
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion–π interaction guided switchable TADF and low-temperature phosphorescence in phosphonium salts for multiplexed anti-counterfeiting†
Jun-Hua Wei,
Yao Xiao,
Jian-Bin Luo,
Zi-Lin He,
Jing-Hua Chen,
Qing-Peng Peng and
Dai-Bin Kuang *
*
Key Laboratory of Bioinorganic and Synthetic Chemistry of Ministry of Education, LIFM, School of Chemistry, IGCME, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: kuangdb@mail.sysu.edu.cn
First published on 17th March 2025
Abstract
Anion–π+ interactions have gained continuous attention in diverse organic aggregates, as they can effectively alter emission behavior. Herein, the anion–π+ interaction is introduced to phosphonium salts, which exhibit tunable thermally activated delayed fluorescence and phosphorescence emission. Intriguingly, the emission spectra evolve from deep-blue to yellow emission by regulation of the anion–π+ interaction strength through varying the anions, such as BF4−, CF3SO3−, PF6−, and NO3, accompanied by adjustable luminescent decay times from milliseconds to several seconds. Notably, bright blue emission with a high photoluminescence quantum yield near 100% is achieved when substituting the iodide ions with larger counter anions. The phosphonium iodide with strong anion–π+ interaction and heavy atom effect shows a high inter-system crossing rate, which inhibits the direct and prompt fluorescence emission. The anion–π+ interaction and twisted structure strongly suppress π–π stacking and afford ultra-high photoluminescence yields. Furthermore, the participation of polar solvent molecules results in the solvation and bathochromic-shift phenomenon of the solid-state phosphonium iodide due to the ionic polarized host–guest structure. This work provides new insights into the anion–π+ interaction in luminescent phosphonium aggregates.
Introduction
Organic luminescent aggregates have attracted tremendous research interest due to their rich structures, remarkable photophysical properties, and abundant element reserves.1–5 These advantages make them widely used in the fields of anti-counterfeiting, biomedicine, cell imaging, light-emitting diodes, and fluorescence sensors. The various weak interactions (e.g. π–π, CH–π, ion–π, and hydrogen bonding) in fluorescent organic aggregates largely influence the emission behavior.6,7 For instance, the weak interaction of CH–π or hydrogen bonding in aggregates inhibits the non-radiative recombination induced by π–π stacking, giving rise to aggregation-induced emission (AIE).8–11 Among the diverse weak interactions, ion–π interactions, including anion–π+ and cation–π− interactions, have gained growing attention in ionic liquids and supramolecular chemistry.12–16Anion–π+ interaction is a category of non-covalent interaction that usually exists between electronegative anions and electron-deficient aromatic systems.17–20 The anion–π+ interaction was first experimentally verified in 1993 and plays a substantial role in the design of ionic fluorescent materials.21 In 2017, Tang and coauthors investigated the role of anion–π+ interaction in inherently charged AIE molecules.22 It was found that the anion–π+ interaction inhibits π–π stacking and actuates room-temperature phosphorescence.23,24 In particular, the anion–π+ interaction exerts a heavy atom effect and promotes the population of triplet states in bromide and iodide salts.
Thermally activated delayed fluorescence (TADF) molecules are an important class of functional optical materials, showing promise in organic light-emitting diodes (OLED), anti-counterfeiting, and scintillators.25–33 TADF emitters require high intersystem (ISC) and reverse intersystem crossing (RISC) rates. Introducing the heavy atom effect through anion–π+ interaction in charged aggregates would be a promising route to construct efficient TADF molecules. However, reports of anion–π+ interaction in TADF emitters are still quite rare.
The field of inherently charged aggregates is limited to electron-deficient protonated nitrogen-containing molecules.22,23,34–37 Though the positive phosphonium core is a strong electron acceptor and meets the requirements in designing host–guest molecules with a small energy gap between S1 and T1 states, the exploration of phosphonium derivatives is rather deficient.38–47 In phosphonium salts with aromatic substituents, the sp3 hybridization of P(V) leads to a twisted molecular framework, which inhibits π–π stacking and promotes a higher radiative recombination rate. Furthermore, the special positions between anions and the electron-deficient π aromatic system connected with P(V) would regulate the strength of anion–π+ interactions and further adjust the emission behavior.48–50 It is also conceivable that the insertion of polar molecules and alteration of counter anions would disturb the anion–π+ interaction of phosphonium salts, providing the feasibility of constructing stimuli-responsive and emission-tunable TADF molecules.
Motivated by the insufficient research into the anion–π+ interaction in TADF phosphonium salts, we aimed to investigate the role of anion species and solvent molecules on the anion–π+ interaction of host–guest phosphonium salts. Herein, we developed a class of host–guest phosphonium salts with dimethylamine-substituted phenyl and phosphonium serving as the electron donor and acceptor, respectively. Spectroscopy and first-principles calculation reveal that the anion–π+ charge transfer contributes to the TADF emission. The anion–π+ interaction applies a heavy atom effect to the iodide product, promoting the population of triplet states. The complexation energies decrease from C3-I to other C3-X with larger anions, leading to the weaker anion–π+ interaction and heavy atom effect. Therefore, the emission behavior of C3-X is highly dependent on the anion species, and ultralong low-temperature phosphorescence is observed when changing the iodide ions to anions with a weaker heavy atom effect. The host–guest phosphonium salts show solvatochromism when exposed to solvent molecules. Leveraging the solid-state solvatochromism and ultra-long low-temperature phosphorescence, the family of C3-X materials was elaborately designed as anti-counterfeiting patterns. This work provides a promising route to design charged molecules with switchable TADF and long-lived phosphorescence by tuning the anion–π+ interactions.
Results and discussion
Synthesis and electronic transition analysis of the iodide salts
The propylated phosphonium iodide (C3-I) was synthesized through a conventional reaction between tertiary phosphine and iodopropane (please see the Experimental section in the ESI† file for details). As displayed in Fig. 1a, C3-I powders exhibit bright blue emission under UV light. 1H and 31P NMR spectra verify the successful synthesis of the target molecules. The organic composition of C3-I was further proved by the elemental analysis results (Table S1†). Single-crystal X-ray diffraction suggests that a new P–C bond is formed in C3-I, where the I− anions distribute among the phosphonium cations (Fig. S1†). The coincidence of the experimental and simulated PXRD patterns certifies the purity of the C3-I powders (Fig. S2†). The phosphonium cations stack in a head-to-tail arrangement, and one phosphonium cation is paired with a counter anion, suggesting abundant weak inter- and intra-molecular interactions. The π–π stacking is suppressed due to the twisted structure of tetrahedral phosphonium.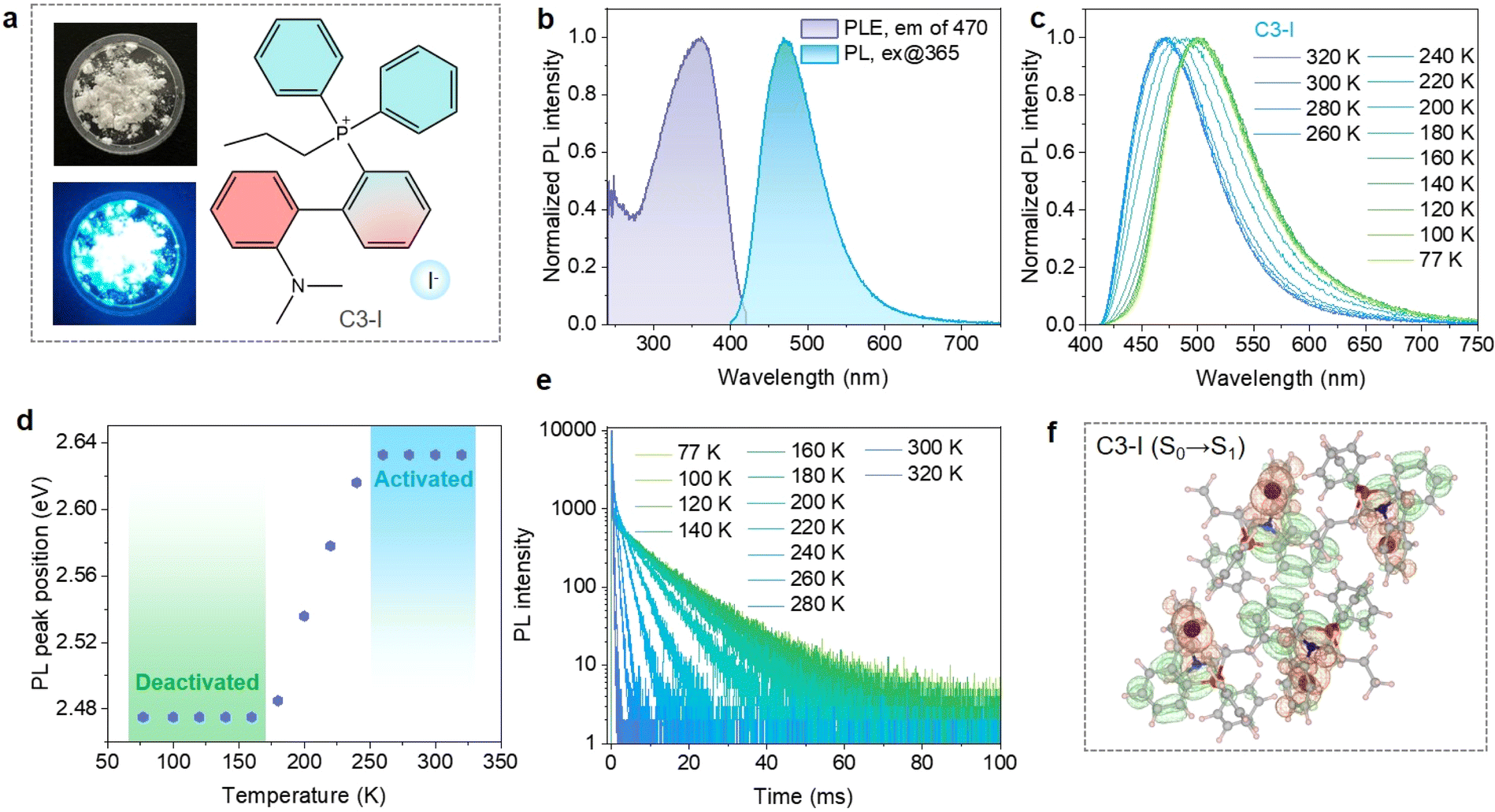 | ||
| Fig. 1 Photophysical properties of C3-I. (a) Photographs of C3-I powders under visible and UV light. (b) PLE and PL spectra of C3-I. (c) Temperature-dependent PL spectra of C3-I. (d) PL peak positions of C3-I versus temperature. (e) Temperature-dependent CIE coordinate diagram TRPL decay curves of C3-I. (f) Charge density difference of S0 → S1 excitation for C3-I. The electron density decreases in the orange area and increases in the green area. | ||
The photophysical properties of C3-I were further investigated. As shown in Fig. 1b, C3-I powders exhibit broad-band blue emission at 470 nm, delivering a high PLQY value of 75.3% when excited at 365 nm. Excitation-wavelength-dependent PL spectra demonstrate that the blue emission originates from the same energy level (Fig. S3†). Temperature-dependent PL spectra show a redshift and broadening trend as the temperature decreases (Fig. 1c). The PL peak position remains unchanged within the temperature range of 77 K to 160 K, which can be ascribed to the spin-forbidden phosphorescence emission from T1 to S0 (Fig. 1d). The triplet excitons can overcome the energy barrier between S1 and T1 states, giving rise to TADF emission from S1 to S0. The emission color transforms from blue to green as the sample is cooled down to 77 K (Fig. S4†), providing one of the encryption channels for designing anti-counterfeiting patterns. The temperature-dependent TRPL decay curves of C3-I display a thermally activated acceleration trend, agreeing well with the TADF mechanism. The TRPL spectrum of C3-I at 300 K gives an overwhelming proportion of TADF (97.34%) compared to prompt fluorescence (2.66%), suggesting that prompt fluorescence is suppressed in C3-I due to the high ISC rate induced by I−.
Time-dependent density functional theory (TDDFT) calculation was performed to reveal the charge transfer dynamics of excited states. As shown in Fig. S5,† the highest occupied molecular orbitals (HOMO) are distributed on the iodide ions and dimethylamine-substituted phenyl fragments, while the lowest unoccupied molecular orbitals are located on the phenyl groups connected with the phosphonium core, showing a spatially separated pattern. Furthermore, the hole–electron analysis demonstrates that the S0 → S1 excitation mainly involves the n–π* and iodide-anion-to-π* transitions (Fig. 1f). Therefore, the intramolecular donor–acceptor interaction within organic cations and intermolecular anion–π+ stacking would play an important role in the electronic transition of solid-state C3-I.
Our group previously synthesized a family of novel phosphonium salts (BzDPP-X) by adopting substituted phosphine and phosphonium as the electron donor and acceptor linked by the polarized π-spacer. The prepared phosphonium bromide and iodide salts exhibit intriguing green TADF emission with much lower PLQY values of 26.9% and 9.8%.26 The higher PLQY for the present C3-I than the previous BzDPP-X is ascribed to the stronger electron-donating ability of dimethylamine and more effective charge transfer in C3-I.
Mechanism investigation of the anion-dependent emission
To investigate the role of the counter anions, we performed ion exchange from parental C3-I and obtained a series of C3-X (X= BF4−, ClO4−, NO3−, CF3SO3−) salts. As displayed in Fig. 2a and b, the obtained C3-X salts show anion-dependent emission under UV light. Intriguingly, C3-BF4 delivers pure blue emission at 458 nm, while C3-NO3 shows green to yellow emission at 507 nm (Fig. 2c and S6a†). Notably, C3-CF3SO3 delivers an ultra-high PLQY value of 99.1% (Fig. S6b†). The TRPL decay curves of C3-X with different anion components (Fig. 2d) can be fitted by double-exponential functions, which can provide more information about the emissive mechanism. The TRPL decay curves suggest that C3-I has the shortest lifetime due to the heavy atom effect of iodide (Fig. 2e). The role of heavy atom effect in the accumulation of triplet excitons in TADF phosphonium salts is consistent with previous reports. For instance, the previous BzDPP-X reported by our group shows anion-dependent emission, where the Cl-based salt is a pure fluorescent material due to the weaker heavy atom effect and low intersystem crossing rate.26 | ||
| Fig. 2 Role of counter anions. (a) Structures of C3-X with different counter anions. (b) Photographs and (c) PL spectra of C3-X under 365 nm UV light. (d) TRPL decay curves of C3-X at 300 K. (e) Long-lived PL lifetimes of C3-X with different anions. | ||
The crystal structure of C3-NO3 could not be obtained due to its poor crystallization ability. Therefore, C3-BF4, C3-I, and C3-CF3SO3 were chosen as the models to investigate the emission mechanism.
SCXRD analyses reveal that the counter anions of BF4− and CF3SO3− successfully substitute the iodide ions (Fig. S7†). Temperature-dependent PL spectra of C3-X were collected to get more in-depth insights into the influence of counter anions. As shown in Fig. 3a and S8,† C3-BF4 exhibits an emission peak at 458 nm that originates from the coexistence of short-lived prompt fluorescence and long-lived TADF emission of the S1 → S0 transition. Intriguingly, as the temperature decreases, a shoulder peak at 516 nm appears, while the intensity of the peak at 458 nm decreases. C3-BF4 shows dual emission at 78 K, and the peak at 516 nm arises from the phosphorescence emission. The TRPL decay curve probed at 516 nm gives an ultra-long average lifetime of 357.18 ms, suggesting the phosphorescence nature at 78 K (Fig. 3b). The phosphorescence spectra of C3-BF4 at 78 K were obtained using the time-gate module (Fig. 3c), which is located at 516 nm with a broadened full width at half maximum (FWHM) compared with 300 K. For C3-CF3SO3, a similarly broadened trend was observed at 77 K (Fig. 3d and S9†). The TRPL decay curve of C3-CF3SO3 at 77 K gives an average lifetime of 995.15 ms, stemming from the phosphorescence emission (Fig. 3e). As displayed in Fig. 3f, the phosphorescence spectrum obtained from the time-gate module shows a broad band profile at 517 nm. Apart from C3-BF4 and C3-CF3SO3, C3-NO3 powders also show green and ultra-long phosphorescence with an average lifetime of 309.76 ms at 77 K (Fig. S10†).
 | ||
| Fig. 3 Electronic transition behavior of C3-X. (a) Temperature-dependent PL spectra of C3-BF4. (b) TRPL decay curve of C3-BF4 at 78 K. (c) The prompt and delayed PL spectra of C3-BF4 at 78 K. (d) Temperature-dependent PL spectra of C3-CF3SO3. (e) TRPL decay curve of C3-CF3SO3 at 77 K. (f) The prompt and delayed PL spectra of C3-CF3SO3 at 77 K. Charge density difference plots of the S0 → S1 excitation in (g) C3-BF4 and (h) C3-CF3SO3. (i) Illustration of the emission mechanism of C3-X. | ||
TDDFT calculations were also performed on C3-BF4 and C3-CF3SO3. It turned out that C3-BF4 and C3-CF3SO3 exhibit larger ΔEST of 0.2240 and 0.2557 eV than that of C3-I (0.1794 eV). The larger ΔEST values are responsible for the lower kRISC of C3-BF4 and C3-CF3SO3 than C3-I (Table S3†). Different from C3-I, the counter anions in C3-BF4 and C3-CF3SO3 are not involved in the HOMO and LUMO orbitals. Hole–electron analyses of C3-BF4 and C3-CF3SO3 reveal that the electron density decreases on the dimethylamine-substituted the phenyl fragment and increases on the phosphonium fragment for the S0 → S1 excitation, which lacks the participation of counter anions (Fig. 3g and h).
Previous reports have highlighted the importance of intermolecular and intramolecular interactions in the anion-dependent emission of donor–acceptor phosphonium salts.43,51 NCI (non-covalent interaction)52 and IGMH (independent gradient model based on Hirshfeld partition of molecular density)53,54 analyses prove the abundant intramolecular and intermolecular interactions in C3-I, C3-BF4, and C3-CF3SO3. The analyses were performed using the Multiwfn package.55,56 As shown in Fig. S11,† NCI analyses for C3-I, C3-BF4, and C3-CF3SO3 indicate there is weak intramolecular interaction between the dimethylamine-substituted phenyl group and phenyl group connected with the phosphonium core. The intramolecular interaction within the host–guest phosphonium cations promotes the electronic transition of n → π*, which is proved by the TDDFT calculation. Furthermore, Fig. S12† presents the IGMH results of C3-I, C3-BF4, and C3-CF3SO3; there exist intermolecular CH⋯π, CH⋯CH, and CH⋯I (or CH⋯F, CH⋯O) of cation–cation and cation–anion interactions. The anion–cation complexation energy of C3-I between the positively charged phosphonium and electronegative iodide anion is calculated as −66.06 kcal mol−1, demonstrating stronger anion–cation interaction than C3-BF4 and C3-CF3SO3 (Fig. S13†). The contribution of iodide ions to the frontier orbitals and effective excited state charge transfer from iodide ions to phosphonium cations reduce the energy gap between S1 and T1 for C3-I, accounting for the higher ISC and RISC rates. The anion–cation complexation energy of C3-BF4 and C3-CF3SO3 between phosphonium and the anions are calculated as −41.54 and −63.42 kcal mol−1. The more compact stacking in C3-CF3SO3 compared with C3-BF4 restricts the motion and non-radiative recombination of organic cations, accounting for the longer decay time of C3-CF3SO3 than C3-BF4.
Based on the above results, the emissive mechanism of C3-X can be described by the model shown in Fig. 3i. The electrons are pumped to the excited singlet states and then relax to the lowest excited singlet state (S1) or undergo an ISC process to form triplet excitons. The direct recombination from S1 to the ground state (S0) gives rise to the short-lived prompt fluorescence. The triplet excitons can overcome the splitting energy between S1 and T1 to populate the S1 state, and then TADF emission with a micro-second lifetime was observed within the activated temperature range. The recombination from T1 to S0 generates ultra-long phosphorescence (afterglow) at low temperatures. The weaker spin–orbital coupling effect and larger ΔEST compared with C3-I lead to the pronounced prompt fluorescence recombination and longer phosphorescence decay time of C3-BF4 and C3-CF3SO3.
Solvation effect in C3-X
To explore the emission behavior in polar solvents, C3-I powders were dissolved in different kinds of solvents. As shown in Fig. 4a, since the dimethylamine-substituted phenyl and the phosphonium fragments afford the host–guest structure, the solutions of C3-I exhibit a strong solvatochromism effect, with green to yellow emission under UV light.57,58 The PL spectrum of the C3-I solution in CH2Cl2 is located at 512 nm and redshifts to 561 nm in water due to the increase of solvent polarity (Fig. 4b, c, and Table S4†). The TRPL decay curve of the CH2Cl2 solution gives an average lifetime of 4.65 ns, lacking a long-lived component (Fig. S14†). It is conceivable that the cation–anion pair dissociates into isolated ions, leading to the disappearance of anion–π stacking. Furthermore, the solvent molecule would bring non-radiative recombination channels to the phosphonium cations and inhibit the population of triplet states, leading to the absence of TADF emission in solution.59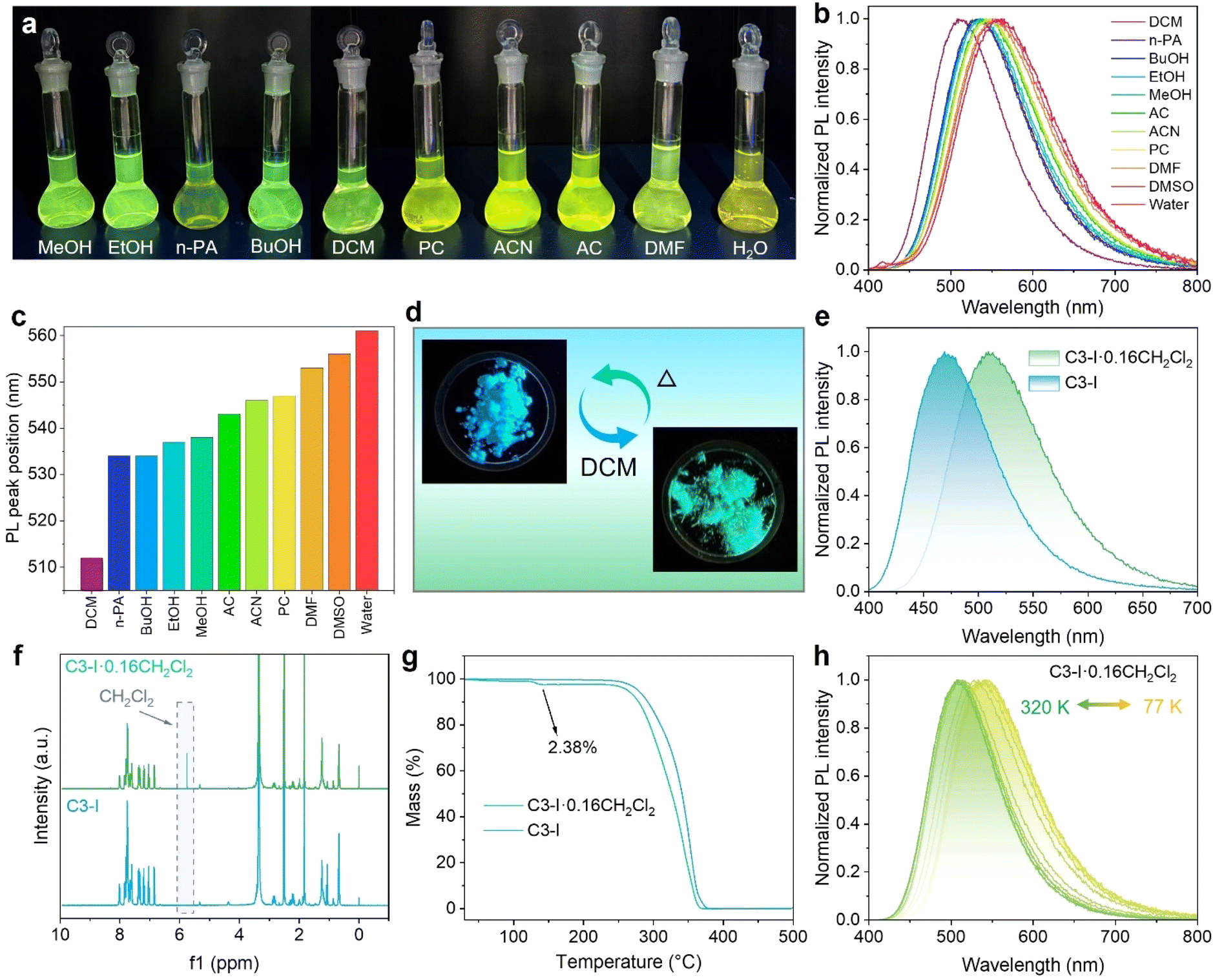 | ||
| Fig. 4 Solid-state and solution-phase solvatochromism of C3-I. (a) Photographs under UV light of C3-I solutions in different polar solvents. (b) PL spectra and (c) PL peak positions of C3-I solutions in different polar solvents. (d) The conversion between C3-I and C3-I containing CH2Cl2 molecules through heating and CH2Cl2 fuming treatment. (e) The PL spectra of C3-I and C3-I·0.16CH2Cl2. (f) TGA plots of C3-I and C3-I·0.16CH2Cl2. (g) 1H NMR spectra of C3-I and C3-I·0.16CH2Cl2. (h) Temperature-dependent PL spectra of C3-I·0.16CH2Cl2. | ||
Meanwhile, C3-CF3SO3, C3-BF4, and C3-NO3 were also dissolved in CH2Cl2, exhibiting green emission under UV light and showing consistent PL profiles (Fig. S15†). The emission of C3-X in solution originates from the phosphonium cations, which are independent of the anions. The dipole–dipole interaction between phosphonium cations and polar solvent reduces the excited singlet state and affords the solvatochromic green emission at room temperature. More information about the solvent effect can be obtained from the solvent-dependent absorption spectra of C3-I. As shown in Fig. S16,† the absorption bands of C3-I are independent of the solvent. The induced structural relaxation of donor–acceptor organic cations results in the rearrangement of solvent molecules. Such phenomena are consistent with the reported donor–acceptor molecules, which possess larger excited-state dipole moments than the ground state.60,61 Intriguingly, the frozen CH2Cl2 solutions of C3-CF3SO3, C3-BF4, C3-NO3, and C3-I exhibited unexpected blue prompt emission and green afterglow when they were cooled to 77 K. As shown in Fig. S17,† the phosphonium cations in the frozen CH2Cl2 solutions of C3-X show coincident blue fluorescence emission and green afterglow at 475 and 535 nm, respectively. The reemerging blue fluorescence emission and ultra-long green phosphorescence emission in the frozen phosphonium cations stem from the suppressed vibration-induced non-radiative process. Besides, the low-temperature phosphorescence of C3-I in different polar solvents was also investigated. As shown in Fig. S18,† blue fluorescence and green emission were also observed in the frozen EtOH and DMSO solutions. Notably, the EtOH solution shows the longest phosphorescence at 77 K (Fig. S19†), originating from the hydrogen bonding of organic cations with EtOH molecules, which further increases the rigidity of organic cations. Therefore, the emission behavior of isolated organic cations in frozen polar solvents resembles that of solid-state salts. The compact packing of the cation–anion pair in the solid state and when frozen inhibit the vibration of the organic moiety, accounting for the population of triplet excitons through ISC.
Considering the solvation response to polar solvents, we attempted to synthesize solid-state materials containing solvent molecules. In detail, C3-I powders were dissolved in CH2Cl2, and then ethyl acetate was added to serve as the anti-solvent. Finally, polycrystals with bright-green emission were precipitated from the mixed solution as the solvents evaporated (Fig. 4d). Fig. 4e demonstrates that the green-emissive polycrystals show a broad-band PL spectrum at 510 nm. As shown in Fig. 4f, NMR analysis verifies the existence of CH2Cl2 molecules. The content of CH2Cl2 is calculated as 2.38% (Fig. 4g), and the ratio of CH2Cl2 to C3-I is derived as 0.16. Temperature-dependent PL spectra were carried out to investigate the emission behavior in solid-state C3-I containing CH2Cl2 molecules. As shown in Fig. 4h, C3-I·0.16CH2Cl2 shows green emission at 300 K, while the emission peak exhibits a redshifted trend and delivers yellow emission at 77 K (Fig. S20a†). Meanwhile, the temperature-dependent TRPL decay curve also shows a thermally activated acceleration trend, which gives an ultra-long lifetime of up to milliseconds at 77 K (Fig. S20b†). These results reveal that both the pure C3-I and C3-I·0.16CH2Cl2 powders exhibit TADF emission behavior, indicating the solvation effect also works in the solid state.62,63
The analogous butylated iodide (C4-I) powders were prepared through a similar reaction between tertiary phosphine and iodobutane.25 C4-I powders exhibit bright sky-blue emission under UV light excitation and give an ultra-high PLQY of nearly 100%. As shown in Fig. S21a,† the steady-state PL spectra of C4-I exhibit a broad emission band at 473 nm and then redshift to 499 nm as the temperature decreases from room temperature to 78 K. The temperature-dependent TRPL spectra present a thermally activated trend, demonstrating the TADF emission mechanism (Fig. S21b†). Furthermore, TDDFT calculation was carried out on C4-I. Fig. S21c† provides the charge density difference (CDD) profile of S0 → S1 excitation for C4-I. The lowest absorption of S0 → S1 involves the charge transfer from I to π* and dimethylamine-substituted phenyl fragment to π*. The photophysical properties and electronic transitions of C3-I and C4-I are similar, where the quaternary phosphonium group is sandwiched by dimethylamine and iodide ion, promoting effective charge transfer. Transparent C4-I single crystals were precipitated from the mixed solutions. C4-I single crystals show bright blue emission under UV light, agreeing with the parental C4-I powders (Fig. S21d†). NMR analysis indicates that the recrystallized C4-I single crystals are devoid of solvent molecules. It is hypothesized that although the structures of C3-I and C4-I are very similar, the small difference in chain length of the substituent groups induces C3-I to recrystallize in the solvent-containing structure.
Construction of the anti-counterfeiting system
Considering the sophisticated luminescence response of C3-X at room temperature and liquid nitrogen temperature, we aimed to construct multiplexed anti-counterfeiting systems. As displayed in Fig. 5a, C3-I and C3-I·0.16CH2Cl2 powders show blue and green emissions at room temperature, while exhibiting green and yellow emissions at 77 K due to the TADF emission mechanism. C3-CF3SO3, C3-BF4, and C3-NO3 feature blue or green emission at room temperature, which also exhibit red-shifted emission at 77 K. Benefiting from the weaker SOC endowed by the loose anion–cation interaction, C3-CF3SO3, C3-BF4, and C3-NO3 exhibit ultra-long green phosphorescence at 77 K, which can even be observed by the naked eye. Notably, C3-CF3SO3 delivers the longest phosphorescence, with a duration time of nearly 10 seconds, showing the feasibility of constructing a lifetime-encrypted anti-counterfeiting system. Fig. 5b demonstrates the operating mechanism of the logic gate. An 8 × 8 dot matrix was built by combining C3-I, C3-I·0.16CH2Cl2, C3-CF3SO3, and C3-BF4 (Fig. 5c). If we denote the green emission dot as “1” and the non-emissive or blue-emission dot as “0”, a series of binary codes can be obtained at different temperatures and delay times (Fig. S22†). The binary codes obtained at room temperature and 77 K give invalid information. The binary code at 77 K with a delay time of 1 s is translated as “WJ_uSYmo.” The real decryption procedure is only available to the authorized staff, and the final code is derived as “SYSUCHEM” (Sun Yat-sen University, Chemistry). A traditional Chinese landscape painting was drawn by using C3-I, C3-CF3SO3, C3-NO3, and C3-BF4 as the pigments (Fig. 5d). The pattern shows dynamic changes at room temperature and 77 K (delay time = 0 s, 1 s, and 10 s). Therefore, by combining the solvation effect in solid state, TADF emission, and anion-controlled low-temperature afterglow, this family of phosphonium salts can be used as multiplexed anti-counterfeiting material. | ||
| Fig. 5 Anti-counterfeiting application. (a) The prompt and afterglow photographs of C3-X at room temperature and liquid nitrogen temperature. (b) The operating mechanism of the logic gate. (c) Anti-counterfeiting patterns at (i) room temperature (UV on), (ii) liquid nitrogen temperature (UV on), and afterglow images at (iii) t = 1 s, (iv) 10 s. (d) The dynamic images of a Chinese landscape painting. | ||
Conclusions
In summary, we successfully prepared a class of host–guest phosphonium salts with bright blue TADF emission. The phosphonium iodide exhibits high ISC and RISC rates due to the heavy atom effect of iodide. The spin–orbital coupling effect can be tuned by changing the anion components, affording a series of phosphonium salts with tunable emission and decay times. The solid-state salts with different anions give ultra-long phosphorescence at low temperatures, allowing the feasibility of designing anti-counterfeiting tags. Investigation of the emission behavior in polar solvents revealed that the anion–π stacking in the solid state plays a vital role in the population of the triplet state. Intriguingly, a solid-state phosphonium iodide containing CH2Cl2 molecules was prepared, which shows solvatochromism and TADF emission in the solid state. With the merits of temperature-dependent TADF emission, solvatochromism effect, and tunable phosphorescence, C3-X and C3-I·0.16 CH2Cl2 were elaborately designed as an anti-counterfeiting system with a high security level.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for C3-I, C3-BF4, and C3-CF3SO3 have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under 2409659, 2409827, and 2409828, and can be obtained from http://www.ccdc.cam.ac.uk/structures/.Author contributions
D.-B. K. and J.-H. W. conceived and designed the experiments. J.-H. W. and Y. X. carried out material synthesis, measurements, and data analysis. J.-B. L., Z.-L. H., Q.-P. P., and J.-H. C. assisted with the DFT calculations and crystal structure analysis. All authors participated in discussing the data and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (22375220, 22405298), the Fundamental Research Funds for the Central Universities (24qnpy052), and the China Postdoctoral Science Foundation (2023M743980).Notes and references
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar, Chem. Rev., 2015, 115, 11718–11940 CAS.
- Q. Xia, Y. Zhang, Y. Li, Y. Li, Y. Li, Z. Feng, X. Fan, J. Qian and H. Lin, A historical review of aggregation-induced emission from 2001 to 2020: A bibliometric analysis, Aggregate, 2022, 3, e152 CrossRef CAS.
- H. Sun, S. Shen, C. Li, W. Yu, Q. Xie, D. Wu and L. Zhu, Stimuli-Responsive Dual-Emission Property of Single-Luminophore-Based Materials, Adv. Funct. Mater., 2024, 35, 2415400 Search PubMed.
- H. Sun, Z. Yu, C. Li, M. Zhang, S. Shen, M. Li, M. Liu, Z. Li, D. Wu and L. Zhu, Single-Luminophore Molecular Engineering for Rapidly Phototunable Solid-State Luminescence, Angew. Chem., Int. Ed., 2025, 64, e202413827 Search PubMed.
- X. Ma, M. Zhou, L. Jia, G. Ling, J. Li, W. Huang and D. Wu, High-contrast reversible multiple color-tunable solid luminescent ionic polymers for dynamic multilevel anti-counterfeiting, Mater. Horiz., 2023, 10, 107–121 RSC.
- Y. Shen, B. Wang, P. Wang, Y. Chen, Z. Xu, W. Huang and D. Wu, Achieving Dual Emission of Fluorescence and Phosphorescence from Anti-Kasha’s Metal–Organic Halides for Information Encryption, Inorg. Chem., 2024, 63, 12073–12080 CrossRef CAS.
- P. Wang, H. Miao, K. Sheng, B. Wang, F. Feng, X. Cai, W. Huang and D. Wu, Efficient blue-light-excitable copper(I) coordination network phosphors for high-performance white LEDs, Chin. Chem. Lett., 2024, 35, 108600 Search PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole, Chem. Commun., 2001, 18, 1740–1741 RSC.
- S. Ma, S. Du, G. Pan, S. Dai, B. Xu and W. Tian, Organic molecular aggregates: From aggregation structure to emission property, Aggregate, 2021, 2, e96 CrossRef CAS.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: New Vistas at the Aggregate Level, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Aggregation-induced emission, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- L. Liu, C. Li, J. Gong, Y. Zhang, W. Ji, L. Feng, G. Jiang, J. Wang and B. Z. Tang, A Highly Water-Soluble Aggregation-Induced Emission Luminogen with Anion-π+ Interactions for Targeted NIR Imaging of Cancer Cells and Type I Photodynamic Therapy, Angew. Chem., Int. Ed., 2023, 62, e202307776 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, The Cation−π Interaction, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Cation−π Interaction: Its Role and Relevance in Chemistry, Biology, and Material Science, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- D. A. Dougherty, The Cation−π Interaction, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- D.-X. Wang and M.-X. Wang, Exploring Anion−π Interactions and Their Applications in Supramolecular Chemistry, Acc. Chem. Res., 2020, 53, 1364–1380 CrossRef CAS PubMed.
- M. Giese, M. Albrecht and K. Rissanen, Anion−π Interactions with Fluoroarenes, Chem. Rev., 2015, 115, 8867–8895 CrossRef CAS PubMed.
- H. T. Chifotides and K. R. Dunbar, Anion−π Interactions in Supramolecular Architectures, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed.
- P. Molina, F. Zapata and A. Caballero, Anion Recognition Strategies Based on Combined Noncovalent Interactions, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Applications of Supramolecular Anion Recognition, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS.
- H.-J. Schneider, F. Werner and T. Blatter, Attractive Interactions Between Negative Charges and Polarizable Aryl Parts of Host–Guest Systems, J. Phys. Org. Chem., 1993, 6, 590–594 CrossRef CAS.
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Ionization and Anion−π+ Interaction: A New Strategy for Structural Design of Aggregation-Induced Emission Luminogens, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, A Facile Strategy for Realizing Room Temperature Phosphorescence and Single Molecule White Light Emission, Nat. Commun., 2018, 9, 2963 Search PubMed.
- S. Garain, S. M. Wagalgave, A. A. Kongasseri, B. C. Garain, S. N. Ansari, G. Sardar, D. Kabra, S. K. Pati and S. J. George, Anion−π-Induced Room Temperature Phosphorescence from Emissive Charge-Transfer States, J. Am. Chem. Soc., 2022, 144, 10854–10861 CrossRef CAS PubMed.
- J. H. Wei, J. B. Luo, Z. L. He, Q. P. Peng, J. H. Chen, Z. Z. Zhang, X. X. Guo and D. B. Kuang, Phosphonium Iodide Featuring Blue Thermally Activated Delayed Fluorescence for Highly Efficient X-Ray Scintillator, Angew. Chem., Int. Ed., 2024, 63, e202410514 CrossRef CAS PubMed.
- J. H. Wei, J. B. Luo, Z. L. He, Z. Z. Zhang and D. B. Kuang, A New Class of Donor-π-Acceptor Phosphonium Salt Exhibiting Intriguing Thermally Activated Delayed Fluorescence, Adv. Opt. Mater., 2023, 11, 2300328 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Thermally Activated Delayed Fluorescence Materials Towards the Breakthrough of Organoelectronics, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- W. Ma, Y. Su, Q. Zhang, C. Deng, L. Pasquali, W. Zhu, Y. Tian, P. Ran, Z. Chen, G. Yang, G. Liang, T. Liu, H. Zhu, P. Huang, H. Zhong, K. Wang, S. Peng, J. Xia, H. Liu, X. Liu and Y. M. Yang, Thermally activated delayed fluorescence (TADF) organic molecules for efficient X-ray scintillation and imaging, Nat. Mater., 2022, 21, 210–216 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Efficient Blue Organic Light-Emitting Diodes Employing Thermally Activated Delayed Fluorescence, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- W. Yang, C. Xie, T. Chen, X. Yin, Q. Lin, S. Gong, Z. Quan and C. Yang, Dynamic Reversible Full-Color Piezochromic Fluorogens Featuring Through-Space Charge-Transfer Thermally Activated Delayed Fluorescence and their Application as X-Ray Imaging Scintillators, Angew. Chem., Int. Ed., 2024, 63, e202402704 CrossRef CAS PubMed.
- W.-F. Wang, M.-J. Xie, P.-K. Wang, J. Lu, B.-Y. Li, M.-S. Wang, S.-H. Wang, F.-K. Zheng and G.-C. Guo, Thermally Activated Delayed Fluorescence (TADF)-active Coinage-metal Sulfide Clusters for High-resolution X-ray Imaging, Angew. Chem., Int. Ed., 2024, 63, e202318026 CrossRef CAS PubMed.
- K. Han and Z. Xia, Coinage Metal Cluster Scintillator for X-ray Imaging, ACS Cent. Sci., 2023, 9, 1263–1265 CrossRef CAS PubMed.
- T.-T. Wang, G. Xie, H.-C. Li, S.-Y. Yang, H. Li, Y.-L. Xiao, C. Zhong, K. Sarvendra, A. Khan, Z.-Q. Jiang and L.-S. Liao, π-Stacked Thermally Activated Delayed Fluorescence Emitters with Alkyl Chain Modulation, CCS Chem., 2020, 3, 1757–1763 CrossRef.
- X. Liu, C. Zhu and B. Z. Tang, Bringing Inherent Charges into Aggregation-Induced Emission Research, Acc. Chem. Res., 2022, 55, 197–208 CrossRef CAS PubMed.
- Y. Hu, L. Barbier, Z. Li, X. Ji, H. Le Blay, D. Hourdet, N. Sanson, J. W. Y. Lam, A. Marcellan and B. Z. Tang, Hydrophilicity-Hydrophobicity Transformation, Thermoresponsive Morphomechanics, and Crack Multifurcation Revealed by AIEgens in Mechanically Strong Hydrogels, Adv. Mater., 2021, 33, 2101500 CrossRef CAS PubMed.
- J. H. Wei, J. F. Liao, L. Zhou, J. B. Luo, X. D. Wang and D. B. Kuang, Indium-Antimony-Halide Single Crystals for High-Efficiency White-Light Emission and Anti-Counterfeiting, Sci. Adv., 2021, 7, eabg3989 CrossRef CAS PubMed.
- J.-H. Wei, W.-T. Ou, J.-B. Luo and D.-B. Kuang, Zero-Dimensional Zn-Based Halides with Ultra-Long Room-Temperature Phosphorescence for Time-Resolved Anti-Counterfeiting, Angew. Chem., Int. Ed., 2022, 61, e202207985 CrossRef CAS PubMed.
- X.-L. Chen, X.-D. Tao, Y.-S. Wang, Z. Wei, L. Meng, D.-H. Zhang, F.-L. Lin and C.-Z. Lu, Phosphonium-Based Ionic Thermally Activated Delayed Fluorescence Emitters for High-Performance Partially Solution-Processed Organic Light-Emitting Diodes, CCS Chem., 2022, 5, 589–597 CrossRef.
- A. Belyaev, B.-K. Su, Y.-H. Cheng, Z.-Y. Liu, N. M. Khan, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, Multiple Emission of Phosphonium Fluorophores Harnessed by the Pathways of Photoinduced Counterion Migration, Angew. Chem., Int. Ed., 2022, 61, e202115690 CrossRef CAS.
- I. Partanen, O. Al-Saedy, T. Eskelinen, A. J. Karttunen, J. J. Saarinen, O. Mrózek, A. Steffen, A. Belyaev, P.-T. Chou and I. O. Koshevoy, Fast and Tunable Phosphorescence from Organic Ionic Crystals, Angew. Chem., Int. Ed., 2023, 62, e202305108 CrossRef CAS PubMed.
- A. Belyaev, Y.-H. Cheng, Z.-Y. Liu, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, A Facile Molecular Machine: Optically Triggered Counterion Migration by Charge Transfer of Linear Donor-π-Acceptor Phosphonium Fluorophores, Angew. Chem., Int. Ed., 2019, 58, 13456–13465 CrossRef CAS PubMed.
- H.-T. Qu, I. Partanen, K.-H. Chang, Y.-D. Lin, I. O. Koshevoy, A. Belyaev and P.-T. Chou, Insights into the Photoinduced Anion Translocation of Donor–π–Acceptor+ (ion)− Molecules, Chem. Sci., 2024, 15, 20045–20055 RSC.
- Y.-S. Wang, X. Lu, J.-H. Song, X. Li, X.-D. Tao, L. Meng, X.-L. Chen and C.-Z. Lu, Highly Efficient Ionic Thermally Activated Delayed Fluorescence Emitters with Short Exciton Lifetimes Towards High-Performance Solution-Processed OLEDs, Chem. Eng. J., 2024, 482, 148865 CrossRef CAS.
- A. W. K. Law, T. S. Cheung, J. Zhang, N. L. C. Leung, R. T. K. Kwok, Z. Zhao, H. H. Y. Sung, I. D. Williams, Z. Qiu, P. Alam, J. W. Y. Lam and B. Z. Tang, Sergeant-and-Soldier Effect in an Organic Room-Temperature Phosphorescent Host-Guest System, Adv. Mater., 2024, 36, 2410739 CrossRef CAS PubMed.
- G. Zhou, Y. Mao, J. Zhang, Q. Ren, M. S. Molokeev, Z. Xia and X.-M. Zhang, Dynamic Phosphorescence/Fluorescence Switching in Hybrid Metal Halides Toward Time-Resolved Multi-Level Anti-Counterfeiting, Adv. Funct. Mater., 2024, 35, 2413524 CrossRef.
- J.-B. Luo, J.-H. Wei, Z.-L. He, J.-H. Chen, Q.-P. Peng, Z.-Z. Zhang and D.-B. Kuang, Bisphosphonium cation based metal halide glass scintillators with tunable melting points, Chem. Sci., 2024, 15, 16338–16346 RSC.
- H. Li, X. Li, H. Su, S. Zhang, C. Tan, C. Chen, X. Zhang, J. Huang, J. Gu, H. Li, G. Xie, H. Dong, R. Chen and Y. Tao, Highly Stable Color-Tunable Organic Long-Persistent Luminescence from a Single-Component Exciplex Copolymer for in Vitro Antibacterial, Chem. Sci., 2024, 15, 14880–14887 RSC.
- Y.-H. Liu, T.-Y. Yan, M.-H. Dong, F.-J. Yu, H. Cao, L. Xiao, Y.-F. Han, X.-W. Kong and X.-W. Lei, Hybrid Metal Halide Family with Color-Time-Dual-Resolved Phosphorescence for Multiplexed Information Security Applications, J. Colloid Interface Sci., 2025, 678, 141–151 CrossRef CAS PubMed.
- T. F. Manny, T. B. Shonde, H. Liu, M. S. Islam, O. J. Olasupo, J. Viera, S. Moslemi, M. Khizr, C. Chung, J. S. R. V. Winfred, L. M. Stand, E. F. Hilinski, J. Schlenoff and B. Ma, Efficient X-Ray Scintillators Based on Facile Solution Processed 0D Organic Manganese Bromide Hybrid Films, Adv. Funct. Mater., 2025, 35, 2413755 CrossRef CAS.
- A. A. Karluk, S. Thomas, A. Shkurenko, B. E. Hasanov, J. Mahmood, M. Eddaoudi and C. T. Yavuz, Single Crystals of Organometallic Manganese Halides as Sustainable High-Luminescence Materials for X-ray Scintillation, J. Mater. Chem. C, 2025, 13, 2165–2171 RSC.
- Y.-S. Wang, T. Zhao, J.-H. Song, X.-D. Tao, D.-H. Zhang, L. Meng, X.-L. Chen and C.-Z. Lu, Realizing high-performance bluish-green ionic thermally activated delayed fluorescence materials through counterion regulation, Chem. Eng. J., 2023, 460, 141836 CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CAS.
- T. Lu and Q. Chen, Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems, J. Comput. Chem., 2022, 43, 539–555 CAS.
- C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 CAS.
- T. Lu, A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn, J. Chem. Phys., 2024, 161, 082503 CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CAS.
- H. Deng, Z. Guo, Y. Wang, K. Li, Q. Zhou, C. Ge, Z. Xu, S. Sato, X. Ma and Z. Sun, Modular synthesis, host–guest complexation and solvation-controlled relaxation of nanohoops with donor–acceptor structures, Chem. Sci., 2022, 13, 14080–14089 CAS.
- K. Isayama, N. Aizawa, J. Y. Kim and T. Yasuda, Modulating Photo- and Electroluminescence in a Stimuli-Responsive π-Conjugated Donor–Acceptor Molecular System, Angew. Chem., Int. Ed., 2018, 57, 11982–11986 CAS.
- W. Zhang, J. Kong, D. Hu, M. Tao, X. Niu, S. Vdović, D. Aumiler, Y. Ma and A. Xia, Solvation-Dependent Excited-State Dynamics of Donor–Acceptor Molecules with Hybridized Local and Charge Transfer Character, J. Phys. Chem. C, 2020, 124, 5574–5582 CrossRef CAS.
- B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, Direct Visualization of Excited-State Symmetry Breaking Using Ultrafast Time-Resolved Infrared Spectroscopy, J. Am. Chem. Soc., 2016, 138, 4643–4649 CrossRef CAS PubMed.
- T. Kim, J. Kim, H. Mori, S. Park, M. Lim, A. Osuka and D. Kim, Symmetry-breaking charge transfer in the excited state of directly linked push–pull porphyrin arrays, Phys. Chem. Chem. Phys., 2017, 19, 13970–13977 RSC.
- B. L. Cotts, D. G. McCarthy, R. Noriega, S. B. Penwell, M. Delor, D. D. Devore, S. Mukhopadhyay, T. S. De Vries and N. S. Ginsberg, Tuning Thermally Activated Delayed Fluorescence Emitter Photophysics through Solvation in the Solid State, ACS Energy Lett., 2017, 2, 1526–1533 CrossRef CAS.
- T. Northey, J. Stacey and T. J. Penfold, The role of solid state solvation on the charge transfer state of a thermally activated delayed fluorescence emitter, J. Mater. Chem. C, 2017, 5, 11001–11009 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2409659, 2409827 and 2409828. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00522a |
| This journal is © The Royal Society of Chemistry 2025 |