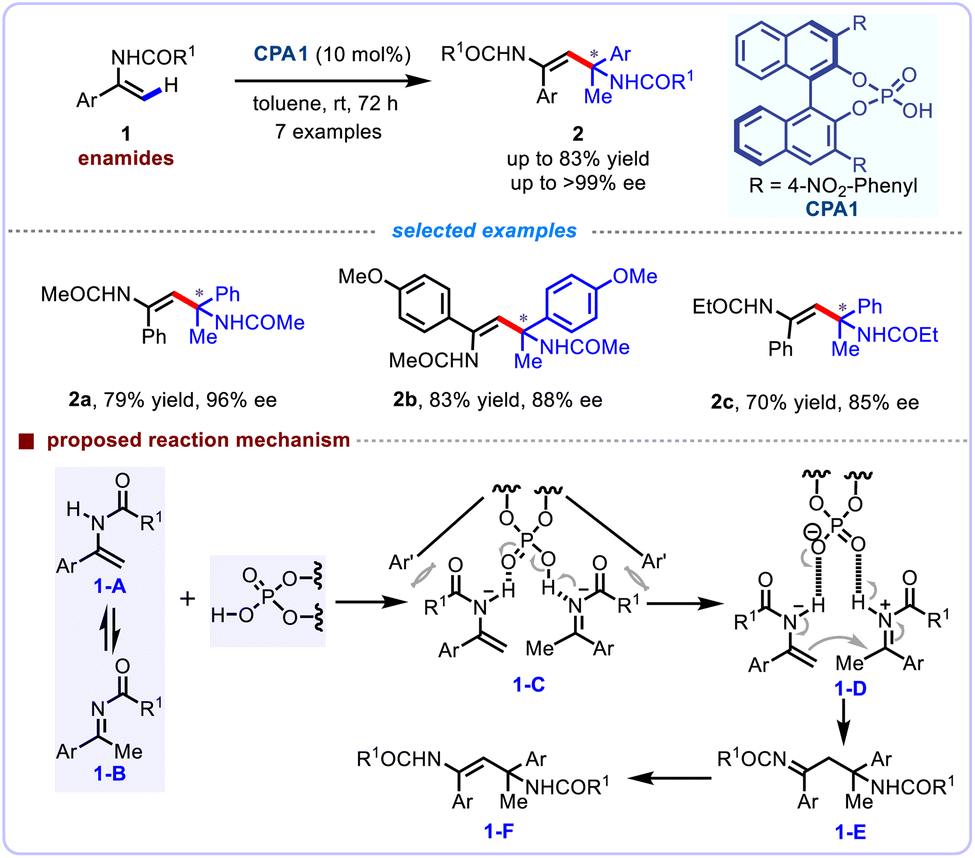
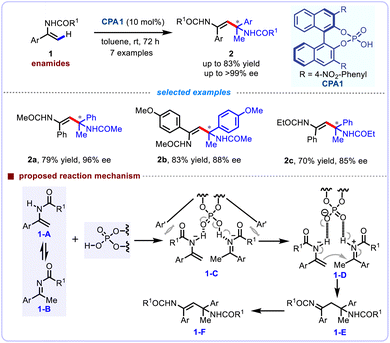
The self-coupling of readily available alkenes is one of the most effective strategies for the rapid assembly of C–C bonds. Nevertheless, the enantioselective C–C bond forming reactions for the construction of quaternary carbon centers through the self-coupling of alkenes via alkenyl C(sp2)–H functionalization strategy remains a challenging task in synthetic chemistry. In 2008, Tsogoeva and co-workers first described an elegant asymmetric self-coupling reaction of various enamides 1, thereafter enabling the enantioselective assembly of quaternary stereocenters feartuing a nitrogen-containing substituent (Scheme 2).18 The bifunctional character of BINOL-derived phosphoric acids, which simultaneously act as Lewis acid (activating electrophilic components through hydrogen-bonding interactions) and Lewis base (coordinating to nucleophilic species via the phosphoryl oxygen), enable dynamic control over reaction pathways by stabilizing transition states through dual activation mechanisms. This unique duality of the BINOL-derived phosphoric acid moiety greatly facilitated this self-coupling, which could readily isomerize enamides into imines under acidic condition. Notably, the corresponding products were obtained in excellent enantioselectivities (up to 99% ee), and their synthetic applications for the asymmetric synthesis of β-aminoketones have been demonstrated. The authors tentatively proposed a mechanism to illucidate this self-coupling reaction. Initially, the equilibrium existed between enamide 1-A and ketimine 1-B in the presence of a chiral Brønsted acid. Through a hydrogen-bonding network, the transient intermediate 1-C was generated. Subsequently, the tight ion pair 1-D was formed owing to the dual function of the phosphoric acid groups, the ketimine can be readily protonated to afford an iminium cation intermediate (electrophile), and the NH group of enamide can form a hydrogen bond as a nucleophile. The coupling reactions occurred smoothly to deliver intermediate 1-E, which further undergoes tautomerization to afford the desired product 1-F, and release the BINOL-phosphate.
 |
| | Scheme 2 Chiral Brønsted acid catalyzed self-coupling of enamides. | |
Later in 2022, Shibata and colleagues investigated the tail-to-tail dimerization reaction of aryl methacrylates 3, and reported the enantioselective self-coupling of methacrylates through a cationic iridium(I)-catalyzed olefinic C(sp2)–H activation, allowing access to a variety of highly useful chiral adipic acid derivatives 4, which are synthetically important feedstock chemicals for the synthesis of functional polymers (Scheme 3).19 Enantioselectivities were generally modest and inferior outcomes of the substrates were documented. However, the utilization of expensive transition-metal catalyst, specific substrate, and the requirement for relatively high temperature and longer reaction time significantly limited the synthetic appeal of this reaction. Mechanism studies revealed that the reductive elimination step initially leads to Z-alkene products, which further undergoes iridium-catalyzed isomerization to afford more stable E-alkene products.
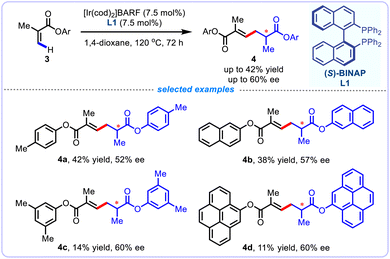 |
| | Scheme 3 Enantioselective dimerization of acrylate derivatives. | |
2.2 Enantioselective alkenyl C–H alkylation reactions
Transition metal-catalyzed asymmetric alkenyl C(sp2)–H bond activation/addition sequence of alkenes have emerged as chemically attractive processes and have enabled the highly efficient synthesis of diverse types of functionalized chiral systems in an atom-economic manner.20 In 2019, Cramer et al. presented an efficient CpxRh(III)-catalyzed alkenyl C–H functionalization/ring-opening reaction of α,β-unsaturated oxime ethers 5 with diazabicycles 6, leading to a diverse array of highly functionalized chiral cyclopentenylamines bearing two stereogenic centers in moderate to good yields (48–96%) and high enantioselectivities (Scheme 4).21 The transformation is also suitable for the enantioselective C–H activation of aryl ketoxime ethers, delivering 2-arylated cyclopentenyl amines with high levels of enantiocontrol. In this report, the chiral CpxRhIII catalyst system is generated in situ by CpxRhI(cod) precatalyst and bis(o-toluoyl) aroyl peroxide. It is worth noting that both the aroyl peroxide additive and the cyclooctadiene group exhibited significant impact on reaction efficiency and chiral induction, thus providing new options for catalyst modification in asymmetric rhodium catalysis.
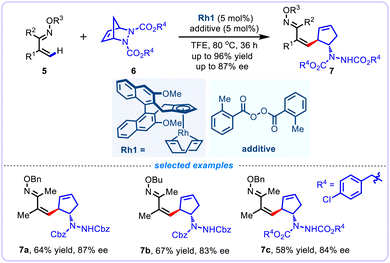 |
| | Scheme 4 Asymmetric CpxRh(I)-catalyzed alkenyl C–H activation/ring-opening reaction. | |
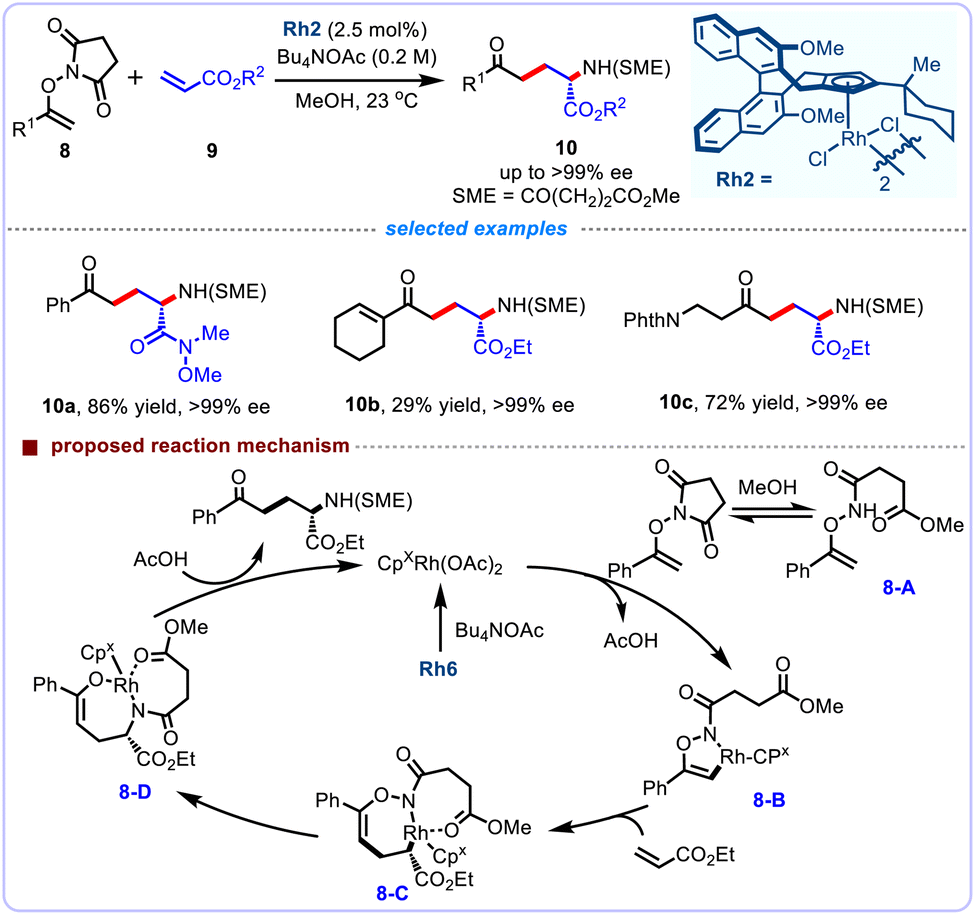
Subsequently, Cramer and co-workers further reported an enantioselective intermolecular carboamination of acrylates 9 through CpxRh(III)-catalyzed vinylic C–H activation of enoxysuccinimides 8, and delivering numerous nonnatural α-amino esters 10 (Scheme 5).22 This stereospecific strategy occurs smoothly under typically mild conditions, and features broad substrate scope, excellent enantioselectivities (up to >99% ee). The chiral rhodium catalyst bearing a tailored sterically hindered chiral Cpx ligand was found crucial for the carboamination chemoselectivity and high levels of enantioinduction. Mechanistically, the authors proposed a plausible mechanism for this enantioselective carboamination. Initially, in the presence of solvent, the N-enoxyphthalimide can reversibly ring-open to form the methyl ester 8-A, which was not successfully prepared and separated due to its low stability. The resulting activated substrate combines with the CpxRh(III) catalyst, and subsequently undergoes vinylic C–H activation to give the rhodacycle 8-B. Subsequent coordination and migratory insertion of alkene delivers the intermediate 8-C. This migration insertion was postulated to be the enantiodetermining step of this transformation. In turn, the product 8-D of carboamination is generated through the C–N reductive elimination and subsequent the N–O oxidative addition. Finally, protonation turns the rhodium(III) catalyst over and releases the expected products.
 |
| | Scheme 5 Asymmetric CpxRhIII-catalyzed vinylic C–H activation of N-enoxysuccinimides. | |
In 2021, Shibata's group disclosed an efficient cross-coupling reaction between α-substituted α,β-unsaturated amides 11 and β-substituted acrylates 12 through an atom-economic Ir(I)-catalyzed alkenyl C–H bond alkylation, furnishing highly functionalized chiral compounds (Scheme 6).23 The enantioselective cross-coupling of two different acrylamides was typically carried out in 1,4-dioxane in the presence of 10 mol% Ir(I) catalyst and 10 mol% Tol-BINAP ligand, giving rise to the formal conjugate adducts with overall good yields and high enantioselectivities (up to 99% yield, and up to 95% ee). Using (S)-Tol-BINAP as a ligand, the enantioselective cross-coupling of cyclic acrylamides with ethyl acrylate has been also realized by this strategy. Mechanistically, the catalytic cycle initiates with reversible oxidative addition of the vinylic C–H bond into the Ir(I) center, generating the hydroiridium species 11-A. Subsequent regioselective carboiridation of the crotonate affords intermediate 11-B, culminating in C–C bond formation through a reductive elimination step that simultaneously releases the alkylated product and regenerates the Ir(I) catalyst.
 |
| | Scheme 6 Enantioselective Ir-catalyzed alkenyl C–H alkylation of acrylamides. | |
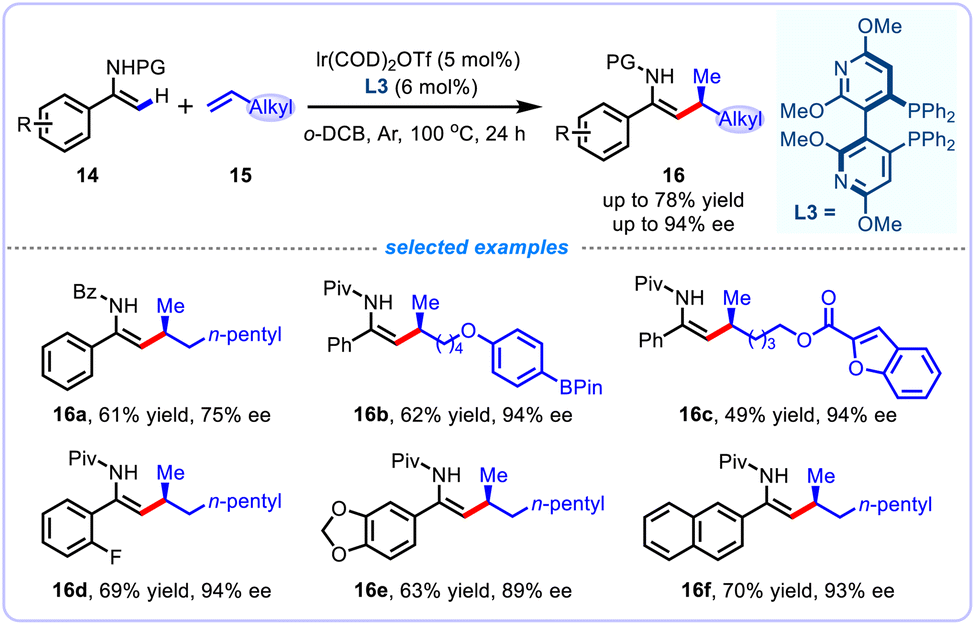
The enantioselective alkenyl C–H functionalization with unbiased aliphatic α-olefins as coupling partners to achieve branch-selective hydrofunctionalization is of particular interest in asymmetric synthesis. To advance this area, Li and colleagues in 2022 reported an unprecedented asymmetric hydroalkenylation of electronically unactivated α-olefins through an efficient iridium(I)-catalyzed chelation-assisted alkenyl C–H activation of enamides, delivering a diverse range of densely trisubstituted enamides 16 bearing an allylic stereocenter with high stereoselectivity (Scheme 7).24 Notably, this atom-economic protocol features high branch-selectivity and enantioselectivity, and exclusive Z-selectivity control. Further derivatization of the thus-obtained enantioenriched enamides can readily afford diverse value-added chiral compounds. DFT calculations were conducted to elucidate the reason for the high branch selectivity and enantioselectivity, and the results demonstrated that the chiral iridium catalyst was postulated to determine the stereochemistry of this transformation during both migratory insertion and reductive elimination steps.
 |
| | Scheme 7 Branch-selective iridium-catalyzed asymmetric alkenyl C–H alkylation of enamides with α-olefins. | |
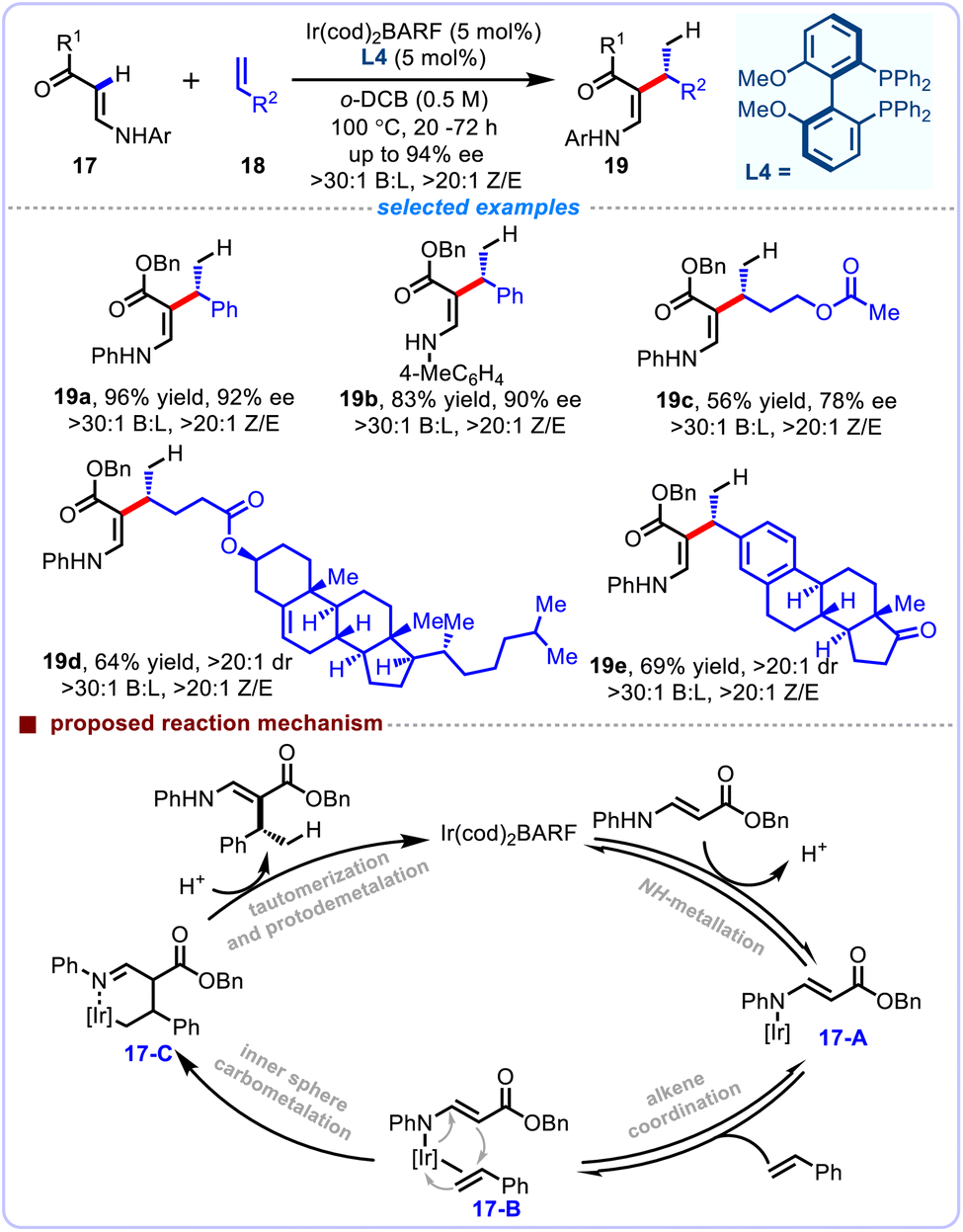
Quite recently, Bower et al. disclosed a conceptually new strategy that generated Ir-aza-enolates compounds through NH-metallation of diverse enamines, enabling highly branch-selective and enantioselective C–H addition reactions of both monosubstituted styrenes 18 and electronically unbiased aliphatic olefins 17 (Scheme 8).25 This distinctive C(sp2)–C(sp3) cross-coupling process tactfully utilized the potential of Ir-aza-enolate species to devise and realize the enantioselective C–H addition reactions. Impressive branch-selectivity and enantioselectivity were observed with complete atom-economy. Upon diastereocontrolled reduction, the resulting products could be derivatized into β2-amino acids with contiguous stereocenters. Control experiments supported a N–H metalation initiating process. The plausible pathway was tentatively proposed as follows: in the presence of Ir(I) catalyst, the substrate undergoes NH-metallation to produce 17-A, which then coordinates with the olefin coupling partner to form 17-B. Subsequent inner sphere and enantiodetermining carbometalation delivers the intermediate 17-C, which finally goes through protodemetalation and tautomerization to afford the hydroalkenylation products. Considering the widespread presence of NH enamines and especially as intermediates in organocatalysis, this newfound strategy may be applicable to the development of new tandem catalytic cross-coupling reactions.
 |
| | Scheme 8 Iridium-catalyzed enantioselective alkenyl C–H alkylation of minimally polarized alkenes. | |
2.3 Enantioselective alkenyl C(sp2)–H bond functionalization and annulation
The enantioselective alkenyl C(sp2)–H bond functionalization and annulation reaction provides straightforward access to a large variety of structurally complex and diversely functionalized chiral cyclic compounds, and a handful of impressive approaches have been achieved in this field. In 2019, Rovis and co-workers successfully introduced an innovative artificial metalloenzyme platform based on monomeric streptavidin (mSav), and further demonstrated its application in the enantioselective annulation of acrylamides 20 and styrenes 21, enabling the asymmetric synthesis of diverse enantioenriched δ-substituted lactams through a tandem alkenyl C–H activation/[4 + 2] annulation sequence (Scheme 9).26 This stereospecific protocol proved broadly tolerant to both acrylamides and styrenes under water-compatible conditions, delivering a diverse set of chiral δ-lactams 22 with excellent yields, and high enantioselectivities. Under exceedingly mild reduction conditions, the resulting annulation products could be readily converted into synthetically appealing enantioenriched piperidines. Remarkably, this new fruitful mSav metalloenzyme platform showed superior reactivity relative to its well-established tetrameric forms. Mechanistically, the authors proposed that metalation of the amide substrate by rhodium catalyst results in the formation of intermediate 20-A. Subsequently, the reversible C–H activation step occurs presumably via a concerted-metalation deprotonation (CMD) mechanism to afford the five-membered rhodacycle 20-B, which further undergoes alkene coordination to produce intermediate 20-C. Subsequent migratory insertion gives rise to the seven-membered rhodacycle 20-D, which then readily undergoes N–O bond cleavage and further reductive elimination to furnish transient Rh(III) intermediate 20-E. Finally, protodemetalation releases the expected annulation products, and regenerates the Rh(III) catalyst.
 |
| | Scheme 9 Enantioselective [4 + 2] annulation with a monomeric streptavidin artificial metalloenzyme. | |
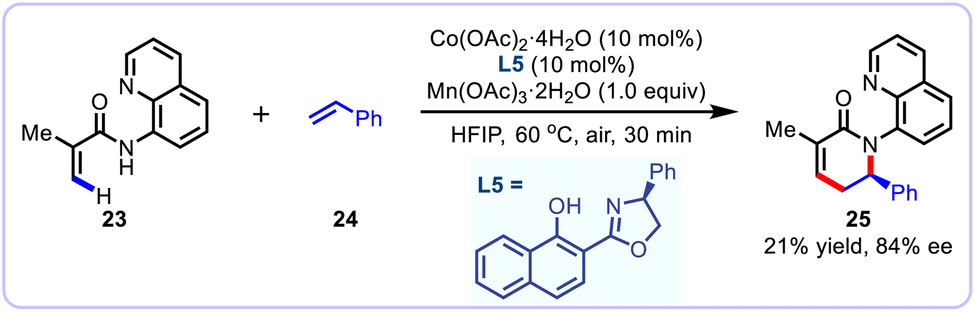
A prominent recent report by the group of Niu and Song documented a cost-effective cobalt-catalyzed enantioselective C–H activation/annulation of benzamides 23 with alkenes 24, allowing access to a diverse array of enantioenriched dihydroisoquinolone derivatives with good functional group compatibility and a high level of enantiocontrol. The reaction occurred smoothly within 10 to 30 min by making use of the readily prepared salicyl-oxazoline (Salox) as the chiral ligand under mild conditions. Notably, this asymmetric annulation reaction was also compatible with more challenging acrylamide substrate, albeit with 21% yield and 84% ee (Scheme 10).27 DFT calculations supported a Co(III)/Co(I) catalytic cycle, wherein the reductive elimination is confirmed as the stereo-determining step.
 |
| | Scheme 10 Cobalt-catalyzed enantioselective C–H annulation with styrenes. | |
Needless to say, cyclopropanes are increasingly encountered in a diverse variety of natural products and biologically related pharmaceuticals,28,29 and they are also highly versatile intermediates for the synthesis of synthetically useful building blocks through ring-opening strategy.30 Accordingly, remarkable efforts have been devoted toward the synthesis of functionalized cyclopropanes through the alkenyl C(sp2)–H functionalization strategy in recent years. In this regard, Rovis and co-workers first reported the cyclopropanation of N-enoxyphthalimides with electron-deficient alkenes using a new Rh(III) catalyst bearing a monosubstituted isopropylcyclopentadienyl ligand (Cpi-Pr),31 and they further continued to expand this strategy to include a broad scope of substrates.32–35 To explore the asymmetric version of this Rovis-cyclopropanation, Cramer and co-workers disclosed a chiral cyclopentadienyl Rh(III)-catalyzed enantioselective and diastereoselective cyclopropanation of electron-deficient alkenes (Scheme 11).36 This stereospecific transformation employed N-enoxysuccinimide 26 as the one-carbon unit to couple with electron-deficient olefins 27, which provided expeditious access to diversely functionalized trans-cyclopropanes 28 with excellent enantioselectivities (up to 95% ee), and high diastereoselectivities (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). Notably, the reaction proceeds uneventfully under open-flask reaction and typically mild conditions. The synthetic appeal of this alkenyl C–H transformation was underscored by the formal synthesis of the KMO inhibitor UPF-648 and the oxylipin family of natural products.
:1 dr). Notably, the reaction proceeds uneventfully under open-flask reaction and typically mild conditions. The synthetic appeal of this alkenyl C–H transformation was underscored by the formal synthesis of the KMO inhibitor UPF-648 and the oxylipin family of natural products.
 |
| | Scheme 11 CpxRh-catalyzed enantioselective vinylic C–H activation of N-enoxysuccinimides with electron-deficient olefins. | |
In 2022, Cramer and colleagues further developed a flexible two-step strategy for the highly enantioselective synthesis of diverse 3-azabicyclo[3.1.0] hexanes 32 featuring a rigid saturated nitrogen-containing skeleton (Scheme 12).37 The chemistry involves the CpxRh(III)-catalyzed alkenyl C(sp2)–H activation of N-enoxysuccinimides 29 to access cis-selectivity of cyclopropanes 31 with high enantio- and diastereoselectivity, which is enforced by a fine-tuned chiral CpxRh catalyst bearing a bulky group in C4 substituent of Cpx ligand scaffold. The resulting enantioenriched cis-cyclopropane dicarbonyls efficiently engaged in the subsequent Cp*Ir(III)-catalyzed iterative reductive cyclization with diverse primary amines, leading to a multitude of potential pharmacologically relevant 3-azabicyclo[3.1.0] hexanes with generally high yields and exclusive diastereoselectivity.
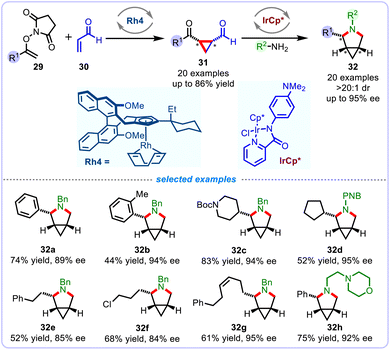 |
| | Scheme 12 Enantioselective synthesis of disubstituted 3-azabicyclo[3.1.0]hexanes. | |
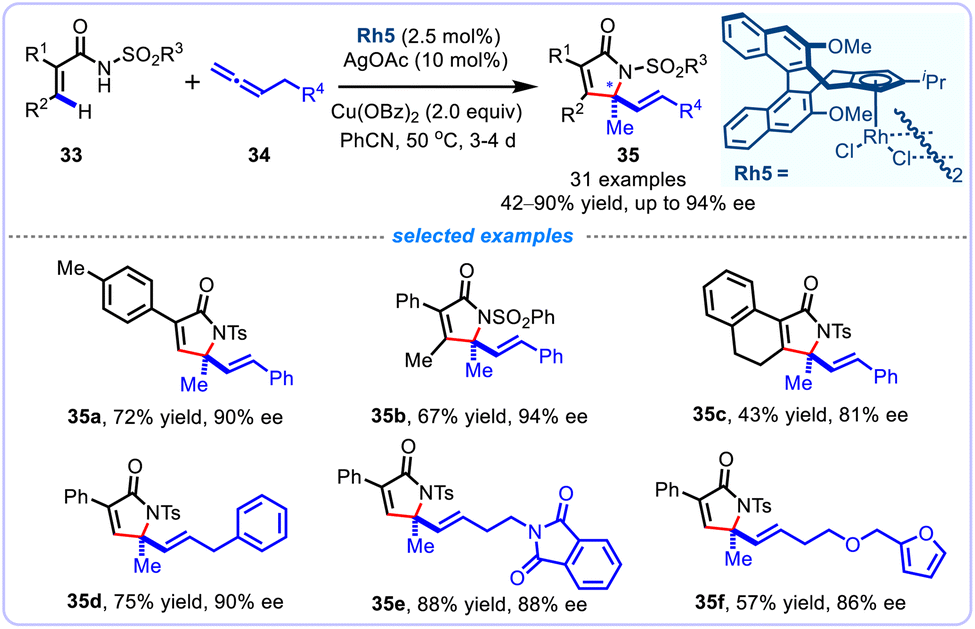
Encouragingly, Cramer's group further realized an enantioselective alkenyl C(sp2)–H functionalization/cyclization of diverse acryl amides 33 with varied allenes 34 enabled by a chiral iPr-bearing trisubstituted CpxRhIII catalyst, allowing the straightforward synthesis of enantioenriched α,β-unsaturated γ-lactams 35 bearing a quaternary stereocenter in excellent enantioselectivity of up to 94% ee (Scheme 13).38 Combination of CpxRhIII catalyst, AgOAc additive, Cu(OBz)2 oxidant in PhCN at 50 °C, a broad range of acryl amides bearing diverse synthetically useful functional groups could be annulated smoothly to produce chiral functionalized 2H-pyrrol-2-ones in generally satisfactory yields (42–90%). Remarkably, the challenging cyclic 2,3-disubstituated acrylamide was also compatible substrate with this stereospecific protocol, and could be converted into a tricyclic lactam in a moderate 43% yield. Noteworthy, the allene substrates serve as C1 fragments to undergo this formal [4 + 1] annulation reaction.
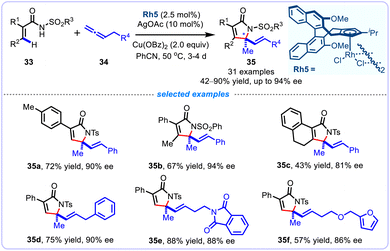 |
| | Scheme 13 Asymmetric CpxRh(III)-catalyzed [4 + 1] annulation of acryl amides with allenes. | |
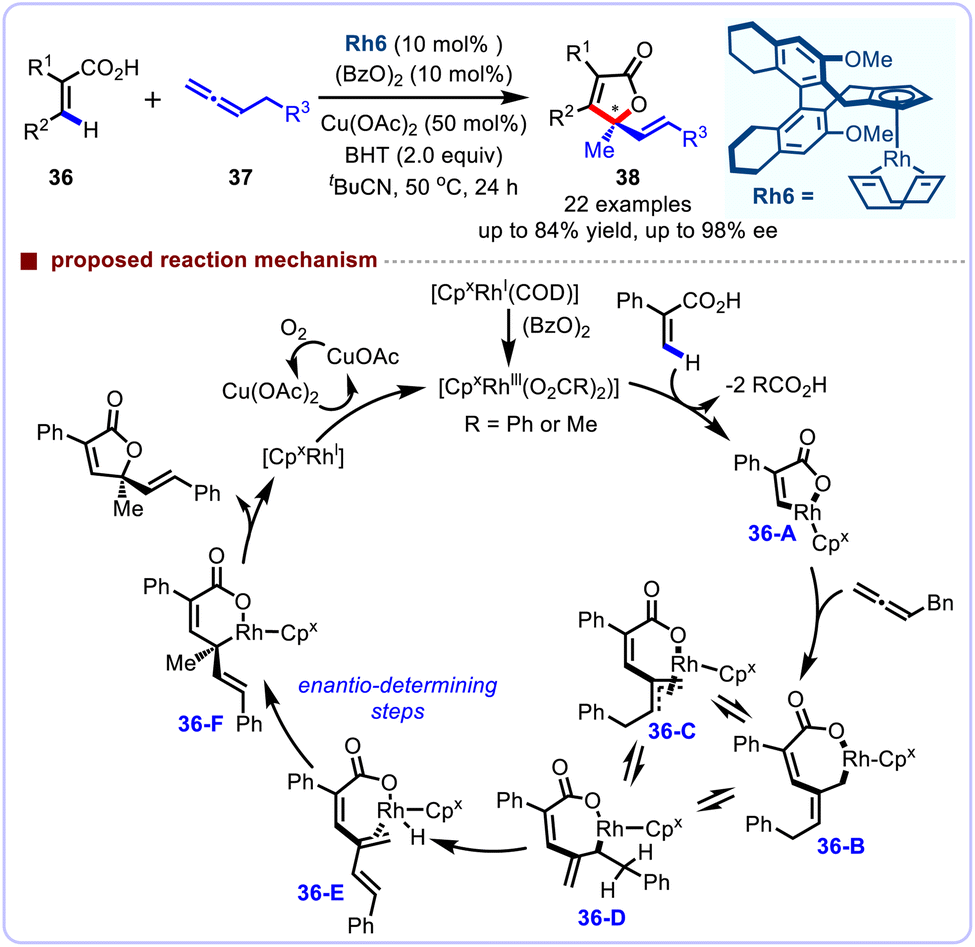
This C–H functionalization/annulation chemistry was further enriched by the precise design of a novel chiral cyclopentadienyl (Cpx) ligand bearing a semi-saturated H8-binaphthyl scaffold. In 2020, Cramer and co-workers illustrated its potential for the enantioselective CpxRh(III)-catalyzed [4 + 1] annulation reaction of acryl acids 36 with allenes 37 (Scheme 14).39 Compared to the existing binaphthyl-derived Cpx ligands, this well-defined, rigid axially chiral H8-binaphthyl scaffold exhibited excellent catalytic performance. Noteworthy, this enantioselective alkenyl C–H activation process nicely tolerates a broad substrate scope, affording the products with overall excellent enantioselectivities (up to 98% ee). The addition of BHT may to some extent mitigate competitive reaction pathways, including conjugate additions, radical reactions and mainly polymerization of the starting materials. Mechanistically, the CpxRhI precatalyst reacts with (BzO)2 to form the active CpxRhIII species, which further undergoes acid exchange and the subsequent alkenyl C–H activation, leading to the five-membered rhodacycle 36-A. Subsequently, allene insertion generates allyl rhodium intermediates (36-B, 36-C, 36-D), and subsequent β-H elimination results in triene intermediate 36-E, then the enantio-determining hydrorhodation to furnish allyl rhodium species 36-F. Finally, reductive elimination produces the desired chiral lactone, and releases a CpxRhI species, which is reoxidized to form the CpxRhIII complex in the presence of CuII(OAc)2. Finally, CuII is regenerated under an air atmosphere.
 |
| | Scheme 14 CpxRh(III)-catalyzed asymmetric alkenyl C–H functionalization of acrylic acids. | |
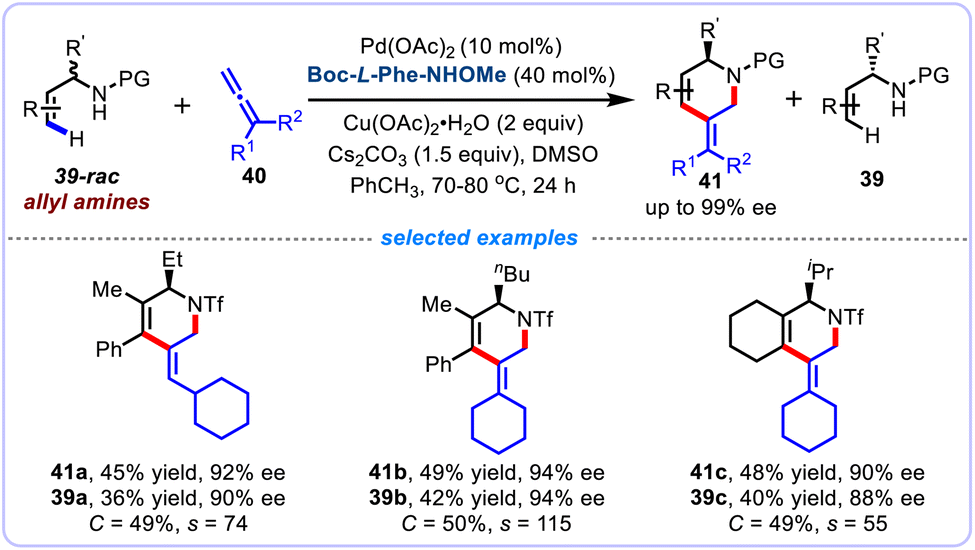
The direct and practical synthesis of chiral products through asymmetric C–H functionalization reactions can also be carried out using kinetic resolutions (KRs) strategy. In 2014, Yu et al. pioneered to investigate the feasibility of such conceptually innovative strategies, and reported an unprecedented palladium(II)-catalyzed enantioselective C–H iodination of racemic benzylic amine substrates.40 They further expanded to establish the highly efficient kinetic resolution processes for the enantioselective C–H arylation of racemic nosyl-protected benzylamines,41 and enantioselective C–H olefination of racemic α-hydroxy and α-amino phenylacetic acids.42 Later in 2021, Gulías and co-workers investigated the asymmetric alkenyl C(sp2)–H activation reactions using kinetic resolution strategy, and elegantly established an efficient atom- and step-economical protocol for the asymmetric Pd(II)-catalyzed alkenyl C(sp2)–H activation/[4 + 2] annulation of racemic α-branched allyltriflamides 39-rac with allenes 40 in the presence of Boc-L-Phe-NHOMe as the ligand (Scheme 15).43 This kinetic resolution strategy features efficient kinetic resolutions (selectivity values of up to 127) and excellent enantioselectivities (up to 99% ee), providing access to a series of enantioenriched chiral piperidine derivatives 41 associated with optically active allyl amines. The value of this strategy has been reinforced by further derivatizations of the thus-obtained chiral tetrahydropyridines, which could be readily converted into a variety of value-added azacycles and different nitrogenated compounds.
 |
| | Scheme 15 Kinetic resolution of allyltriflamides through alkenyl C–H functionalization with allenes. | |
Beyond the above-mentioned intermolecular approaches, relatively rare examples were reported on the intramolecular version of these asymmetric alkenyl C–H functionalization strategies to assemble highly appealing chiral cyclic products. In 2017, Gulías, Mascareñas and López pioneered to investigate the intramolecular asymmetric alkenyl C–H functionalization/cyclization sequence, and disclosed the seminal example of iridium(I)-catalyzed enantioselective hydroalkenylation of 1,1-disubstituted alkenes 42 (Scheme 16).44 With the assistance of carboxamide directing group, this alkenyl C–H functionalization strategy follows an exo-cyclization pathway. Markedly, this reaction showed considerable generality for both alkenyl derivatives and aromatic substrates, giving rise to diverse enantioenriched cyclic and polycyclic scaffolds embodying quaternary carbon stereocenters. Further treatment of the products with RuCl3/NaIO4 can readily prepare a variety of cyclic ketones with α-quaternary stereocenters. The authors postulated a plausible mechanism involving chelation-assisted alkenyl C–H activation, exo-migratory insertion, and reductive elimination steps.
 |
| | Scheme 16 Asymmetric iridium(I)-catalyzed intramolecular C–H activation of alkenes. | |
3. Alkenyl C(sp2)–H bond functionalization for the construction of axially chiral compounds
Axially chiral compounds are frequently encountered in diverse natural products and biologically active compounds, and have also been recognized as broadly useful privileged chiral ligands and organocatalysts that have witnessed wide applications in asymmetric catalysis.45 A growing number of C–H functionalization strategies have been accomplished to the enantioselective synthesis of numerous structurally diverse optically active atropisomers.46–48 In this section, we summarized the recent advances on the atroposelective olefinic C(sp2)–H functionalization of alkene feedstocks to precisely fabricate axially chiral compounds. Chelation-assisted and transient directing group (TDG) strategies are discussed in detail. Special emphasis has been given to more challenging organocatalytic atroposelective alkenyl C–H functionalization reactions. Moreover, the construction of C–N and N–N axially chiral atropisomers via the direct atroposelective alkenyl C–H functionalization/annulation strategies are also covered.
3.1 Atroposelective alkenyl C–H olefination
Although considerable advances have been achieved in alkenyl C–H functionalizations of styrenes,5–10 the atroposelective synthesis of axially chiral styrenes through the asymmetric alkenyl C–H functionalization strategy has been a long-standing challenge, presumably due to the relatively low configurational stability of the obtained products.45,49 By incorporation of chiral spiro-phosphoric acid ligand CPA2 and thioether directing group, in 2021, Shi and co-workers pioneered an innovative palladium-catalyzed atroposelective alkenyl C–H olefination of styrenes, allowing access to diverse atropisomeric styrenes 46 containing a synthetically attractive conjugated 1,3-diene scaffold (Scheme 17).50 This thioether-directed alkenyl C–H olefination reaction occurred smoothly under the conditions, and a wide substrate scope was documented with satisfactory yields of 27–99% and exclusive Z-selectivity. Moreover, this strategy also opens up a new opportunity for the atroposelective synthesis of atropisomers with two stereogenic axes. Under slightly modified conditions, a series of diaxially chiral styrenes were prepared in generally high yields (62–79%) and excellent enantioselectivities (94–99% ee). Treatment with m-CPBA at −78 °C, the thus-obtained chiral styrenes could be readily oxidized into axial chiral sulfoxides, which are highly valuable chiral S–olefin ligands exhibiting impressive efficiency in the Rh-catalyzed asymmetric addition reactions. This dramatically enhanced the synthetic appeal of this transformation.
 |
| | Scheme 17 Atroposelective thioether-directed alkenyl C–H olefination. | |
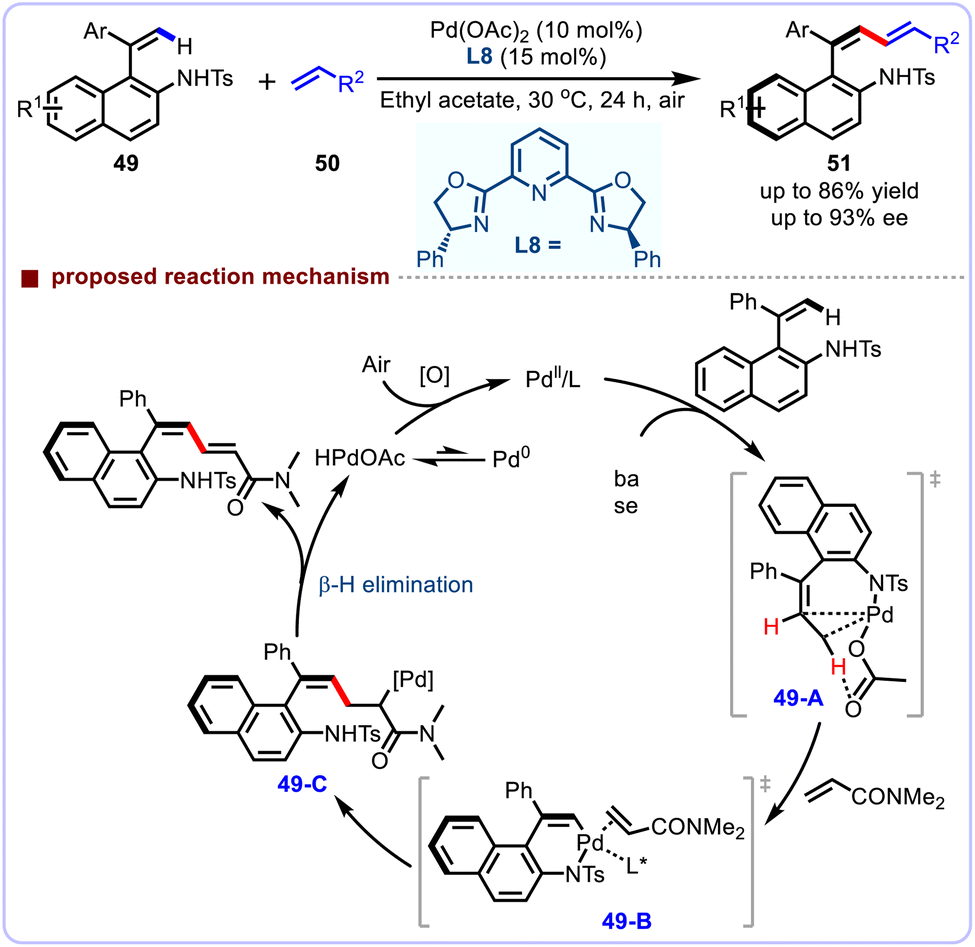
Subsequently, Xu and co-workers also accomplished a similar tosylamine-group-directed strategy for the atroposelective alkenyl C–H olefination under aerobic conditions (Scheme 18).51 With chiral tridentate Pybox L8 as the ligand, a broad range of substrates bearing diverse electronically differentiated substituents on the phenyl ring were well accommodated, furnishing the atropisomeric styrenes in overall high yields and enantioselectivities. It was worth emphasizing that the sterically hindering ortho-substituted substrates do not participate efficiently, a moderate yield and enantioselectivity were observed. The scope of this methodology was further expanded to include acrylates bearing natural product fragments as coupling partners where the atroposelective alkenyl C–H olefination proceeded with high enantioselectivities. Impressively, this reaction can be scaled up without erosion of enantioselectivity. A plausible mechanistic pathway has been proposed to elucidate the current atroposelective protocol. First, the alkenyl C–H bond is regioselectively activated by the chelation assistance of sulfonamide directing group to produce intermediate 49-A. Next, a coordination process with N,N-dimethylacrylamide substrate generates the intermediate 49-B, which then undergoes migratory insertion to afford intermediate 49-C. Subsequently, the intermediate 49-C undergoes β-H elimination to afford the expected cross-coupling products, and release the palladium(0) intermediate, which is finally regenerated by the oxygen in air.
 |
| | Scheme 18 Atroposelective tosylamine-group-directed alkenyl C–H olefination. | |
Despite these impressive achievements, the widespread use of chelation-assisted strategies for alkenyl C–H functionalization reaction is inevitably related to some shortcomings such as the tedious pre-installation and subsequent removal of the directing group, which undoubtedly leads to compromise the overall efficiency of these reactions. To this end, an increasing number of transient directing group (TDG) protocols have been successfully devised for the atroposelective alkenyl C–H functionalization reactions. For instance, Zhang and co-workers in 2022 disclosed an aldehyde derived chiral transient directing group (CTDG) strategy for the Pd-catalyzed atroposelective β-C–H alkenylation of styrenes to afford diverse axially chiral aryl dienes 54 (Scheme 19).52 This work exemplified successful use of an amino acid-derived as the transient chiral auxiliary to enable this atroposelective alkenyl C–H functionalization through a challenging seven-membered endo-cyclopalladation process. A broad range of substrates were documented in high yields (up to 93%) and excellent enantioselectivities (up to >99% ee). Noteworthy, the resulting axially chiral 1,3-dienes generated from this process offered a synthetic handle for the synthesis of valuable axially chiral carboxylic acids (CCAs), which were found to be promising ligands in the Cp*Co(III)-catalyzed asymmetric C–H alkylation reactions.
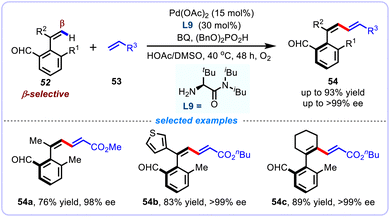 |
| | Scheme 19 Atroposelective Pd-catalyzed transient group directed β-C–H alkenylations. | |
Engle and co-workers described an analogous transient directing group (TDG) strategy for the α-selective alkenyl C(sp2)–H olefination of 2-alkenyl benzaldehydes 55, affording a series of densely functionalized chiral 1,3-dienes 57 with excellent regio- and E/Z-selectivity (Scheme 20).53 In this report, a broad range of alkenyl benzaldehyde 55 with different substitution patterns were competent substrates to couple with tert-butyl acrylate 56. Aside from electronically activated alkenes such as N,N-dimethylacrylamide, acrylonitrile, and vinyl sulfonate, unbiased aliphatic alkenes were equally competent coupling partners, albeit with modest yields. Using L-tert-leucine as the chiral transient directing group, the authors were able to synthesize enantioenriched axially chiral 1,3-dienes through an atroposelective version of this strategy. Mechanistically, this transformation utilizes reversible condensation between alkenyl aldehyde and amino acid TDG to promote the coordination of Pd catalyst, and facilitate subsequent chelation-assisted C–H activation process through a tailored carboxylate base. To elucidate the catalytic mechanism, catalytically related alkenyl palladacycle complexes were synthesized and further characterized through X-ray crystallography. DFT calculations were conducted to reveal the energy distribution of the alkenyl C–H activation and the origin of the atroposelectivity.
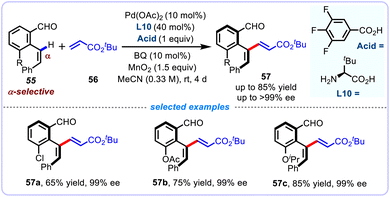 |
| | Scheme 20 Atroposelective Pd-catalyzed alkenyl C–H olefination by a transient directing group. | |
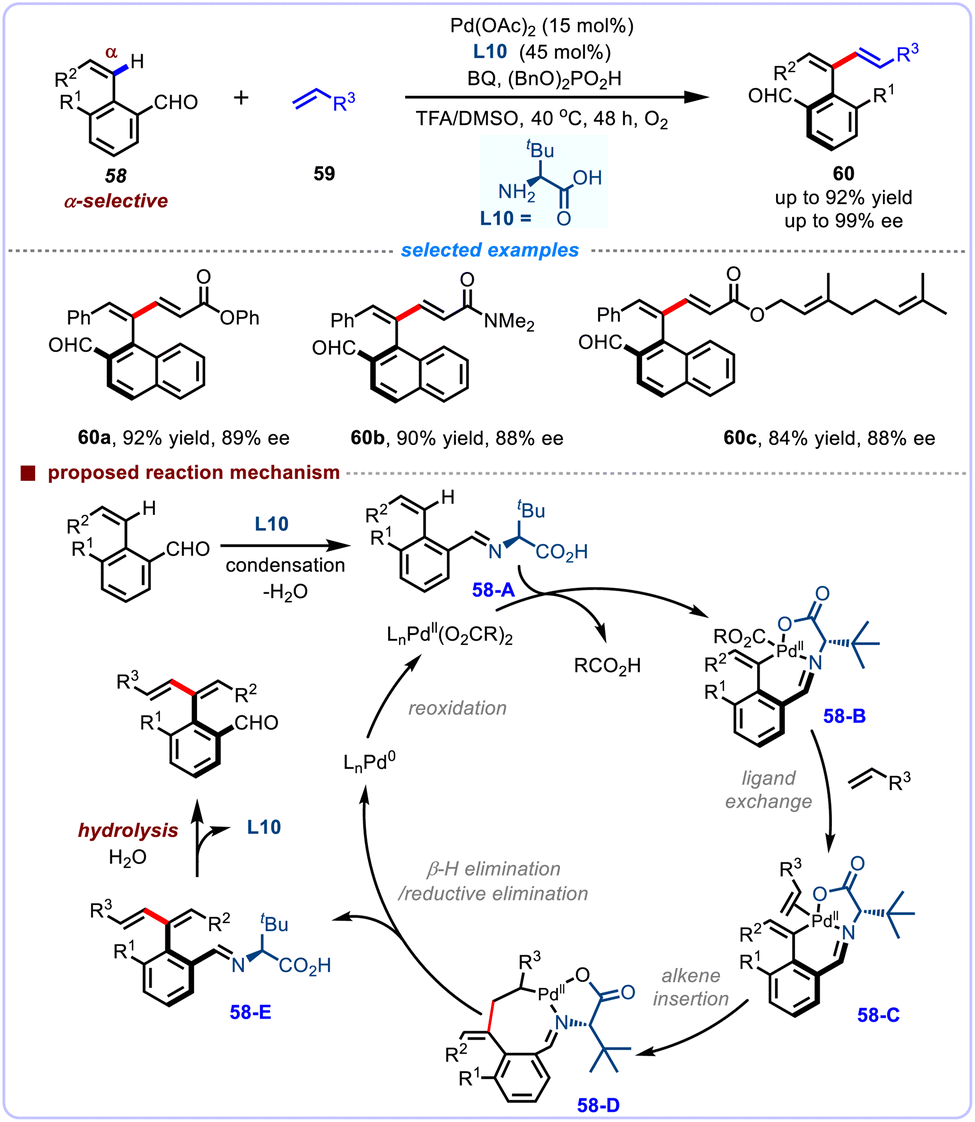
With the efficacious assistance of the aldehyde/L-t-leucine derived chiral transient directing group, Zhang and co-workers also developed an efficient cTDG strategy for the atroposelective α-selective alkenyl C–H olefination of 2-vinyl benzaldehydes through a six-membered endo-cyclometalation process (Scheme 21).54 Using similar catalyst combination of Pd(OAc)2 catalyst, benzoquinone (BQ) oxidant, MnO2 co-oxidant, and (BnO)2PO2H additive in TFA/DMSO at 40 °C, a series of 2-vinyl benzaldehydes 58 bearing diverse functional groups olefinated smoothly, affording diversely functionalized 1,3-dienes 60 in up to 92% yield and up to 99% ee. Of special interest is the observation that acrylate coupling partners bearing sensitive (+)-dehydroabietylamine, geraniol and cholesteryl moieties were generally compatible with this transformation. When treated with NaClO2/NaH2PO4, the thus-obtained axially chiral aryl dienes could be readily oxidized to the corresponding chiral carboxylic acids, which illuminated applicability as new type of ligands in the CoIII-catalyzed asymmetric 1,4-addition of indoles with maleimide. Mechanistically, the initial in situ condensation of aldehyde with amino acid L10 generates imine 58-A, which then coordinates to palladium species and facilitate the subsequent chelation-assisted C–H cleavage, leading to the formation of the six-membered exo-palladacycle 58-B. Next, the intermediate 58-B undergoes ligand exchange and subsequent migratory insertion with olefin substrates to furnish the eight-membered exo-palladacycle 58-D. Then, the intermediate 58-D undergoes β-H elimination and imine hydrolysis to afford the axially chiral product, and releases amino acid L10. Meanwhile, the intermediate 58-D undergoes reductive elimination and subsequent β-H elimination to produce the Pd(0) species, which is finally reoxidized to the active Pd(II) species.
 |
| | Scheme 21 Atroposelective Pd-catalyzed transient group directed α-C–H alkenylation. | |
3.2 Organocatalytic atroposelective alkenyl C–H functionalization
Over the past decades, asymmetric organocatalysis has emerged rapidly as a fruitful platform for the atroposelective synthesis of axially chiral compounds.55 Due to inherently low configurational stability and lack of suitable ligands or catalysts to control the atroposelectivity, it is a formidable challenge for chemists to efficiently assemble the distinctive axially chiral acyclic 1,3-diene atropisomers, which are potentially attractive yet underutilized scaffolds. Quite recently, Tan and co-workers expanded their asymmetric organocatalysis strategy56,57 and elegantly reported the seminal work on the challenging atroposelective synthesis of axially chiral 1,3-butadienes via the organocatalytic functionalization of alkenyl C–H bonds (Scheme 22).58 This protocol was particularly strategic to enable the facile synthesis of diverse highly stable atropisomeric 1,3-butadienes 62 in typically satisfactory yields and enantioselectivities. Remarkably, the thus-obtained products exhibited complete Z/E selectivity (>20:1). The author speculated that the chiral enamine with completely retention of enantiomeric purities might be the intermediates that can couple with various electrophilic reagents. The in situ Grignard exchange reaction with PhMgCl could convert the stereoisomerically enriched 1,3-butadienes back into the reactive nucleophilic intermediates, which greatly unlocked subsequent reaction with different electrophiles in one-pot fashion. It should be noted that this asymmetric transformation of 1,3-dienes with other electrophiles such as tosyl azide 62d afforded the corresponding functionalized products in one-pot, 2-step process. Mechanistic studies revealed that the reaction occurred through a sequence of phosphate formation, cation exchange, bromination, deprotonation, and 1,3-Br migration, which not only bypasses the traditional indirect process of olefinic C–H bond functionalization reactions, but also differs from transition-metal-catalysed C(sp2)–H activation. These findings are expected to open up new alternative methods for further investigation into axially chiral olefin chemistry and to stimulate more research on the organocatalytic activation of other inert structures.
 |
| | Scheme 22 Synthesis of atropisomeric 1,3-butadienes via organocatalytic alkenyl C–H bond functionalizations. | |
3.3 Atroposelective annulation reactions
Despite these impressive advances, practical approaches for the atroposelective synthesis of C–N axially chiral atropisomers through the enantioselective alkenyl C–H functionalization and annulation strategy indubitably remain very rare, presumably due to the remarkably low rotational barrier and weak conformational stability of the resulting atropisomers.59,60 In this regard, the group of Yang and Niu first investigated the cobalt-catalyzed alkenyl C–H activation and annulation between vinylamide 63 and diphenylacetylene 64 for the synthesis of C–N axially chiral atropisomers (Scheme 23).61 Despite an indisputable high level of stereoselectivity (97% ee), the particularly low efficiency greatly inhibited the synthetic appeal of this reaction. The low efficiency of vinylamide may be due to the effect of electronic effects. Compared with benzamide, the conjugation effect between the double bond and carbonyl group in vinylamide is significantly weaker than that between the benzene ring and carbonyl group in benzamide.
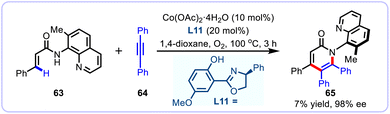 |
| | Scheme 23 Atroposelective cobalt-catalyzed C–H activation and annulation of vinylamide with diphenylacetylene. | |
To address this issue, the same group further expanded their cobalt-based strategy, and established an efficient atroposelective C–H activation/[4 + 1] annulation strategy between benzamides 66 and isonitriles 67, providing an efficient and practical route to a series of diversely functionalized C–N axially chiral 3-iminoisoindolinones 68 (Scheme 24).62 The 8-aminoquinoline auxiliary bearing a steric substituent served as both the efficacious directing group and integral part of the C–N axially chiral atropisomers, whereas isonitriles functioned as the C1 synthon to undergo this formal [4 + 1] annulation reaction. Both aliphatic and aromatic isonitriles were proved to be equally effective, affording the atropisomers bearing a five-six heterobiaryl skeleton. Satisfactorily, a variety of substrate scopes were documented with generally high reactivities and enantioselectivities. Apart from (hetero)aromatic benzamides, the alkenyl substrates were also applicable for this distinctive protocol. Under the aerobic conditions, the atroposelective [4 + 1] annulation of vinylamides with isonitrile occurred smoothly to furnish the atropisomerically enriched products in good yields (74–92%), and moderate enantioselectivities (70–84% ee).
 |
| | Scheme 24 Atroposelective cobalt-catalyzed alkenyl C–H activation and annulation with isonitriles. | |
As a particularly attractive yet challenging chiral system, the N–N atropisomers are widely present in numerous bioactive natural products, drug molecules and functional materials.63 Due to the inherently low rotational barrier, short length as well as weak nature of the N–N bond, the N–N axis construction through alkenyl C(sp2)–H activation remains a daunting challenge.64 To this end, Niu and colleagues successfully established an efficient cobalt/chiral Salox catalyzed atroposelective aryl C(sp2)–H activation/annulation protocol, affording a series of N–N axially chiral isoquinolinones 71 with overall high yields and enantioselectivities (Scheme 25).65 In this report, cost-effective Co(OAc)2·4H2O was employed as the catalyst in the presence of a chiral salicyloxazoline (Salox) ligand with environmentally friendly O2 as the terminal oxidant. Under electrochemical cobalt/Salox catalysis, the N–N axially chiral isoquinolinones were obtained with comparable yields and enantioselectivities. Remarkably, vinylamides were also proved to be compatible with this newfound protocol, yielding the annulation products with excellent level of enantiocontrol (98 and 99% ee). A plausible mechanism that involves the Co(III)-species formation, chelation-assisted alkenyl C–H activation, migratory insertion, and final reductive elimination was proposed, wherein the reductive elimination was postulated to serve as the enantio-determining step.
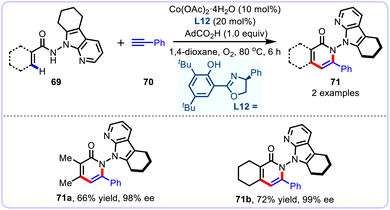 |
| | Scheme 25 Cobalt-catalyzed C–H activation/annulation to construct N–N axially chiral skeletons. | |
4. Conclusions and outlook
The asymmetric alkenyl C–H functionalization has emerged rapidly as an attractive strategy to fabricate highly functionalized chiral compounds from readily available alkenes. As detailed in this review, a diverse number of enabling strategies with broad applicability have been summarized. Specifically, the enantioselective self-coupling protocols afforded the chiral products in a highly atom-economic manner. Kinetic resolution strategies have been successfully performed to access highly functionalized chiral compounds associated with optically active precursors. Meanwhile, the intramolecular version of asymmetric alkenyl C–H functionalization has also been established to synthesize highly valuable chiral cyclic products. Using chelation-assisted or transient directing group (TDG) strategies, the alkenyl C–H functionalization of styrenes enabled the atroposelective synthesis of structurally diverse axially chiral styrenes featuring an attractive 1,3-diene scaffold. Moreover, the robust atroposelective alkenyl C–H functionalization/annulation strategies allowed an efficient route to precisely fabricate C–N and N–N axially chiral atropisomers. Needless to say, these methodologies offered incredibly powerful and versatile synthetic tools that greatly complement traditional asymmetric catalysis for the facile synthesis of structurally diverse enantioenriched alkenes and their derivatives.
These impressive advances have taken this field significantly forward, but challenges lie ahead need be addressed for future development of more efficient enantioselective alkenyl C–H functionalization reactions. For example, the vast majority of these reactions required the use of precious transition metals such as Pd, Rh, and Ir catalysts to achieve high efficiency, which remarkably posed scalability and environmental concerns of these reactions, the use of less-toxic, cost-effective 3d metals such as cobalt catalyst is still very rare. Additional work to broaden metal-free and organocatalytic methods will provide opportunities to enhance the synthetic appeal of these strategies. Although sporadic examples of kinetic resolution strategy have been investigated, related methodological improvements remain highly intriguing. Intramolecular strategies that are capable of synthesizing macrocycles have thus far remained elusive. Moreover, in-depth mechanistic studies are still highly sought to better understand the underlying mechanism of these reactions. Meeting these challenges will require continuous efforts toward the thriving development of conceptually innovative strategies, and the precise design of new catalysts as well as novel catalytic pathways. With these issues addressed, we can expect new breakthroughs and improvements in this burgeoning field. Overall, we hope that this review will stimulate new development of more practical and broadly applicable strategies with respect to the asymmetric alkenyl C–H functionalization reactions, and expand their applications for the construction of diverse synthetically useful chiral compounds as well as the total synthesis of complex natural products in the future.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Author contributions
X. J. S., T. C. W., M. Z. L. and T. P. L. conceived the idea and contributed to the writing, review and editing of the manuscript. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We gratefully acknowledge the financial support from the China Postdoctoral Science Foundation (2024M750790), and the Start-up Grant of Henan University of Technology (2023BS066). The authors also thank the financial support from the National Natural Science Foundation of China (no. 22101095).
Notes and references
-
J. Wang, Stereoselective alkene synthesis, Springer Verlag, Heidelberg, New York, 2012 Search PubMed.
- K. Wang, F. Hu, Y. Zhang and J. Wang, Sci. China: Chem., 2015, 58, 1252–1265 CrossRef CAS.
- S. Tang, K. Liu, C. Liu and A. Lei, Chem. Soc. Rev., 2015, 44, 1070–1082 RSC.
- M. Maraswami and T.-P. Loh, Synthesis, 2019, 51, 1049–1062 CrossRef CAS.
- J. Zhang, X. Lu, C. Shen, L. Xu, L. Ding and G. Zhong, Chem. Soc. Rev., 2021, 50, 3263–3314 RSC.
- B. Liu, L. Yang, P. Li, F. Wang and X. Li, Org. Chem. Front., 2021, 8, 1085–1101 RSC.
- M.-Z. Lu, J. Goh, M. Maraswami, Z. Jia, J.-S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef CAS PubMed.
- V. K. Maikhuri, J. Maity, S. Srivastava and A. K. Prasad, Org. Biomol. Chem., 2022, 20, 9522–9588 RSC.
- X. Lu, Y. Wang, K. Xie and J. Zhang, Synlett, 2024, 34, 2262–2292 Search PubMed.
- Y. Zhu, Y. Liao, S. Jin, L. Ding, G. Zhong and J. Zhang, Chem. Rec., 2023, 23, e202300012 CrossRef CAS PubMed.
-
(a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC;
(b) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, 759–770 CrossRef CAS PubMed.
- B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS.
- K. Yang, M. Song, H. Liu and H. Ge, Chem. Sci., 2020, 11, 12616–12632 RSC.
- Ł. Woźniak, J.-F. Tan, Q.-H. Nguyen, A. M. Vigné, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516–10543 CrossRef.
- C.-X. Liu, S.-Y. Yin, F. Zhao, H. Yang, Z. Feng, Q. Gu and S.-L. You, Chem. Rev., 2023, 123, 10079–10134 CrossRef CAS PubMed.
- For selected reviews on radical-type asymmetric C–H bond functionalizations, see:
(a) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed;
(b) Z. Zhang, P. Chen and G. Liu, Chem. Soc. Rev., 2022, 51, 1640–1658 RSC;
(c) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS;
(d) X. Wang, J. He, Y.-N. Wang, Z. Zhao, K. Jiang, W. Yang, T. Zhang, S. Jia, K. Zhang, L. Niu and Y. Lan, Chem. Rev., 2024, 124, 10192–10280 CrossRef CAS PubMed.
- C. Baudequin, A. Zamfir and S. B. Tsogoeva, Chem. Commun., 2008, 4637–4639 RSC.
- M. Kojima, M. Sasaki, M. Ito and T. Shibata, Adv. Synth. Catal., 2022, 364, 1666–1670 CrossRef CAS.
- Y. Zhang, J.-J. Zhang, L. Lou, L. Lin, N. Cramer, S.-G. Wang and Z. Chen, Chem. Soc. Rev., 2024, 53, 3457–3484 RSC.
- S.-G. Wang and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 2514–2518 CrossRef CAS.
- C. Duchemin and N. Cramer, Angew. Chem., Int. Ed., 2020, 59, 14129–14133 CrossRef CAS PubMed.
- T. Shibata, M. Kojima, S. Onoda and M. Ito, Org. Lett., 2021, 23, 8158–8162 CrossRef CAS.
- X. Sun, E.-Z. Lin and B.-J. Li, J. Am. Chem. Soc., 2022, 144, 17351–17358 CrossRef CAS.
- F. Hong, C. M. Robertson and J. F. Bower, J. Am. Chem. Soc., 2024, 146, 22923–22929 CrossRef CAS PubMed.
- I. S. Hassan, A. N. Ta, M. W. Danneman, N. Semakul, M. Burns, C. H. Basch, V. N. Dippon, B. R. McNaughton and T. Rovis, J. Am. Chem. Soc., 2019, 141, 4815–4819 CrossRef CAS.
- D. Yang, X. Zhang, X. Wang, X.-J. Si, J. Wang, D. Wei, M.-P. Song and J.-L. Niu, ACS Catal., 2023, 13, 4250–4260 CrossRef CAS.
- D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631–4642 RSC.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS.
- V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS.
- T. Piou and T. Rovis, J. Am. Chem. Soc., 2014, 136, 11292–11295 CrossRef CAS PubMed.
- T. Piou and T. Rovis, Nature, 2015, 527, 86–90 CrossRef CAS PubMed.
- T. Piou, F. Romanov-Michailidis, M. A. Ashley, M. Romanova-Michaelides and T. Rovis, J. Am. Chem. Soc., 2018, 140, 9587–9593 CrossRef CAS.
- E. J. T. Phipps and T. Rovis, J. Am. Chem. Soc., 2019, 141, 6807–6811 CrossRef CAS PubMed.
- E. J. T. Phipps, T. Piou and T. Rovis, Synlett, 2019, 30, 1787–1790 CrossRef CAS.
- C. Duchemin and N. Cramer, Chem. Sci., 2019, 10, 2773–2777 RSC.
- O. Lahtigui, D. Forster, C. Duchemin and N. Cramer, ACS Catal., 2022, 12, 6209–6215 CrossRef CAS.
- S.-G. Wang, Y. Liu and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 18136–18140 CrossRef CAS.
- S.-G. Wang and N. Cramer, ACS Catal., 2020, 10, 8231–8236 Search PubMed.
- L. Chu, K.-J. Xiao and J.-Q. Yu, Science, 2014, 346, 451–455 CrossRef CAS.
- K.-J. Xiao, L. Chu, G. Chen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 7796–7800 CrossRef CAS.
- K.-J. Xiao, L. Chu and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 2856–2860 CrossRef CAS.
- J. M. González, B. Cendón, J.
L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2021, 143, 3747–3752 CrossRef.
- D. F. Fernández, M. Gulías, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2017, 56, 9541–9545 CrossRef.
- J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 Search PubMed.
- G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC.
- L. Jin, Q.-J. Yao, P.-P. Xie, Y. Li, B.-B. Zhan, Y.-Q. Han, X. Hong and B.-F. Shi, Chem, 2020, 6, 497–511 Search PubMed.
- D. Parmar, R. Kumar and U. Sharma, Org. Biomol. Chem., 2024, 22, 5032–5051 RSC.
- C.-X. Liu, W.-W. Zhang, S.-Y. Yin and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed.
- L. Jin, P. Zhang, Y. Li, X. Yu and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 12335–12344 Search PubMed.
- D.-T. Dai, M.-W. Yang, Z.-Y. Chen, Z.-L. Wang and Y.-H. Xu, Org. Lett., 2022, 24, 1979–1984 Search PubMed.
- C. Shen, Y. Zhu, W. Shen, S. Jin, G. Zhong, S. Luo, L. Xu, L. Zhong and J. Zhang, Org. Chem. Front., 2022, 9, 2109–2115 Search PubMed.
- M. Liu, J. Sun, T. G. Erbay, H.-Q. Ni, R. Martín-Montero, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2022, 61, e202203624 CrossRef CAS PubMed.
- C. Shen, Y. Zhu, W. Shen, S. Jin, L. Zhong, S. Luo, L. Xu, G. Zhong and J. Zhang, Org. Chem. Front., 2022, 9, 4834–4839 RSC.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS.
- J. K. Cheng, S.-H. Xiang and B. Tan, Acc. Chem. Res., 2022, 55, 2920–2937 CrossRef CAS.
- Q.-H. Wu, M. Duan, Y. Chen, P. Yu, Y.-B. Wang, J. K. Cheng, S.-H. Xiang, K. N. Houk and B. Tan, Nat. Catal., 2024, 7, 185–194 CrossRef CAS.
- O. Kitagawa, Acc. Chem. Res., 2021, 54, 719–730 Search PubMed.
- P. Rodríguez-Salamanca, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem.–Eur. J., 2022, 28, e202104442 CrossRef.
- X.-J. Si, D. Yang, M.-C. Sun, D. Wei, M.-P. Song and J.-L. Niu, Nat. Synth., 2022, 1, 709–718 CrossRef CAS.
- T. Li, L. Shi, X. Zhao, J. Wang, X.-J. Si, D. Yang, M.-P. Song and J.-L. Niu, Org. Lett., 2023, 25, 5191–5196 CrossRef CAS.
- G. Centonze, C. Portolani, P. Righi and G. Bencivenni, Angew. Chem., Int. Ed., 2023, 62, e202303966 CrossRef.
- S. M. Verma and R. Prasad, J. Org. Chem., 1973, 38, 1004–1010 CrossRef CAS.
- T. Li, L. Shi, X. Wang, C. Yang, D. Yang, M.-P. Song and J.-L. Niu, Nat. Commun., 2023, 14, 5271–5281 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac and
Ming-Zhu
Lu
*ac and
Ming-Zhu
Lu