
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic 1,3-difluoroalkylcarboxylation of alkenes by triple kinetic-controlled radical self-ordering†
Hong
Fu
,
Zuo-Shuai
Wang
,
Si-Jia
Li
,
Lin-Yuan
Zhu
,
Xiao-Jian
Wang
,
Hong-Chen
Wang
and
Bing
Han
 *
*
State Key Laboratory of Applied Organic Chemistry (SKLAOC), College of Chemistry and Chemical Engineering, Lanzhou University, 222 South Tianshui Road, Lanzhou, 730000, People's Republic of China. E-mail: hanb@lzu.edu.cn
First published on 19th February 2025
Abstract
A transition-metal-free protocol for the unsymmetrical radical 1,3-difunctionalization of alkenes has been established for the first time in the form of 1,3-difluoroalkylcarboxylation by a photocatalytic radical three-component reaction of allyl formates, trifluoroacetanilides, and cesium formate. This reaction employs formate as the carboxylating reagent and trifluoroacetanilide as the difluoroalkylating reagent via C–F bond activation. As a result, a series of previously inaccessible unsymmetrical difluorinated adipic acid derivatives can be easily and efficient prepared. Mechanism studies reveal that triple kinetic-controlled radical self-ordering is the key to this unique reaction. This radical sorting involves the fast initiation of a CO2 radical anion and its chemoselective addition and reduction, followed by the slow generation of a fluoroalkyl radical and its chemo-/regioselective addition. Notably, this strategy is also suitable for the 1,3-difluoroalkylcarboxylation of unsymmetrical and cyclic alkenes through diastereoselectively constructing two or three consecutive stereocenters.
As a common class of chemicals with a wide range of sources, the diverse conversion reactions of olefins provide a convenient and practical way to obtain high-value-added functional molecules. Among these conversions, 1,2-difunctionalization is the one of the most representative modes, as it affords a convenient method for simultaneously introducing two functional groups and has received extensive attention.1 Both symmetrical and unsymmetrical 1,2-difunctionalization reactions have been fully developed by introducing two identical or different functional groups, respectively (Scheme 1A).2,3 Recently, the 1,3-difunctionalization of olefins has become a hot research topic because it provides a facile method to obtain useful 1,3-difunctionalized moieties through an unusual remote functionalization of alkene (Scheme 1B).4 Several practical strategies have been established, which can be divided into two-electron and one-electron processes. The positional isomerization of olefins mediated by transition metal hydride intermediates is the dominant method of the former, and the precious metal Pd and the coinage metal Ni have proven to be the most effective catalysts in these conversions.5 Metal-free 2e processes have also been developed using high-valence iodine as the oxidizing reagent, which involve ionic rearrangement facilitated by I(III), leading to the 1,3-difluorination of olefins.6 Conversely, with the emergence of increasingly efficient and controllable radical initiation methods, olefin 1,3-difunctionalization reactions involving one-electron processes have gradually been developed. One elegant approach is to use radical 1,2-functional group (FG) migration as a key link to realize a variant 1,3-difunctionalization in the form of a 1,2,3-trifunctionalization.7 Some successful examples have employed 1,2-boron,7a 1,2-acyl,7b–d and 1,2-aryl7e migrations. In addition, the radical addition/elimination/re-addition (AERA) strategy has also proven to be a promising protocol.8 Consequently, some symmetric 1,3-difunctionalizations are realized, such as dicarboxylation,8a difluroalkylation,8b diaminoalkylation8c and diphosphination.8d Although these approaches widen the range of olefin 1,3-difunctionalizations, most are limited to the symmetrical pattern, while their unsymmetrical counterparts, especially for the 1e-process, remain an unrealized goal.
 | ||
| Scheme 1 1,3-Difunctionalization of olefins. | ||
As the radical AERA strategy affords a viable method for symmetrical 1,3-difunctionalization, it is expected to be applicable to unsymmetrical reactions as well. Despite this expectation, it presents a formidable challenge, as the process is bound to involve multiple competitive reactions due to the employment of two different radicals. These competitions result in the disordered radical additions of the two radicals to the substrate alkenes and the derived alkene intermediates, as well as undesired radical cross-coupling.9 Thus, both symmetrical and unsymmetrical 1,3-difunctionalizations and the unwanted cross-coupled adducts would be produced as a complex mixture. To obtain the desired 1,3-unsymmetrical product while avoiding the formation of other byproducts, a triple kinetic-controlled process was hypothesized; this process depends on several factors (Scheme 1C). First, a significant rate difference is necessary for the generation of two kinds of radicals to be applied (kinetic I). Secondly, the radical species produced first should have a faster addition rate to the substrate alkene than the other, slow-forming radical species to obtain a single intermediate alkene (kinetic II). Thirdly, the slow-forming radical should have kinetic selectivity toward preferential addition to the derived intermediate alkene rather than the substrate alkene (kinetic III). Finally, the cross-coupling between these two radicals should be much slower than their addition to the corresponding alkenes.
Herein, we realize this hypothesis in the form of the radical 1,3-difluoroalkylcarboxylation of olefins by a photocatalytic three-component reaction of allyl formates, trifluoroacetanilides, and cesium formate (Scheme 1D). To the best of our knowledge, this protocol represents the first successful example of unsymmetrical 1,3-difunctionalization via a 1e-process. This approach initiates from the photocatalytic oxidation of formate to generate a carbon dioxide radical anion (CO2−˙),10 which preferentially reacts with allyl formates through fast radical addition. The nascent tertiary radical would be converted to the key intermediate but-3-enoate by subsequent reduction and further elimination of a formate anion.11,8b With the consumption of allyl formate and the accumulation of but-3-enoate, CO2−˙ begins to reduce trifluoroacetanilide to a difluoroacetanilide radical via C–F bond activation.12 The latter preferentially adds to but-3-enoate rather than allyl formate by taking advantage of the kinetic rate difference. Finally, the radical 1,3-difluoroalkylcarboxylation is achieved by this triple kinetic amplification process. As a result, a series of previously inaccessible unsymmetrical difluorinated adipic acid derivatives can be easily prepared using this convenient photocatalytic multi-component reaction. Adipic acid derivatives are very useful structural units and play important roles in organic synthesis and polymerization.13 In this context, this protocol not only successfully extends the radical AERA sequence to 1,3-unsymmetrical difunctionalization of olefins but also provides a simple method toward significant moieties.
The study was commenced by stirring allyl formate a1, trifluoroacetanilide b1, cesium formate, and cesium carbonate in dimethylsulfoxide (DMSO) under irradiation by blue LEDs (425 nm, 30 W) at room temperature under an argon atmosphere. When 4CzIPN was used as the photocatalyst, the 1,3-difluoroalkylcarboxylation took place smoothly and gave the desired product c1 in 60% yield (Table 1, entry 1). Formate proved to be the best leaving group (LG); when a2–a7, which contained OAc, OBz, Ts, trimethylphenylsulphonyl (Mts), thiophenyl, and Br groups, were tested, the reaction gave unsatisfactory yields (entries 2–4).14 When sodium formate was used instead of cesium formate as the reactant and reductant, the reaction yield was reduced to 41% (entry 5). Cesium carbonate was essential for efficient reaction, as the yield of c1 dropped dramatically in its absence (entry 6). When DMF was used instead of DMSO, c1 was obtained in only 28% yield (entry 7). Other photocatalysts, namely, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (PC2), N-Ph-Mes-Acr-BF4 (PC3), and 4DPAIPN (PC4), were also tested, but they were ineffective except for PC4, which provided c1 in 52% yield (entries 8–10). The amount of b1 used also influenced the reaction efficiency, as when it was decreased to 3 equiv. and 1.5 equiv., the yield of c1 was reduced to 51% and 23%, respectively (entries 11 and 12). Additionally, the yield of c1 was slightly increased to 68% when the reaction was performed under a CO2 atmosphere (entry 13), whereas no reaction took place when the reaction was conducted under a CO2 atmosphere without the addition of cesium formate (entry 14), implying that cesium formate is crucial for efficient reaction, and a CO2 atmosphere is merely conducive to promoting the reaction, but not a decisive factor. Notably, no reaction occurred when the reaction was performed without PC1 or without light irradiation (entries 15 and 16).
|
|
||
|---|---|---|
| Entry | Variation of conditionsa | Yieldb |
| a Reaction conditions: a1 (0.2 mmol, 1 eq.), b1 (4 eq.), HCO2Cs (2 eq.), Cs2CO3(0.5 eq.), 4CzIPN (PC1, 2 mol%), DMSO (3 mL), Ar, blue LEDs (425 nm, 30 W), rt, 12 h; then MeI (0.3 mL) and K2CO3 (1.0 mmol), rt, 4 h. b Isolated yield. | ||
| 1 | None | 60% |
| 2 | a2 instead of a1 | 23% |
| 3 | a3, a4, a6, a7 instead of a1 | <10% |
| 4 | a5 instead of a1 | 26% |
| 5 | HCO2Na instead of HCO2Cs | 41% |
| 6 | Without Cs2CO3 | 45% |
| 7 | DMF instead of DMSO | 28% |
| 8 | PC2 instead of PC1 | Trace |
| 9 | PC3 instead of PC1 | Trace |
| 10 | PC4 instead of PC1 | 52% |
| 11 | 3 eq. of b1 | 51% |
| 12 | 1.5 eq. of b1 | 23% |
| 13 | Under CO2 atmosphere | 68% |
| 14 | CO2 atmosphere instead of HCO2Cs | NR |
| 15 | Without PC1 | NR |
| 16 | In the dark | NR |

With the optimal conditions in hand (entry 13 in Table 1), the reaction scope was investigated, and the results are shown in Scheme 2. The allyl formate scope was first investigated (Scheme 2A). 2-Phenylallyl formates bearing para-substituents with a wide range of electronic properties, such as trifluoromethyl, fluorine, chlorine, bromine, methyl, phenyl, and methoxy groups, were all converted smoothly in the reaction, providing the desired products c2–c8 in good yields. When the para-bromophenyl functionalized allyl formate was used in the reaction, in addition to the normal product c5, which was obtained in 40% yield, c1 was also formed in 16% yield. The formation of c1 can be attributed to the fact that c5 is prone to further reductive debromination under the reaction conditions.15 In addition, meta-substituted 2-phenylallyl formates bearing electron-deficient or electron-donating groups, such as trifluoromethyl, chlorine, methyl, methoxy, and acetamido groups, on the phenyl ring exhibited good compatibility with the protocol, delivering products c9–c13 in moderate-to-good yields. Similarly, this approach was also suitable for ortho-substituted phenyl counterparts, as demonstrated in the cases of c14 and c15, which produced the corresponding products in good yields. Notably, olefins containing disubstituted phenyl moieties, such as 2,3-dichlorophenyl, 3,4-piperonyl, and 3,4-ethylenedioxyphenyl moieties, were also good candidates for the transformation, affording products c16–c18 in good yields. In addition to phenyl, olefins incorporating other aromatic and heteroaromatic groups, such as naphthalene, furan and thiophene, were also well tolerated in the method, giving c19–c21 in moderate yields. Significantly, when unsymmetrical alkenes with different chain lengths were subjected to the reaction, the regio- and diastereoselective 1,3-difluoroalkylcarboxylation took place smoothly, providing products c22–c24 in moderate yields as a single diastereomer. The structure and the anti-configuration of c22 were confirmed through an X-ray diffraction study. The excellent regioselectivity of the unsymmetrical olefins indicates that the addition of CO2−˙ rather than the difluoroalkyl radical onto the allyl formate is the key step. In addition, allyl formates involving cyclic alkenes, such as cyclohexene and cyclooctene, were also suitable for this protocol. Through regio- and diastereoselective processes, c25 and c26 were produced as a single diastereomer with three continuous chiral centers in 34% and 33% yields, respectively. The structure and the adjacent trans-configuration of the three continuous stereocenters of c25 were confirmed through an X-ray diffraction study. It is noteworthy that when the aromatic ring moiety of the allyl formates was replaced by electron-deficient groups such as cyano, ester and amide groups, the reaction still proceeded smoothly, providing the corresponding products c27–c33 in moderate-to-good yields. The functional group tolerance of this approach is also fully demonstrated by the incorporation of functional groups such as esters, amides, and alkynes into the allyl moiety.
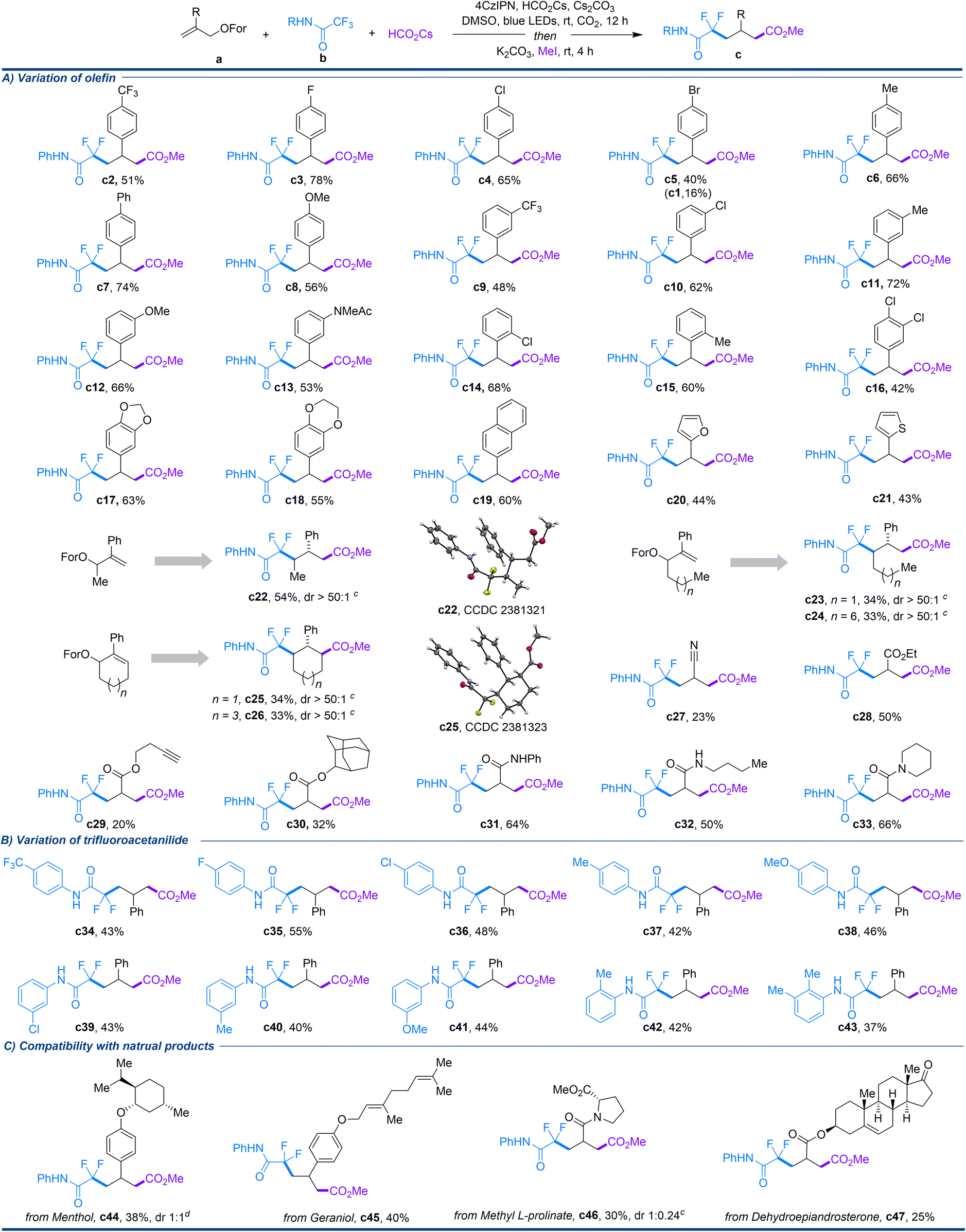 | ||
| Scheme 2 Exploration of substrate scope.a,b aReaction conditions: a (0.2 mmol, 1 eq.), b (4 eq.), HCO2Cs (2 eq.), Cs2CO3(0.5 eq.), 4CzIPN (2 mol%), DMSO (3 mL), CO2 (1 atm), blue LEDs (425 nm, 30 W), rt, 12 h; then MeI (0.3 mL), K2CO3 (2.0 mmol), rt, 4 h. bIsolated yield. cThe diastereomer ratio was determined by 1H NMR. dThe diastereomer ratio was determined by 19F NMR. | ||
Next, the scope of the trifluoroacetamides was tested, as shown in Scheme 2B. N-Phenylamides containing para-substituents with a variety of electronic properties, such as CF3, F, Cl, Me, and MeO, were all suitable for the conversion, providing the corresponding products c34–c38 in moderate yields. In addition, N-phenylamides containing meta-/ortho-substituents were also good partners for this method, as illustrated by c39–c43. To investigate the compatibility of the protocol for complex skeletons, allyl formates containing natural products, including terpenes, an amino acid, and a steroid, were tested in the approach as well. As illustrated in Scheme 2C, the scaffolds of menthol, geraniol, L-proline, and dehydroepiandrosterone were all compatible in the reaction, producing the corresponding products c44–c47 in moderate yields.
To further prove the practicability of this strategy, a gram-scale preparation reaction was conducted, as shown in Scheme 3. Reaction on a 6 mmol scale using a1 proceeded successfully and gave 1.19 g of the product c1 (57% yield). The usefulness of the product was also demonstrated by subsequent derivatization to provide various difluoro-containing key intermediates, as shown in Scheme 3. When c1 was treated with different amounts of LiAlH4 at room temperature or under heating, the gradient reduction of c1 could be easily achieved by generating the full and partial reductive deoxygenation products d and e in 56% and 70% yields, respectively. The latter could be further converted to α,α-difluoro-N-phenylcaprolactam f in 54% yield under Mitsunobu reaction conditions. Hydrolysis of c1 under basic conditions gave α,α-difluoro-adipic acid g in 70% yield, which could be further reduced to α,α-difluorohexanediol h in 61% yield.
 | ||
| Scheme 3 Gram-scale synthesis and follow-up conversions. | ||
To further understand the mechanism of this reaction, control experiments, fluorescence quenching experiments and kinetic experiments were performed, as shown in Scheme 4. When the reaction time was shortened to 4 h under the standard conditions, c1 was obtained in only 8% yield and a1 was recovered in 35% yield, accompanied by the formation of methyl but-3-enoate i and difluoroacetanilide k in 32% and 5% yields, respectively (Scheme 4A-1). In addition, when the aforementioned reaction was treated with HCl as a work-up process, the corresponding but-3-enoic acid i′ was also obtained in 28% yield. By treating ester i or acid i′ with b1 under the standard conditions, the desired product c1 was obtained in 85% yield in both reactions (Scheme 4A-2 and 3). Apparently, but-3-enoate is the key intermediate of the reaction, which is produced by the fast addition of the generated CO2−˙ to allyl formate. In addition, the formation of a tiny amount of k and c1 also implies that the fluoroalkyl radical is slowly produced and preferentially added to the intermediate alkene but-3-enoate rather than to allyl formate. This view is also confirmed by the lack of detection of possible intermediate j. However, when ethyl trifluoroacetate and N,N-disubstituted trifluoro-acetanilide were used instead of b1 for the reaction under standard conditions, they were totally recovered, and only intermediate i was obtained (Schemes 4A-4 and 5). These results suggest that the N–H moiety of b is critical for the reaction, as it is probably involved in the spin-centre shift promoted reductive defluorination of trifluoroacetanilide12 and final intramolecular proton transfer.16 This triple kinetic-controlled reaction is apparently crucial for the efficient unsymmetrical 1,3-difunctionalization, which is also confirmed by the reaction rate curves as illustrated in Scheme 4B. Moreover, fluorescence quenching experiments showed that the excited PC1* could be significantly quenched by cesium formate, but was hardly quenched by a1 and b1 (Scheme 4C). These results not only indicate that the reaction begins from the photoredox of formate by the excited state of PC1 but also verifies that CO2−˙ is produced first in the reaction. Unlike in Yu's strategy, in which formate derived in situ from the reduction of CO2 by sacrificing an equivalent of Ph3SiH acts as a carboxylation reagent,7e formate is used directly as the carboxylation regent as well as the reductant in this study. Although the cyclic voltammogram illustrated in Scheme 4D implies that a1 (E1/2 = −2.13 V vs. SCE) could also be continuously reduced by CO2−˙ (E1/2 = −2.2 V vs. SCE) to an allylic carbanion to further add to CO2 to form the key intermediate but-3-enoate (see ESI† for a possible alternative path), this process is obviously not a significant path, because, as shown in entry 1 of Table 1, the reaction could also occur efficiently without CO2. This result indicates that the formation of but-3-enoate is more likely to involve the addition of CO2−˙ to allylic formate rather than reduction by it. In addition, the obtained reductive potentials of b1 (E1/2 = −2.18 V vs. SCE) and but-3-enoate (E1/2 = −2.30 V vs. SCE) suggest that the former could indeed be reduced by CO2−˙, whereas the reduction of the latter does not seem easy. In addition, DFT (density functional theory) calculation results also confirm that the proposed kinetic-controlled process of the addition of CO2−˙ to allyl formates is barrierless, but its addition to the intermediate but-3-enoate is a very difficult process with an extremely high energy barrier of 57.7 kcal mol−1. In contrast, the addition of a fluoroalkyl radical to but-3-enoate is favorable and only requires crossing an energy barrier of 11.6 kcal mol−1, while its addition to allyl formates is unfavorable and requires crossing an energy barrier of 20.4 kcal mol−1 (see ESI† for details of DFT calculations).
 | ||
| Scheme 4 Mechanistic studies. | ||
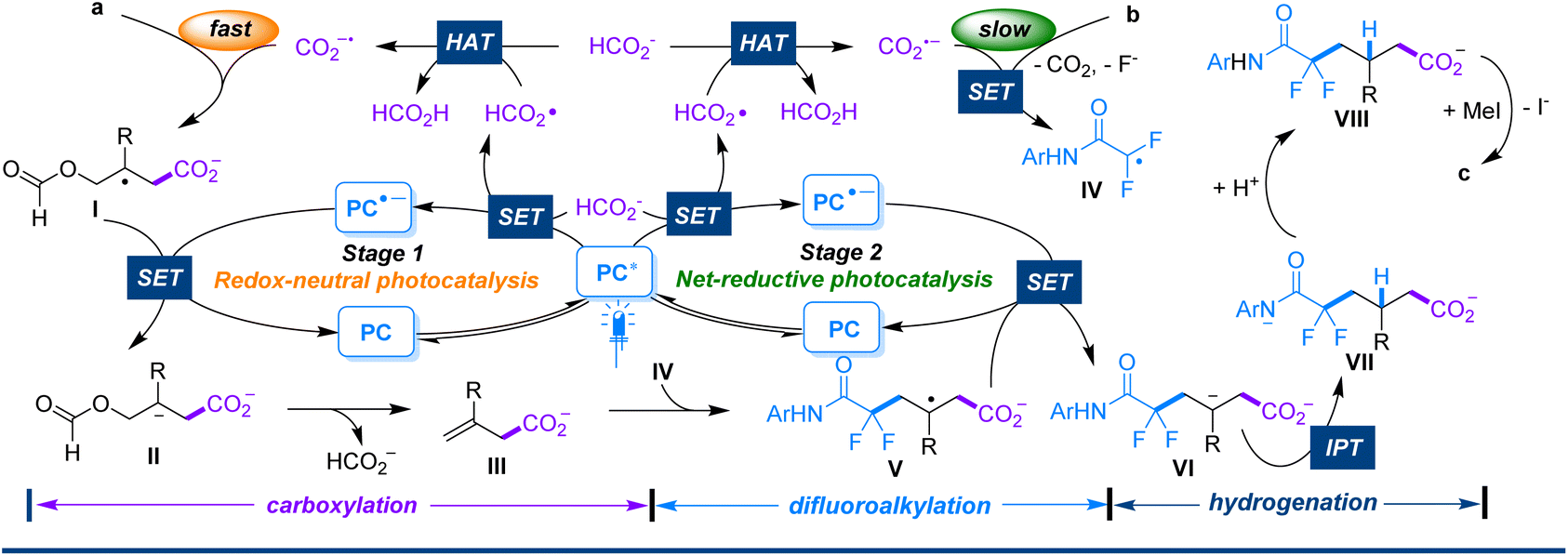 | ||
| Scheme 5 Proposed mechanism. | ||
Based on our experimental observations and previous reports in the literature,16,17,8b a plausible mechanism involving a two-stage photocatalytic cycle is illustrated in Scheme 5. The reaction is initiated by the photooxidation of formate (E1/2 = +0.93 V vs. SCE for HCO2−/HCO2˙)10c by the excited PC1* (E1/2 = +1.35 V vs. SCE for PC*/PC−˙)18 to produce PC−˙ and a formoxyl radical via a single-electron transfer (SET) process. Hydrogen atom transfer (HAT) between the formoxyl radical and formate would give formic acid and CO2−˙. The latter adds rapidly to substrate avia a barrierless process to provide the radical intermediate I. I would be further reduced by PC−˙ to the carbanion intermediate II, which immediately eliminates formate through C–O bond cleavage to yield intermediate III accompanied by the regeneration of PC (stage 1). Conversely, with the consumption of substrate a and accumulation of intermediate III, CO2−˙ begins to reduce substrate b. Through SET reduction and subsequent defluorination-facilitated spin-centre transfer, b is converted to the radical IV. IV tends to add to the key intermediate III to provide radical V, which is further reduced by PC˙− to give the intermediate VI (stage 2). VI immediately undergoes intramolecular proton transfer (IPT)16 from the tethered N–H to the carbanion center to form the intermediate VII, which subsequently abstracts a proton from the surroundings to yield the intermediate VIII. Finally, after treatment with MeI, VIII is methylated to produce the desired product c.
In summary, a transition-metal free protocol for the 1,3-difluoroalkylcarboxylation of alkenes has been established for the first time via a photocatalytic three-component reaction of allyl formates, trifluoroacetanilides, and cesium formate. This approach employs 4CzIPN as a cost-effective organic photocatalyst, formate as a carboxylating reagent, and trifluoroacetanilides as the difluoroalkylating reagent. Consequently, a series of structurally useful difluorinated adipic acid derivatives were efficiently produced. This chemistry represents the first successful example of the unsymmetrical 1,3-difunctionalization of alkenes via a 1e-process as well as the first unsymmetrical extension of the radical AERA strategy. The triple kinetic-controlled radical self-ordering is the crucial key to the success of the protocol, which involves the sequential production of CO2−˙ and difluoromethylaniline radicals and their chemo-/regioselective additions to alkenes. This strategy not only broadens the boundaries of radical AERA strategy but also provides a new tactic for the unsymmetrical 1,3-difunctionalization of olefins. Other 1,3-unsymmetrical difunctionalizations of olefins are being explored in our groups.
Data availability
The ESI† includes all experimental details, including synthesis and characterization of all starting materials and products reported in this study, and mechanistic studies. NMR spectra of all products reported are included as well.Author contributions
B. H. and H. F. conceived and directed the project. H. F., S. L., L. Z., X. W. and H. W. performed experiments. H. F., S. L., L. Z. and X. W. prepared the ESI.† Z. W. performed the DFT calculations and drafted the DFT parts. B. H. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 22471109, 22171118, and 21873041) and the Science and Technology Major Program of Gansu Province of China (22ZD6FA006, 23ZDFA015, and 24ZD13FA017) for financial support.Notes and references
- For selected reviews, see: (a) H.-M. Huang, P. Bellotti, J. Ma, T. Dalton and F. Glorius, Nat. Rev. Chem., 2021, 5, 301–321 CrossRef CAS PubMed; (b) H. Jiang and A. Studer, Chem. Soc. Rev., 2020, 49, 1790–1811 RSC; (c) L. M. Wickham and R. Giri, Acc. Chem. Res., 2021, 54, 3415–3437 CrossRef CAS PubMed; (d) S. Zhu, X. Zhao, H. Li and L. Chu, Chem. Soc. Rev., 2021, 50, 10836–10856 RSC; (e) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC; (f) X. Bao, J. Li, W. Jiang and C. Huo, Synthesis, 2019, 51, 4507–4530 Search PubMed.
- For selected works, see: (a) N. Fu, G. S. Sauer and S. Lin, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef CAS PubMed; (b) Q. Wu, A. Roy, E. Irran, Z.-W. Qu, S. Grimme, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 17307–17311 Search PubMed; (c) B. Tian, P. Chen, X. Leng and G. Liu, Nat. Catal., 2021, 4, 172–179 CrossRef CAS; (d) T. Ju, Y.-Q. Zhou, K.-G. Cao, Q. Fu, J.-H. Ye, G.-Q. Sun, X.-F. Liu, L. Chen, L.-L. Liao and D.-G. Yu, Nat. Catal., 2021, 4, 304–311 CrossRef CAS; (e) L. Liu, M. C. Aguilera, W. Lee, C. R. Youshaw, M. L. Neidig and O. Gutierrez, Science, 2021, 374, 432–439 CrossRef CAS PubMed; (f) X. Hu, I. Cheng-Sánchez, W. Kong, G. A. Molander and C. Nevado, Nat. Catal., 2024, 7, 655–665 CrossRef CAS PubMed; (g) J. Z. Wang, W. L. Lyon and D. W. C. MacMillan, Nature, 2024, 628, 104–109 CrossRef CAS PubMed.
- Our work for difunctionalization of olefins. (a) S.-Q. Lai, B.-Y. Wei, J.-W. Wang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21997–22003 CrossRef CAS PubMed; (b) D. Wei, T. Liu, Y. He, B. Wei, J. Pan, J. Zhang, N. Jiao and B. Han, Angew. Chem., Int. Ed., 2021, 60, 26308–6313 CrossRef CAS PubMed; (c) D.-T. Xie, H.-L. Chen, D. Wei, B.-Y. Wei, Z.-H. Li, J.-W. Zhang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202203398 Search PubMed; (d) S.-P. Hu, C.-H. Gao, T.-M. Liu, B.-Y. Miao, H.-C. Wang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2024, 63, e202400168 CrossRef CAS PubMed.
- (a) R. K. Dhungana, R. R. Sapkota, D. Niroula and R. Giri, Chem. Sci., 2020, 11, 9757–9774 RSC; (b) D.-K. Wang, L. Li, Q. Xu, J. Zhang, H. Zheng and W.-T. Wei, Org. Chem. Front., 2021, 8, 7037–7049 RSC; (c) X. Ma, Q. Zhang and W. Zhang, Molecules, 2023, 28, 3027–3072 CrossRef CAS PubMed.
- (a) H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153–165 CrossRef CAS PubMed; (b) Y. Li, D. Wu, H. Cheng and G. Yin, Angew. Chem., Int. Ed., 2020, 59, 7990–8003 CrossRef CAS PubMed; (c) H.-Y. Tu, S. Zhu, F.-L. Qing and L. Chu, Synthesis, 2020, 52, 1346–1356 CrossRef CAS; (d) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600–607 RSC; (e) Z. Wu, J. Meng, H. Liu, Y. Li, X. Zhang and W. Zhang, Nat. Chem., 2023, 15, 988–997 CrossRef CAS PubMed.
- (a) H. A. Sharma, K. M. Mennie, E. E. Kwan and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 16090–16096 CrossRef CAS PubMed; (b) A. C. Dean, E. H. Randle, A. J. D. Lacey, G. A. M. Giorio, S. Doobary, B. D. Cons and A. J. J. Lennox, Angew. Chem., Int. Ed., 2024, 63, e202404666 CrossRef CAS PubMed.
- (a) K. Jana, A. Bhunia and A. Studer, Chem, 2020, 6, 512–522 CrossRef CAS; (b) R. Liu, Y. Tian, J. Wang, Z. Wang, X. Li, C. Zhao, R. Yao, S. Li, L. Yuan, J. Yang and D. Shi, Sci. Adv., 2022, 8, eabq8596 CrossRef CAS PubMed; (c) H. -Z. Xiao, B. Yu, S. -S. Yan, W. Zhang, X. -X. Li, Y. Bao, S. -P. Luo, J. -H. Ye and D. -G. Yu, Chin. J. Catal., 2023, 50, 222–228 CrossRef CAS; (d) G. Zhao, S. Lim, D. G. Musaev and M.-Y. Ngai, J. Am. Chem. Soc., 2023, 145, 8275–8284 CrossRef CAS PubMed; (e) H.-Z. Xiao, B. Yu, S.-S. Yan, W. Zhang, X.-X. Li, Y. Bao, S.-P. Luo, J.-H. Ye and D.-G. Yu, Chin. J. Catal., 2023, 50, 222–228 CrossRef CAS.
- (a) Z. Yao, G. Li, Y. Zhou, L. Xu and D. Xue, Adv. Synth. Catal., 2023, 365, 224–229 CrossRef CAS; (b) B. Yu, Y. Liu, H.-Z. Xiao, S.-R. Zhang, C.-K. Ran, L. Song, Y.-X. Jiang, C.-F. Li, J.-H. Ye and D.-G. Yu, Chem, 2024, 10, 938–951 CrossRef CAS; (c) X.-Y. Wang, P. Xu, W.-W. Liu, H.-Q. Jiang, S.-L. Zhu, D. Guo and X. Zhu, Sci. China: Chem., 2024, 67, 368–373 CrossRef CAS; (d) H. Xin, L. Zhang, J. Liao, X.-H. Duan, X. Yang and L.-N. Guo, Org. Lett., 2024, 26, 6030–6034 CrossRef CAS PubMed; (e) J.-X. Yu, Y.-Y. Cheng, X.-Y. Zeng, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2024, 26, 6809–6813 CrossRef CAS PubMed; (f) W. Shan, Z. Wang, C. Gao, X. Li, W. Zhuang, R. Liu, C. Shi, H. Qin, X. Li and D. Shi, Green Chem., 2024, 26, 9749–9756 RSC.
- (a) M. Uno, S. Sumino, T. Fukuyama, M. Matsuura, Y. Kuroki, Y. Kishikawa and I. Ryu, J. Org. Chem., 2019, 84, 9330–9338 CrossRef CAS PubMed; (b) H. Li and S. Chiba, Chem Catal., 2022, 2, 1128–1142 CrossRef CAS; (c) E. L. Saux, M. Zanini and P. Melchiorre, J. Am. Chem. Soc., 2022, 144, 1113–1118 CrossRef PubMed.
- (a) J. Majhi and G. A. Molander, Angew. Chem., Int. Ed., 2024, 63, e202311853 CrossRef CAS PubMed; (b) W. Xiao, J. Zhang and J. Wu, ACS Catal., 2023, 13, 15991–16011 CrossRef CAS; (c) Y. Huang, J. Hou, L.-W. Zhan, Q. Zhang, W.-Y. Tang and B.-D. Li, ACS Catal., 2021, 11, 15004–15012 CrossRef CAS; (d) S. N. Alektiar, J. Han, Y. Dang, C. Z. Rubel and Z. K. Wickens, J. Am. Chem. Soc., 2023, 145, 10991–10997 CrossRef CAS PubMed; (e) S. N. Alektiar and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 13022–13028 CrossRef CAS PubMed.
- (a) W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS PubMed; (b) F.-L. Haut, R. S. Mega, J. V. Estornell and R. Martin, Angew. Chem., Int. Ed., 2023, 62, e202304084 CrossRef CAS PubMed; (c) S. Senapati, S. K. Parida, S. S. Karandikar and S. Murarka, Org. Lett., 2023, 25, 7900–7905 Search PubMed.
- (a) Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS PubMed; (b) S.-S. Yan, S.-H. Liu, L. Chen, Z.-Y. Bo, K. Jing, T.-Y. Gao, B. Yu, Y. Lan, S.-P. Luo and D.-G. Yu, Chem, 2021, 7, 3099–3113 Search PubMed; (c) M. W. Campbell, V. C. Polites, S. Patel, J. E. Lipson, J. Majhi and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 19648–19654 Search PubMed; (d) J.-H. Ye, P. Bellotti, C. Heusel and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202115456 CrossRef CAS PubMed.
- (a) Y. Wan and J.-M. Lee, ACS Catal., 2021, 11, 2524–2560 CrossRef CAS; (b) J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 Search PubMed; (c) J. Yang, J. Liu, H. Neumann, R. Franke, R. Jackstell and M. Beller, Science, 2019, 366, 1514–1517 Search PubMed.
- (a) J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 12619–12623 CrossRef CAS PubMed; (b) K. Sun, M. Ueno, K. Imaeda, K. Ueno, M. Sawamura and Y. Shimizu, ACS Catal., 2021, 11, 9722–9728 Search PubMed; (c) Y. Zhou, Y. Qin, Q. Wang, Z. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202110864 CrossRef CAS PubMed; (d) X. Chen, X. Luo and P. Wang, Tetrahedron Lett., 2022, 91, 153646 Search PubMed.
- (a) A. J. Boyington, C. P. Seath, A. M. Zearfoss, Z. Xu and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 4147–4153 CrossRef CAS PubMed; (b) A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS PubMed; (c) C. M. Hendy, G. C. Smith, Z. Xu, T. Lian and N. T. Jui, J. Am. Chem. Soc., 2021, 143, 8987–8992 CrossRef CAS PubMed.
- P. Lei, B. Chen, T. Zhang, Q. Chen, L. Xuan, H. Wang, Q. Yan, W. Wang, J. Zeng and F. Chen, Org. Chem. Front., 2024, 11, 458–465 RSC.
- (a) A. M. Vasquez, J. A. Gurak Jr, C. L. Joe, E. C. Cherney and K. M. Engle, J. Am. Chem. Soc., 2020, 142, 10477–10484 CrossRef CAS PubMed; (b) Z. Wu, S. N. Gockel and K. L. Hull, Nat. Commun., 2021, 12, 5956 Search PubMed; (c) J.-T. Li, G. Fang, G.-Y. Wu and C.-X. Zhuo, ACS Catal., 2023, 13, 12648–12655 CrossRef CAS.
- Y. Liu, X.-L. Chen, X.-Y. Li, S.-S. Zhu, S.-J. Li, Y. Song, L.-B. Qu and B. Yu, J. Am. Chem. Soc., 2021, 143, 964–972 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2381321 and 2381323. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08607d |
| This journal is © The Royal Society of Chemistry 2025 |