
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStatus and outlook of solid electrolyte membrane reactors for energy, chemical, and environmental applications
Liangdong
Fan
 *a,
Wanying
Luo
a,
Qixun
Fan
a,
Qicheng
Hu
a,
Yifu
Jing
b,
Te-Wei
Chiu
*c and
Peter D.
Lund
*d
*a,
Wanying
Luo
a,
Qixun
Fan
a,
Qicheng
Hu
a,
Yifu
Jing
b,
Te-Wei
Chiu
*c and
Peter D.
Lund
*d
aShenzhen Key Laboratory of New Lithium-ion Batteries and Mesoporous Materials, Department of New Energy Science and Technology, College of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518060, Guangdong, China. E-mail: fanld@szu.edu.cn
bDepartment of Materials Science, Shenzhen MSU-BIT University, Shenzhen 517182, Guangdong, China
cDepartment of Materials and Mineral Resources Engineering, National Taipei University of Technology, Taipei, Taiwan. E-mail: tewei@ntut.edu.tw
dNew Energy Technologies Group, Department of Applied Physics, Aalto University School of Science, FI-00076 Aalto, Finland. E-mail: peter.lund@aalto.fi
First published on 17th February 2025
Abstract
Solid electrolyte membrane reactors (SEMRs) can be operated at high temperatures with distinct reaction kinetics, or at lower temperatures (300–500 °C) for industrially relevant energy applications (such as solid oxide fuel/electrolysis cells, direct carbon fuel cells, and metal–air batteries), chemical (such as alkane dehydrogenation, C–C coupling, and NH3 synthesis), environmental (De-NOx, CO2 utilization, and separation), as well as their combined (one-step coupled CO2/H2O co-electrolysis and methanation reaction, power and chemical cogeneration) applications. SEMRs can efficiently integrate electrical, chemical, and thermal energy sectors, thereby circumventing thermodynamic constraints and production separation issues. They offer a promising way to achieve carbon neutrality and improve chemical manufacturing processes. This review thoroughly examines SEMRs utilizing various ionic conductors, namely O2−, H+, and hybrid types, with operations in different reactor/cell architectures (such as panel, tubular, single chamber, and porous electrolytes). The reactors operate in various modes including pumping, extraction, reversible, or electrical promoting modes, providing multiple functionalities. The discussion extends to examining critical materials for solid-state cells and catalysts essential for specific technologically important reactions, focusing on electrochemical performance, conversion efficiency, and selectivity. The review also serves as a first attempt to address the potential of process-intensified SEMRs through the integration of photo/solar, thermoelectric, and plasma energy and explores the unique phenomenon of electrochemical promotion of catalysis (EPOC) in membrane reactors. The ultimate goal is to offer insight into ongoing critical scientific and technical challenges like durability and operational cost hindering the widespread industrial implementation of SEMRs while exploring the opportunities in this rapidly growing research domain. Although still in an early stage with limited demonstrations and applications, advances in materials, catalysis science, solid-state ionics, and reactor design, as well as process intensification and/or system integration will fill the gaps in current high temperature operation of SEMRs and industrially relevant applications like sustainable clean chemical production, efficient energy conversion/storage, as well as environmental enhancement.
 Liangdong Fan | Dr Liangdong Fan is a Research Professor in the Department of New Energy Science and Technology, College of Chemistry and Environmental Engineering, Shenzhen University, China. He received two PhD degrees, one in Energy Technology from the Royal Institute of Technology (KTH, Sweden) in 2014 and another in Chemical Technology from Tianjin University (China) in 2012. He was a postdoctoral Research Fellow at Nanyang Technological University (2014.06–2015.11). At the end of 2015, Dr Fan joined Shenzhen University as a Lecturer/Research Associate Professor (Principal Investigator) and was promoted to Research Professor in Sept. 2023. His current research interests are developing inorganic solid materials for high temperature solid oxide cells for small molecule conversion into sustainable fuel, chemicals, and energy. He has published more than 100 works including in Nature Catalysis, Energy & Environmental Science, Chemical Science and Electrochemical Energy Reviews with more than 6600 times overall citations and an H index of 47. |
 Te-Wei Chiu | Prof. Te-Wei Chiu is presently working as a distinguished professor and department head at the Department of Material and Mineral Resources Engineering, National Taipei University of Technology, Taiwan. He received a BS in chemistry in 1995 from National Cheng-Kong University and an MS in chemistry in 1997 from National Chong-Chen University, Taiwan. He obtained a PhD degree in Materials Science Engineering from the Tokyo Institute of Technology, Japan in 2003. Later, he worked as a post-doctoral researcher at Tsukuba Research Centre for Interdisciplinary Materials Science, Tsukuba University (2003–2005). He worked at the National Institute of Advanced Industrial and Science Technology (AIST), Japan from 2005 to 2008. His areas of research interest include the application of nanomaterials and functional oxides for energy technology such as the synthesis of nanoparticles (0D), and fiber (1D), thin film (2D), porous (3D), and tubular architectures for environmental and energy device applications. He has published more than 170 research articles in international journals. |
 Peter D. Lund | Peter D. Lund is Professor in Advanced Energy Systems at Aalto University with past visiting positions in Germany and China. His main interest is in sustainable energy, including different nanomaterial systems. He is active in senior roles with EU energy initiatives: he chaired the Advisory Group Energy of European Commission and Energy Steering Panel of European Academies Science Advisory Council, and co-chaired the European Academies' Science Advice for Policy on the Energy Transition. He is past vice-chair of the Finnish Climate Panel, member of the Swedish Engineering Academy in Finland and the Finnish Academy of Science and Letters. He is Honorary Professor at Southeast University (Nanjing) and received the prestigious China Friendship Award in 2021. He is Editor-in-Chief of Oxford Open Energy and co-EIC of Wiley Interdisciplinary Reviews on Energy & Environment. |
1. Introduction
Environmental issues such as carbon emissions from fossil fuel-based energy production necessitate efficient energy conversion and use of clean energy sources. Intelligent process innovations will be important to achieve these goals. Electrification through ‘green’ electricity from renewable sources (e.g., wind and solar) linked to electrochemical processes is becoming economically attractive with a major potential to lower key greenhouse gas CO2 and CH4 emissions and even convert them to valuable chemicals and fuels.1–3 Likewise, direct conversion of carbon-based fuel into electricity employing the electrochemical reaction path can lead to reduced CO2 emissions.4 In parallel, chemical synthesis can also more smartly supply reaction energy through electrochemistry using ‘green electrons’.5–7 In some cases, this has been prompted by new regulations that encourage electricity and electrode/catalyst use. For example, the emerging power-to-X conversion relies mostly on renewable power, where the X stands for H2, CO, NH3, CH4, CH3OH, C2H4, etc., or, liquid fuels such as methanol and ethanol from abundant small molecules, such as H2O, CO2, N2, and CH4, and also depends on the choice of reaction and reactor operation as well as their thermodynamics.8–12 Farr and Vayenas first realized this concept in 1980 by producing NO from NH3 feedstock in a solid oxide fuel cell (SOFC) using a noble-metal catalyst.13 Electrification as such has already been applied in chemical industries, e.g., in chloralkali industries, and similar success stories could be envisioned assuming that cheap renewable power would be abundantly available in the future.5,6,14To realize both efficient energy conversion and storage, reliable electrochemical technology is needed, in which a tailored combination and coupling of consumption and production of electrical, chemical, and thermal energy is preferred.15 Moreover, these technologies should be capable of directly coupling electrical and chemical processes to make their industrial use attractive. Furthermore, other benefits of the electrochemical process are as follows.5,6
(i) Avoiding the reverse reaction balance limitation since the reactants are fed separately, not competing for the same catalytic sites, and have no thermodynamic limitation, which leads to efficient usage of reactants since they can be recirculated.
(ii) Electrochemical cells often result in natural product separation due to the use of a membrane that keeps reactants and products generated at the cathode and anode separately. For example, in water electrolysis, the reaction products H2 and O2 already appear at the opposite electrode sides of the membrane, and one of the products may be mixed with steam but it can be easily purified/recovered through condensation.
(iii) Electrochemical methods enable bond-formation steps using electricity, which offers an alternative/controllable and safe driving force namely voltage that can enable operation at lower temperature and pressure out of reach by the conventional catalytic process such as the thermal catalysis process.
(iv) Through intentional specific potential adjustment, the selectivity and conversion rate of the reaction are well controllable when combined with a proper choice of electrode catalyst.
Among the electrification technologies, fuel cells, and their reverse process, electrolysis cells, possess a unique capability of reversible operation, i.e., directly converting the chemical energy in fuels such as hydrogen and hydrocarbons to electricity, or in the electrolysis mode storing the energy from renewable electricity in chemicals, characterizing by high efficiency and large-scale capability since no intermediate process such as combustion/mechanical conversion process is involved. Among the different types of fuel cells, the solid oxide cell (SOC) operates at elevated temperatures as an all-solid-state cell showing the highest priority for such a purpose, also because of fuel flexibility.16,17 In the all-solid-state form, there is no need for liquid cell materials to avoid corrosion and easy separation on the opposite electrode. Elevated temperature operation also brings multiple advantages vs. low temperature reaction:9,18 (i) significantly improved reaction kinetics by improving exchange current density (j0) at a high temperature which is essential for a high reaction conversion rate, leading to considerably improved technological electrical/energy efficiency; the total energy efficiency can be further improved to use highly valuable heat energy under fuel cell conditions and joule heat in the electrolysis mode; (ii) much reduced reliance on rare precious metal catalysts/active components; (iii) fuel flexibility, avoiding highly pure hydrogen needed in polymer exchange membrane fuel cells under close to room temperature operational conditions; a flexible bridge from current fossil fuel to future hydrogen economy can therefore be expected; (iv) reduced electrical energy consumption under solid oxide electrolysis operational conditions; moreover, by utilizing waste heat from different sources, the electrolysis process can be tuned into a chemical-electricity co-generation mode, significantly enhancing the energy utilization efficiency and realizing alternative purposes; (v) achieving homogeneous distribution of the reactant in the form of ions such as O2− and H+ from the electrolyte lattice instead of O2 or H2 molecules direct contacting to the catalytic reaction under ideal conditions (such as temperature), debarring undesired side reactions such as over-oxidation of CH4 activation products, improving the reaction selectivity and yields, holding economic and environmental significance; (vi) the same device can be reversibly operated in the fuel cell or electrolysis cell mode, which enables quick deployment of current joint fossil fuel and renewable power sources.
2. Fundamentals of SEMRs
Solid electrolyte membrane reactor (SEMR) development has advanced considerably in recent years and continues to progress rapidly due to the abovementioned characteristics.19 A general diagram of a SEMR is shown in Fig. 1. The reactor has two chambers that are separated by a gas-tight membrane in most cases, but sometimes a porous membrane is also applied. The membrane is made of a solid oxide material that is capable of transporting O2− or H+ at elevated temperatures (typically 873–1273 K but extended to 573–1273 K with the evolution of cell materials and fabrication technology). The SOC can be operated in either SOFC or SOEC mode, depending on whether a current/potential is applied or not or it is operated using various ionic conductors like alkali ions and CO32−etc.20,21 In fuel cell mode, fuel conversion takes place simultaneously with the power generation, whereas in the electrolysis mode, additional energy like electricity is supplied to the system to sustain nonspontaneous reactions. In other words, the same unit can make fuel or value-added chemicals through electrolysis during periods of excess power production like abundant solar energy storage in the form of electricity during the daytime, and then convert the fuel back to electricity when the grid needs to support the large electricity requirement. One of the unique characteristics of SEMR is its possibility to operate under transient load conditions over a broad range of feed gas compositions while maintaining faradaic efficiency of nearly 100%, which renders this particularly suitable in future scenarios of intermittent availability of renewables.22 The transfer of O2−/H+ through the solid membrane is essential for the function of SEMRs, generating a flux of ions to balance the electrons which are transferred through the external circuit, and either oxidize or reduce the reactant to produce fuels or value-added chemicals.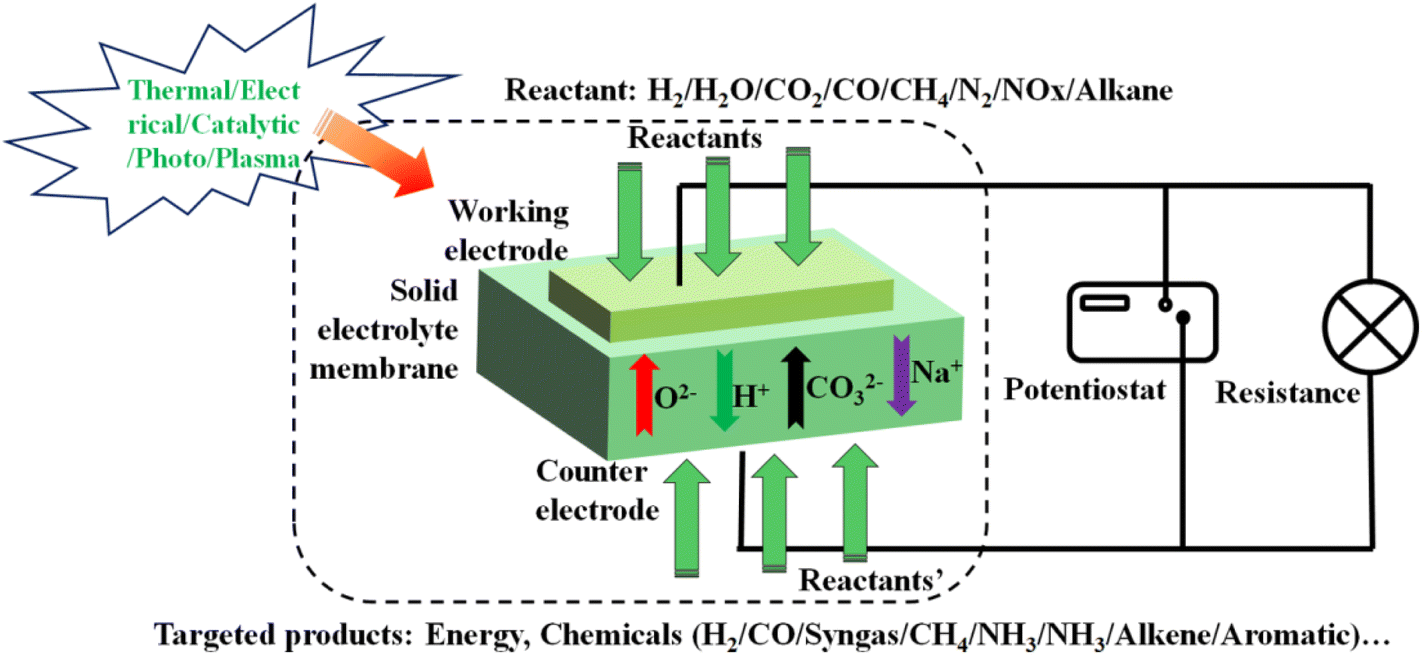 | ||
| Fig. 1 The working principle of a SEMR for versatile energy, chemical, and environmental applications. | ||
The increasing availability and affordability of renewable energy, mostly from solar and wind sources, has a large impact on the industrial process and transport sectors, which makes electrification and de-carbonization of the chemical industry reasonable and affordable.5 In this context, advanced technologies for energy storage, transportation, and direct conversion of electricity into chemical products are required. SOCs or SEMRs, enabling interconversion between chemical energy and electrical energy, can be therefore seamlessly integrated to overcome the challenges associated with the intermittent nature of renewable energy, combined with downstream chemical processes to produce various liquid fuels, rendering it a highly capable all-rounder.23,24
The SOC is more than a simple power source or electrolysis device. The high operation temperature together with the closed electrode chamber with the catalyst turns the SOC into an ideal chemical reactor or SEMR. Various reaction parameters, such as gas atmosphere, conversion, and selectivity can easily be tuned by adjusting the operational mode, temperature, potential bias, and so on. This provides an attractive advantage compared to thermal and other conversion reactors. Moreover, such a unique characteristic may encourage the possibility of chemical–electrical coupling in SEMRs and open alternative avenues toward electrocatalytic synthesis of chemicals at higher temperatures.25 SEMRs have been employed in many important chemical processes,20,21,26,27 as follows:
(1) Highly efficient generation of green hydrogen through water electrolysis using renewable power sources or electricity through fuel oxidation
(2) Direct splitting of CO2 to produce CO fuel and O2 for de-carbonization and neutrality purposes space exploration
(3) Combining H2O electrolysis or H2O/CO2 co-electrolysis to yield advanced alkanes
(4) Steam or dry-reforming or partial oxidation of hydrocarbons to form compositionally well-controllable syngas
(5) Oxidative coupling of methane (OCM) to form alkanes, olefins and even arenes
(6) Ammonia NH3/oxynitride synthesis and de-NOx technology
(7) Oxidative and non-oxidative dehydrogenation of alkanes, olefins and even arenes
(8) Gas separation for a highly purified gas product
(9) Power and chemical co-generation
(10) High temperature metal–air batteries
(11) Direct carbon fuel cells
(12) Many other integrated processes such as combined electrolysis with F–T synthesis in one step or one device…
Even though the water electrolysis cell using a SEMR was reported already in 1968,28 limited interest was shown due to the insufficient performance. However, since the pioneering work in 1979,29 there has been huge research progress on SEMRs for versatile chemical processes. Many excellent studies, review papers, and monographs that report, discuss, and analyze the applications and understand SEMR processes were reported. A broad range of aspects concerning fundamental operating principles and the applications of these solid-state electrolytes and these types of reactors in heterogeneous catalysis and chemical cogeneration (chemical products and energy) have well been documented.1,30–33 However, most of the previous work focus on particular aspects of the fields like CO2, H2O, and H2O/CO2 co-electrolysis34–36 methane coupling to produce C2 hydrocarbon1,26,31,34,37–39 or two wide coverage,27,40–42 and needed significant literature update31,43,44 since considerable progress has been made in the last decades with the positive evolution of the key electrolyte material,20,45,46 and catalyst/electrode components,47 the cell geometries from typical construction from SOC with dense electrolyte and porous anode and cathode (alternative functional layer on/in electrode even porous electrolyte based),48–50 never mentioned the progress in the system combined operation and coupling/integration.51–53 Therefore, a timely and comprehensive review of the solid electrolyte membrane is needed.
Membrane reactors operating at low temperatures (300 °C) will not be discussed in this article. The focus will be on membrane reactors operating at a temperature between 300 and 800 °C because the majority of industrially important catalytic processes occur in this temperature range with favorable reaction thermodynamics and kinetics. The present work refers to SEMR-related scientific fields such as electrochemistry, materials science and technology, chemical reaction engineering, solid-state physics, and heterogeneous catalysis as well as calculation/quantum chemistry. The aim of the present review is not only to provide a survey of relevant studies but also to present the characteristic features, methods, and techniques used in this class of reactors. Hence a classification of SEMRs based on different ionic conductors (O2−, H+, mixed O2− and H+, carbonate ionic composite conductor) and working modes (fuel cell or electrolysis, pumping or extracting mode) is presented, with case study demonstrations (Fig. 2). Discussion is moved from the typical reactions based on different ionic conductors to SEMRs with featured multi-functionality and/or specific geometrics. A particular section on the use of SEMR for gas separation is also highlighted using the specific properties of the solid electrolyte materials. The system and device integration or coupling or process intensification with SEMRs is also included. In addition, a typical phenomenon or an additional functionality regarding SEMRs, i.e., electrochemical promotion of catalysis (EPOC) effect is also included to highlight the fundamentals of accelerated reaction rate under the electrical/thermal coupling conditions. Finally, the conclusion and future outlook of this interesting yet important field are made and recommendation of those reactors for industrial application purposes is suggested.
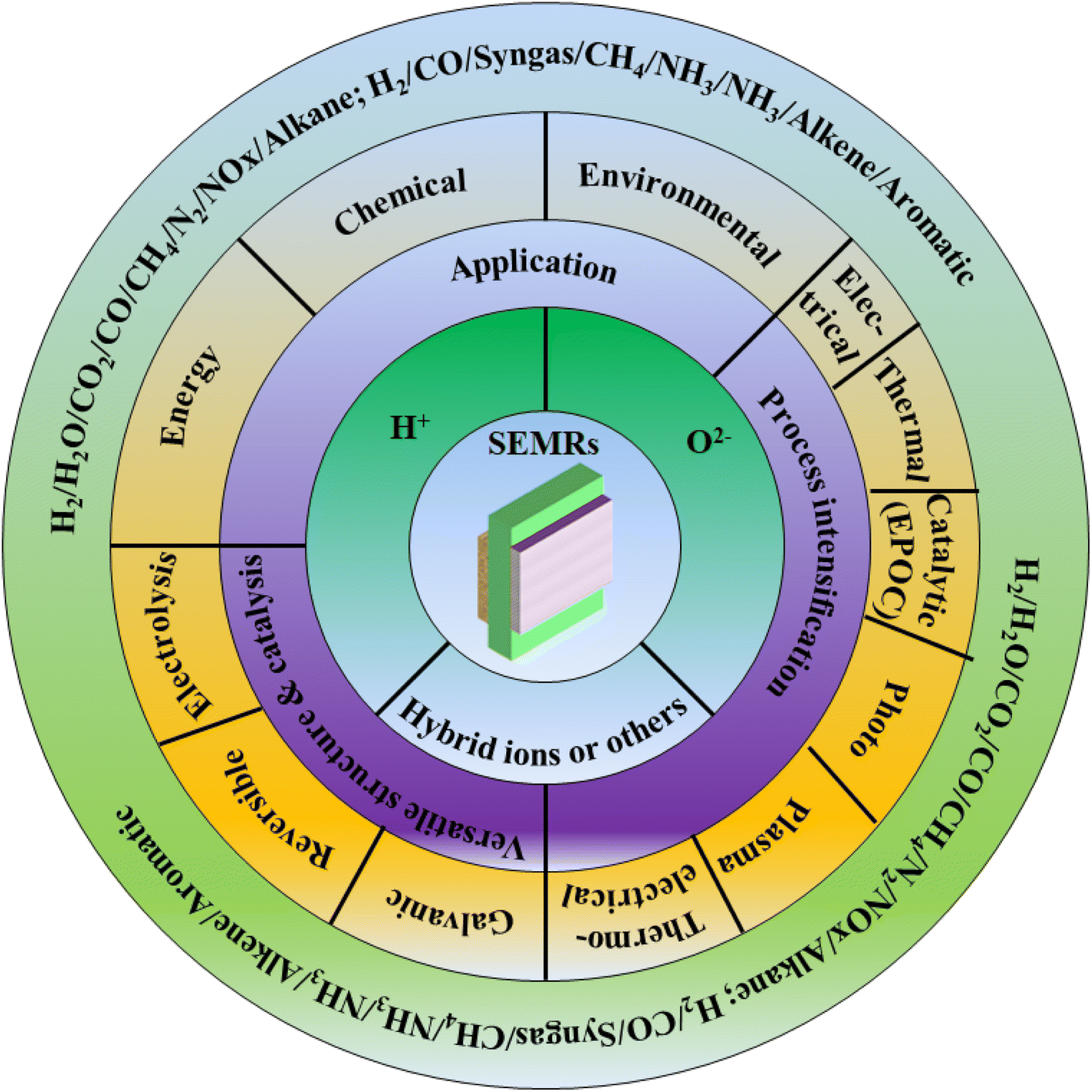 | ||
| Fig. 2 A schematic illustration of outlining the focus of this work. | ||
3. Advances and challenges of SEMRs with various ionic conductors
Similar to the SOC, the types and compositions of the ionically conductive electrolytes in the SEMRs play key roles in reactor temperature, electrode/catalyst materials, detailed reactor classification, as well as reactor thermodynamics and kinetics. To ensure high faradaic efficiency, the ionic transport number of electrolytes should be close to unity and remain stable under operational conditions such as a harsh reducing atmosphere and high applied voltage, under which a number of electrolyte materials, such as the partially electron-conductive doped ceria electrolyte, cannot be employed even in a low temperature range. Currently, the typical oxygen ionic conductor is still the state-of-the-art yttrium-stabilized zirconia (YSZ). However, the high activation energy of ionic conduction requires an operating temperature above 700 °C. High temperature results in high reaction kinetics and favorable thermodynamics in the electrolysis mode. However, it also results in a deep reaction to produce untargeted chemicals, and the quick degradation of reactor performance due to cell component instability, incompatibility, and mechanical property issues. To reduce the reactor temperature, LaGaO3 electrolytes typically doped with strontium and magnesium (denoted as LSGM), proton conductive BaCeO3, and BaZrO3 perovskite oxide and their solid solution state are developed. Particularly, proton conduction not only reduces the reactor temperature but also widely expands the reactor diversity, with the non-oxidative dehydrogenation of alkanes to olefins as a typical example.54 In addition, the mixed conduction of the Ba(Zr,Ce)O3 material is also used for functional purposes,55 like coke removal47 and double-chamber hybrid electrolysis.56 In this section, the discussion will be on SEMRs with different ionic conductors and operational modes, like pumping and extractor modes.57 A typical example will be given to show the advances.3.1 SEMRs with oxygen ionic conductors
3.1.1.1 Water electrolysis for green hydrogen production. In terms of excellent electrical efficiency and decoupling direct reaction process by electrolyte, and direct adaptation of SOC devices with improved reliability62–64 and efficiency,8 green hydrogen generation based on SOECs is already entering the market. There are already a lot of excellent experimental studies and reviews to cover information on SOECs including steam electrolysis thermodynamics, SOEC electrolyte/electrode material/catalyst development, electrode structure design, cell fabrication technology, reaction kinetics/mechanism, as well as cell durability for long-term operation.8,38,42,62,65–71 The longest steam electrolysis operational time based on SOEC is 23
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h (reaching the targeted period) using Ni-GDC/6Sc1CeSZ/GDC/LSCF cells with a voltage degradation rate of 0.57%/1000 h based on a 45 cm2 cell study at 0.9 A cm−2.62 The current research interests focus on the performance and durability improvement for commercial applications including the anode Ni-based cermet electrode's redox and poisoning issue and the anode/electrolyte interface delamination issue61 due to the high oxygen concentration and aggregation in this area. Strategies such as the application of mixed ionically and electronically conductive electrode materials,61 especially for materials with high oxygen capacity,72,73 and modification of cell electrode microstructure, like using transitional functional layers74 or straight pores75 and the smart strategy of reversible operation between fuel cells and electrolysis mode71 have been proposed to alleviate these issues.
000 h (reaching the targeted period) using Ni-GDC/6Sc1CeSZ/GDC/LSCF cells with a voltage degradation rate of 0.57%/1000 h based on a 45 cm2 cell study at 0.9 A cm−2.62 The current research interests focus on the performance and durability improvement for commercial applications including the anode Ni-based cermet electrode's redox and poisoning issue and the anode/electrolyte interface delamination issue61 due to the high oxygen concentration and aggregation in this area. Strategies such as the application of mixed ionically and electronically conductive electrode materials,61 especially for materials with high oxygen capacity,72,73 and modification of cell electrode microstructure, like using transitional functional layers74 or straight pores75 and the smart strategy of reversible operation between fuel cells and electrolysis mode71 have been proposed to alleviate these issues.
3.1.1.2 CO2 utilization. Similar to steam electrolysis, extensive studies on the efficient reduction and conversion of CO2 using SOECs were reported in the literature.76 These include, e.g., the pioneering work on CO2 electrolysis with SOECs for use on Mars to produce O2 for life-support.77 As a notorious greenhouse gas, its utilization and conversion to valuable chemicals are considered the most crucial approach to realizing carbon neutrality.26 In this context, the industrial generation of carbon-based molecules—such as drugs, commodities, and polymers, in addition to chemical fuels—from CO2 is subject to intense research using different conversion technologies including SEMRs, electrochemical reduction in aqueous solution, and photocatalytic reduction (Table 1), among which electrochemical reduction using SEMRs shows the most promising characteristics like low energy consumption, quick reaction, high Faraday efficiency, and energy efficiency, even though its product is only CO because of the thermodynamic limitation.78,79 CO2 reduction in the cathode of an O-SOEC generally follows three steps:80–84 (1) adsorption of CO2 on the surface of the electrocatalyst with lattice oxygen ions and formation of carbonate species or with an oxygen vacancy to form bicarbonate intermediates, (2) decomposition of the intermediates by electrons in sequence, which is normally considered as the overall rate-determining step, and (3) desorption of the CO molecule. However, the exact reduction process is highly dependent on the applied catalyst, for example, for a ceria-based cathode electrocatalyst, Ce3+/4+ participates in all the elementary steps of CO2 reduction.85 Generally, CO is the thermodynamically favored product for CO2 reduction at elevated temperatures. Recently, there has also been interest in adopting PCECs for CO2 reduction, in which CO2 acts as the precursor to react with H+ or H2 at the electrode/electrolyte interface yielded by steam electrolysis at the cathode and transported through the electrolyte layer. Under such conditions, CO2 could be electrochemically or thermally activated at the cathode to produce CO or upgrade to interesting low-carbon hydrocarbon fuels.86 It should be mentioned that different from H2O splitting, CO2 reduction should overcome a larger energy barrier (750 kJ mol−1) because of the large C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O binding energy. Moreover, CO2 is a linear molecule; combined with its larger molecular volume, its diffusion, adsorption, and activation attract particular attention and require higher energy input and higher reduction temperature. Therefore, direct CO2 electro-reduction in SOECs normally works above 700 °C. Under such harsh conditions, only stabilized zirconia and doped LaGaO3 can be used, while ceria is normally used as a functional layer to overcome chemical compatibility, and electrode active species to increase the triple-phase boundary to improve reaction kinetics.
O binding energy. Moreover, CO2 is a linear molecule; combined with its larger molecular volume, its diffusion, adsorption, and activation attract particular attention and require higher energy input and higher reduction temperature. Therefore, direct CO2 electro-reduction in SOECs normally works above 700 °C. Under such harsh conditions, only stabilized zirconia and doped LaGaO3 can be used, while ceria is normally used as a functional layer to overcome chemical compatibility, and electrode active species to increase the triple-phase boundary to improve reaction kinetics.
| SEMRs | In aqueous electrolytes | Photocatalytic reduction | |
|---|---|---|---|
| Energy consumption | Low | High | Low, sustainable |
| Conversion rates | Quick | Voltage dependent | Slow rate |
| Slow rate | |||
| Product selectivity | Sole product CO | Rich products like CO/CH4/CH3OH/C2H5OH… | |
| Others | High faradaic efficiency | Poor energy efficiency | Poor energy efficiency |
| Good stability | Low yield | Insufficient stability | |
| High energy efficiency | Insufficient stability | ||
Considerable attention has been paid to the development of active cathode electrocatalysts to replace Ni, an active but fragile catalyst.87–89 Alternative cathodes are related to perovskite oxides, a series of compositionally and structurally flexible complex oxides, because of the phase durability and high resistance toward the thermodynamically favored carbon deposition issue. However, their bulk CO2 reduction activity is far way inferior to the Ni-based cathode. Therefore, current efforts focus on modifying the perovskite oxide through doping, constructing a hybrid catalyst, and optimizing the microstructure to adjust the electronic structure and increase the electrode adsorption capability, the active site, and reduce the gas diffusion barrier. For example, Xi et al.90 reported that replacing Mo with Mg in Sr2FeMoO6−δ not only improved the electrode redox stability but also enhanced the electrolysis efficiency by 100%. The improved cell performance was ascribed to the reduced formation energy of oxygen vacancies induced by reduced d–p band coupling which is believed to be caused by the shortage of d electrons of the Mg element. In other words, the electronic structure of the active transition metal is modified by the doping approach which favors the CO2 reduction rate-determining step, the activation, and the dissociation step. The same group also demonstrated that a proper increase of the Fe and Mo ratio in the same Sr2FeMoO6−δ double perovskite led to exceptional CO2 reduction activity, comparable to the state-of-the-art Ni-based electrode. They confirmed that the Fe-ion oxidation state increases with a slight increase of Fe element that favors the metal–oxygen hybridization and shifts its bulk O p band energy toward the Fermi level. Such electronic structure variation reduces the O-vacancy formation and migration energy, resulting in more oxygen vacancy defects and favoring O ion transport, promoting the catalytic reaction kinetics.91 Different from the above cation replacement method, anion doping is also demonstrated to favor CO2 adsorption and activation. For example, the replacement of O with F ions could reduce the CO2 reduction electrode area polarization resistance from 1.130 to 0.656 Ω cm2 at 800 °C.92 Such improved electrochemical performance is induced by the increased surface reaction rate constant and bulk diffusion coefficient by factors of 2–3 as reflected by the approximately doubly increased CO2 adsorption capability, plus 35–37% oxygen vacancy concentration. A more interesting work reported adopted a single-site anchored perovskite electrocatalyst for CO2 reduction in SOECs for the first time.93 Through compositional control of the content of Pd raw materials, Pd single-site perovskite oxide materials instead of homogeneously doping are formed by a simple mechanical mixing and pyrolysis. The single site enables structurally Pd coordinative unsaturation, which results in plenty of oxygen vacancies. The latter facilitates CO2 dissociative adsorption, electron transfer, and mass transport, thus leading to improved CO2 electrolysis performance.
Doping generally leads to elevating the elementary reaction rate of the CO2 reduction reaction. However, with the single-phase perovskite cathode, the final performance is limited by the bulk properties of perovskite oxide. The low electrical conductivity under the cathode atmosphere and the limited surface area and catalytic activity normally lead to insufficient activity. A common method is to prepare hybrid catalysts for CO2 reduction reactions. For example, a composite material containing (La0.75Sr0.25)0.97Cr0.5Mn0.5O3 (LSCM) and Ce0.6Mn0.3Fe0.1O2 (CMF) was prepared using a simple mechanical mixing method and applied as a cathode on LSGM for CO2 reduction.94 The targeted SOECs show excellent CO2 reduction performance of 2.64 A cm−2 at 1123 K and applied voltages of 1.5 V in a 50% CO2/50% CO atmosphere, which is the ever-best single-cell performance reported in the literature. The much-improved CO2 reduction activity is ascribed to the enhanced CO2 adsorption ability, surface oxidant catalytic activity, and electrocatalytic activity induced by Fe and Mn-doped ceria. It should be noted that singly doping Mn or Fe into ceria has limited solid solution capability. On the other hand, co-doping can unexpectedly increase the solid solution content, up to 40% in this case, which significantly changes the material's chemical and physical properties, leading to exceptional performance in this work. Besides the mechanical mixing to form the hybrid catalyst, recent self-assembly including exsolution methods also showed promising performance in SOECs for CO2 reduction.95–101 For example, recent work reported adopting the self-assembly method to prepare Pr(Ca)Fe(Ni)O3−δ perovskite and Ca2Fe2O5 brownmillerite dual-phase composite in a nominal perovskite phase of Pr0.2Ca0.8Fe0.8Ni0.2O3−δ for CO2 reduction.102 The Ca2Fe2O5 in the double-phase material with a large highly ordered oxygen vacancy concentration is used to adsorb CO2 while the perovskite oxide functions as the catalyst for CO2 dissociation. The composite cathode with a synergistic effect has led to improved CO2 reduction performance. Recently, a redox exsolution strategy has been widely used for CO2 reduction in SOECs. The exsolution led to a well-manipulated nanoscale metal/oxide interface with enriched oxygen vacancy for CO2 high temperature adsorption and conversion. Xie et al.103,104 reported that the decomposition of oxide precursors Ni/Nb1.33(Ti0.8M0.2)0.67O4 (M = Mn, Cr) yielded redox MnOx with large oxygen vacancy concentration and Ni/Cu metal or alloy on ceria/CrOx which are active for CO2 reduction. The strongly anchored functional phase and oxygen vacancy at the interfaces not only improve the electrolysis performance but also lead to better durability under the redox cycle and long-term operation. Another exsolution example is the preparation of active metal or alloy nanoparticles on phase structural redox stable perovskite oxides. The metal/oxide interface formed by exsolving CoFe or RuFe alloy on the parent perovskite oxide can significantly improve the CO2 electrolysis performance through enhanced adsorption and activation under SOEC conditions.97,98,105 They also observed in situ the atomic scale exsolution of metal nanoparticles in the order of Co ions, Fe ions, and the final CoFe alloy nanoparticles using STEM. Moreover, the CO2 reduction reaction performance can be retrieved during 12 redox cycles. More recently, considering the sluggish diffusion process of dopant cations and limited driving force for metal/alloy nanoparticle exsolution, they proposed to promote the exsolution of RuFe alloy nanoparticles via repeated redox operation which enriches the Ru ions underneath the perovskite oxide surface. With the repeated redox operation, more RuFe nanoparticles aggregated and resulted in 74.6% current density improvement as well as 1000 h durability at 1.2 V and 800 °C. Both the suggested exsolution method and in situ exsolution process observation are important steps for understanding and application of this promising technology. A similar study demonstrated that the exsolved metal/alloy nanoparticles on an oxide parent showed higher performance and electrolysis durability than the infiltrated one with a similar composition and operational conditions.106 When used as the cathode for SOECs, NiFe nanoparticles exsolved (La0.75Sr0.25)0.95Cr0.5Fe0.35Ni0.15O3−δ (LSCrF as), NiFe infiltrated LSCrF and the one without any nanoparticles showed the corresponding current densities of 1.15, 0.8 and 0.59 A cm−2 at 800 °C and 1.5 V, and the former showed a degradation rate of 0.012% h−1 during a tested period of 260 h while the intermediate one degraded in a rate of 0.15% h−1 in 26 h due to the severe agglomeration of the infiltrated nanoalloy, suggesting the nanoparticle exsolved LSCrF as promising cathode material for efficient CO2 utilization.
Apart from the development of active and reliable cathode materials for CO2 reduction, electrode microstructure optimization in SEMRs is also an important approach to improving cell performance and durability by increasing gas diffusion and exposing more active sites for CO2 reaction.75,107,108 For example, microchannel structural cathode-supported SOECs were constructed for CO2 reduction.107 The microchannel not only allows quick CO2 gas diffusion, reducing the concentration polarization but also facilitates high-content catalyst loading and distribution. The SOEC based on deposited Sr2Fe1.5Mo0.5O6−δ (SFM) cathode showed an electrode polarization resistance of 0.25 Ω cm2 and a current density of 1.1 A cm−2 at 1.5 V in the dry CO2 without safety gas as well as promising stability; these performances reached the highest level at that time.
Much progress has been made in SOECs for CO2 reduction. However, the research and applications still lag behind steam electrolysis. Many perovskite oxide cathodes have already demonstrated comparable performance to the Ni-based cermet electrode. Moreover, the current CO2 reduction in SOECs is still limited to high-temperature operation (≥700 °C), to maintain sufficient current output, and an intrinsic large electrical input, which make the conditions severe for system stability for practical applications. A reduction of the operational temperature to 300–600 °C will benefit the system stability, involve low investment, as well as extend the product range, and not just limit to CO at the elevated temperature.36
3.1.1.3 CO2/H2O co-electrolysis.
3.1.1.3.1 Compositionally tailor-made syngas by co-electrolysis. Syngas is an important feedstock for various important industrial applications. Since SOECs can reduce both steam and CO2, the co-electrolysis of H2O and CO2 to obtain syngas has received increased attention in recent years.9,70,109 Generally, under the SOEC conditions, both the water splitting and CO2 reduction reaction take place, and the water-splitting rate is considerably quicker than that of CO2 reduction due to the low activation energy barrier and consequent higher kinetics,110,111 which leads to the advantage of low thermo-neutral voltage, i.e., lower electrical consumption. For example, Torrell et al.110 reported that with H2 safety gas, the electrolysis current densities at 1.7 V with pure steam and with H2O
:CO2 ratios of 1:1 were 750 and 620 mA cm−2. This suggested that the main reaction in the mixed gas is steam electrolysis. However, the presence of the reverse water shift reaction (RWSR): CO2 + H2 = CO + H2O (ΔH298 = 41 kJ mol−1) at the elevated temperature makes the final gas composition complex. In other words, both the electrochemical and thermal-catalysis processes contribute to the final products. Workable strategies to improve the individual CO2 electrolysis rate with the H2O/CO2 mixed inlet gas are highly desirable. Either the application of a higher electrical field111 or improving the operating temperature112 was suggested to boost CO2 electrolysis performance by considering the high energy barrier of CO2 activation. Differently, recent work reported that a BaCO3 nanoparticle infiltrated Ni-YSZ fuel electrode improved the electrolysis performances of SOECs with H2O, CO2, and H2O–CO2 inlet gas, respectively, because of the enhanced surface charge transfer process, while it degrades the SOFC performance.113 The highest improvement is achieved in CO2 reduction, which largely contributes to the improved chemical adsorption of CO2 with BaCO3 infiltration.
Syngas with a controllable (diverse ratio range) or ideal composition that favors downstream applications like the Fischer–Tropsch (F–T) process to synthetic fuel production at an H2/CO ratio of 2:1 is preferred. To reduce the RWSR process, co-electrolysis without safety gases like H2 and CO is preferred. This can be achieved by using non-metal-based fuel electrocatalysts that do not require safety gases,107,110 even the state-of-the-art Ni-cermet electrodes could be electrochemically reduced and activated.114 For example, syngas with an H2/CO ratio close to 2 can be obtained from an efficient 20% CO2–80% H2O electrolysis without a safety gas on an Sr2Fe1.5Mo0.5O6 nanostructure infiltrated YSZ scaffold with a finger channel and a modified phase-inversion tape-casting method.107 Such a nanostructure electrode also can directly use CO2 as the fuel, yielding a current density of 1.1 A cm−2 at 1.5 V at 800 °C, comparable to a Ni-based cermet electrode. Another work proposed adjusting appropriate inlet gas fraction, temperature, and electrolysis current to obtain a controllable H2/CO ratio of approximately 2 in a symmetric SOEC using the same electrode materials as the above case for H2O/CO2 co-electrolysis.115 With an appropriate H2/CO ratio, the upgradation to synthetic fuel will be facilitated in the commercialized F–T plants.
3.1.1.3.2 In situ methanation using O-SOECs with a higher round-trip efficiency. Another important reason to develop co-electrolysis technology is its capability to produce higher hydrocarbon fuels like CH4 accompanied by the electrolysis process in the one-step pathway, instead of first producing syngas that is then processed downstream into chemicals. Compared with syngas, one step pathway to hydrocarbon fuel can store renewable power sources in higher energy density and use the existing infrastructure such as natural gas pipelines and end-use appliances.116 Generally, the methanation process was taken placed through the following reactions: CO + 3H2 =
CH4 + H2O (ΔH298 = −206 kJ mol−1) and CO2 + 4H2 = CH4 + 2H2O (ΔH298 = −165 kJ mol−1) with the obtained syngas from H2O/CO2 co-electrolysis. The second reaction is also known as the Sabatier reaction. Therefore, the methanation reaction does not require the reduction of CO2, which can reduce significant energy. Instead, according to the literature, compared with the co-electrolysis in SOECs to obtain syngas and then integrating with the F–T process to produce synthetic fuel, the one-step process has multiple benefits:117,118 (1) the energy utilization efficiency can be significantly improved in the one step process since it balances the heat in situ between the exothermic methane production reaction and the endothermic H2O/CO2 electrolysis reaction in a single device. Taking H2O electrolysis to obtain H2 and then combining with CO2 to produce CH4 as an example, the thermodynamic maximum round-trip efficiency of H2O electrolysis is around 76% at 800 °C, which is already much lower than that required for direct CH4 synthesis, both under the thermal neutral conditions; (2) reduced industrial facilities, free-from the F–T process and the corresponding instruments and investment; (3) improvement in system reliability. Moreover, this process could be economically competitive with the biomass-to-liquid process if the provided electricity is from nuclear power and surplus renewable power.119,120
The methanation process through the co-electrolysis technology has been widely investigated using the O-SOEC, in which CO2 and H2O are co-fed into the cathode chamber, and the O element is extracted to form O2− which is then transported through the electrolyte layer to the anode layer, where it is released electrons and moved to the cathode through an external circuit, forming O2 gas. In the cathode chamber, CO and H2 are produced which can react with the remaining H2O and CO through the above equation to produce methane under an electric field or thermal–catalysis conditions. It was initially proposed by NASA in the manned space colonization on Mars to convert the Martian CO2 atmosphere and H2O into methane fuel and build a astronaut life-support system. CO2 hydrogenation is first carried out using the YSZ solid electrolyte and Rh electrode in the configuration of a single-chamber reactor.103,104,122 It is found that both CO and CH4 were produced at temperatures of 346–477 °C and their yield rates have an inverse relationship with each other with the application potential, i.e., the rates of CH4 were enhanced with positive potentials while the rate of CO formation with negative potentials,122 suggesting different electrochemical promotion effects. Later work developed a monolithic SOC (Rh/YSZ/Pt or Cu/TiO2/YSZ/Au) reactor to investigate the hydrogenation of CO2 at atmospheric pressure.123 For the former cell, it is found that the selectivity of CH4 can reach 12%, while in the case of Cu/TiO2/YSZ/Au cells, the selectivity of CH4 could reach ∼100% under open-circuit polarization conditions at temperatures of 220–380 °C. Those pioneering research studies highlighted the important roles of cell structure design and electrode catalyst selection.
It should be noted that the H2O or CO2 electrolysis reaction preferably takes place at the elevated temperature with high reaction kinetics, while the exothermic methanation reaction is more favored at the reduced temperature of recommended value of 300–500 °C. The mismatch in temperature makes the heat balance of those two reactions quite difficult. In 2010, Bierschenk et al.118 experimentally observed that methane-containing fuels are produced during electrolysis operation, which decreased the thermal-neutral voltage (SOEC applied voltage) and thereby allowed improved round-trip storage efficiency. They observed that the CH4 yields are more favorable at reduced temperature (∼600 °C), enabled by the LSGM electrolyte to replace the conventional YSZ electrolyte, and/or increased pressure (∼10 atm) based on their thermodynamic calculations and preliminary experimental analysis. The obtained CH4 yield can be improved from trace to 14.3% with the reduction of SOEC operational temperature from 750 °C to 600 °C based on the same material system, and it can be further improved to 26.7% by properly applying the operational pressure. This opened a new avenue for methanation R & D. Almost at the same time, Xie et al.124 reported the use of a novel Fe–La0.2Sr0.8TiO3 fuel electrode for direct synthesis of methane by combining the co-electrolysis of CO2–H2O in an oxygen-ion conducting SOEC and in situ Fischer–Tropsch-type synthesis in a synchronous process. A CH4 rate of 0.0047 mL min−1 with a faradaic yield of 2.8% at 650 °C with a feedstock of 2 mL per min H2O and 1 mL per min CO2 were obtained. The methanation yield is limited by the heterogeneous catalysis process which is against the thermodynamic behavior. Following these pioneering studies, Chen et al.121 smartly designed long enough tubular cells with two distinct temperature zones: one for H2O–CO2 co-electrolysis at elevated temperature with a high conversion rate and one for the F–T synthesis process at the reduced temperature (down to 250 °C) for the one-step methanation reaction through co-electrolysis (Fig. 3c). The yield of CH4 was substantially improved to 11.84% and an overall CO2 conversion ratio of 64.1% at atmospheric pressure. Based on a similar cell design, an increased CH4 yield of 23.1% was demonstrated by optimizing the H:C ratio in the inlet gas by adding H2 into the cathode chamber with H2O–CO2 mixed gas.125 However, the split-zone approach engenders thermo-mechanical stress to the super-long cell and delicate electro-ceramic cell components and brings reliability concerns. A combination of efficient co-electrolysis and the F–T process at the reduced temperature is preferred.
 | ||
| Fig. 3 The proposed cell structure design for coupling the low temperature F–T process and high temperature co-electrolysis process.124 Reproduced with permission. | ||
Further work tried to improve CH4 yield through reducing temperature and the addition of H2 gas.126 It was observed that without the H2 gas, the CH4 yield increased negligibly from OCV to the applied voltage of 1.5 V, while with the 20% H2 fed into the cathode, a clear increase of the CH4 yield by 3–4% is observed at 550 °C. With the application of the higher ionic conductive ScSZ electrolyte layer and Ni-YSZ cathode catalyst, a maximum CH4 yield of 9.94% was achieved. Based on the two-dimensional multiscale electro-thermal tubular SOEC model calculation with parameter optimization on the materials, flow modes, and operating conditions, the CH4 production ratio can be improved from 13% to 50% at 550 °C and 1.3 V for the same inlet gas composition, even though the temperature is considered still too high to obtain acceptable CO2 to CH4 yield. Furthermore, the applied model calculation results revealed that the H2O/CO2 co-electrolysis and methanation reactions can be simultaneously enhanced by feeding a cold gas in counter-flow mode and under pressurized conditions. At 29 bar, the tubular LSGM-based SOEC could realize a conversion ratio of 98.7% and an electricity-to-gas efficiency of 94.5% in the thermal neutral mode.127 Such a theoretical calculation suggested that developing methane-integrated SOECs operating at 300–600 °C can efficiently convert CO2/H2O into more valuable chemicals, which will be a new research direction in the future. However, the concept of in situ methanations in SOECs is still at a nascent stage and requires significant advancements in SOEC materials, F–T catalysts, and cell/reactor geometric design, along with optimization of cell operating conditions (temperature, pressure, and applied potential) to maximize CH4 yield below 550 °C. Another interesting but important branch for the one-step methanation process is realized by the PCECs due to the superior proton conductivity at low temperatures that are crucial to realizing high reaction kinetics and CH4 yields, which will be discussed later.
3.1.1.3.3 Fuel-assisted electrolysis (FAE). Under electrolysis conditions, though the major voltage loss is mainly ascribed to the electrode polarization resistance of the fuel reduction reaction for both steam and CO2 electrolysis, the polarization loss for the oxygen evolution reaction (OER) in the anode chamber should not be ignored. In the initial study, the OER was used to prepare oxygen sources for life support in space. It can be employed in further applications considering the high value-added pure and fresh O2 and provides the opportunity for process intensification. An important application is reforming CH4 or other less purified/valuable solid fuels like carbon128,129 and coal;130 gaseous fuels such as CO,131 natural gas,132,133 and biofuel & biomass134 were also used to obtain syngas feedstock or achieve chemical upgradation, besides the hydrocarbon fuel. This comprises an important and hot research topic, fuel-assisted electrolysis (FAE). A distinguishing characteristic of FAE is its strong coupling of the endothermic electrolysis reaction with the exothermic fuel reforming or upgradation reaction, and the uphill potential barrier for the OER is also avoided, which has consequently led to significantly reduced electrical power input (both cell voltage and electricity are reduced) while increasing the production of valuable gases. Moreover, with the increase in temperature to a certain point, such an FAE device can be turned into a combined chemical and power co-generation device.135 According to the one-dimensional elementary reaction kinetic model analysis,131 the CO-assisted steam electrolysis cell could save 80% of electricity and produce better steam electrolysis performance. It was also found that the assisted electrolysis cell based on different fuels showed different anode polarization behaviors. Another study even proposed that such a cell can be operated at room temperature and can realize a higher hydrogen generation rate, suggesting promising application in wide cell temperature windows for chemical and power generation with low carbon emission advantage.136 Although the overall reaction of the FAE process is the same as that for fuel steam/CO2 reforming, it is more advantageous to perform such a process by an electrical-to-chemical energy conversion route based on the solid electrolyte membrane reactor, which is promising to serve as a load lever for energy storage between renewable electricity and fuel. In addition, FAE can be implemented as distributed syngas generator under atmospheric pressure, which is different from traditional reforming reactor that typically operates as a large chemical plant in pressurized operation.
3.1.1.3.3.1. Solid carbon/coal-assisted electrolysis reaction . In 1979, carbon-assisted electrolysis of water was first reported at room temperature in a H2SO4 electrolyte solution,29 showing that the carbon-assisted process only required a thermodynamic energy of about 9.5 kcal and a reversible potential of only 0.21 V to take place. The coal provided the additional electron required for H2O splitting and consequently CO generation. Moreover, the authors hinted at a more rapid and steadier oxidation rate at the temperature range of 200–600 °C. A more recent study demonstrated that a steam–carbon electrochemical cell with YSZ as an electrolyte and Pt as an electrode can produce carbon-free hydrogen and positive OCV at the temperature of 800–900 °C under a carbon/(H2O–H2) atmosphere.137 The higher the H2O concentration in the cathode chamber, the larger the cell potential. At a negative voltage of 0.6 V, the electrolysis current density could reach 17 mA cm−2. Lei et al.138 also reported a carbon-assisted CO2–H2O co-electrolysis cell using LSGM-supported SOECs. The thermodynamic calculation and experimental results (Fig. 4) demonstrated that the co-electrolysis potential can be reduced by about 1 V by adding carbon to the anode, and the electricity input can be reduced to less than 10% of that without carbon. For example, a potential of 1.5 V is required to reach an electrolysis current density of 1.2 A cm−2 without carbon fuel, while it is reduced to 0.8 V with carbon (Fig. 4c). It was also found that CO was mainly formed through a so-called “CO shuttle” mechanism: simultaneous production of CO on the anode and syngas CO/H2 on the cathode side through the well-known Boudouard reaction (C + CO2 = 2CO) at elevated temperatures (Fig. 4d). Moreover, as seen in Fig. 4b, the theoretical electrical demand (ΔG) decreases with increase in temperature. When the temperature is higher than 900 K, ΔG becomes a negative value, that is, carbon reacts with H2O to produce CO2 and H2, spontaneously. Such an integration of carbon gasification and SOECs provides a promising method for the efficient storage of renewable electricity using coal/biomass and CO2/H2O, and producing clean fuel.
 | ||
| Fig. 4 Thermodynamic calculation of the required energy for CO2–H2O co-electrolysis without (a) and with (b) carbon fuel in the anode, (c) comparison of the I–V curves of the electrolysis cell, and (d) temperature-dependence of anode outlet gas composition.138 Reproduced with permission. | ||
Another interesting study attempted to mitigate the large polarization resistance of H2O electro-splitting using the carbon-assisted method.139 Different from the above approach, they first decomposed CH4 in the anode compartment to H2 and carbon under OCV conditions; the deposited carbon penetrated the porous Pt/YSZ electrode and had intimate contact with the electrode catalyst, which is different from that of the conventional design where carbon fuel is on the top of the fuel electrode. After the decomposition period, steam is introduced to the other electrode with the Pt catalyst, which is electrolyzed to H2 and O2− under the much lower polarization conditions, the O2− is then moved to the carbon electrode allowing the removal of the previously deposited carbon and regenerating the Pt/YSZ electrode. Such a coupling catalysis and electrocatalysis process enabled efficient H2 production (CH4 pyrolysis and H2O electrolysis) and cell electrode regeneration with a much lower energy input compared with the general CH4 steam reforming process. 18 cycles of durable operation of the applied cell materials and operational mode demonstrated the feasibility of such a novel process and device. Moreover, ethane and ethylene were observed during the regeneration step because of the electrocatalytic methane oxidative coupling reaction on the Pt/YSZ porous catalyst film.
3.1.1.3.3.2. Gaseous chemical-assisted electrolysis . Besides the solid fuel, gaseous chemicals, like CH4, natural gas, and biogas with a higher diffusion capability and reactivity are also used in fuel-assisted SOECs. In 2000, the first kind of patent on a natural gas-assisted electrolyzer for hydrogen and syngas co-generation was filed.140 An oxygen ionic conductor is used for the electrolyte, water is introduced into the cathode chamber where it is reduced to H2, and the oxygen ions are then transported through the electrolyte and reach the anode chamber where they react with the supplied natural gas mainly CH4 to form CO and H2; with further injection of H2O, the gas–steam shift reaction takes place and converts CO and H2O to CO2 and H2. Moreover, the oxygen transportation rate can be accelerated by 1–2 orders of magnitude in the presence of a gaseous fuel at the anode.141 The natural gas-assisted process reduced the electricity input by 65% and concurrently improved the H2 yield. However, such a technology has not received sufficient attention, probably due to the immature SOFC technology and the low hydrogen price of the mature technologies. In 2008, Jiang et al.142 reported adopting a perovskite oxide hollow-fiber membrane for the simultaneous production of hydrogen and synthesis gas by combining water splitting with partial oxidation of methane. Pure H2 was obtained in the inner tube of the hollow fiber by H2O decomposition and the oxygen ion is transported to the extern of the fiber to react with CH4 to synthesize syngas due to the oxygen partial pressure difference. In fact, such a reactor is an evolution of the steam-reforming process setup. However, it showed a much higher H2 yield due to the avoidance of the equilibrium reaction and presented higher system controllability since the reactants are separated in the two chambers. Following this work and the increased attention to the energy crisis, extensive efforts have been made in the last decade with a focus on the development of novel electrode materials, like redox stable Sr2Fe1.5Mo0.5O6−δ perovskite, with high activity and durability and alternative fuels to intensify the oxygen transport process to reduce the electricity demand.132,143–145 For example, a hybrid catalyst Ru-LSM/YSZ, in which Ru was used for the oxygen evolution reaction and LSM/YSZ catalytically oxidized the diluted CH4 to form CO/H2, was developed as a bifunctional anode electrode in a CH4-assisted CO2 electrolyzer.143 Such an anode catalyst enables a higher CH4 single pass conversion rate of 75% at only an applied electrolysis current density of 200 mA. Moreover, the products from each electrode chamber can be collected separately and/or accurately mixed to prepare compositionally adjustable syngas, allowing great flexibility for various final products. To further improve the electrode activity and overcome the potential carbon deposition issue under the above conditions, a metal/oxide hybrid was developed through the in situ exsolution process as a symmetrical electrode catalyst for electrochemical reforming of CH4/CO2 in an SOEC (Fig. 5a–d).146 A NiCu alloy was precipitated on the surface of (La0.75Sr0.25)0.9(Cr0.5Mn0.5)0.9(NixCu1−x)0.1O3−δ which greatly favored the chemical adsorption of CO2 (Fig. 5b) and oxidizing reforming activity of CH4, and the exsolved catalyst with a nominal composition of Ni0.5Cu0.5 gave the optimized performance. The exsolved NiCu alloy, the in situ formed active interface (Fig. 5c and d), and the application of CH4 in the anode compartment in FAE reduce the required voltage and the electrode polarization resistance; a maximum electrolysis current density of 2.5 A cm−2 could be achieved at the applied voltage of 1.5 V with the introduction of CH4 gas into the anode chamber, which is double that of the electrolysis cell without the assistance of CH4 fuel (Fig. 5e). Moreover, the H2/CO molar ratio in gas products and the CO2/CH4 conversion rate are highly dependent on the electrode composition, the applied electrolysis current density as well as the composition of inlet gas. An optimized ratio of 2 can be obtained by adjusting the electrode material composition and the cell operational parameters, accompanying CO2/CH4 conversion rate above 90% (Fig. 5f). The constructed electrochemical reforming cells also showed stable performance in a 300 h period and were kept inert for 10 redox cycling tests due to the strong interaction of the exsolved metal with the perovskite oxide parent.
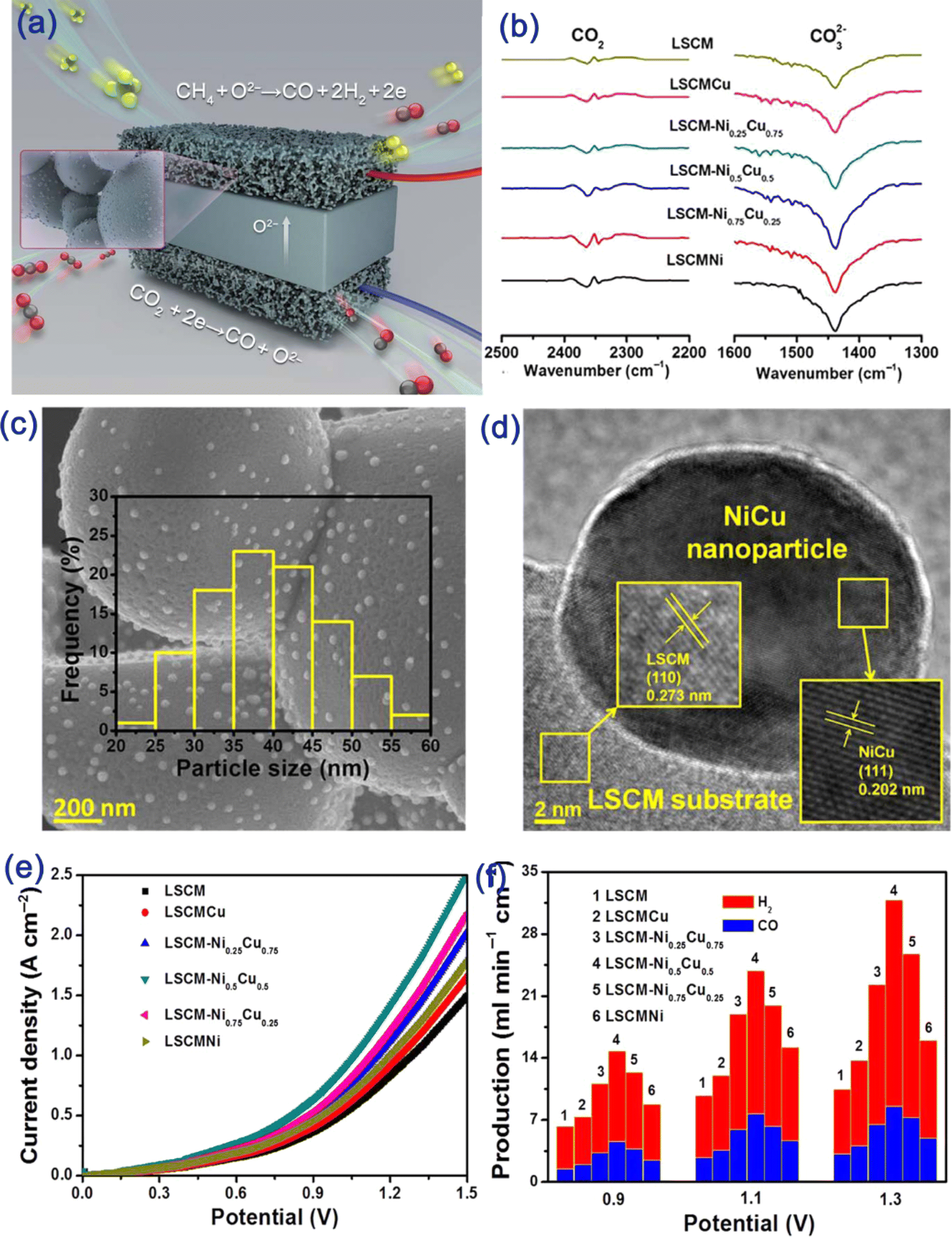 | ||
| Fig. 5 (a) Schematic illustration of the CH4-assisted CO2 electrolysis based on the SOECs with the symmetric electrode, (b) comparison of the CO2 chemical adsorption at 800 °C using in situ FTIR spectroscopy on Ni and Cu co-doped LSCM materials, (c and d) SEM and TEM images of the NiCu alloy precipitation on the LSCM substrate, (e) comparison of the I–V curves and (f) H2/CO flowing rate of the outlet gas in FAE with various electrode materials.146 Reproduced with permission (open access). | ||
The in situ exsolution idea was also employed and optimized for both electrodes in CH4-assisted CO2 electrolyzer cells135 considering that both anode and cathode chambers are in a reducing atmosphere. Rh nanoparticles with a particle size of 2.1 nm and coverage density of 11000 μm−2 were precipitated on the surface by treating under 5% H2/N2 and 600 °C for 10 h, which led to a higher CH4 conversion rate by 3.7–13.4% and gave a molar ratio of 1.8–2.0 for H2/CO. Moreover, such a reactor can be operated in the power and chemical co-generation mode. For instance, when the applied voltage is −0.15 V at 850 °C, the syngas yield is 5.6 mL min−1 cm−2, and enables a power density of 30 mW cm−2; all are remarkably better than that without the Rh nanoparticle precipitation.
Another interesting work adopted Ni-based quasi-symmetrical electrodes for biofuel-assisted water electrolysis.134 The application of biofuel not only increases energy conversion efficiency but also stores abundant biomass energy into valuable fuels. Such an FAE enabled a thermal-neutral operation at the electrolysis potential of 0.36 V, which is above 1 V lower than that of the steam electrolysis alone. To facilitate the biofuel, typically the ethanol, reforming (CH3CH2OH + 3H2O → 2CO2 + 6H2), a channeled anode support was prepared by the mesh-assisted phase-inversion process and 5 wt% Ru/GDC fibrous catalysts were loaded into the well-aligned channeled pores. A recorded high electrolysis current density of 3.0 A cm−2 was obtained at 1.3 V, which were well preserved in 300 h of testing, and no carbon deposition is observed under the electrochemical oxidation conditions, especially with a high O2−/ethanol ratio at the largely applied electrolysis current density.
3.1.1.3.3.3. Electrolysis in tandem with fuel upgradation . The extracted oxygen from CO2/H2O splitting is mainly used for the fuel partial oxidation reforming in the above cases, i.e., the oxygen from H2O/CO2 electrolysis is added to the fuels or converted to the oxygenated chemicals (electrolysis in tandem with fuel upgradation). There is also another typical reactor in which the fuels are converted to upgraded chemicals in tandem with the H2O/CO2 electrolysis, such as from oxygenated alkanes to form alkenes and water. A typical example is the oxidative coupling of methane (OCM) and oxy-dehydrogenation of an alkane to an alkene with CO2/H2O electrolysis for producing C2 compounds (ethane and ethene) at an elevated temperature. In this type of reactor, the cathode activated gaseous H2O/CO2 into O2− species, which permeated through the solid electrolyte; selective oxidation reactions then take place in the anode with permeated oxygen species. Integrating the OCM and CO2/H2O reduction processes in one electrolysis apparatus with full application of valuable O2 and heat management can provide economic and environmental benefits.1 Besides that, the transportation of oxygen species could help in C–H bond cleavage in CH4 and suppress the carbon deposition issues that are not achievable by other methods. Therefore, the effective utilization of methane as a chemical resource has been strongly desired and extensive studies have been performed after the pioneering work by Keller and Bhasin.147 However, it should be noted that a deep oxidation reaction seems to be a more favorable route both from the thermodynamic and kinetic aspects. A key to achieving high C2 yields was to control the type and state of oxygen species1,148–151 for the oxygenation process. Different approaches like a sequential feed of oxygen and methane prefer lattice oxygen involved processes using metal oxide instead of gaseous oxygen and using SEMRs. It has been demonstrated that the electrochemically permeated oxygen showed higher activity and selectivity for the production of C2 compounds over other forms.152–157 For example, the highest C2 yields of 16.7% (11.5% C2H4 + 5.2% C2H6) and 82.2% C2 selectivity, as well as exceptionally high durability in 100 h operation for O2 electrolysis in conjunction with the OCM process using an O-SEMR based on the intentionally constructed Fe-oxide interface in the anode by the exsolution approach, were reported.158 However, when in conjunction with the CO2 electrolysis, a C2 yield of only 5.08% was obtained at 850 °C, which is far from the standard required for commercialization. Recent studies reported adopting a CeO2 single crystal or Ni-doped single crystal as the anode for CH4 oxidation coupling or ethane dehydrogenation with CO2 electrolysis at the cathode.151,157 The application of a single crystal effectively improved the hydrocarbon selectivity without deep oxidation by selectively using the low active lattice oxygen instead of highly active adsorption oxygen.157 The exsolution of Ni in the anode significantly improves the catalytic activity and C2 selectivity.151 With 2.25% Ni doping into the single crystal CeO2, the electrolysis current density reaches 1.2 A cm−2 at 1.6 V. Moreover, a C2 selectivity of up to 99.5% was reported with a CH4 single pass conversion of 7%. To further improve the C2 yield, the focus should also be given to the conversion of alkanes. For example, an active La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF)–Sm0.2Ce0.8O2−δ was developed as the anode catalyst for ethane conversion. The conversion of ethane can be well adjusted by the applied current/voltage. However, the pristine anode showed a little bit of deep oxidation to yield a lot of CO2. By intentionally adding a thin layer of Al2O3, ethylene selectivity can be improved over 90% (Fig. 6a–d). The addition of Al2O3 not only effectively reduced the amount of adsorbed oxygen species, leading to improved ethylene selectivity and stability, but also altered the electronic structure of the Fe active center by forming an Al–O–Fe interface, which increased the density of states around the Fermi level and downshift of the empty band, leading to the enhanced ethane adsorption and conversion. The suggested Al2O3 modified anode shows high durability under 200 h testing under a current of 20 mA and ethane flow rate of 4 mL min−1 (Fig. 6e).
 | ||
| Fig. 6 (a) Schematic illustration of the coupling of electrochemical oxidative dehydrogenation of ethane and CO2 electrolysis into an SOEC and TEM image of the LSCF–SDC anode infiltrated with γ-Al2O3, and electrochemical performance of SOEC under different operational conditions: (b) with different anodes at a current of 20 mA and ethane flow rate is 4 mL min−1, (c) at different currents with the ethane flow rate of 4 mL min—1, (d) at different ethane flow rates and a current of 60 mA on the LSCF–SDC + Al2O3 anode and (e) stability test at a current of 20 mA and ethane flow rate of 4 mL min−1.156 Reproduced with permission. | ||
Different from the above two-chamber configurations, Caravaca et al.152 employed a single-chamber SMER of Pt/YSZ/Ag for water electrolysis in conjunction with an OCM reaction. The influence of the applied current (± values), reaction temperature, and feeding composition on the H2 and C2 production were systematically evaluated. They demonstrated that the possibility of controlling the rate of O2− supply enables the controllability and optimization of the production rate of the desired compounds (H2 and C2). A maximum C2 yield of 8% could be achieved under negative current and an inlet CH4 concentration of 0.6–0.8% at 820 °C. Recently, Kyun Kim and their colleagues further proposed a novel design for the integration of CO2 electrolysis and the OCM process.154 A successive but two-stage zone reactor was designed (Fig. 7a). The first zone of the reactor is a typical SOEC using LSGM as an electrolyte, a SrTi0.3Fe0.55Co0.15O3−δ (STFC) anode electrode, and an STFC-GDC cathode. In this stage, CO2 is split into CO and O2 at 850 °C. The latter is mixed with CH4 to reach the second zone of the reactor, where CH4 conversion and C2 selectivity are favored with the applied Mn–Na2WO4/SiO2 catalyst at 825 °C. The assembled SOEC showed an electrolysis current density of 550 mA cm−2 at the applied voltage of 1.5 V, which enables the production of O2 at the rate of 1.9 mL min−1 cm−2 (Fig. 7b). A C2 yield of 25.4% and a stable short-term (100 h) performance were achieved through the designed reactor capable of independently controlling the operating conditions of two reactions in one apparatus. A combination of efficient SOEC and active catalysts would be a promising method to further improve the C2 yield of such an integrated process.
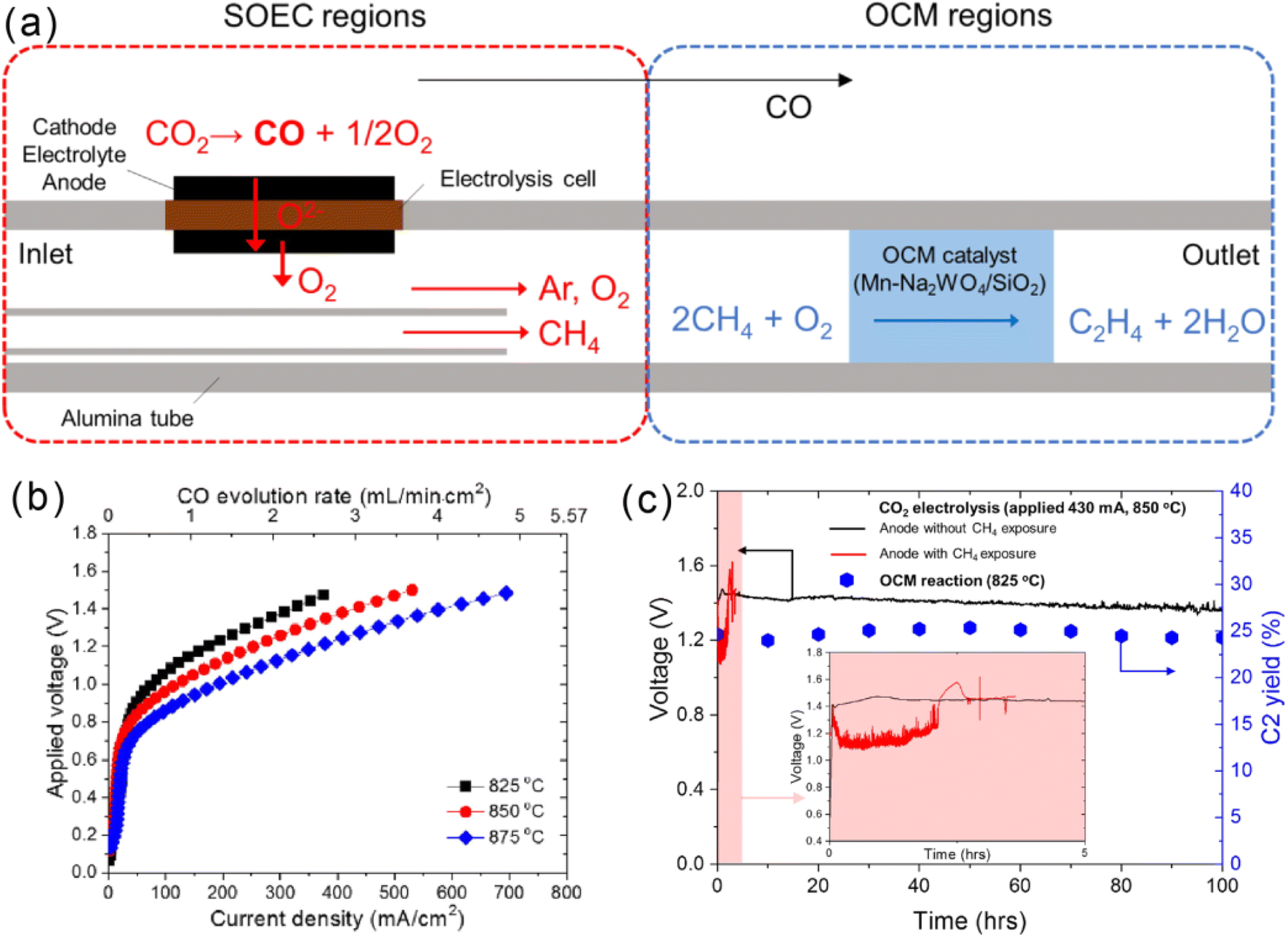 | ||
| Fig. 7 (a) Schematic diagram of integrating the CO2 electrolysis with the OCM process in a successive two-stage reactor design, (b) SOEC performance and (c) durability comparison.154 Reproduced with permission. | ||
3.1.1.4 Other reactions. Besides the aforementioned reactions based on the O-SEMR, the unique separated chambers also present many opportunities for other advanced synthesis.159–162 Recent work reported synthesizing ammonia through the electrochemical reduction of nitric oxide and water using the oxygen-conducting (La0.9Sr0.1Ga0.8Mg0.2O3−δ) SEMRs.160 Generally, ammonia synthesis is achieved through the Haber process at elevated temperatures and chamber pressure. Recently, the low temperature electrochemical process using N2 and H2O precursors was also demonstrated. However, the competitive hydrogen evolution reaction and the strong triple bonds lead to low faradaic efficiency and low yields. In addition, the sluggish reaction kinetics at room temperature hinder its wide application.161 Inspired by those studies, Kwon et al.160 innovatively proposed to use the SOEC with a perovskite oxide cathode catalyst SrTi0.3Fe0.55Co0.15O3−δ-GDC and BSCF-GDC anode to synthesize ammonia using NO and H2O as the reactants at 650 °C. Benefiting from the improved kinetics at the elevated temperature, a recorded electrochemical NH3 production rate of 1885 μmol cm−2 h−1 and a faradaic efficiency of 34.8% were achieved at 1.5 V (Fig. 8), both are significantly larger than that of the low temperature electrochemical approach. This work potentially provides a new approach to electrolysis-based NH3 production from environmentally harmful NO gas. One potential barrier is that the enriching NO with high concentration should be performed since most NO sources from exhaust gas are diluted gas.
 | ||
| Fig. 8 Schematic illustration of the electrochemical synthesis of NH3 from NO and H2O using oxygen-conducting SEMRs and the comparison of NH3 yield with other studies operated at low to higher temperatures.160 Reproduced with permission. | ||
There is also intensive interest in developing SEMRs for the removal of nitric oxide (NOx, including NO and NO2, such a process is called De-NOx),163 one of the typical contaminants in exhaust gas emissions, especially for coal-based power plants and a lean-burn gasoline engine for automotives. Different operational modes use SEMRs for De-NOx, including (1) operating under open circuit conditions, the reducing gas (fuel) is only applied for construction of the electrical field which favors the NOx decomposition to N2 and O2, while no fuel is consumed in this process.164 It is interesting to see that complete emission control for zero pollution of the automotive engines was achieved165 because of the electrochemical promotion of the catalysis (EPOC) effect or non-faradaic electrochemical modification of catalytic activity (NEMCA, will be detailed in Section 6); (2) NOx is reduced in the cathode side to form N2 and O2−, then the O2− will be transported to the anode and oxidized to O2 (ref. 166) and (3) similar to case 2, the obtained O2− in NO reduction in the cathode will involve the anodic fuel oxidizing reaction of chemical fuels like H2, CO, ethane and propane that results in power generation.167 Since there is already excellent review work on the typical De-NOx process,166,168 attention will be given to the NEMCA effect later.
 | ||
| Fig. 9 Publication numbers vs. time in Web of Science database by searching for “Cogeneration” and “solid oxide fuel cells” both in “Topic”, up to Jun. 01, 2024. | ||
3.1.2.1 Advanced fuel electrodes and novel design for fuel oxidation. A distinct advantage over other SEMR technologies, more exactly SOFCs in this section, is their fuel flexibility because of the high temperature operation. The technology is more attractive in the current fossil fuel era, where sufficient conversion of hydrocarbon fuel with reduced CO2 emission is required. SOFCs can directly use hydrocarbon fuel without transitional reforming, with high efficiency, and the CO2 emission can be significantly reduced compared to other technologies. However, for the typical SOFCs, the Ni-based cermet anode cannot work properly due to the super-catalytic activity toward hydrocarbon cracking, leading to the carbon deposition and deactivation of the anode. An alternative anode catalyst is therefore urgently needed.
3.1.2.1.1 Modified Ni-cermet electrode. One of the first approaches is to modify the Ni-based anode catalysts.181,182 For example, the mixture of Ni metal with other metals can reduce the crack capability while not reducing the total oxidation activity.183 The addition of the transition metal to form an alloy can reduce the degree of crystallization of formed carbon, which could be easily removed through reacting with the products from hydrocarbon oxidation reactions, like CO2 and H2O. However, there is always a compromise between activity and anti-coke capability. It is found that catalysts with good water maintenance/adsorption capability can effectively promote the removal of carbon. For example, in 2009, Yang et al.169 reported that the Ni catalyst anode composited with BaZr0.1Ce0.7Y0.2−xYbxO3−δ (BZCYYb), a kind of claimed mixed O2− and H+ conductor, worked effectively with wet C3H8 under OCV conditions and with dry C3H8 under ‘discharging’ conditions, but not with dry C3H8 under OCV conditions. Such unique performance response and experimental phenomena suggested that BZCYYb with the water adsorption capability can effectively remove the thermodynamically unstable carbon through the equation: H2O + C → CO/CO2 + H2 reaction. Moreover, the same functionality also helps improve the sulfur tolerance to 20 ppm. The same group also intentionally loaded BaO islands on the Ni surface through an evaporation deposition method, which formed numerous nanostructure BaO/Ni interfaces to readily adsorb water and facilitate carbon removal reaction with the adsorbed water.184 They experimentally demonstrated that the designed BaO/Ni interface could work stably in dry C3H8 and CO as well as gasified carbon with a current density of 500 mA cm−2 at 750 °C for 100 h, while the Ni-YSZ anode cannot work properly within 1 h. The density functional theory calculations showed that the dissociated OH from the adsorbed H2O on BaO reacts with thermodynamically formed carbon on Ni near the BaO/Ni interface to produce CO and H species, which are then oxidized at the triple-phase boundaries to form CO2 and H2O, achieving the purpose of carbon removal. It is interesting to see that the newly formed BaO/Ni gave higher peak power density over the pristine one when using humidified CO, suggesting another role of the modified anode, promoting the reforming of CO to yield easily oxidized fuels, like H2. To accelerate the application of SOFCs and use the current public infrastructure, not hydrogen, Zhan et al.185 reported attaching an additional catalyst layer of Ru–CeO2 on the Ni-YSZ anode to internally reform octane, a typical high-purity compound similar to gasoline. The resulting cell gave a stable power density of 0.3–0.6 W cm−2 at 770 °C for 50 h. The application of Ru–CeO2 internally reforms 5% iso-octane/9% air/86% CO2 fuel mixtures resulting in a large content of CO/H2/CH4, which is closer to direct oxidation under fuel cell operating conditions and avoids the carbon deposition issue. With the worldwide research tendency to reduce the operating temperature of SOFCs to intermediate to low temperature, the direct application of hydrocarbons meets even challenges, especially for CH4 activation. In 2018, Chen et al.180 adopted a similar idea by coating the Ce0.90Ni0.05Ru0.05O2 (CNR) catalyst on Ni-BZCYYb, which enabled the synergistic thermal catalytic and electrocatalytic reforming of CH4 to H2 and CO (Fig. 10a and b). Integrating the highly active nanofiber-like PrBa0.5Sr0.5Co1.5Fe0.5O5+δ cathode, the assembled SOFCs showed 0.37 W cm−2 peak power density at 500 °C, which is higher than that reported in the literature work under similar conditions, and was comparable to the cell performance fueled by H2. Moreover, the designed cell showed stable cell performance with CH4 fuel for more than 380 h. Both the presence of Ni and Ru elements favor the improved CH4 conversion and H2/CO selectivity (Fig. 10c–e). The application of an additional layer with improved thermal catalytic and electrocatalytic activity enables low temperature high-performance with hydrocarbon fuel, which may accelerate the commercialization of the SOFC technology. The only issue of such cell design is the challenge of current collection if integrated into the fuel cell stack.
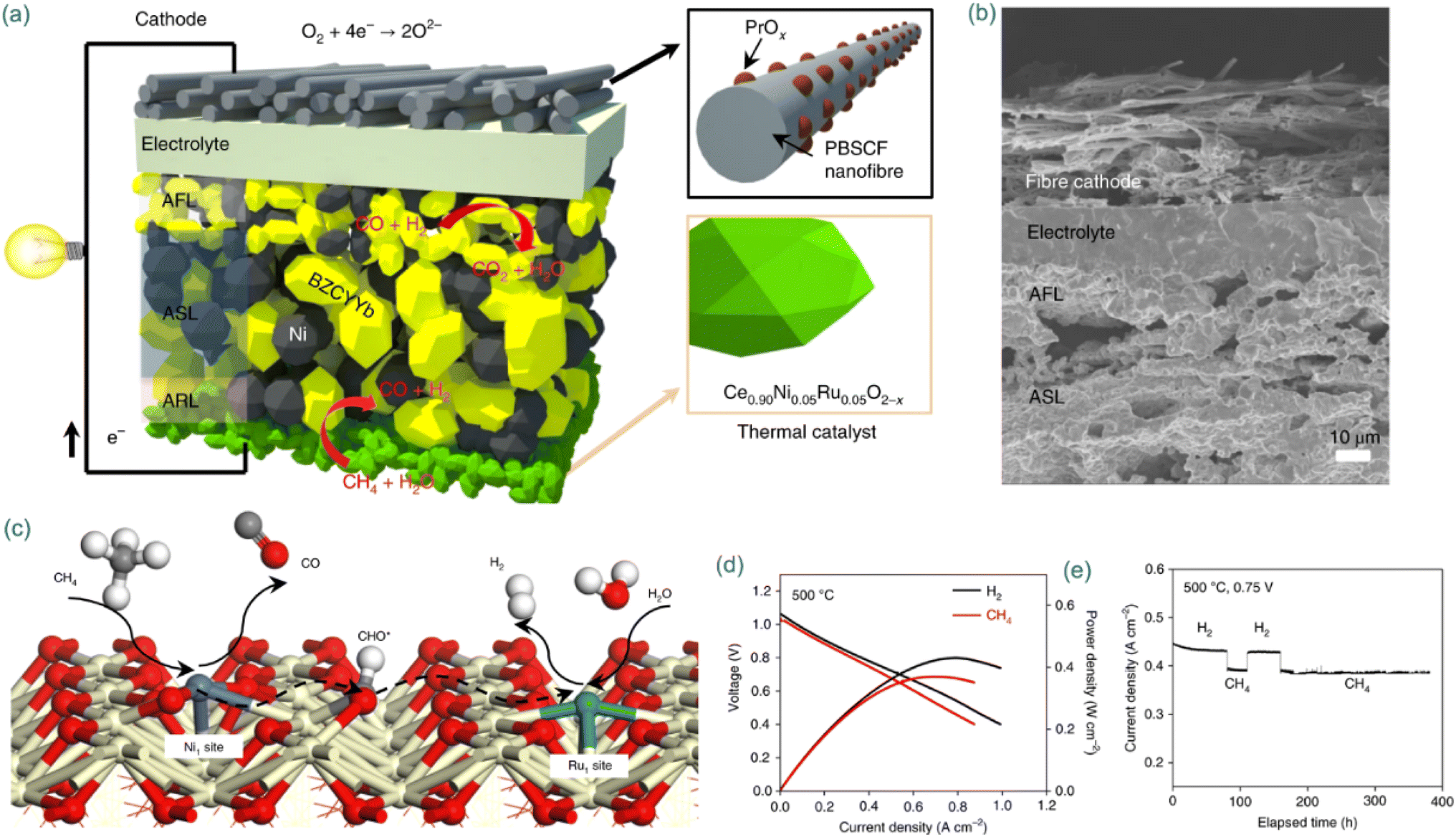 | ||
| Fig. 10 (a) Schematics of a Ce0.90Ni0.05Ru0.05O2 catalytic layer modified classic BZCYYb SOFC, (b) cross-sectional SEM image, (c) the synergistic effect of Ni and Ru element for CH4 and CO2 activation using DFT calculations, (d) fuel cell performance, and (e) durability.180 Reproduced with permission. | ||
Another effective strategy to overcome the carbon deposition issue is to use hydrocarbon + air fuel in a single chamber. For example, Hibino et al.186 reported the operation of a typical SOFC cell unit (Ni-YSZ anode/SDC electrolyte/Sm0.5Sr0.5CoO3) with a mixture of C2H6 or C3H8 and air. It was found that the peak power density reached 403 and 101 mW cm−2 at 500 and 350 °C without the carbon deposition issue. Such an encouraging performance is enabled by the selectively catalytic activity of Ni and Sm0.5Sr0.5CoO3 for fuel oxidation and oxygen reduction, the presence of air in the fuel, and the resulting H2O/CO2 from the electrochemical products help remove the formed carbon. The authors also compared the influence of electrolyte type (YSZ, SDC, and LSGM) on the electrochemical performance. Among these, SDC showed the highest activity toward hydrocarbon oxidation since ceria also acts as a catalyst for electrochemical reactions and YSZ showed the lowest electrochemical performance due to the lowest ionic conductivity.
3.1.2.1.2 Alternative electrode catalyst. One of the solutions to avoid the carbon deposition issues in SOFCs with methane as the fuel is to replace Ni with other metals that favor the oxidation of hydrocarbon without carbon formation. In 1999, Perry Murray first reported adopting a layer of (Y2O3)0.15(CeO2)0.85 (YDC) between Ni-YSZ (current collection layer) and YSZ electrolyte and selecting a suitable temperature in the range of 500–700 °C;187 the carbon deposition issue induced both by the direct CH4 pyrolysis and CO disproportionation reaction is well-treated, and the reduced YDC gave high catalytic activity toward CH4 direct oxidation, which is comparable to the performance of hydrogen oxidation but with a distinct reaction mechanism based on the EIS responses. Such pioneering work demonstrated the feasibility of CH4 direct oxidation with superior performance while being free from coke deposition issues. Following this work, Park et al.178 developed a composite anode of Cu and ceria for the direct electrochemical oxidation of CH4, C2H6, 1-butene, n-butene, and toluene without reforming and they did not result in any carbon formation in the anode. The much-reduced hydrocarbon pyrolysis capability of Cu combined with active ceria yielded the direct oxidation of the abovementioned hydrocarbon into CO2 and H2O as final products. No carbon formation when the cells was operated at 0.12 W cm−2 in dry butane at 700 °C for 48 h. Besides transition metals with reduced activity that are composited with ceria catalyst, a big group of active catalysts, perovskite oxides, that can replace Ni-cermet are developed. Perovskite oxides are famous for their compositional & structural flexibilities and their unique mixed ionic and electronic conductivity, which introduce multiple benefits for direct/non-direct electrochemical oxidation of hydrocarbons. They also showed interesting anti-sulfur poisoning capability, which is more attractive with real hydrocarbon fuels, like municipal natural gas. One of the distinct advantages of perovskite oxides is their redox stability in an anode atmosphere, where the introduced fuel results in a super low oxygen partial pressure, i.e., the reducing atmosphere, while the oxidation reaction products like H2O and CO2 significantly improve oxygen partial pressure at the gas/electrode or active site surface, which leads to the redox cycling of the active site. For a typical Ni-based cermet anode, the big volume change cycle between metallic Ni and NiO causes large strain stress, as well as redox-induced aggregation, reducing the electronic conduction and active site and consequently the anode activity and cell performance degradation. The application of redox-stable perovskite oxide well addresses the above challenges. In 2003, Tao et al.188 first reported that La0.75Sr0.25Cr0.5Mn0.5O3, a chromate perovskite oxide, showed a comparable H2 oxidation activity (ASR of 0.2 cm2 at 900 °C) and stable structure in anode and cathode atmospheres. Such anode catalysts also demonstrated a maximum power density of about 0.2 W cm−2 at 0.5 V at 900 °C for wet (3% H2O) CH4 oxidation with only a trace of carbon detected. Exceptional work by Huang et al.189 identified Sr2Mg1−xMnxMoO6−δ (SMMO), a double perovskite oxide material, as an active and long-term stable anode catalyst for methane fuel. Such a double perovskite oxide shows interesting oxygen ion conduction, much improved electronic conductivity (10 S cm−1) in H2, and well-balanced activity and stability with the highly coordinated Mo in the lattice compared with other reported perovskite oxides. Thus, SOFCs based on this anode gave a peak power density of 438 mW cm−2 at 800 °C in fry CH4 fuel, which is reduced to 338 mW cm−2 with the addition of 3% steam, suggesting the direct oxidation capability of SMMO for CH4 direct oxidation. Moreover, such a double perovskite oxide showed high resistance toward sulfur poisoning and no performance degradation with 5 ppm H2S in fuel. Another typical oxide is the layered perovskite which generally showed higher promise for hydrocarbon oxidation due to the intrinsic charge and structure capability. Work by Sengodan et al.190 fully used those capabilities and first reported the application in direct hydrocarbon SOFCs. They obtained the double-phase perovskite oxide PrBaMn2O5+δ (PBM) by in situ annealing of the oxide precursor of Pr0.5Ba0.5MnO3 in a reducing atmosphere. At 800 °C, layered PBM shows high electrical conductivity of 8.16 S cm−1 in 5% H2 and demonstrates peak power densities of 1.3 W cm−2 at 850 °C using propane fuels; the latter was one of the highest performances at that time. Such an anode also showed encouraging sulfur resistance up to 50 ppm, and the PBM with the Co–Fe catalyst could run stably with C3H8 fuel for 500 h under a current density of 0.2 A cm−2 at 700 °C. In another study, Yang et al.191 used a similar method to prepare a layered perovskite oxide but with a newly formed Co–Fe nano alloy during phase conversion. The resulting Co–Fe/perovskite oxide gave 0.6 W cm−2 in CH4 and 0.94 W cm−2 in C3H8 at 850 °C and showed an anti-sulfur capability up to 100 ppm without a clear poisoning effect. The constructed cells could work stably at 800 °C and 0.6 A cm−2 in H2 with 50 ppm H2S for more than 500 h and 0.2 A cm−2 in CH4 and 0.4 A cm−2 in C3H8 for a total of 150 h as well as 20 times H2–N2–air cycles. The results demonstrated showed that the combination of active transition metal/alloy with phase stable perovskite oxides will be the future approach for high efficiency and direct utilization of hydrocarbon fuel54,192–194 and other functional purposes, and bridging the gap between the current fossil fuel era and future hydrogen economic society.
3.1.2.1.3 Novel cell structure design. The applications of other than Ni based anode catalysts do bring improved anode anti-coking capability, but generally suffers from low fuel cell performance and requires a dedicated cell assembly procedure. As one may see the application of both perovskite oxide and Cu cermet as the anode supporter to the fabrication of a single cell is difficult or impossible because of the coarsening and evaporation issue during high temperature cosintering with electrolyte. A novel cell configuration design is therefore required. Moreover, the application of a porous while active catalytic layer on the Ni-based cermet anode is a good option. However, both the suspicions of carbon deposition under low current conditions and the difficulty of the current collection remain. Different from previous work, Barnett's group reported adopting a conducting oxide material as the cell anode support and built-in barrier layer to construct an anode supported cell with a Ni-cermet active layer for efficient natural gas (85% CH4, 10% C2H6, and 5% C3H8) operation.195 The cell gave a peak power density of over 560 mW cm−2 at 800 °C and ran stably at the current density of 0.2, 0.4, and 0.8 A cm−2 for 9 h, while the conventional cell failed in 6 h. With such a conducting oxide supporter, the single cell also showed high cell operational stability during 7 times redox cycling (first in H2 for 45 min, then in air for 30 min). Sullivan et al.196 also confirmed such results in tubular SOFCs with simulated biogas over 12 days of continuous operation. They explained that the increase in the local concentration of electrochemically produced H2O and CO2 throughout the anode resulted in higher local H2O-to-C ratios which helped remove the formatted deleterious carbon.
3.1.2.2 Hydrocarbons to H2/CO and power. As shown in Fig. 9, the application of SEMRs attracted increased attention during those years. More specifically, as the effective area and the resident time of the reactant in the anode chamber are quite short, the overall conversion of the chemical energy into electricity is impossible. In the practical operation, the fuel utilization efficiency is kept at 70–90% to ensure good electrical efficiency. By the way, electrochemical fuel oxidation is generally a highly exothermic reaction, the produced heat can be well managed to reform the remaining fuel with the produced H2O and CO2, or the remaining fuel can be catalytically partially oxidized in the anode active site, or downstream process to form syngas, with an oxygen ionic conductor, such a power and chemical cogeneration is frequently reported in the single chamber SOFCs (SC-SOFCs). The oxygen or air mixed with the hydrocarbon was co-fed into the SC-SOFCs. The anode and cathode layers selectively catalyze fuel oxidation reaction and oxygen reduction reaction for power generation, and the remaining fuel and oxygen, as well as the resultant H2O/CO2, react further to generate CO and H2. For example, in 2014, Shao et al.197 fabricated and tested the performance and syngas production of a flow-through (first outside of cell then inner of cells) tubular SC-SOFC with a mixed CH4/O2 inlet gas. SC-SOFC presented an open circuit voltage of 1.02–1.08 V at the temperature of 650–800 °C, a peak power density of 300 mW cm−2, and a total power output of 1.5 W was achieved at 750 °C. Single-cell can be operated stably at 700 °C for 10 h at a constant current density of 250 mA cm−2. With an additional GdNi/Al2O3 catalyst on the two sides of the inner tube, the system can realize a CH4 conversion rate of 90.6%, CO selectivity of 95.4%, and H2/CO ratio of 2.04 at a furnace temperature of 800 °C. The same group50 reported the integration of SC-SOFC with a downstream thermal catalytic partial oxidation system for power and syngas cogeneration without greenhouse gas emission (Fig. 11a). A cell configuration of Ni/YSZ/YSZ/SDC/BSCF-SDC was assembled, in which the BSCF cathode showed high activity toward the ORR while poor activity for hydrogen oxidation. The peak power density of SC-SOFCs can reach an impressive value of 1500 mW cm−2 at 700 °C using a CH4–O2 gas mixture. With the integration of the downstream catalytic layer, a methane conversion >95%, CO and H2 selectivity higher than 98%, and a polarization current density independent H2
:CO ratio = 2 were obtained, without any waste gas emission. Though the efficiency of SC-SOFCs is slightly lower than that of double chamber SOFCs, the ideal syngas production efficiency offsets 100%. Both the utilization rate and the yield of H2/CO with a ratio close to 2 are very promising for future deployment.
 | ||
| Fig. 11 Different strategies for CH4-fed SEMRs for power and syngas cogeneration: (a) single chamber-SEMR integration with a downstream catalytic partial reformer50 and (b) thermal and catalytic coupling within the SEMR anode.198 Reproduced with permission from ref. 50 and 198. | ||
The application of SC-SOFCs for CH4 cogeneration resulted in interesting performance and fuel utilization efficiency. However, careful gas (fuel/oxidant mixture) injection should be rationalized to avoid the possible explosion of the CH4/O2 mixture. In contrast, double chamber SOFCs (general one) have distinct advantages; the fuel and oxidant are separated in two chambers; the oxygen can be electrochemically pumped from the cathode to the anode and severed as the oxidant for CH4 reforming in the form of partial oxidation, both the reaction rate and safety can be well controlled. One difficulty in using traditional SOFCs is their limited CH4 gas diffusion capability over H2 (ref. 199) in the well-sintered anode layer. A well-designed anode structure without a CH4 gas diffusion limitation should be developed. In 2020, Dong et al.198,200,201 at Jinan University developed microchannel SOFCs with a loaded catalyst for CH4 fuel conversion to electricity and syngas co-generation through catalytic partial oxidation (Fig. 11b). The microchannel anode was prepared by a mesh-templating phase-inversion method with one end open on the anode surface and the other end terminated by a porous layer, the latter is well-tailored for electrolyte coating and co-sintering. To improve the anode catalytic activity, a CeO2 nanocrystal,200 or a dedicated nano fibrous Ni/CeO2–Al2O3 catalyst198,201 prepared by the electrospinning process is impregnated into the microchannel by the capillary force. The adoption of micro-channeled pores significantly improved the SOFC power output (increased to 2.5 times) with the feeding of 30% CH4/Ar at 600 °C due to significantly improved gas diffusion, i.e., reduction of the concentration polarization. It is demonstrated that a minimum CH4 concentration of 0.35 atm and 0.80 atm should be used in microchannel SOFCs and no-channel SOFCs, respectively. Moreover, they found that the introduction of the catalytic layer into the microchannel remarkably improved the syngas yields by almost by 6 times, and CH4 conversion from less than 40% to above 75% and increase with the increase of CO2 concentration, as well as H2/CO ratio. The final one reached 2 but decreased with increase of CO2 concentration in the anode gas mixture. The micro-channeled SOFC with the catalyst also showed impressive operational stability at a fixed current density of 1.05 A cm—2. The voltage, ohmic and electrode polarization resistance, H2 yield and CH4 conversion rate were stable during 120 h testing.
3.1.2.3 Oxidative dehydrogenation of alkanes. In Section 3.1.1.3, we saw that ethylene is one of the most important building blocks in the chemical industry; the application of solid oxide electrolysis with ethane dehydrogenation brings multiple benefits like overcoming the thermodynamic limitation and the possibility of over-oxidation compared with the conventional steam reforming, high temperature pyrolysis, and direct oxidative hydrogenation methods, respectively. However, additional electrical energy is still required even though it can be adjusted to realize the power and chemical cogeneration at the much-improved temperature. Alternatively oxidative dehydrogenation of alkanes to light olefins is an attractive, low-energy, alternative route which could reduce the carbon footprint of its production.202,203 The direct application of oxygen ions which are pumped from the cathode side with the source from oxygen molecules could still realize such functionality according to the following equations (taking C2H6 fuel as an example):
| Cathode: O2 + 4e− = 2O2− |
| Anode: C2H6 + O2− = C2H4 + H2O + 2e− |
| Overall reaction: O2 + 2C2H6 = 2C2H4 + 2H2O |
Both selectivity and yield will be enhanced by this “oxygen buffer” approach since it can avoids deep oxidation. Oxygen enhances the forward reaction by reducing H2 content, therefore shifting the reaction equilibrium, reducing the side reaction of coking and cracking, and transforming the reaction from an endothermic process into an exothermic one. In 2002, Akin and Lin204 reported an ethene reaction yield of 56% with a selectivity of 80% at 875 °C by using a dense Bi1.5Y0.3SmO3 tubular membrane reactor without catalyst over 11% yield at a steady state in a fixed bed reactor. Bi-based oxide with hetero-elemental doping is a good oxygen ionic conductor with partial electronic conduction under real reaction conditions, which allows the transport of O2− by converting O2 outside of the tubular SEMR reactor and then reacting with the inner C2H6 to form C2H4. The effect of the operational temperature and C2H6 partial pressure on the C2H4 selectivity and yield are carefully investigated. The SEMR operation is much more beneficial because of high selectivity and yield.205 Some other type of membrane reactors based on perovskite oxides with a mixed ionic and electronic conductor is also used and showed interesting C2H4 yield due to the catalytic function of the applied materials.206,207 However, an additional gas separation process is required and most perovskite oxides cannot be stably operated under real conditions, and the total energy efficiency could be further improved. For example, the chemical energy during a partial oxidation reaction which is converted to heat can be managed to produce highly valuable electricity in SEMRs while wasted in other reactors. Therefore, Dogu et al.208 developed such SEMRs and exploited a La and Cl co-doped SrTiO3 anode catalyst for improving the oxidative dehydrogenation of ethane to ethene at 550 and 600 °C. The reactor La0.2Sr0.8TiO3±δClσ/YSZ/LSM-YSZ showed a one-time ethane conversion of 4–14% and an ethene yield of 4–10% at the current density of 0–3.5 mA cm−2, which are 4–10 times higher than that with SrTiO3±δ and 2–5 times better than that of La0.2Sr0.8TiO3±δ. The improved catalytic activity was ascribed to the improved oxygen mobility, an abundance of Lewis acid sites, as well as more Brønsted acid sites as demonstrated by the multiple characterization techniques including DRIFTS, XPS, TPO-CO2, and laser Raman spectroscopy. However, high ethene selectivity is ensured in this work; low ethane conversion due to the limited SOFC efficiency with quite low current density is observed, and optimization of the SOFC efficiency should be managed. Moreover, a high-C compound conversion that easily induces coking at the SOFC operating temperature other than C1 and C2 compounds should be developed. A “best of both worlds” scenario by simultaneously obtaining excellent power output and a high yield of high-C chemicals could be realized. This was addressed recently by Yan et al.25 In their work, thin film O-SEMRs with a configuration of Co7W6@WOx core–shell catalyst loaded (La,Sr)TiO3-YSZ|YSZ|YSZ-La0.6Sr0.4FeO3 (LSF) are prepared. The cell was fed with a H2 and n-butane mixture. Under the optimized cell operating parameters and catalyst design, the reactor reached a peak power density of 212 mW cm−2 at 650 °C, and a C4 alkene (butenes and 1,3-butadiene) yield higher than 50% under a butane conversion higher than 80% was achieved; coke free, are limited CO/CO2 are produced in this SEMRs. The reactor performance can be further improved by employing higher ionic conductive and thinner electrolytes. Similar work by Tan et al.203 reported the adoption of NiFe alloy nanoparticles (NPs) exsolved Pr0.8Sr1.2Ni0.2Fe1.3Mo0.5O6−δ as the electrode for oxidative dehydrogenation of propane. With the gradually exsolved NiFe alloy, the SEMRs reached a propane conversion of 71.4% and light olefin (C2H4 + C3H6) yield of 70.1% under a current density of 0.3 A cm−2 (750 °C) with a propane flowing rate of 40 mL min−1. The cell showed high resistance toward carbon deposition under constant current mode during 100 h of testing.
Another interesting study by Zhang et al.209 reported operating propane fuel in SOFC mode for power and chemical cogeneration. They developed an active CoFe alloy-perovskite oxide catalyst for proton oxidation with LSGM electrolyte. An excellent power density of 0.92 W cm−2 has been achieved with C3H8 fuel (3 vol% H2O) and the SOFC worked stably under OCV and constant current density of 0.4 A cm−2 at 850 °C. Both the 1H nuclear magnetic resonance (NMR) spectrum and infrared (IR) spectrum demonstrated that polycyclic aromatic hydrocarbons were produced in the exhaust stream of the anode, which successfully demonstrated the co-generation system and promising application of SEMRs.
3.1.2.4 Oxidative coupling of CH4 to C2 compounds. The oxidation coupling of methane (CH4 + O2 → C2H4 + 2H2O, ΔG = −69 kcal mol−1) was first proposed by Keller and Bhasin in 1982 using a thermal catalysis method with a metal oxide supported on a-Al2O3 as a catalyst, in which a CH4 conversion of 4% was achieved.147 Later, different groups tried to develop active catalysts to improve the CH4 conversion and selectivity of C2 compounds.210,211 However, as mentioned in the previous sections, significant levels of oxidation parallel products are formed regardless of the kind of oxidant, like CO2 and O2, since the total oxidation of CH4 is thermodynamically favored at high temperatures.212 Compared with thermal catalysis, the application of SEMRs enables the supply of reactants at the desired rate to improve selectivity, shift the equilibrium, and remove the undesired products during the reaction by controlling the electrode potential and external electron flow.213 The controllability of oxygen partial pressure in SEMRs enables higher selectivity of C2 or higher hydrocarbons since the latter follows the half-order dependence on O2 partial pressure while deep oxidation shows first-order dependence. To improve the C2 selectivity, SEMRs can be operated at a low partial pressure of oxygen.214 In particular, for SEMRs, the oxidation of CH4 not only produces electrical power but also yields highly valuable chemicals of C2 compounds,1 which make methane coupling reactions more attractive. In 2005, Kiatkittipong et al.153 found that the SEMRs showed improved C2 selectivity over heterogeneous catalysis. However, the yield of CH4 to C2 compounds is low because of the poor fuel cell efficiency which should be largely ascribed to the low activity of cell components even though the electrode selectivity could reach 96.5%.215 With the quick evolution of novel fuel cell materials and technology, the CH4 conversion has been remarkably improved. In ref. 216, a SOFC tubular design using a Scandia stabilized zirconia (ScSZ) electrolyte, LSM cathode multilayer cell structure with the Na–W–Mn–Ce/SiO2 catalyst integrated was applied. The author demonstrated a 60.7% CH4 conversion and a high C2 selectivity (41.6%), which is close to a C2 yield threshold of 50%.217 Such results demonstrate considerable advantages over conventional heterogeneous catalysis. The results are much higher than that of Appamana's work based on a similar material system.218 Both the application of tubular cells with extended CH4 residence time and high ionic conductive ScSZ electrolyte over classic YSZ electrolyte enables such a much-improved energy conversion and C2 compound yield. It is also interesting to see that a high ethylene-to-ethane ratio of 5.8 was obtained, which highlighted the best performance among SEMRs for the OCM reaction. However, further improvement of the C2 yield looks difficult as the desired products are far more reactive with oxidants than with methane, i.e., the C2 products are easily oxidized to CO2/CO. Also because one-pass CH4 conversion is hard to improve with a limited residence time of the CH4 in the pores of the anode, an integrated gas recycle electrocatalytic reactor and separator system was proposed by Jiang et al.217 in 1994. The C2 products of SEMRs with YSZ electrolyte and Ag-based catalyst were extensively sieved (up to 100%) using an appropriate molecular sieve trap in the cycling loop, then the unreacted CH4 and undesired ethane were recycled to the SEMRs for further conversion. Such a recycling operation with efficient removal of C2 products resulted in an ethene selectivity of up to 88% and CH4 conversion of 97%, i.e., the yield of ethene reached 85% in the final sample.
Besides the experimental operating condition optimization such as temperature and/or pressure, researchers also performed techno-economic analysis on the OCM process in membrane reactors.219,220 Based on the developed 1D membrane reactor model, Cruellas et al.220 found that the increased yield from the optimized operation led to a positive impact on the economics and performance of the downstream separation, which resulted in the cost of ethylene production being 595–625 € per ton C2H4, 25–30% lower than the benchmark naphtha steam cracking process. Moreover, the CO2 emissions of the OCM studied processes are also 90% lower than with the reference, exhibiting better environmental benefits. Those techno-economic analysis results confirm the competition of the OCM process based on membrane reactors over conventional technology, encouraging the efforts to achieve a larger prototype. The authors also mentioned that the price of the membrane is crucial for the OCM scale-up process. In addition, the operational durability as well as the system durability are also important factors in determining the large-scale application potential. Therefore, an effective integration of experimentation, modeling, and techno-economic analysis is of great importance for the future implementation of SEMRs.
3.1.2.5 Other SEMRs for co-generations. Interest is also given to methanol synthesis from CH4 using the SEMRs since methanol is an important industrial chemical, energy carrier, and intermediate product. It should be noted that methane activation should be performed at elevated temperatures, while methanol may be subjected to decomposition at this temperature. The key to realizing methane-to-methanol conversion is to develop an active and selective transition metal oxide catalyst. Torabi et al.221 from Fuel Cell Energy Inc. developed an intermediate temperature anode-supported SOFCs based on sprayed GDC-Li2CO3, LiNiO2, and doped SrTiO3 as the electrolyte, cathode, and anode, respectively. The composite electrolyte showed 100 times higher ionic conductivity over doped ceria alone. To screen active catalysts, they employed the tubular fixed reactor. The initial results demonstrated that the main product using SrTiO3 is methanol with selectivity over 90% over the temperature range of 300–600 °C. When the CH4 flow rate is in the range of 70–100 mL min−1, the complete conversion of CH4 is achieved within 1 minute. The application of lattice O2− instead of molecular O2 is believed to induce a superior performance. Another interesting application of SEMRs for fuel and electricity co-generation from a C1 precursor is reported by Neophytides et al.222 They developed an Ag symmetric cell based on YSZ electrolyte at atmospheric pressure and temperature of 547–697 °C. Both the cell current density, the methanol partial pressure with He on the CH3OH conversion, and CO, CO2, and HCOH were investigated. The cells gave an open circuit over 1.0 V. It was demonstrated that the selectivity to HCOH is around 90% at methanol conversions ≥30%. These results confirmed that Ag is a selective catalyst for the targeted reaction and SEMRs can serve as a good platform for high-efficiency conversion C1 chemistry.
Similar to the previous discussion, the application of SEMRs is also highly interesting for higher hydrocarbon conversion even though the selectivity to the targeted chemical is much lower than that of low carbon-containing hydrocarbons due to the mismatched operating temperature for reaction and missing active catalyst for the selective reaction.223 In 2009, Ji et al.224 reported adopting MoV0.3Te0.17Nb0.12O as an active and conductive anode catalyst for propane selectivity to acrylic acid at the temperature of around 400 °C. When combined with the low temperature high ionic conductive Bi4Cu0.2V1.8O11−δ electrolyte for SEMRs, the cell gave an open circuit voltage of 0.7 V and peak power density of 10 mW cm−2 at 420 °C using the C3H8:12H2O:15He mixed gas. Moreover the C3H8 conversion was around 3.0–4.7% with an acrylic acid selectivity of 67–72.9%. Such results demonstrated the feasibility of SEMRs for application in high hydrocarbon conversion and combination with electricity conversion. One more interesting work reported by Zhang et al.209 shows that ever macromolecular compounds such as polycyclic aromatic hydrocarbons (PAHs) can be synthesized by accompanying high fuel-to-electricity conversion efficiency. A Co–Fe nano alloy exsolved Pr0.4Sr0.6Co0.2Fe0.7Nb0.1O3 perovskite material developed by the authors' group was used as a catalyst and LSGM as the electrolyte, operating at 750–850 °C using 3% H2O humidified C3H8 as fuel. The cell presented a peak power density of 0.33–0.92 W cm−2 and worked stably under both conditions of OCV and current density of 0.4 A cm−2 for a total of more than 80 h. The analysis of liquid by-products in the exhaust stream by combined gas chromatography-mass spectrometry, IR, and NMR showed that it is a mixed PAH.
Previous attention is mainly given to low carbon-based fuels or chemicals for efficient energy conversion and storage. SEMRs are also widely adopted for other related chemicals like NH3, HCN, NO, H2S, and SO2 synthesis or abandonment or used for environmental treatment, which has a significant role in economic and environmental impacts, which we think will promote the commercialization of such a promising technology. Since the pioneering work by Vayenas and Farr et al. on the use of SEMRs (Pt/YSZ/Pt) to directly convert NH3 to NO with yields ≥60% and simultaneously generate electric energy at 900–1200 K, this field has received increased attention with much-improved progress.225 Another example of the use of SEMR is the synthesis of HCN from the methanol/methane ammonia steam mixtures (2CH4 + 2NH3 + 3O2 → 2HCN + 6H2O).226–228 The SEMR adopted an active Ni–CGO–iron antimony oxide catalyst and was operated in the temperature range of 500–650 °C. Under the optimized cell operational conditions, a maximum yield of 40% (to methanol input) and selectivity for conversion of methanol to HCN of 47.5% was achieved with CO2, N2, H2, and CH4 as the side products. It was also found that the HCN was obtained by ammonolysis of methanol instead of electrochemical ammonia oxidation; the H2 produced by the side reactions was oxidized to provide the electricity, and the electrochemical supply of oxygen to the fuel side had a positive effect on the cell selectivity to HCN. In those studies, the electricity output was relatively low. However, their promoted role in targeted sample synthesis is quite huge, as claimed by EPOC or NEMCA, which will be discussed in Section 6. The H2S, SO2, and/or NOx are also used as oxidants for simultaneous greenhouse gas treatment and clean energy generation.229–232 In 2016, Li Kang developed multi-channel tubular SOFCs and ran them with N2O instead of air as the oxidant.229 An increase of 50% in power density was observed over that with air oxidant, such an integration suggested techno-economic feasibility to eliminate the cost penalty for N2O abatement and yield highly valuable electrical energy.
3.1.2.6 Direct carbon fuel cells. One of the advantages of SOFCs is their fuel flexibility. SOFCs fueled with solid state carbon instead of gaseous and liquid fuel received particular attention because of the widely available carbon-based fuel and the >100% theoretical thermodynamic efficiency as well as the high energy density.233,234 The major challenge in using carbon-based fuel is to improve the fuel–catalyst contact sites or continuous fueling like gaseous or liquid fuel. Another challenge is the reduced real efficiency because of the emission of thermodynamically favored CO in the anode direct oxidation reaction as well as the Boudouard reaction (C + CO2 → 2CO), even though the performances of most DCFCs rely on CO oxidation, instead of the direct carbon oxidation. Nevertheless, this may suggest the chemicals (CO) and electric energy co-generation from this point of view (Fig. 12a). Prof. Liu's group from the South University of Technology performed a systematical investigation of such a co-generation system from active electrode material development,235 carbon fuel precursor selection, electrochemical and thermal catalytic mechanism, small stack construction, demonstration,233,236 and modeling.237 Tubular DCFC single cells that unite with one close end or 2-cell stack were constructed by the dip coating technique (Fig. 12b). Then they were assembled into DCFC devices using LSM-YSZ as the cathode and Ag-GDC as the anode. Ag is an active catalyst for CO oxidation. Activated carbon with a 5% Fe catalyst to catalyze the Boudouard reaction is used as fuel. Such a DCFC 2-cell stack gave an open circuit voltage of 2.11 V and a peak power density of 319 mW cm−2 at 800 °C, which is close to that with hydrogen as fuel (Fig. 12c), suggesting the high activity of the applied catalyst and suitable cell structure. When the cell is operated at a fixed current of 2 A, the 2-cell stack can be operated continuously over 1.1 h. During the oxidation period, the peak CO yield can reach 25 mL min−1 though it gradually reduced with time. In contrast, CO2 concentration increased with the applied time (Fig. 12d). The authors also carefully calculated the electrical energy conversion and overall conversion efficiency for three cells in single-cell configurations that were operated at different applied currents. Generally, the former is proportional to its operating voltage while inversely proportional to its operating current. When the current is 1 A, the electrical conversion efficiency could reach 40% and the overall efficiency could be over 70% (Fig. 12e).235
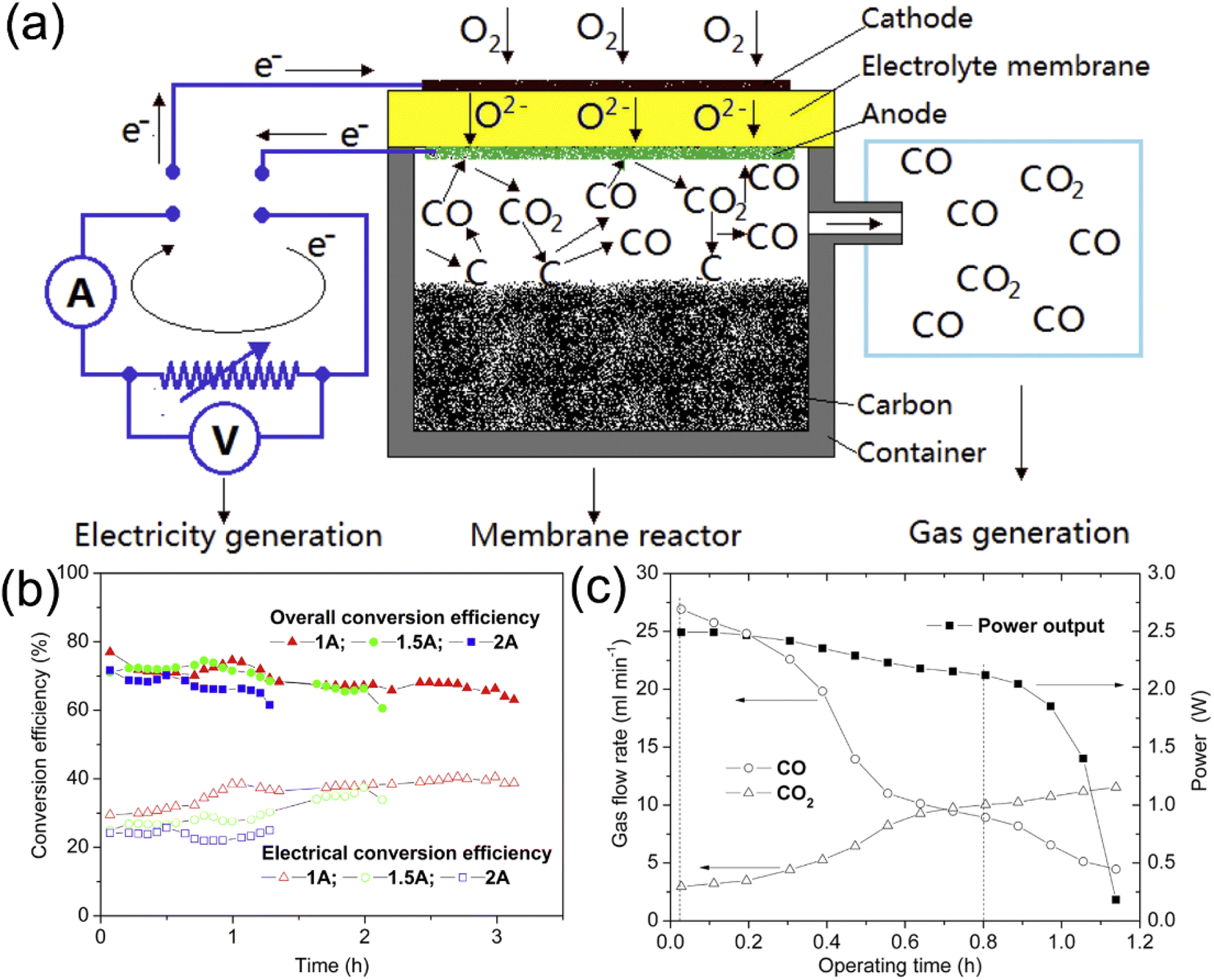 | ||
| Fig. 12 Schematic illustration of electricity–gas cogeneration in DCFC: (a) single cell and (b) the corresponding exhausted gas composition and power response to the operating time of 2-cell stack operated at 2 A, at 800 °C and (c) electrical efficiency and overall conversion efficiency of the cells operated with different current at 800 °C.235 Reproduced with permission from ref. 235. | ||
Considering that the electrochemical performance of DCFCs operated at elevated temperatures like 800 °C is contributed by the CO oxidation nature and currently most DCFCs are constructed on small size cells or tubular cells with special sealing, the same group invented a compact and seal-less direct carbon SOFC stack for potential application.236 The SOFC stack is composed of a 12-array cell on one side of a large-scale single YSZ electrolyte plate in series, leaving the anode exposed but without direct contact with the carbon fuel. Each opposed cathode and anode constitute a cell unit, thus there are 12 unit cells on the single electrolyte. When the cells are operated using activated carbon or wood powder (or sawdust) made from a Bauhinia tree branch as carbon-rich fuel with Fe loading, an OCV value of 10.8 V, peak power of 10.2 W and a discharge energy of 29 W h were achieved on the activated carbon fuel, and they were increased to 11.6 V and 13.1 W with the Bauhinia tree branch wood powder fuel. A similar cell design but in the form of a tubular structure was also used by the same group to make a rechargeable carbon air battery,233 which makes DCFC more adaptive in practical applications. Furthermore, numerical modeling was also performed to understand the physical/chemical processes of the CO and electricity co-generation in DCFC.237 The 2D model confirmed the feasibility of a non-contact design of carbon/anode, and a large distance between the carbon fuel and the porous anode still could give a good electrochemical performance. The DCFC gave a reduced performance at the reduced temperature mainly ascribed to the low Boudouard reaction kinetics. With careful control of the operational parameters, like the temperature and applied cell current, the molar fraction of CO can be well controlled. It is also found that the higher the cell current, the lower the CO generation. Thus, a commonly used anode configuration resulted in improved DCFC performance but decreased CO fraction in the exhausted gas. Thus, such a DCFC showed promising application in CO-electricity application.
The DCFC performance is contributed by CO oxidation; how to effectively utilize the CO from the oxidation reaction and Boudouard reaction is therefore investigated since most CO is released during the testing. In this regard, Prof. Shao from the Nanjing University of Technology developed a combined solid oxide YSZ electrolyte type based DCFC with CO2 permeating membrane to improve electrochemical performance and carbon utilization efficiency. The CO2 permeating membrane is made up of the doped ceria–carbonate composite, which keeps CO2 at a suitable concentration but confines all the unreacted CO in the anode chamber, which favors the CO oxidation kinetics and utilization efficiency and subsequently improves DCFC performance. Such a DCFC presented an OCV of 1.056 V and a peak power density of 279.3 mW cm−2 at 850 °C. Moreover, a 2-cell stack was also built and delivered continuous operation for 200 min at a constant current density of 300 mA. However, it should be noted that the carbon utilization efficiency is still only 14.36%, which is mainly ascribed to the de-activation of catalysts.238 The same group then tried to fix the active Boudouard reaction catalyst FemOn (active component)–K2O (promoter) first on Al2O3 and pre-sintered at high temperatures. The modified DCFC showed similar performance to the previous one but with a much improved continuous operational time of 314 min at 750 °C and a high carbon utilization efficiency of up to 98.7%, which is ever the highest one in the literature.239
People also tried to improve the DCFC performance by increasing the contact area of the carbon fuel with the anode catalyst by creating a nanoporous electrode238,240–244 or using a redox mediator,245,246 especially at the reduced temperature where the effect of the Boudouard reaction is not so significant. The poor point-like solid–solid contact not only restricts the electrode kinetics but also does not favor electronic conduction. In this context, hollow nanofibers of Ce0.6Mn0.3Fe0.1O2 by the electrospinning method,244 Cu-modified Ni foam,240 and honeycombed porous, carbon fuel size-matching architecture anode243 with a three-dimensional open structure were developed. These 3D anodes not only increased the contact area with carbon and CO but also facilitated mass and electron transport, which significantly improved the DCFC electrochemical performance. A peak power density of 765 mW cm−2 was obtained based on the carbon fuel size-matching architecture anode at 800 °C, which is one to two times higher than the previous cases. Besides the development of an active electrode with optimized cathode microstructure, in 2011, the Gorte group from the University of Pennsylvania reported adopting a molten Sb2O3/Sb redox mediator for DCFCs.245 In such a novel design, fuel cell performance is realized through the anode oxidation of metallic Sb at the electrolyte interface, producing Sb2O3, that is then reduced by the fuel in a separate step in the anode. In other words, the chemical energy of carbon is first stored in molten Sb, then further oxidized by the O2− from the cathode. Therefore, the overall cell open circuit voltage is ascribed to the Sb oxidation, not to carbon oxidation anymore. It is worth noting that the Nernst potential of the Sb–Sb2O3 mixture is only 0.75 V, which is a little bit lower than that of carbon oxidation. However, the electrode resistance associated with Sb fuel is impressively low, approximately 0.06 Ω cm2 because of the molten state of fuels, which has close contact with the anode catalyst. Therefore, the peak power density reaches 350 mW cm−2 with an electrolyte-supported cell made from ScSZ on different carbon fuels, like sugar char, rice starch, carbon black, and graphite, and the redox moderated DCFC showed improved operating stability and service life compared with previous cases. Another interesting method reported by Jiang et al.246 who proposed and demonstrated a hybrid molten carbonate/solid oxide direct carbon fuel cell, in which molten carbonate is mixed with carbon and refined in the anode chamber, the liquefied “fuel in molten” effectively reduces the electrode polarization resistance of the anode. With an optimized carbonate–carbon composition, the hybrid DCFC showed a maximum power density of 390 mW cm−2 using an LSM cathode, which was further improved to 878 mW cm−2 using an LSC cathode under flowing air, comparable to extant fuel cell technologies. The exceptional DCFC electrochemical performance makes a solid step toward the wide application of DCFC.
Exceptional cases of the direct oxidation of carbon in DCFCs should be highlighted. Inspired by the nanoporous/size matching anode catalyst and microstructure development, as well as the “liquid fuel” concept, Ding et al.241,242 developed a highly efficient, 3D solid-state architectured anode which was templated by fabric textile coupons and combined with ceria–carbonate, a low temperature superior ionic conductive electrolyte to directly oxidize carbon fuel below 600 °C. DCFCs showed maximum power densities of 143, 196, and 325 mW cm−2 at 500, 550, and 600 °C, respectively. The cell also operated steadily with a rated power density of ≈0.13 W cm−2 with a superior carbon utilization efficiency of over 85.5%. Moreover, the peak power density was continuously improved to 392 mW cm−2 at 600 °C based on the same cathode catalyst but the optimized microstructure of 3D ceramic textiles. The DCFCs with direct oxidation of carbon to CO2 while without going through intermediate fuel CO can remarkably improve energy conversion efficiency, which should become a future research interest in the academic and industry field.
3.1.2.7 Solid oxide metal–air batteries. Similar to the fuel cell, metal–air batteries received increased attention in recent years due to their high energy storage density and capability to perform fast and deep charge/discharge cycles.247 This has positioned the metal–air battery as a front-runner in the commercial development of large-scale energy storage devices. However, the current metal–air battery system is mainly investigated at room temperature or in the liquid electrolyte solution. The large electrode polarization resistance induced by the oxygen reduction remains the major challenge and it requires high loading of the precious metal catalyst. In the previous section, we showed that the SOCs operated at elevated temperatures showed much-reduced electrode and total polarization loss. So, metal–air batteries operated at an elevated temperature should hold a higher potential for energy conversion and storage. In recent years, metal–air batteries, like Fe–air,247–251 Mo–air,252 Si–air,253 W–air,254 Mg–air,255 Li–air,256 and Sb–air batteries257 have been reported, in which Fe–air batteries received particular attention because of the abundant and non-toxic nature of Fe fuel. In 2011, a redox flow battery concept based on the solid oxide membrane was first demonstrated.247 Such a battery comprises two key decoupled components: a reversible solid oxide cell (RSOC) and a metal/metal oxide redox couple. In a typical operation, Fe is first loaded on the surface of the anode, when water/steam is introduced and it reacts with Fe to form/release H2, and Fe is oxidized to FeOx. The resulting H2 is then electrochemically oxidized to generate electricity in SOFC mode, i.e., the discharging process in a battery. When all Fe is oxidized, the discharging process of battery is stopped; then the discharging process is initiated. H2O is recycled and electrochemically split into H2, which is fed into the anode chamber to reduce FeOx, regenerating metallic Fe to close the redox cycle. When considering the whole process, the overall reaction is the reverse reaction 2M + xO2 ⇔ 2MOx (Fig. 13). Since the flow of reaction gas resembles the flow of electrodes in a conventional liquid Redox Flow Battery (RFB), this is termed a “Solid Oxide RFB (SORFB)”.247 Inoishi et al.248 also demonstrated such an RFB with H2O/H2 as a redox mediator. Such a novel RFB can produce an energy capacity of 348 W h per kg-Fe and a round-trip efficiency of 91.5% over twenty reliable charge/discharge cycles, demonstrating the feasibility and promising further deployment. One critical issue with SORFBs, similar to that of DCFCs, is the limited O2− transportation from the electrode to FeOx. If directly oxidized, the reaction product FeOx is a semiconductor with low electronic conductivity, which will hinder further reaction of the remaining Fe. So similar to the redox media-assisted DCFC, gas-mediated SORFBs received increased attention.249–251,258,259 Another consideration of a solid oxide Fe–air battery is improving reversibility and durability. Huang proposed multiple strategies in this aspect.249 For example, iron carbide replaced the metallic Fe. The reaction product while using iron carbide is CO and CO2, which will create a unique porous structure to improve the reaction kinetics; the excess carbon in the anode can act as additional active fuel to engage in the redox reaction for additional storage capacity. With H2O/H2 mediation, the battery was continuously cycled at a current density of 10 mA cm−2 for 10 consecutive cycles (each for 10 min), producing a constant discharge specific energy of 1258 W h per kg of Fe, which is 93% of the theoretical specific energy. The calculated round-trip efficiency is 83.3%. The cell also showed good stability for more than 100 cycles at 550 °C and j = 10 mA cm−2. To further address the slow kinetics in iron/iron oxide redox couples, the Pd catalyst259 and a combined proton-conducting BaZr0.4Ce0.4Y0.1Yb0.1O3 (BZC4YYb) and reduction-promoting catalyst Ir251 were developed to improve the H+ diffusion and FeOx-reduction kinetics. When the battery operated under 550 °C, 50% Fe-utilization, and 0.2C, it exhibited a discharge-specific energy density of 601.9 W h per kg-Fe with a round-trip efficiency of 82.9% for 250 h of a cycle duration of 2.5 h; at a reduced temperature of 500 °C, the same battery exhibits 520 W h per kg-Fe discharge energy density with an RTE of 61.8% for 500 h. Such exceptional durability and reversibility are much higher than those of a 10 h duration energy storage target that can only be marginally achieved using the most dominated energy storage technologies, like pumped-hydro storage, redox flow batteries, lithium-ion batteries, and sodium–sulfur batteries.251 Further work from the same group recently demonstrated the feasibility of such a novel rechargeable battery for applications requiring both large current density and fast charge/discharge cycle capability through improving the FeOx-to-Fe reduction kinetics and reducing the RSOC high electrode potential.258 With the adoption of the classic three-electrode configuration, the polarization resistance contribution of the oxygen evolution reaction and other sources are well deconvoluted and determined. Combining all the improvements made in battery materials including the FeOx–Fe reduction kinetics, the group can realize 12.5 cycling with a discharging specific energy density of 625 W h per kg-Fe and a round trip efficiency of 87%, such a big step may move this RFB a step closer to practical applications. Further scaling of the solid oxide Fe–air battery may realize longer stability, like daily, weekly, monthly and even seasonal cycles.
 | ||
| Fig. 13 (a) Cell structure, working principle and cycling performance with different anode fuel materials at UFe = 50% and 0.2C (10 mA cm−2) at 550 °C (b) and 500 °C (c) of a solid oxide iron–air battery.251 Reproduced with permission (open access). | ||
 | ||
| Fig. 14 (a) Schematic illustration of an RSOC, comparison of (b) voltage and (c) impedance spectra responses of an SOC during a constant-current electrolysis test, and a reversible cycling test.71 Reproduced with permission. | ||
3.2 SEMRs with proton conductors
SEMRs with oxygen ions are characterized by high temperature operation due to the large ionic radius, atom weight, and two negative electrons. The high temperature operation leads to quick kinetics. However, it also brings the challenges of strict material requirements and subsequent high operational costs. Another additional drawback is the low degree of coupling with other thermal, chemical, and electrochemical processes. For example, the co-electrolysis cell with H2 and CO co-generation with optimized volume ratio could be further in situ utilized to produce hydrocarbon fuel in one step through the F–T process. However, the thermodynamically favored temperature range for F–T process is 300–500 °C, much lower than that of the efficient electrolytic temperature. Direct integration of the exothermal F–T process and endothermic electrolysis is favored and the integration will also mean reduced reactor and space occupation.In 1981, Iwahara first observed proton conduction in oxide materials and demonstrated their application in solid oxide cells.30,262 In 2002, the principle of an H2 pump to purify the H2 gas using proton conductive SrCe0.95Yb0.05O3−δ at 800 °C was demonstrated by Matsumoto.263 Those studies open a new research field.264 However, in the following years, the research progress on H-SEMRs (Fig. 15) was much lower than that on its counterpart, the oxygen ionic conductor, due to the lack of stable electrolytes and efficient cathode materials,66 even though it has higher electrochemical conversion efficiency since water is produced in the cathode in the power generation mode or water is mixed with the oxidant in the electrolysis mode, while the fuel, hydrogen, will not be diluted and it can be directly recycled if properly pressurized and electrochemically compacted.15,265 However, the research activity of H-SEMRs is comparable because of the significant room for process integration, especially for the combined heat and power cogeneration through the hydrogenation or dehydrogenation reaction by adding or stripping hydrogen from organic compounds (CH4 to C2H4–6; C–C to CC; CC to C–C),47 and electrochemical ammonia synthesis,266 CO2 conversion267 and electrochemical hydrogen pressurization15 because of the matched operating temperature and reaction kinetics. Remarkable progress in last two decades has been made in the advanced proton conductor development,169,268 active electrode materials,269,270 in particular, the triple conductive materials,20,21,171,269,271 quick evolution of the cell fabrication technology,171 and cell interface optimization,173,272 which made the H-SEMRs more reliable and more efficient;66,273,274 these related research areas have become hot subjects/topics in the energy, environmental and chemical industrial field.
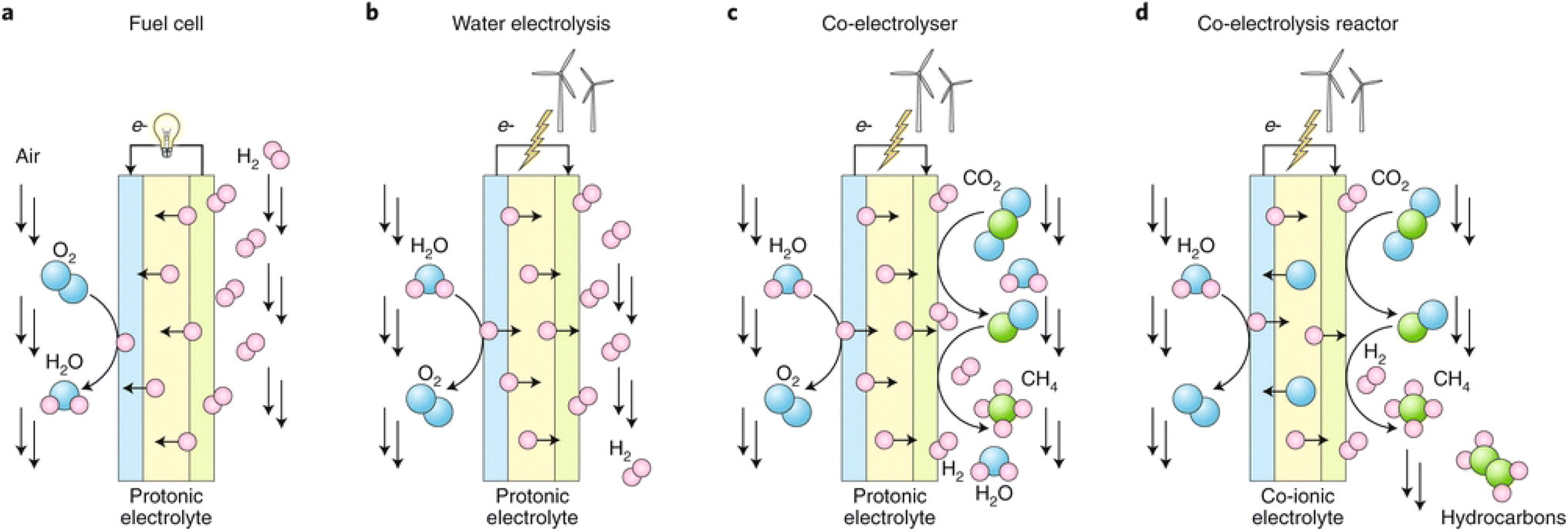 | ||
| Fig. 15 The different working principles of H-SEMRs: (a) fuel cell, (b) electrolyzer cell, (c) CO2/H2O co-electrolysis, and (d) co-electrolysis reactor based on co-ionic conductor.66,265 Reproduced with permission. | ||
It should be noted that efficient proton conductors and active electrode materials are pre-conditions of efficient SEMRs. Much progress has been made in those aspects.275–277 Prof. Liu Meilin's group from Georgia Institute of Technology contributed enormously to the development of active oxide proton conductors. In 2006, they first reported BaZr0.1Ce0.7Y0.2O3−δ as an active proton conductor for low temperature SOFCs which showed the highest ionic conductivity among known electrolyte materials.277 The same group later revealed the doping of Yb in the above composite and realized hybrid ion (H+/O2−) conducting materials in a single-phase oxide. The unique hybrid ionic conduction property grants the SOFC high anti-coking and sulfur poisoning capability.169 Another exceptional case of active electrolyte material exploration is revealed by Zhou and his colleagues, they found that a typical electronically conductive material SmNiO3 was changed with active proton conductor materials through H+ filling-controlled Mott transition.268 However, the compromise of the electrolyte materials' conductivity and stability and the lack of active electrode material reduce their potential. Until recently, O'Hayre developed a combined strategy of developing a reactive active sintering technique and infiltrated active electrode material BaCo0.4Fe0.4Zr0.1Y0.1O3−δ for superior performance and durable PCFCs, reaching 455 and 142 mW cm−2 at 500 °C in the case of hydrogen fuel and CH4, respectively, as well as more than 1400 h performance durability.171 Such a breakthrough work renewed the interest and activity of H-SEMR fields. With further extensive work on scaling up cell fabrication273 and addressing the interface properties, fuel flexibility,21 poisoning-induced durability issues173,272,276 as well as reversible operation,278 H-SEMR or electrifying chemistry with protonic cells23 made significant progress in superior energy conversion efficiency20,21 and targeted reactions or chemical generation yields.47,279 Since high-performance PCFCs are not the major concern of this work, readers are suggested to refer to the related reviews on this subject, we will focus more on the chemical generation or system integration in SEMRs using proton conductors.
3.2.1.1 Chemical and/or power cogeneration.
3.2.1.1.1 Alkane to alkene. Light olefins such as propylene and ethylene have drawn worldwide attention since they are the most important raw materials in the petrochemical industry. For example, they are the major intermediate chemicals for polymers, their global demand growth is estimated to be 4–5% per annum. While the typical manufacture is based on the steam cracking of hydrocarbons from fossil fuels on a large scale, both the low yield and large CO2/NOx gas emissions as well as the energy-intensive process are the major barriers. The oxidative dehydrogenation of alkanes has been researched intensively.280 However, it is not easy to control the degree of deep oxidation of the precursors to CO2 and thereby achieve high ethylene selectivity. The application of H-SEMRs allows easy control of the reaction rate and overcomes the deep oxidation issue as well as the chemical equilibrium limitation, which has become one of the hot research topics although it is still in the early scaling stages (Table 2). Since 2007, a research group led by Prof. JingLi Luo performed extensive work on the dehydrogenation process from ethane and propane to propylene and ethylene using H-SEMRs by developing active anode catalysts, studying the carbon deposition phenomenon and dehydrogenation mechanism, to improve the process efficiency and operational stability with the final target of putting them into application. In their first run C3H8–O2 H-SEMRs with BaCe0.85Y0.15O3 and Pt symmetric electrode at the temperature range of 550–650 °C281 achieved a selectivity of C2H4 and C3H6 higher than 60% and 20%, respectively at a conversion rate of up to 40%. Moreover, the migrated H+ can react with cathodic O2, generating a peak power density of 35 mW cm−2 at 650 °C. Such pioneering work demonstrated the feasibility of H-SEMRs for light olefin production from an alkane. In parallel, they investigated the possible carbon deposition issue during propane dehydrogenation with the newly developed electrode catalyst, Cr2O3.282 They found that the carbon deposition under fuel cell ‘discharging’ conditions was much less than that under OCV conditions, the former can be further reduced with the addition of the K promotor. Careful adjustment of the ionic conductivity of the electrolyte through optimization of sintering parameters, precursor particle size, and application of active catalysts, like nano size Cr2O3,283 MoCx,284 Cu–Cr2O3,285 Cr3C2-WC,286 and Co2CrO4 (ref. 287) or optimizing the anode/cell microstructure/configuration283,288 was beneficial for the efficiency and fuel conversion.289,290 A typical example is that this group developed a metal/alloy-perovskite oxide composite anodic catalyst for H-SEMR feeding with ethane.194,291 The CoFe alloy nanoparticles were well socketed on the surface to perovskite oxide parents after being annealed in a reducing atmosphere. The SEMR based on such a novel anode catalyst gave a peak power density of 348.84 mW cm−2 at 750 °C with ethane fuel. Moreover, high ethylene yields of up to 41.5% and a selectivity of 91% as well as no CO2 emission were achieved. Durable testing over 100 h with a current density of 0.65 A cm−2 was also demonstrated. A critical challenge to such high temperature H-SEMRs is their low efficiency, as you may see from the above case that the yields are mostly lower than 50%. To address the above challenge, Yan presented an ingenious combined approach of nano engineering of TPB by loading Co7W6@WOx core–shell nanoparticles on an anode matrix and co-feeding of hydrogen with the chemical feedstocks.25 They reported that the yield of butenes and 1,3-butadiene from the n-butane dehydrogenation reaction at 650 °C reached 52% with a selectivity of 93.7%. Besides the simple dehydrogenation, a deep alkane crack reaction was also observed using Ni-exsolved perovskite oxide as a catalyst.292 Conversion selectivity to propylene and ethylene increased with the applied current densities, reaching 36% and 32%, respectively, at 300 mA cm−2 and 700 °C using the H-SEMRs. In their work, the peak power density of the reactor could reach 450 mW cm−2 at 750 °C, suggesting a promising approach for electricity and light olefin cogeneration. In a quite recent study, Lei coupled the nitrous oxide decomposition into the above alkane to alkene process, in which NOx was used to replace oxygen as the oxidant.291,293 Both ethane conversion (from 44.9% to 45.2%) and fuel cell performance (from 200 mW cm−2 to 208 mW cm−2 at 750 °C) were improved with NOx utilization efficiency of 19.0%. Thus, such a multi-win technology is promising for fuel upgradation and greenhouse gas reduction.
| Anode | Electrolyte | Fuel | Flowing rate (mL min−1) | Conversion | Selectivity | Yield | Power density (mW cm−2) | Ref. | Duration |
|---|---|---|---|---|---|---|---|---|---|
| Pt | BaCeY0.15O3 | C3H8 | 20–80 | 5–30% (550–650 °C) | 90–95% for CH4 + C2H4 + C3H6 | — | 17–35 (600–650 °C) | 281 | — |
| Cr2O3 + Ag | BaCe0.8Y0.15Nd0.05O3−δ | C2H6 | 150 | — | — | 8–31% for C2H4 | 51–118 | 283 | 50 h (650–750 °C) |
| MoCx | BaCe0.7Zr0.1Y0.2O3−δ | 100 | 7.7–42.6% | 97.5–87.2% | — | 65–215 | 284 | 100 h | |
| Cu–Cr2O3 | 100 | 10–40% | 90–100% | 81–170 | 285 | 10 h | |||
| Cr3C2 and WC | 75 | 10–40% | 91–96% | — | −185 | 286 | 80 h | ||
| Co–Cr2O3 | BaCe0.8Y0.15Nd0.05O3−δ | 100 | — | 91.6% | 31.2% | 173 | 287 | No CO2 | |
| La0.6Sr0.4Fe0.8Nb0.1Cu0.1O3−δ | BaCe0.7Zr0.1Y0.2O3−δ | 30 | 43.3% (750 °C) | 94% | — | 90 | 288 | 12 h | |
| Co/Fe-(Pr0.4Sr0.6)3 (Fe0.85Mo0.15)2O7 | 100 | 15–50% (650–750 °C) | 90–100% | 13.2–41.5% (650–750 °C) | 349 (750 °C) | 291 | 750 °C for 100 h | ||
| FeNi3-(PrBa)0.95(Fe0.8Ni0.2)1.8Mo0.2O6−δ | BaZr0.1Ce0.7Y0.2O3 | 30 | 50.9% | 92.1% | 46.9% | 318 (750 °C) | 295 | 50 h | |
| Au | BaCe0.7Zr0.1Y0.2−xNdxO3−δ | 100 | 3–23% (600–700 °C) | 100–90% | 23% (700 °C) | 123 (700 °C) | 296 | — | |
| Pr0.6Sr0.4Fe0.8Nb0.1Cu0.1O3−δ | BaZr0.1Ce0.7Y0.1Yb0.1O3−δ | 30 | 45.2 | 92.8 | — | 209 (750 °C) | 293 | N2O as oxidant/24 h | |
| Co–SrMo0.8Co0.1Fe0.1O3−δ | BaCe0.7Zr0.1Y0.2−xNdxO3−δ | 100 | 12.5–41.3 at 650–750 °C | >91% | 11.9–37.8% | 258 (750 °C) | 194 | 50 h | |
| Aligned carbon nanotube forests | BaZr0.4Ce0.4Y0.1Yb0.1O3−δ | 20 | 37.6 (650 °C) | >90 | — | 111 (650 °C) | 297 | 90 h | |
| PtGa/ZSM-5-(Pr0.3Sr0.7)0.9Ni0.1Ti0.9O3 | C3H8 | 50 | 15% | >90 | — | 172 (550 °C) | 298 | 20 h | |
| (Pr0.3Sr0.7)0.9Ni0.1Ti0.9O3 catalyst layer | BaZr0.3Ce0.5Y0.2O3−δ | C3H8 + H2O | 10 | 20–70% | 68% for C3H6 + C2H4 (700 °C) | — | 94–450 (650–750 °C) | 292 | 50 h |
| Co7W6@WOx-La0.4Sr0.6TiO3 | YSZ | n-C4H8 | — | 80% (650 °C) | 65% for C4 alkenes | 50% | 212 | 25 | 7 h |
3.2.1.1.2 C–C coupling/upgradation. Direct conversion/upgradation of light olefins like methane to C2 aromatics through the electrochemical method using the “green electrons” from renewable sources received increased interest because of the largest production volume and energy consumption based on the conventional thermal catalysis process.5,148 One more additional benefit is the electrochemical approach can well avoid the severe thermodynamic constraint. The application of proton conductors compared with the mixed ionic and electronic conductors or pure oxygen ionic conductors in SEMRs also holds advantages through a non-oxidative dehydrogenation process, and the product selectivity is much higher than in other processes.
In light olefin conversion, methane is the most investigated molecule. One of the key issues is the high chemical stability of CH4 due to the absence of a dipole moment and the small polarizability (2.84 × 10−40 C2 m2 J−1), high temperature is preferred.1 While the direct conversion of CH4 to CxHy in the absence of oxidants is thermodynamically unfavorable in a large range of temperatures.12 In factor, CH4 can be non-oxidatively activated to the couple as C2–C6 hydrocarbon based on the following equations, the exact products are dependent on the applied temperature, the operational mode,149 and the last but most important one, the catalyst.294
| 2CH4 → 2H2 + C2H4, ΔH025°C = +202 kJ |
| 6CH4 → 9H2 + C6H6 |
As one may see from the above equations, these processes are not spontaneous, so SEMRs are normally operated in the electrolytic or pumping mode at elevated temperature to efficiently force the H+ to move to the low concentration side, while too high temperature may result in carbon deposition through the CH4 pyrolysis reaction. Therefore, maintaining high H+ conductivity and high stability of the CH4 coupling reaction is the key to realizing high efficiency. Similar to the previous discussion, the main proton conductor in this section is still the perovskite oxide. In 1992, Hibino was the first to adopt SrCe0.92Yb0.05O3−x perovskite oxide H-SEMRs with Ag catalyst for CH4 activation and conversion to C2 hydrocarbon.299 As expected, under the OCV conditions, the conversion rate of CH4 was limited, which was significantly improved when the galvanostatic current was used; the formation rate of C2H6 and C2H4 increased with the increase of the applied current, and the evolved hydrogen in the cathode was in agreement with the theoretical value, suggesting that the proton transfer number of the applied electrolyte is close to unity. Moreover, no CO2/CO was observed in the products. The same group later showed that a similar material system reached C2-compound formation rates of 0.23 to 0.52 μmol min−1 cm−2 when the cathode Ar gas was replaced by air since H2 was oxidized to water which speeded up the transportation of proton.300 The results demonstrate the feasibility of the application of H-SEMRs for CH4 coupling. A similar MAE system was also utilized by adding a composite of Au–5Ce5Na2WO4/SiO2 to promote the conversion of CH4 and selectivity of C2 hydrocarbon.301 Moreover, a CH4–steam–O2 mixture was introduced to prevent the problem of carbon formation. The reaction products were C2H4, C2H6, H2, CO, and CO2. Such an anode fuel composite also allowed the SEMRs to be operated under the fuel cell conditions, which showed higher resistance toward carbon deposition induced by pyrolysis. It was found that 95% of hydrogen was transported and a 20% increase of C2 production was achieved.
In 2016, Morejudo and colleagues47 reported one of the most significant developments using mainly proton conducting solid-state electrolytes for the direct conversion of CH4 to aromatics with continuous H+ removal while being free from carbon deposition issues (Fig. 16a). The SEMRs were assembled with a 25 μm-thick BaZr0.7Ce0.2Y0.1O3−d (BZCY) supported on a Ni-BZCY cathode, Cu anode plus a critical shape-selective Mo/zeolite catalyst (Fig. 16b). When CH4 was introduced into the tubular anode chamber, it was catalytically converted to aromatics with a constant yield of 10% at 710 °C, a current density of 40 mA cm−2, and a space rate of 150 mL g−1 h−1 at 1 bar and a methane conversion rate of ∼11% (Fig. 16c and d). The yield can be further improved to over 20% with the increase of applied current density, or the H2 extraction rate, while it reduced from 12% to 1.5% under the fixed bed reaction conditions after 45 h operation, against 9% under SEMR condition (Fig. 16e). In other words, the degradation rate of CH4 to aromatics in SEMRs was one order of magnitude lower than that observed in the conventional fixed bed reactor; improved resistance to carbon deposition was ascribed. Such an exceptional anti-degradation capability was ascribed to the mixed H+ and O2− (0.3% of H2 conductivity in this case) conductivity of BZCY at the elevated temperature.169 Minor CO formation in the anode chamber could be a good indicator of such O2− conduction property, since the scavenging of reactive carbon from the catalyst surface occurred via the steam reforming reaction C + H2O → CO + H2, in which H2O is formed from O2− with H2 in the anode dehydrogenation process.
 | ||
| Fig. 16 Co-ionic conductive SEMRs for CH4 to aromatics conversion: (a) tubular cell configuration and material system, (b) SEM image of MEA based on BZCY electrolyte, (c) percentage of H2 extracted and O2 injected versus current density at 700 °C, (d) aromatic yields vs. time and (e) CH4 conversion.47 Reproduced with permission. | ||
One may see that the one-pass conversion of CH4 was ∼11% in the above case, which resulted in a relatively low aromatics yield. Recently, Sakbodine et al.302 designed a new tubular H-SEMR for CH4 conversion to C2 and aromatic products. The SEMRs are composed of a mixed ionic–electronic SrCe0.7Zr0.2Eu0.1O3−δ thin film (≈20 μm) supported on the outer surface of a one-end capped porous SrCe0.8Zr0.2O3−δ tube. And the internal tube was filled with a coke-resistant Fe©SiO2 catalyst.303 In this case, the residence time of CH4 in the reactor is extended so that the conversion rate and C2 and aromatics product yields are much improved. Under the tested conditions, a stable ≈30% C2+ single-pass yield, with up to 30% CH4 conversion and 99% selectivity to C2 (C2H4 and C2H2) and aromatic (C6H6 and C10H8) products were obtained. Furthermore, the selectivity of C2 and aromatics could be intentionally manipulated by adding H2 or changing the H2 extraction rate during the cell operation. The same group later demonstrated that replacing N2 at the outside of the reactor with O2 can realize the energy balance between the endothermic dehydrogenation reaction and exothermic H2 combustion on opposite sides of the membrane and overcome the common carbon deposition issue for CH4 pyrolysis reaction at the elevated temperature.304
3.2.1.1.3 Hydrocarbon to syngas. The direct application of hydrocarbon fuel on PCFCs is limited due to the quick carbon deposition issue even though currently most proton conductors possess mixed proton and oxide ionic conduction and hydration capability.169 To effectively utilize hydrocarbon fuel, sufficient reforming chemicals like CO2, H2O, and/or O2 are required to overcome the carbon deposition issue.21 In this case, the reaction of in-site reforming hydrocarbon fuel takes place in the anode chamber to produce H2 and CO mainly, in which H will be oxidized to form H+ and transported through the electrolyte layer and reduce O2 in the cathode in the presence of electrons from the external circuit, yielding electricity. During the reforming process, H2O and CO2 will also be produced due to the presence of limited O2− conduction in the electrolyte, which will react with hydrocarbon to produce CO and H2. In other words, the hydrocarbon/reforming agent co-fed PCFCs may serve as a potential platform for producing syngas. Moreover, with the proper control of the feed gas competition and cell operational parameters, an ideal CO
:H2 molar ratio of 1:2 could be achieved. One of the critical issues to effectively realizing hydrocarbon fuel reforming and hydrogen oxidation reaction while free from carbon deposition issues is the lack of an active anode catalyst. The state-of-the-art anode catalyst Ni generally suffers from carbon poisoning and low redox stability. Precious metals like Rh have high activity but resources are limited resource. Other impurities like H2S may degrade the anode activity with time. To address the above critical issues, Hua et al. proposed two methods of anode engineering to improve PCFC performance and fuel compatibility.305,306 In 2016, they first reported intentionally adding an anodic functional layer, consisting of an Ni0.8Co0.2–La0.2Ce0.8O1.9 (NiCo–LDC) composite on a Ni cermet anode surface.305 Such multiple-twinned bimetallic nanoparticles loaded on LDC proton conductors have superior catalytic activity toward in situ dry reforming and also CO2 resistance, which helps improve the H2 concentration in the anode active layer and electrolyte stability. It was found that the CO2 conversion reached 91.5% at 700 °C and the SOFC operated stably for more than 100 h under galvanostatic conditions (1 A cm−2) in a CH4–CO2 freestream. Moreover, the selectivity and conversion of CO2 and CH4 as well as oxidation of hydrogen resulted in no CO2 but CO-concentrated syngas in the anode, so this process can be considered an important process for CO2 conversion and utilization. Considering the difficulties of the current collection and the complexity of additional layer fabrication induced by the anode catalytic layer, the same group further developed an even simpler method to address the above-mentioned technique's difficulties. The cell is still based on the Ni-based cermet anode but is successively infiltrated with a redox stable and catalytically active PBM (9 wt%, the fuel is named FC-PBM) and Ni4Co bimetal catalyst (1.5%) on the anode surface (denoted as FC-NiCo/PBM).306 It is interesting to see that all infiltrations were selectively deposited on the surface of the BZCYYb grain because of its highly hydrophilic nature (Fig. 17a). The infiltrated nanocatalyst showed exceptional catalytic activity toward CH4–CO2 dry reforming to produce H2 and CO and prevented the corrosion of BZCYY by the H2O/CO2 (Fig. 17b). The fuel cell performance of the CH4–CO2 co-fed PCFCs was improved from 0.4 W cm−2 to 0.8 W cm−2 and then over 1.1 W cm−2 after the successive infiltration of PBM and Ni4Co catalysts even in the presence of 50 ppm H2S (Fig. 17c). Moreover, the cell could be operated stably for 36 h at a constant current density of 1 A cm−2. Nearly 100% CH4 conversion was achieved, and no CO2 but CO-concentrated syngas was continuously obtained (Fig. 17d). These results will benefit the commercialization of PCFCs and SEMRs. Another interesting study by Dr Chuancheng Duan recently further explored the potential of PCFCs by fully using the exclusive proton conducting properties below 650 °C to realize exceptional power generation (0.30–0.94 W cm−2 at 55–650 °C), chemical (syngas) production and CO2/CH4 gas mitigation.307 The authors developed a novel anode catalyst Sm0.2Ce0.7Ni0.1Ru0.05O2−δ which in situ forms exsolved Ni and Ru on SDC. Ni in the hybrid catalyst is responsible for CH4 activation, while Ru favors CO2 activation and coke resistance by changing the reaction pathway, enabling superior fuel cell performance, quick CO2/CH4 reaction, and system durability. The wise design can be fully exploited for application of the catalyst and SOFC reactor in chemical manufacturing and energy sector as well as environmental protection. The same group308 reported that steam-fueled SOECs can selectively convert CO2 to CO without hydrocarbon fuels like methane production at a wide range of temperatures of 400–600 °C and applied potentials. A specific electrode catalyst, in situ exsolved Ni–Fe/Sr2Fe1.4Mo0.5O6−δ nanoparticle electrocatalyst, was developed to replace the typical Ni-based cermet anode which effectively inhibited the formation of formic species, and subsequently selective production of CO. Therefore, this work suggests the importance of the rational design of electrode catalysts for selectively manufacturing chemicals. Recently, a similar system was used for steam reforming CH4 for effectively converting CH4 and greatly improved the fuel cell efficiency at the reduced temperature.309 These processes provide an alternative approach for CH4/CO2 utilization and achieve fuel/chemical feedstock production.
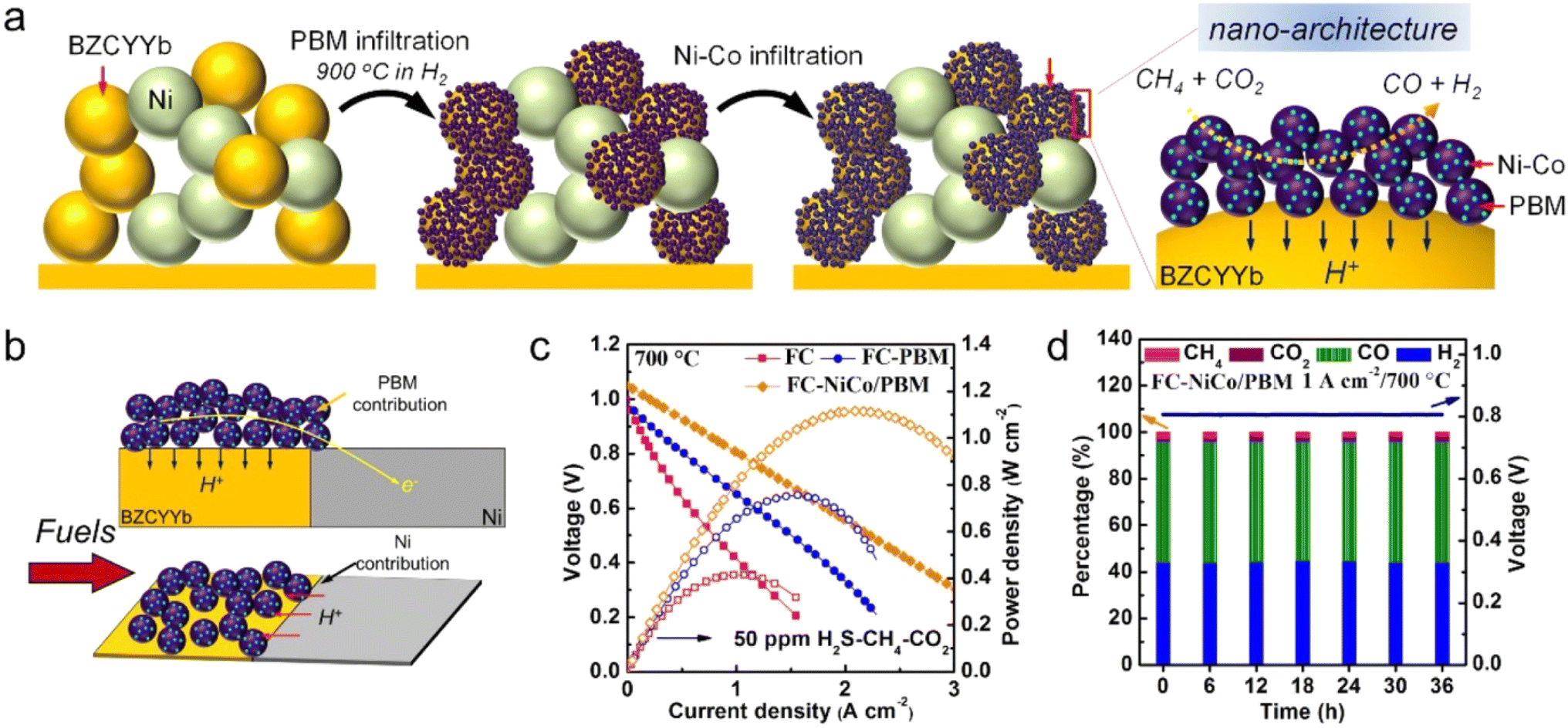 | ||
| Fig. 17 (a) Schematic preparation procedures of NiCo/PBM catalyst selectively loading on BZCYYb in the porous nickel cermet anode, (b) the role of PBM nanocatalyst and Ni in cermet, (c) electrochemical performance of PCFCs fed with 50 ppm H2S–CH4/CO2 fuel using different fuel electrodes and (d) the exhaust gas compositions of FC-NiCo/PBM during continuous 36 h testing.306 Reproduced with permission. | ||
3.2.1.2 Hydrogen co-generation/separation. Different from the case of power and chemical cogeneration in SEMRs during the dehydrogenation process where the H2 is used to produce electricity, the penetrated H2 can utilize/compact directly or be catalytically converted to other chemicals. However, additional energy to promote the dehydrogenation and hydrogenation reaction in the form of electricity or heat energy is required. An additional benefit of such an operation is close to 100% product since only proton conduction takes place in the electrolyte considering the hybrid O2−/H+/hole(electronic) conduction of the widely used BaCeO3-based proton conductors. So under the typical operational conditions, the former cases can be considered as a hybrid oxidative and non-oxidative dehydrogenation process. If the cathode side is the pure hydrogen process, the overall process for the latter case can be considered as a pure non-oxidative process.
3.2.1.2.1 Chemicals and hydrogen co-generation. In 2018, Ding first reported the electrochemical non-oxidative deprotonation of ethane using BZCYYb H-SEMRs with Ni-BZCYYb anode and PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) cathode catalysts (Fig. 18a and b).310 They demonstrated that under an electrochemical overpotential of 140 mV, a constant current density of 1000 mA cm−2 should be obtained for ethane dehydrogenation reaction under SOEC mode (Fig. 18c and d). Nearly 100% ethylene selectivity and a hydrogen production rate of 0.448 mol cm−2 per day could be achieved at 400 °C. Moreover, they further demonstrated that compared with a conventional industrial ethane steam cracker, such an electrochemical dehydrogenation process can save 65% of process energy (Fig. 18e) and reduce 72% of carbon footprint (Fig. 18f). Those values can be further improved and the process can be a net gain energy process if renewable electricity and heat are used and the heating value of produced hydrogen is taken into account, suggesting a disruptive approach for petrochemical manufacturing, shifting the paradigm from thermal chemical practice to a clean electrochemical energy regime with renewable energy. Similar energy saving and process efficiency were also reported by Xie using H-SEMRs but with different electrode components.311 One of the differences is that an active dehydrogenation catalyst Pt/Ga-ZSM5 was used to improve the ethane conversion rate to 40% in a single pass conversion, while only 3.9% ethane was dehydrogenated in Ding's report.310 The C2H4 yield can be improved to over 50% with the applied temperature of 700 °C. Furthermore, a steam electrolysis treatment was proposed to regenerate the catalyst by oxidizing the deposited carbon, such an additional operation alleviates catalytic degradation by 74% compared with the in-treated system. To further improve the ethane conversion and ethylene selectivity, the same group developed an active exsolved metal–oxide NixCu1−x–Nb1.33(Ti0.8Mn0.2)0.67O4−δ hybrid catalyst and applied CO2-assisted H2 oxidation process in the cathode.54 The highest ethane conversion of 75.2% and ∼100% ethylene selectivity at an applied voltage of 0.8 V in this electrochemical reactor were obtained, accompanied by the syngas production in the cathode. This demonstrated that the high-temperature electrochemical dehydrogenation process using SEMRs integrates the advances of electrochemistry and thermal catalysis would be a promising method to replace the classic thermochemical process.
 | ||
| Fig. 18 Ethane dehydrogenation using H-SEMRs. (a) Schematic of applied reactors and cell components, (b) a cross-sectional SEM image of an actual electrochemical cell, (c) the corresponding voltage and (d) durability test at a current density of 1 A cm−2 as a function of time, and a comparison of the (e) process energies and (f) carbon footprint for ethylene production from ethane based on electrochemical and industrial steam cracker processes.310 Reproduced with permission. | ||
3.2.1.2.2 Hydrogen separation. SEMRs allow electrochemical supply of one of the active species/intermediates (H+, O2 in the form of O2−, CO2 in the form of CO32−…) from one electrode to another electrode to finish the electrochemical reactions. When there are no reactants that will react with those active species/intermediates, a gas evolution reaction will take place. With a continuous gas evolution reaction, the gas will be concentrated and even compacted at the sweep side; the whole process can be considered as a gas separation process. With more and more attention paid to future sustainable energy carriers such as H2, research interests increased considerably in recent years. SEMRs are also used as membrane reactors for highly purified hydrogen generation from low-concentration H2, steam, NH3, CH4, and biogas.15,274,312 For example, Zhan reported that the separation rates of highly purified H2 at 0.51 V and 500 °C was 3.3 and 2.4 mL cm−2 min−1 out of the simulated renewable hydrogen blended in natural gas pipelines with 10% H2–90% N2 and 10% H2–90% CH4, respectively, using a 10 μm thick BaZr0.1Ce0.7Y0.2O3−δ (BZCY) electrolyte sandwiched by two Ni-BZCY electrodes.313 The total Faraday efficiency surpassed 96%, suggesting the feasibility of such technology for the on-site purification of hydrogen. Moreover, when the reactor integrates other processes like electrochemical hydrogen compression, it can achieve both super high hydrogen removal/purification efficiency and overall energy efficiency. Rebollo first reported that a dense mixed ionically and electronically conductive BaCe0.65Zr0.20Y0.15O3−δ (BCZ20Y15) and Ce0.85M0.15O2−δ (M = Y and Gd, hereafter referred to as YDC15 and GDC15) all-ceramic composite membrane was applied as a hydrogen separation membrane at temperatures higher than 500 °C.312 In such composite materials, BCZ20Y15 is considered an H+ conductor, and Ce0.85M0.15O2−δ is a typical O2− conductor while presenting mixed O2− and e− conducting behavior under hydrogen reduction conditions, which is frequently used as a composite electrolyte material in SOFCs, so it still can be considered as SEMRs. When membranes with a 50
:50 volume ratio, BCZ20Y15–GDC15, at a thickness of 610 μm were exposed to both 3% hydrated Ar (sweep side) and H2 (feed side) using Pt as a catalyst, the hydrogen separation rate increased with the applied temperature and reached values up to 0.27 mL min−1 cm−2 at 755 °C, one of the highest H2 fluxes obtained for bulk mixed protonic–electronic membranes. When the temperature was further increased to 1040 °C, a temperature high enough for thermal splitting of H2O, the flux remarkably increased to 1.75 mL min−1 cm−2 and 2.40 mL min−1 cm−2 when both sides of the membrane (700 μm thick) or only the sweep side was hydrated, respectively, which is the highest value of H2 fluxes based on bulk mixed protonic–electronic membranes. It is interesting to see that both H2 flux from the feed side in the form of bulk H+ transportation across the membrane and H2 production through the water splitting reaction in the sweep side co-contributed to the superior H2 flow in such a composite membrane. They also showed impressive CO2 and sulfur tolerance under the practical hydrogen separation conditions.312
3.2.2.1 CO2 hydrogenation to methane. Compared with the extremely endothermic H2/CO formation reaction in the electrochemical process, the production of CH4 in the reactor is a less endothermic process, which enables improved efficiency. Moreover, the easy storage capability and higher energy density of CH4 fuel are widely pursued.86,118 On the other hand, as discussed in the previous paragraph, temperature plays a crucial role in the in situ methanation process. H-SOECs, therefore, offer several key benefits to achieve high-efficiency direct CH4 production: (1) better balance between the thermodynamics of the exothermic Sabatier reaction and the chemical kinetics of CO2 conversion because of the much higher proton conductivity below 600 °C than its oxygen ionic conductor counterpart; (2) physical separation of CO2 and H2O allows gases at opposing electrodes, which favors a Le Chatelier's principle-free process, facilitating higher CH4 production. In addition, the principle of proton conductor-based methanation devices is distinct from that of the O-SOECs. Only the electrolysis splitting of H2O at the anode is required, which produces O2 and H+. The former is released as a gas directly at the anode, and the latter passes through the proton conducting electrolyte and reacts with CO2 at the cathode before or after the formation of H2 to form the targeted hydrocarbon fuel (CH4 in this case). The separate supply of H2O and CO2 can effectively improve the CH4 yield by reducing the reverse conversion according to Le Chatelier's principle.117 The research on the development of proton conductors, the compatible electrode materials, and their fabrication and applications in ceramic fuel cell/electrolysis cells for power and chemical fuel production has made considerable progress in recent years,20,21,23,47,171,267,271,276,292,314–317 which favored the research on the CO2 hydrogenation to CH4 efficiency. For example, Xie reported synthesis of CH4 by the electrochemical reduction of CO2 in an SOEC based on the BaCe0.5Zr0.3Y0.16Zn0.04O3−δ electrolyte of 60 μm in thickness and Fe/FeOx composite cathode.317 The application of a proton conductor enabled the cell's operation at 614 °C even though a largely applied over-potential of 2.6 V was required to reach a current density of 1.5 A cm−2, and increased CH4 contents in the outlet gas were observed. The production of CH4 and CO reaches 0.07 and 3.25 mL min−1 cm−2, respectively, under the above operational conditions with a CO2 conversion rate of 65%. The CH4 yield was double that through the control process of F–T synthesis under similar conditions. In addition, the authors also observed that the electrochemical reduction of CO2 is more efficient than the reverse water gas shift reaction (CO2 + H2 → CO + H2O) as follows under the same geometry and compositions.
Under favorable thermodynamic conditions, the challenge to achieve higher CH4 production requires the development of an active electrode and catalyst to realize higher H2 yield efficiency and subsequently improved CO2 hydrogenation process.20,21,171,318 With the quick evolution of new advanced electrode materials and cell fabrication technology and the understanding of the reaction kinetic process, remarkable CH4 yield improvement has been achieved. It was reported that PCECs using BaZr0.8Y0.2O3−δ (BZY) as the electrolyte and SrEu2Fe1.8Co0.2O7−δ as the anode displayed an exceptional current density of 1.23 A cm−2 at 1.5 V and 100 h of smooth operation was achieved at 550 °C.318 The presence of CO2 in the cathode interestingly promoted the electrode reaction by accelerating the proton transportation and simultaneously shifting the equilibrium to boost the formation of synthetic hydrocarbons. Moreover, both the formation of CO and CH4 increased with the increase of electrolysis density. A combined experimental and in situ spectroscopy study confirmed that the yielded proton in the anode continuously replenished to the cathode to react with the intermediate formed by adsorption of CO2 on the electrode surface and finally yield CO and CH4. An impressive study by Duan and O'Hayre reported that a button PCEC with BZCYYb electrolyte and highly active triple conductive electrode BZCFY showed a CH4 yield of 7.5% at 500 °C.20 Further combination of PCEC with a methanation fixed bed (300 °C) using CeO2 with 10 wt% Ni can reach a CO2 conversion of 79.7% and CH4 selectivity of 99.1% at atmospheric pressure, i.e., CH4 yield of 79%. Moreover, with careful control of the operational conditions, the hole conduction of proton conductor which is detrimental to the electrical efficiency of the electrolysis process is effectively suppressed, and a high faradaic efficiency of up to 98% has been achieved. Both the performance metrics were higher than those of the previous reports (by 2–3 times), and the Faraday efficiency and electricity to hydrogen efficiency were the highest in the open literature. Another breakthrough study by Li et al.267 reported selectively producing CH4 (up to 95%) using Ir–ceria-based catalysts at 400 °C in PCECs for CO2 reduction that operates at low overpotential and ambient pressure. Moreover, dedicated control of Ir speciation can be achieved by tuning the Ir–O hybridization through adjusting the Ir cluster size on ceria-based oxides. The Ir speciation in the form of either an ionic state or metallic state with a distinct catalyst surface chemical environment can selectively produce either CO or CH4, significantly demonstrating the techno-economic feasibility of the process. Furthermore, the authors also demonstrated that such a strategy is also workable on other functional oxides like PBM-BZY and when in tandem with electrode hydrogenation of C2H6 to produce C2H4 and CH4. This work highlighted the importance of precise control of the chemical environments of the surfaces of the catalyst but also pointed out the roles of intermediate temperature PCFCs with the combined thermal–electrochemical process for upgrading CO2 for valuable synthetic fuels. More recent work also evaluated the one-step yield to 34.6% at 1 A cm−2 and 450 °C using a designed PCEC unit-cell stack on a large scale (active area of 5 cm2) that enables the efficient utilization of reactants (Fig. 19 and b).117 With the increased applied current density, both the H2 yield and CH4 yield increased (Fig. 19c–e). Moreover, they found that with exhaust H2 recycling, a critical yield of 71.2% could be achieved because of the increased CO2 conversion and reduced hole conduction (Fig. 19f). In addition, the direct comparison of the electrochemical conversion of H2O–CO2 and thermochemical conversion of CO2–H2 under similar conditions confirmed a higher yield for the former process, suggesting that the effect of EPOC effect is also suitable for the CO2 methanation reaction. Therefore, continuous work can focus on the development of active catalysts to achieve acceptable rates and selectivity. Recently, Chen deposited a CeO2 thin film on the surface of Ni-BZCYYb.86 It is found that the decorated CeO2 modulates the adsorption of CO2 and facilitates proton transfer for the hydrogenation process, leading to accelerated CH4 production. Experimentally, an improvement of more than 3 times CH4 selectively (absolute value 17%) at 550 °C and 1205 mA cm−2, i.e., 0.2 mL min−1 cm−2 under typical PCEC conditions, was obtained compared with unmodified cells. The addition of the CeO2 layer modified CO2/CO adsorption, and promoted the formation of surface C–H by accelerating the migration of H+ toward the electrode surface, then improving CH4 synthesis. This work highlighted the importance of SOFC reactors by combining heat, solar–thermal, and thermo-electrochemical processes.
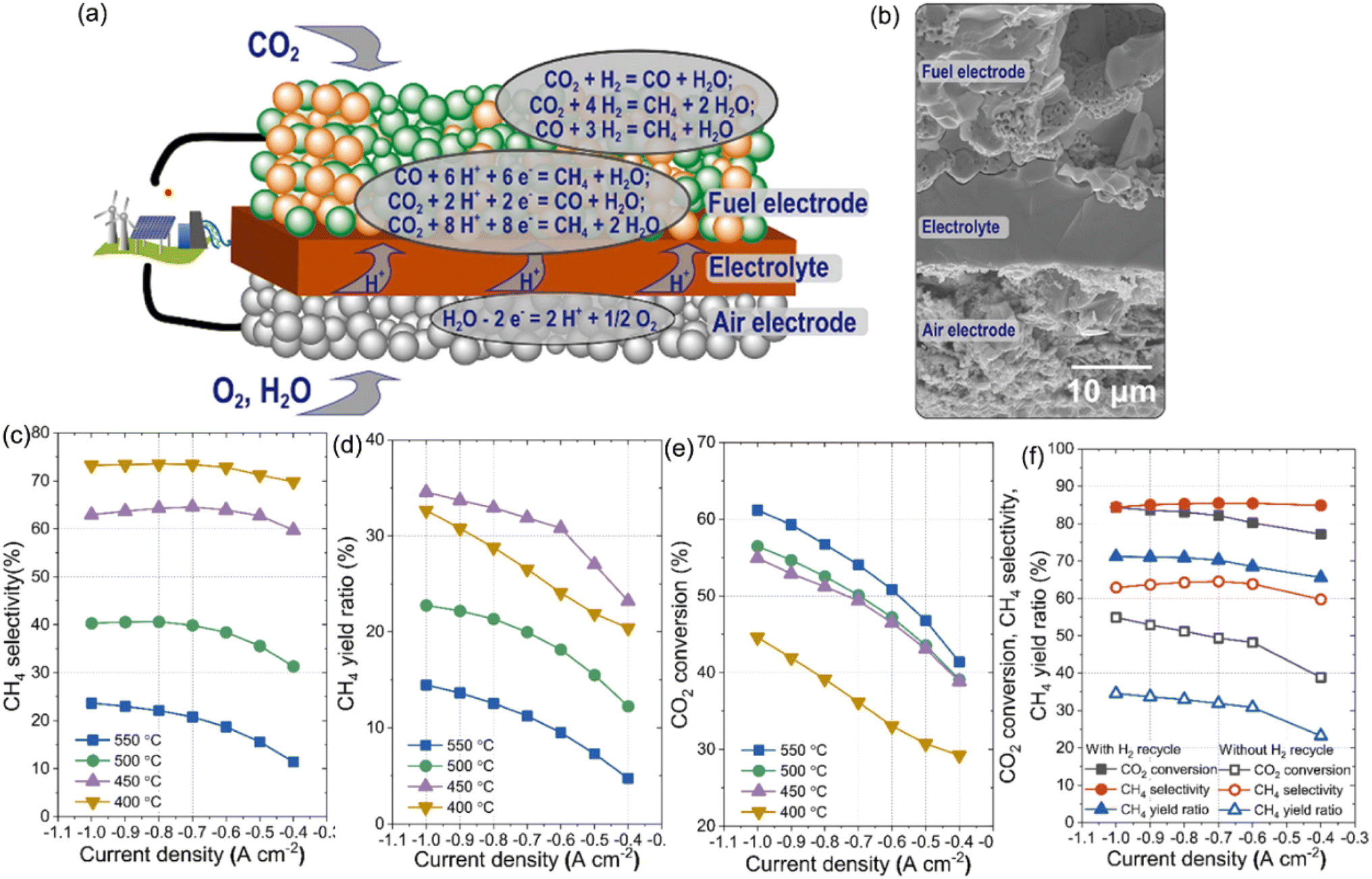 | ||
| Fig. 19 (a) Schematic illustration of the possible electrochemical and chemical reactions during electrolysis and upgrading CO2–H2O into CH4 in the PCEC (b), (c–e) one pass CO2 conversion, CH4 selectivity, and CH4 yields at 400–550 °C, and (f) with H2 recycling at 450 °C.117 Reproduced with permission. | ||
Different from the above cases, Duan reported the selective production of CO instead of CH4 by the CO2 hydrogenation process,308 whereas most of the work in the literature has reported mixed yields of CO and CH4.20,117,267 In their work, novel exsolved NiFe alloy nanoparticles socketed on an Sr2Fe1.4Mo0.5O6−δ negative electrode instead of a classic Ni-based cermet electrode were applied. It is found that selectivity to CO was ∼100% over a wide range of operating temperatures (400–600 °C) and applied potentials/current densities. Further investigation of the CO2 reduction mechanism based on the in situ diffuse reflectance infrared spectroscopy (DRIFTS) showed that the designed electrode inhibited the formation of formic species, which therefore leads to selective production of CO under such operational conditions. Such an exception again demonstrates the crucial rule of tuning product selectivity by engineering the negative electrode. Further improving the yields of CH4 through the CO2 hydrogenation process is still highly desirable. Pressurized SEMRs based on proton conductors under optimized reaction conditions, like total pressure, H2/CO2 ratio, and current density would be a potential solution.315
3.2.2.2 Electrocatalytic NH3 synthesis. Ammonia (NH3) is one of the largest-volume industrial chemicals produced in the world as an agricultural fertilizer and chemical compound, which is also considered an energy carrier in the future hydrogen economy, Hydrogen 2.0, because of carbon-free characteristics and well-equipped infrastructure. However, its synthesis relies on the Haber–Bosch (H–B) process, where high temperatures (400–600 °C) and high pressures (20–40 MPa) with an iron-based catalyst are required. The H–B process is a low temperature-favored exothermic reaction, while high temperature is needed to cleave the strong N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond.5,319 Moreover, this process inevitably produces huge CO2 emissions (1% of global value) both for hydrogen production and the direct synthesis process.320 In contrast to thermal synthesis, electrosynthesis showed multiple benefits:8,321 operation under mild conditions since e− reduction enables a much-reduced temperature and pressure; much reduced decentralized plants; eliminating the hydrogen facility by using water as a hydrogen source; free from carbon sources and energy by integrating renewable power sources.322,323
N bond.5,319 Moreover, this process inevitably produces huge CO2 emissions (1% of global value) both for hydrogen production and the direct synthesis process.320 In contrast to thermal synthesis, electrosynthesis showed multiple benefits:8,321 operation under mild conditions since e− reduction enables a much-reduced temperature and pressure; much reduced decentralized plants; eliminating the hydrogen facility by using water as a hydrogen source; free from carbon sources and energy by integrating renewable power sources.322,323
Versatile electrolytes can be used for electrochemical NH3 synthesis.321 Among these, solid-state proton conductor efficiently operated at 400–700 °C showed the most promise since both suitable temperature and active solid-state proton promote the kinetics of NH3 and are free from subsequent gas purification as well as the utilization of water as a hydrogen source. The major challenges of electrochemical NH3 synthesis are related to the compromise of activity (current density) and selectivity (NH3). Another challenge to the wide commercialization of this process is the limited NH3 synthesis rate, with the majority of the experimental studies reporting rates lower than 10−8 mol cm−2 s−1,324 which is one order of magnitude lower than that of the suggested value of 10−7 mol cm−2 s−1 plus a faradaic efficiency of up to 50%, as suggested by Giddey.320 Therefore, active and selective cell components are highly desirable.
The first experimentally electrochemical NH3 synthesis on a solid-state proton conductor (SrCe0.95Yb0.05O3) was demonstrated by Marnellos in 1998.266 In their work, H2 and N2 were supplied separately to the chamber of the end-closed tubular cell with Pd as the electrode catalyst. Under the polarization voltage conditions, they reported that 80% H+ which is the reaction product of H2 oxidation (H2 − 2e− → 2H+) in the anode was converted to NH3 through the following reaction N2 + 6H+ + 6e− → 2NH3, and the overall conversion rate was limited by the proton supply in the anode to cathode. Although the yield of this work is quite low, it demonstrated and offered an alternative route to permit operation at atmospheric pressures and temperatures, eliminating the thermodynamic requirement on conventional catalytic reactors. The same group also demonstrated the use of a single chamber cell configuration to synthesize NH3.323 Improved performance was further reported through the deployment of an anode supporting cell structure. Wang, therefore, developed Ni-cermet anode-supported PCFCs with perovskite oxide Ba0.5Sr0.5Co0.8Fe0.2O3−α as the cathode.325 A dense BaCe0.85Y0.15O3−α (BCY15) electrolyte was prepared by the modified spin coating method on the anode layer. Under a typical H2/N2 atmosphere, a maximum rate of ammonia formation of 4.1 × 10−9 mol s−1 cm−2 was obtained under a direct current of 1 mA at 530 °C, which is double the value of the previous work, suggesting the promise in improving the NH3 yield by that potential method. A similar method was also employed by Vasileiou to reduce the ohmic resistance of proton electrolytes in a single chamber reactor using Rh film as the cathode to improve the NH3 efficiency.326 Due to the limitation of proton conductivity at the applied temperature range, Tao et al. proposed to use ceria-based composite electrolytes,327 like ceria–Ca3(PO4)2–K3PO4 (ref. 328) and carbonate-oxide329–331 to replace oxide proton conductors because of the improved ionic conductivity at the intermediate temperature range which favors both the cleavage of the triple bond of the N2 molecule and NH3 stability. The hybrid ionic conduction of composite electrolytes also enabled distinct operational modes, proton or oxygen ionic conduction, with input electrical energy using different raw gases, including H2/N2, H2O/N2,331 natural gas,328 and even with wet air332,333 based on different catalysts. It is found that the applied temperature, voltage/current, and catalysts influenced the final N2 conversion rate and NH3 yield. In 2007, Liu's group first demonstrated the synthesis of NH3 from N2 and natural gas with a ceria–Ca3(PO4)2–K3PO4 composite electrolyte.328 Natural gas was used as a proton source because of its wide availability. Ag–Pd was used as the symmetric electrode catalyst. It was found that the NH3 product rate first increased with the applied cell potential, up to 1 V, and then it remained unchanged above 1.0 V. NH3 yield rate also increased first with the applied temperature, and reached the maximum value of 6.95 × 10−9 mol s−1 cm−2 at 650 °C and then reduced with further increase of temperature. Another critical issue for NH3 synthesis is related to the electrode activity, especially for the N2 activation. Efforts were also made to develop active catalysts to improve NH3 efficiency. Fe has been used as an industrial catalyst in the H–B process, therefore, its catalytic activity was also tested in electrochemical systems.322 It was found that an increase of 80% reaction rate was obtained when protons were electrochemically “pumped” to the Fe electrode compared with that under OCV conditions. Ru is a well-known catalyst for NH3 synthesis at relatively low temperatures because of the high activity for the dissociation of the N–N triple bond. However, due to its precious metal nature, effective utilization of Ru is required. Kosaka therefore applied the exsolution method to deposit 2–5 nm nanoparticles that are highly dispersed on the La0.3Sr0.6TiO3 perovskite oxide surface.334 An improved NH3 formation rate was observed in this system. A recent study employed BZCYYb as a proton electrolyte, BZCFY as an air electrode, Ni-based cermet as a cathode, and a novel Ru–(BaO)2(CaO)(Al2O3) NH3 catalytic functional layer;335 a record NH3 yield of 1.2 × 10−8 mol cm−2 s−1 was obtained and it can be further improved to 2.1 × 10−6 mol cm−2 s−1 with the application of a pressurized system (12.5 bar) and the reactor maintained exceptional stability during more than 160 h testing. More interestingly, a hybrid approach to cyclic operation between NH3-fueled power generation and electric power-driven NH3 synthesis was demonstrated in this work, which may find suitable application in a future hydrogen economic society. To this end, Li recently synthesized Ru/La0.25Ce0.75O2−x through hydrothermal treatment with abundant hydroxyl groups as active catalysts for NH3 production integrated with H-SEMRs.338 Both the computational and experimental results demonstrated that Ce3+–OH/Ru sites facilitate both the triple bond decoupling and single bond N–H formation at 400 °C and 1 bar of N2, which resulted in an NH3 rate of 2.92 mol h−1 m−2 and a maximum Faraday efficiency of 25%. The former is about two orders of magnitude higher than the literature value. A more general integration of co-producing NH3 and other chemicals, like alkane dehydrogenation or natural gas reforming process on one device and in one step, is desirable, which will benefit the efficiency and sustainability of such coupling technology. To this end, as can be seen from Table 3, the NH3 yield rate is still lower than that of the benchmarking value, 4.35–8.75 × 10−7 mol s−1 cm−2 for industrial application,320 the faradaic efficiency should also be much improved. Novel N2 activation or ionic conducting electrolyte materials are expected.336
| Anode | Electrolyte | Cathode | Catalyst | Temp. (C) |
Faradaic efficiency | NH3 rate, mol cm−2 s−1 × 109 | Cathode reactant | References |
|---|---|---|---|---|---|---|---|---|
| a SSC: Sm0.5Sr0.5CoO3−δ; CGCO: Ce0.8Gd0.18Ca0.02O2−δ; PNFCO: Pr0.6Ba0.4Fe0.8Cu0.2O3; LSFC: La0.6Sr0.4Fe0.8Cu0.2O3−δ; CGO: Ce0.8Gd0.2O2; BZCYYb: BaZr0.1Ce0.7Y0.1Yb0.1O3−x; BSCF: Ba0.5Sr0.5Co0.8Fe0.2O3; BCTYZ: Ba0.95Ce0.6Tb0.1Y0.2Zr0.1O3. | ||||||||
| Pd | SrCe0.95Yb0.05O3 | Pd | — | 570 | 78% | H2 pump rate dependent | N2 | 266 |
| NiO-BCY | BaCe0.85Y0.15O3−α | BSCF | — | 530 | 58% | 4.1 | N2 | 325 |
| Ni | BaCe0.2Zr0.7Y0.1O2 | Rh | — | 600 | 6.2 | 13 | N2 | 326 |
| Ag–Pd | YDC-Ca3(PO4)2–K3PO4 | Ag–Pd | — | 650 | — | 6.95 | N2 | 328 |
| Ag–Pd | LiAlO2–(Li/Na/K)2CO3 | Ag | CoFe2O4 | 400 | —% | 0.2 | N2 | 329 |
| Co3Mo3N | 450/0.8V | 0.33 | N2 | 337 | ||||
| Ni-SDC | SDC-(Li/Na/K)2CO3 | LSFC-Ce0.8Sm0.2O2−δ | — | 450 | — | 5.39 | N2 | 330 |
| SSC-CGCO | (Li,Na,K)2CO3-CGCO | CoFe2O4-CGCO | — | 400/0.6 V | — | 0.65 | N2 + H2O | 331 |
| LSFC-CGCO | SSC-CGCO | — | 400/1.4 V | 0.1% | 0.05 | N2 + H2O | 333 | |
| PNFCO-CGO | CGO–(Li,Na,K)(2)CO3 | PNFCO-CGO | — | 400/1.4 V | 1% | 1.07 | Wet air | 332 |
| La0.3Sr0.6Ti0.6Ru0.4O3 | BaCe0.9Y0.1O3 | Pt | — | 500 | — | 3.8 × 10−3 | N2 | 334 |
| BZCFY | BZCYYb | Ni-BZCYYb | Ru–B2CA | 400 | — | 12 | N2 | 335 |
| Ag–Pd | BaCe0.7Zr0.2Sm0.1O3 | Ag–Pd | — | 500 | 50% | 2.7 | N2 | 338 |
| Ni-BZCYYb | BZCYYb | PrBa0.5Sr0.5CoFeO5+δ (PBSCF) | Ru/La0.25Ce0.75O2−x | 400/0.6 V | 25% | 81 | N2 | 339 |
| Ag–Pt or LSCF | BaZr0.8Y0.2O3−δ | Ag–Pt or LSCF | 500/0.8 V | 0.5% | 0.085 | N2 | 340 | |
| Pt | BaCe0.9Y0.1O3 | K, Al-modified Fe-BCY | — | 650/1.5 V | 0.1% | 0.6 | N2–H2 | 341 |
| NiO-BCTYZ | BCTYZ | Fe@MXene | — | 650 | 8.4% | 8.24 | N2 | 342 |
| La0.33Sr0.67Cr0.33Fe0.67O3 | GDC | La0.33Sr0.67Cr0.33Fe0.52Ru0.15O3 | — | 500 | 0.25% | 0.473 | N2 + H2O | 343 |
| Pt | La5.5WO11.25−δ | Ru | — | 350 | 43.8% | 3.78 | N2 | 319 |
3.3 SEMRs with hybrid ionic conductors
High temperature ionic conductors generally showed higher single ionic transfer numbers than the low-temperature polymer-based electrolyte. Indeed, most recently employed fluorite-type or perovskite-type oxides were supposed to be either pure proton or oxygen ion conductors, rather than dual-ion conductors, even though in 2009, Liu experimentally demonstrated that the Yb doped conventional BaZr0.1Ce0.7Y0.2O3−δ (BZCYYb) was hybrid oxygen ionic and proton conductors and it demonstrated exceptional activity and anti-coke deposition capability with hydrocarbon fuel even with the Ni-based cermet electrode.169 Such outstanding results demonstrated the advantages of mixed ionic conduction over single ionic conductors. In addition, because of the non-conflicting oxygen ion and proton diffusion channels, i.e., oxygen ion transport through oxygen vacancy, while protons move with the lattice oxygen, each ionic conductivity is an addition to total ionic conductivity, therefore, hybrid ionic conductors show higher ionic conductivity over single ionic conductors344 and avoids the use of a fuel gas humidification system at the anode side.345 Besides the hybrid oxygen ionic and proton conductor, composite electrolytes based on ceria and salt also showed hybrid proton, oxygen ionic, and carbonate conductivity under a typical fuel cell atmosphere, which have also resulted in different SEMR applications in the literature.The co-ionic character, mainly in typical proton ceramics, has been successfully explored for SEMR applications.23,47,348 One typical example is used for methane coupling for the direct production of aromatics,47,302 where the proton conduction is used to extract H+ from methane to shift the balance toward the production of CH* and subsequently to realize non-oxidative methane de-hydro-aromatization in the presence of the catalyst, and the minor oxygen ionic conduction was fully used to distribute oxide ions along the reactor length to activate the coke and reactivate the catalyst for the dehydrogenation reaction. Such unique co-ionic conduction enables an extremely high carbon efficiency, improving the techno-economic process viability. Another exceptional case of a used mixed ionic conductor is the dual-chamber water splitting to produce green hydrogen in “hybrid SOECs” (Fig. 20a–c).56 When combined with highly active electrode materials, Ni as the anode catalyst and NdBa0.5Sr0.5Co1.5Fe0.5O5+δ as the cathode catalyst, the resultant SOEC gave a significantly improved current density of 3.16 A cm−2 at 1.3 V and 750 °C in 10% humidified hydrogen/air atmospheres over single H- or O-SOEC reported in the literature (Fig. 20d and e) and showed much-reduced overpotential than that of the PEM electrolysis operated at the low temperature (Fig. 20f). Such a hybrid SOEC represents a promising alternative option for cost-effective and highly efficient green hydrogen generation. To this end, mixed ionic conduction brings many advantages, which showed impressive electrochemical performance and could find interesting applications in SEMRs. However, since the operational conditions and atmosphere are different from that of a single ionic conduction mode, both the suitable electrode and cell/stack material system should be developed and optimized to maximize electrical efficiency and product selectivity.
 | ||
| Fig. 20 Schematic diagrams of the working principles of (a) O-SOEC, (b) H-SOEC, (c) hybrid-SOEC, and (d–f) electrochemical performance and comparison with the literature work and other electrolysis systems.56 Reproduced with permission. | ||
Generally, when the solid electrolyte is a mono-ion conductor, an external circuit is required to separate gases of a high degree of purity. For the oxide–carbonate composite, the hybrid CO32− and O2− conduction enables highly selective CO2 separation without an external circuit, the electrochemically active species are transported through the membrane under a gradient of electrochemical potential/or active substance partial pressure difference, only CO2 is permeated through the electrolyte in such a pure ceria–carbonate composite membrane, which significantly reduces the cost and complexity of further treatment process. As demonstrated by Wade,354,355 opposing ionic currents of carbonate and oxide ions are presented under the separation conditions on the YSZ–carbonate and ceria–carbonate composite membrane while the membrane with the Al2O3 matrix does not result in strong CO2 permeability or selectivity. Thereby, its selectivity to CO2 is exclusive, which has excellent compatibility with the high-temperature process stream where membrane-operated elevated temperatures in a continuous fashion are needed. When such a ceria–carbonate was used as a membrane for CO2 separation, under the gas atmosphere of 4.8% H2–CO2–N2/He, the CO2 permeation rate reached 1.84 mL min−1 cm−2 at 700 °C when the carbonate content is 50% of the total volume.352 Such an exceptional value is at least one order of magnitude higher than that of the literature value. It was found that the CO2 flux rate is linearly increased with the increase of H2 partial pressure of the feed side that provides increased oxide-ion flux because of increased oxygen chemical gradient across the membrane. Another Bi1.5Y0.3Sm0.2O3/molten Li/Na/K2CO3 (43.5/31.5/25 mol%) composite material gave a CO2 permeation flux of 6.60 × 10−2 mL cm−2 min−1 at 650 °C.356 These results demonstrated the promise of using multi-ionic conductive oxide-carbonate SEMRs for high-flux membrane separation.
Another interesting application of SEMRs for membrane separation is targeting chemical synthesis. With the extraction of CO2 from the reaction zone, the thermodynamic equilibrium of typical reactions may be shifted, or a higher reactant concentration of reactant will be realized which is beneficial to the chemical reaction kinetics. For example, Dong reported highly purified H2 using a catalyst-free ceramic–carbonate dual-phase membrane reactor.357 The gasifier syngas with the composition of H2, CO, CO2, and H2O passes through the tubular ceria–carbonate composite membrane reactor since the membrane serves as a CO2 permeation membrane. When CO2 is removed from the mixed gas the water gas shift reaction H2O + CO → H2 + CO2 is right-shifted, producing a higher H2 partial pressure without any catalyst. When operated at 900 °C, the SEMRs gave a CO2 flux rate of 2.7 × 10−3 mol m−2 s−1, and CO conversion and CO2 recovery of 26.1% and 18.7%, respectively, which are much higher than the conventional fixed bed reactor under the identical conditions. Such catalyst-free WGS SEMRs could result in a substantial reduction in the capital cost and remarkable simplification of highly purified H2 production. Such composite electrolyte-based SEMRs are also used in hydrogen production with carbon dioxide capture during steam reforming of the methane process,358 which shares a similar principle to the above case.
To this end, the CO2 separation functionality of the ceria–carbonate membrane is also used in direct carbon SOFCs (DCFCs). Generally, high performance is mainly contributed by the electrochemical oxidation of CO, which is produced by the oxidation reaction of carbon fuel with the in situ produced CO2, instead of carbon direct oxidation due to the limited solid–solid interfaces. To intensify CO production and remove CO2, the inert carrier gas is frequently used, which, however, reduces the CO utilization efficiency, resulting in low carbon to electricity conversion efficiency. To overcome such a compromise, Shao et al.238 applied a ceria–carbon composite membrane onto the U-shaped DC-SOFCs, both of which served as an anode sealant and CO2 separation membrane. With such a composite membrane separator, sufficient CO2 partial pressure is ensured which facilitates the carbon reforming reaction to produce CO, the latter remains in an anode chamber for electrochemical oxidation. The CO2 pressure gradient between the anode chamber and the atmospheric environment could force CO2 permeation/removal from the anode chamber. With the addition of a ceria–carbonate composite membrane, an attractive peak power density of 279.3 mW cm−2 was achieved for the DC-SOFCs at 850 °C, and a small stack composed of two cells can be operated continuously for 200 min, which is much better than that without the ceria–carbonate membrane. Further work could focus on the fabrication, design, and up-scaling of such promising technology.
4. Advancing SEMRs with multiple architectures
Different from the conventional two-chamber SEMRs (in the disc configuration), frequently in SOFCs, where the reactants are separated by the solid electrolytes that are dense enough to overcome the gas cross-issue, reactants can be co-fed into a chamber and the fuel and oxidant are selectively oxidized and reduced at the anode and cathode. The solid electrolyte disk is suspended in a flow of the reacting mixture and serves as a catalyst supporter. Such a specific reactor configuration is called a single-chamber solid oxide fuel cell (SC-SOFC).49,50,186,359 In 2000, Hibino first reported running SC-SOFCs with propane and air at 500 °C.186 An OCV of 0.9 V and peak power density of 403–101 mW cm−2 at 500–350 °C were obtained. Such a promising electrochemical performance stimulated further development of SC-SOFCs. In 2005, a thermally self-sustaining micro SC-SOFC stack was demonstrated for portable applications.49 Similar fuel cell performances were also obtained at the stack level, but the self-sustaining character, low temperature operation, and hydrocarbon fuel direct usage make SC-SOFCs suitable for potential application to replace other energy conversion technologies. It should be noted that in the initial study, SC-SOFCs were mainly developed for energy conversion, while in 2011, Shao further combined SC-SOFCs with the downstream reforming catalyst to construct a reactor for the co-generation of power and syngas with zero waste gas emission.50 The same group also compared the electrochemical performances of the CH4–O2 gas mixture (2:1) co-fed SC-SOFCs with two different modes and with or without catalysts (Fig. 21): one mixed gas flowed instantaneously to the anode and cathode (Fig. 21a), another mixed gas first to the cathode then to the anode chamber (Fig. 21b). Both the OCV and peak power density of the second mode were higher than those of the first mode, whose peak power density was 2 times higher.197 When the GdNi/Al2O3 catalyst was further added to the gas before the anode chamber (Fig. 21c), 2.6 times higher peak power density was achieved and such a cell can be stably operated at 700 °C for 10 h. With a further addition of the catalyst layer at the outlet of the anode chamber (Fig. 21d), synthesis gas with a H2:CO ratio close to 2 was obtained, suggesting a significant advantage for practical applications.
 | ||
| Fig. 21 Different SEMR design based on SC-SOFCs with/without catalyst components in tubular structures: (a) all flow-through mode, (b) first cathode chamber then anode chamber, (c) similar to mode b but with catalyst at the inlet of anode chamber and (d) similar to mode c with additional catalyst at the outlet of the anode chamber.197 Reproduced with permission. | ||
In the SC-SOFCs, the electrolyte layer acts as the ionic conductor, not a gas separator, so a dense electrolyte layer may not be needed while can still effectively extract energy from chemicals through SOFC functionality. Therefore, in 2013, Guo proposed a all-porous SOFC concept to serve as a bridging technology between dual-chamber SOFCs and SC-SOFCs.48,360 Interestingly, such a porous SOFC not only effectively solves the deactivation issue caused by carbon deposition when using hydrocarbons in conventional dual chamber SOFCs, but also enables higher fuel cell electrical efficiency and overcomes the inflammability issue over SC-SOFCs. By carefully controlling the distribution of gaseous O2 through the cathode chamber partial pressure and the flowing rate at the anode side, all porous SOFCs showed a maximum OCV of 0.855 V and the proof of concept cells operated in a methane-containing atmosphere showed very stable power density without carbon deposition for 2000 h.48 An additional advantage for such novel SOFCs is the fast-start procedure by applying oxygen-rich conditions at the anode, which may promote the SOFCs for portable or mobile applications. In their later work, an anode supported by all porous SOFCs was further developed to replace the electrolyte one with low electrochemical performance. Even though the OCV values are reduced due to a large oxygen partial pressure in the anode chamber, the peak power density of the former one improved to 214 mW cm−2 at 750 °C, which is over 14 times than that observed with the latter case. No visible carbon deposition or anode microstructure change was observed at the anode after operation in CH4 for 220 h.360
Similar to SC-SOFCs and all porous SOFCs, sealant-free direct flame SOFCs (DF-SOFCs) using combustible gases, liquids, and solids were also proposed for combined power and heat co-generation.361–363 In this cell configuration, the anode side is directly exposed to a fuel-rich flame, while the cathode is open to ambient air. The flame serves as the fuel since the insufficient combustion or utilization of chemicals that are partially reformed by air, and heat suppliers enables thermal sustenance of the DF-SOFCs. The initial DF-SOFCs gave an inferior electrochemical performance while, by the slight modification of the anode by creating serially connected cells, the DF-SOFCs showed an OCV over 0.8 V and a peak power density of 318 mW cm−2.361 Moreover, even with Ni-cermet electrodes, the coke issue is hardly observed in DF-SOFCs since the fuels in the flame are homogeneously converted to syngas before reaching the anode, which shows a clear advantage over conventional dual chamber SOFCs.
The disc structural reactor involves mature fabrication technology, and optimal conduction/mass transfer, but requires dedicated sealing and the active area is limited. Differently, the tubular and hollow SEMRs are less sensitive to sealing issues, especially at elevated temperatures, while they suffer from current collection and low power density problems. Thus, further development of strategies to obtain and deploy ion-conducting materials and active electrodes with new configurations/structure design as well as topological optimization and multiscale modeling would be a great advance for the application of SEMRs. To this end, Li et al.364 used commercially available materials to build a ceramic fuel cell having an evolved ‘micro–monolithic’ geometry with droplet-shaped channels (Fig. 22a). Such a unique structure enabled the deliberate spatial separation of the electronic electrochemically active area from the electronically conducting mechanical support, therefore, remarkably reducing the gaseous fuel's diffusional length (Fig. 22b and c). Such a novel device configuration could use low calorific value gases (LCVGs) like methane with concentrations as low as 10% and create an area-specific atmosphere that suppresses coking in the electronic electrochemically active region. When operated with 10% LCVGs, such a design reactor achieved an impressive power density of 1.77 W cm−2 at 750 °C (Fig. 22d). Long-term stability up to 500 h was also demonstrated at 700 °C and a constant voltage of 0.7 V when fueled with a CH4–CO2 mixture (1:1 ratio) fuel and ambient air as the oxidant, respectively.
 | ||
| Fig. 22 Novel SEMR (SOFC) design: (a) the micro–monolithic anode support incorporating 6 sub-channels with droplet shapes, (b) full structure simulation, (c) utilizing LCVGs with reactions at triple-phase boundaries, and (d) power density/voltage–current density characteristics with various LCVGs fed as fuels. With permission (open access) from ref. 364. | ||
5. Process intensified SEMRs
SEMRs operated at high temperatures with redox electrical potential are devices that are integrated thermal and electrical processes, which have already shown high potential over the individual process.365,366 Besides, when integrated with other possible energies, like solar or more general photo energy, or thermoelectric for a single component reaction or system level energy integration, on one hand, the electrochemical reaction kinetics will be further improved, on the other hand, such SEMRs will be operated by the external energy to realize the energy conversion/storage in chemicals or power, which represents the most interesting options for process identification and modularization.180,367–369 With the quick deployment of renewable energy sources, more attention is given to light energy harvesting and conversion since solar energy could be considered as the final source of all kinds of energy with sustainable characteristics. These have stimulated considerable efforts and achieved fundamental progress in recent years.5.1 Photo-intensified solid oxide cell process
In 2013, Ye reported and theoretically demonstrated that by intentionally replacing a liquid or polymeric solid electrolyte in conventional photo-electrochemical cells (PENs) with a solid ionic conductor, the operating temperature could be well extended from the current 60–80 °C to a much higher range.370 Moreover, the results of this work contrast sharply with the conventional view: solar-to hydrogen efficiency decreases with increased temperature. Instead, it reaches a maximum of 17% at 723 K with a non-precious electro-catalyst-based SOEC integrated single-junction light absorber (band-gap of 2.0 eV). Such a case not only demonstrates the feasibility of solid oxide photo-electrochemistry for the dissociation of water to H2, but also proves the potential higher efficiency because of its operating isothermal nature. Another interesting phenomenon of the SEMRs with semiconductor-based electrodes (SrTiO3 with Pt current collector) showed a cell voltage in the 200 mV range at ca. 400 °C upon UV light, which suggests the direct photo energy storage in SEMRs in the form of chemical energy. Moreover, such a cell voltage remains even after switching off the light.371,372 The time-dependent stoichiometry of oxygen in semiconductor materials (SrTiO3,371 TiO2,371,372 CeO2 (ref. 373) as shown in Fig. 23a) upon UV light is ascribed to the changed cell voltage.374,375 When a photovoltaic cell (Sr-doped LaCrO3 thin film on a SrTiO3 single crystal) is in tandem contact with a solid oxide electrochemical cell (porous LaSrFeO3/YSZ/porous LaSrFeO3), a photo-voltage as high as 900 mV at 400 °C was obtained,376 which enables a current of ≈1 mA cm−2 to pump oxygen from low to high partial pressures, i.e., to convert radiation energy to chemical energy. All these cases show the feasibility of achieving high-temperature photo-electrochemical water splitting. | ||
| Fig. 23 Process-intensified SEMRs using different external energy sources: (a) light-induced cell voltage increase in solid oxide photo-electrochemical cells,373 (b) thermoelectric SOFCs,377 (c) SEMRs intensified by H2 energy storage in the anode51 and (d) plasma intensified SEMRs for N2 activation and NH3 synthesis.378 Reproduced with permission from individual reference-related publishers. | ||
Light not only induces the bulk oxygen stoichiometry change with time but also leads to the change of the chemical environment of the grain boundary of polycrystalline ceramics, leading to the change of the conductivity of ceramic materials. For grain boundary conductivity, it is considered as the main limitation of the low ionic conductivity of most ceramic oxide ionic conductors. In 2022, Defferriere and colleagues52 found that the grain boundary conductivity of a 3GDC thin film increased approximately 3.5 times at 250 °C and the conductance activation energy was reduced from 1.12 to 0.68 eV under illumination. After experimentally excluding the contribution of heat and electronic conductivity, photo-generated electrons that decreased the potential barrier heights associated with space charge zones depleted in charge carriers between adjacent grains was contributed; in other words, the phenomenon of light-induced reduction in the grain boundary space charge potential occurred, reducing the grain boundary resistance.
Recent work reported that the hydroxyl radical yielded from reduced TiOx powder under high temperature infrared light excitation,53 a typical condition of SOFCs, can help the removal of deposited carbon at the SOFC anode. Moreover, SOFC electrochemical performance was also improved by 7 times with simulated biogas fuel (70% CH4+ 30% CO2) at 850 °C. Such work provided a novel method to solve the commonly observed carbon deposition issue in hydrocarbon--fueled SOFCs, promoting wide application.379 To this end, Su and Hu recently reported a combined thermal and photo-promoted activation of C–H and CO bonds of CO2/C2H6 drying reforming gas in a novel low temperature carbonate super-structured SOFCs with an additional functional layer of Pt/black TiO2 catalyst at 550 °C.380 Compared with the regular thermal catalytic anode process, the hybrid catalytic anode process induced by light illumination improved both the conversion rate (25%) of CO2/C2H6 and the yield of CO/H2, which improved SOFC peak power density; a record-high PPD of 168 mW cm−2 was achieved at 550 °C, 17% higher than that operated in the dark. Furthermore, the cell exhibited excellent stability without coking and delamination during ∼50 h operation. The joint thermo–photo catalysis was demonstrated to be a unique chemical process for SOFCs in the aspects of improved efficiency and operational stability.
5.2 Integrated H-SEMRs by the palladium membrane
As aforementioned, PCECs have exhibited the most promising characteristics to replace the traditional O-SOFCs for energy conversion at reduced temperatures. With the evolution of SOFC fabrication technology and novel electrode material development especially the triple conducting cathode materials,171,272,276 PCFCs have shown impressive electrochemical performance at temperatures of 500–600 °C, such as 500 mW cm−2 at 500 °C. Further modification of the interface properties of PCECs improves the electrochemical efficiency.173,272 A distinguished example of further improvement of the electrochemical performance of PCFCs was the one called “hydrogen membrane fuel cells (HMFCs)”.381,382 In 2005, the authors smartly deposited a 0.7 μm BaCe0.8Y0.2O3 layer on a Pd film (40 μm in thickness, a exceptional proton transportation and storage metal) by pulsed laser deposition. When integrated with a common perovskite oxide cathode, such a Pd membrane integrated PCECs gave peak power densities of 0.9–1.4 W cm−2 at the temperature range of 400–600 °C, and the corresponding total area specific polarization resistance was about 0.34 and 0.2 Ω cm2, in which electrode polarization resistance occupied about two-thirds of the cell total resistance but still much lower than that of the general case with the same cathode materials. The promoted cathode reaction in such HMFCs was proven by Jeong et al.383 in 2021. They found that, compared with the general Ni-based porous anodic/electrolyte interface, the blocking effect of the Pd membrane and its metal/oxide interface resulted in an accumulation of O2− at the interface, which acquires extra protons to compensate for the negative charge from the accumulated O2−, subsequently resulting in higher proton conductivity at the interface than that in identical PCFC membranes; with the application of overpotential, such an accumulation behavior was further improved, triggering proton pumping to the perovskite oxide cathode side, therefore improving the cathode kinetics and delivering exceptional electrochemical performance at the reduced temperature which is hard to be accessed in normal case.5.3 Thermoelectric SOFCs
In 2012, Huang proposed and demonstrated a concept of thermoelectric SOFCs. The basic concept is illustrated in Fig. 23b.377,384 An elongated thermoelectric p-type Na1−xCuxCo2O4 was used as both a cathode for SOFCs and a thermoelectric generator. Due to the elongated design, the area close to the electrolyte is the hot end (T1), whose temperature is higher than that (T2) of the cold end (Fig. 23b). For such a thermoelectric material, the temperature gradient drives charge carriers from the hot end to the cold end and therefore, appending a positive thermoelectric voltage to the SOFC because they share the same electrical field direction. Therefore, the whole cell voltage consists of two parts: one from the conventional SOFC and other from the thermoelectric generator (Fig. 23b). The total voltage (Vtotal) of the thermoelectric SOFCs is expressed as Vtotal = Vcell + ΔVthemo. It is found that the additional voltage (ΔVthemo) was 13.9 mV for NaCo2O4 and 11.1 mV for Na0.7Cu0.3Co2O4 at 800 °C, respectively, while it was only 0.1 mV for the SOFCs with a typical Ba0.5Sr0.5Co0.8Fe0.2O3 cathode, demonstrating the successful process integration and energy intensification.5.4 Intensified conversion by energy storage
Another interesting example of SEMR intensification is energy storage. Fuel cells are characterized by continuous energy conversion as long as the fuel is provided. However, their power output quickly decreases to zero if the fuel supply is interrupted. The discontinuous power supply may not be desirable for niche applications such as miniature autonomous systems and military technologies that require operational characteristics for short periods.51 Auxiliary energy storage devices like lithium-ion batteries may be coupled for such a purpose. However, the total weight and volume of the system will increase. In this context, storing chemical energy in the fuel cell materials would therefore be a great advantage.Van Overmeere proposed and demonstrated that vanadium oxide would be a good choice.51 Because of its change in oxidation state, vanadium oxide is a potential candidate for fuel oxidation. Moreover, hydrogen can be first inserted into the lattice of VO2 and V2O5 and the nonstoichiometric V6O13 and V3O7,385 leaving a wide H/V ratio in the range of 0.3–1.9 depending on the oxide and the applied conditions with sufficient hydrogen fuel. When integrated into a silicon wafer supported free-standing miniaturized SOFC with YSZ (80–95 nm in thickness) and Pt cathode, the deposited vanadium oxide (VOx) and Pt/VOx anode material-based SOFCs showed inferior performance over the pure Pt-based electrode (Fig. 23c). Moreover, compared to the potential decrease (to 0 V) in 15 s for the Pt anode cells when fuel was intentionally interrupted, the VOx and VOx/Pt-based cells maintained 32–210 s, depending on the vanadium oxide film thickness and the applied current density (Fig. 23c). In other words, the vanadium oxide anode-based fuel cells supply electricity for a 2–14 times longer period after the hydrogen is turned off. Both the change of the vanadium valence state and the hydrogen storage capability are ascribed to the extended power supply period, which may contribute to the ultra-miniaturization of power sources for mobile energy in the future (Fig. 23c). Similarly, Guan showed that the metal hydride could also serve such a purpose for uninterrupted electricity generation.386 With the metal hydride anode, fuel can be oxidized to produce electricity in the fuel cell mode (continuous fuel supply), while the same device would be operated in battery mode, the stored H2 in metal hydride decomposes to metal and H, which continuously serves as fuel for a certain time. Taking TiH2 as an example, the TiH2–air battery shows a continuous 96 h discharge at 10 mA current density, and a flat discharge potential of 0.85 V over 90 h, which is quite impressive for practical applications. Moreover, stable cycling operation between the fuel cell (H2 injection) and battery mode (Ar injection into the anode) was demonstrated for 75 h with each mode being operated for 15 min, showing promise for load leveling in the electricity grid.
5.5 Plasma-activated SEMRs for N2 conversion
Electrochemical NH3 synthesis is a promising approach for N2 fixation. However, in Section 3.2.2.2, we saw that both ammonia formation rate and faradaic efficiency are lower than that of economic targeted values, >10−7 mol cm−2 s−1 and 50%. The low rate of N2 triple bond cleavage is a primary challenge. In this regard, Sharma proposed a plasma-activated H-SEMR (Ni–BaCe0.2Zr0.7Y0.1O3−δ/BaCe0.2Zr0.7Y0.1O3−δ/Pt) for NH3 synthesis (Fig. 23d).378 In the SEMR anode, water is electrochemically splited into O2 and H+. The latter is transported to the cathode chamber to react with the plasma-activated nitrogen to form NH3. A maximum NH3 production rate of 26.8 nmol s−1 cm−2 with an exceptional Faraday efficiency of 88% at 500 °C. Moreover, such plasma-assisted SEMRs proved superior to the individual pure plasma and plasma catalysis approaches. The much-improved N2 activation and the interaction of the N activation intermediate with the catalyst co-contributed to the outstanding N2 reduction and NH3 synthesis reaction. By changing the SEMRs from a proton conducting oxide to an oxygen ionic conducting oxide, the same group by the proof of principle also demonstrated that high faradaic efficiencies up to 93% were achieved for NO production at 650 °C, and NO concentration was 1000 times greater than the reaction equilibrium concentration at the same temperature and pressure.162 These two studies showed the huge scope of SEMRs on integration of plasma energy sources to intensify the targeting reaction.5.6 Single-step hydrogen generation, separation, and compression
SEMRs simultaneously enable chemical reaction, product separation and compression in one step, therefore holding promise for process intensification. As you can see in the above case, both H2 flux and water splitting reaction contributed to the super high H2 production rate at an extremely high temperature. Serra proposed the use of a BaZrO3-based proton-conducting electrolyte membrane for producing high-purity H2 from CH4, NH3, and biogas steam reforming reactions.15,274 The designed tubular H-SEMRs with a Ni particle catalyst enabled the production of a nearly pure stream of CO2 and in situ electrochemically compressed H2 up to 50 bar, which enables effective carbon capture and 99% hydrogen element utilization from reactants (H2O and CH4) (Fig. 24c–e).274 Moreover, the thermal energy was well managed by coupling thermal-chemical processes, i.e., endothermic reforming reactions with heat from electrochemical gas separation and compression by the galvanic operation of the electrochemical cell (Fig. 24a and b). Therefore, a thermally balanced operation regime is also achieved by coupling several thermo-chemical processes with total energy efficiency higher than 78% in a small-scale hydrogen plant according to the simulation result. Besides the H2O splitting reaction, CH4, NH3, and biogas also demonstrated as are sources of H2 energy. Clark demonstrated similar results using NH3 and biogas as sources.15 Moreover, they developed a new nickel-based glass–ceramic composite interconnect to scale up such SEMRs at the single cell level to 36-cell stack level, which allows good balance of the endothermic reaction without producing temperature gradients and entropic effects that lead to the drop of efficiency. Such a counter-flowing stream balanced the heat flow and maintained stable operating conditions, enabling 99% hydrogen recovery and up to 140 bar hydrogen compression, suggesting a high degree of process intensification. Such progress highlights the potential of the practical application of the thermochemical device for efficient hydrogen production, also the realization of carbon neutrality with 100% CO2 capture and utilization. | ||
| Fig. 24 (a) Schematic illustration of H-SEMRs integrated with an electro-chemical H2 compression process, (b) energy balance and system micro-integration for operation at 800 °C for a feed inlet of one mole CH4, (c) tabular SEMRs with Ni catalyst loaded inside the ceramic tube (insets are SEM cross-section images of the overall cell and electrode reactions), (d) H2 production rate versus current density compared with the theoretical value and (d) conversion, yield of CO2 and CO versus hydrogen recovery at 800 °C, 10 bar, S/C = 2.5. Steam pressure on the hydrogen side inlet was 0–2.0 bar. Reproduced with permission from ref. 274. | ||
6. Electrochemical promotion of catalysis (EPOC) in SEMRs
As can be seen from the above discussion, significant research efforts have been made in the application of SEMRs to different processes and reactions by combining heterogeneous catalysis and solid-state electrochemistry at elevated temperatures. Under the typical conditions, SEMR allows the electrochemical supply of one of the reactive, O2−/H+, or mixed conductive ions by a faradaic operation, where the reaction rate is in general proportional to the applied current/ionic transportation rate. Nevertheless, the catalytic reaction rate, in many cases, exceeded the rate of the electrochemical supply of ionic species (non-faradaic operation), even by several orders of magnitude, which is the combinative effect of electric field (faradaic process) and catalytic activity (non-faradaic process). In those cases, the electrochemically supplied ionic species could behave as a catalytic promoter to significantly improve the reaction rate and/or product selectivity.27,40 Such a phenomenon is called the EPOC or NEMCA effect, which was first discovered by Stoukides and Vayenas in 1981.387 They found that the C2H4O selectivity and yield of ethylene oxidation on polycrystalline silver can be affected significantly by electrochemical operational modes. Under the open circuit conditions, the Ag catalyst acts as a normal catalyst on the YSZ electrolyte, like a fixed bed reactor. While under the applied external voltage that pumped O2− to the catalyst, C2H4O's selectivity and yield increased considerably. In particular, the increase in the rate of C2H4O production can exceed the rate of O2−-pumping by a factor of 400. The opposite effect was also observed upon inversion of the voltage polarity. Since the discovery of the EPOC effect, more than 100 reaction systems have been demonstrated.40,116,388–394 The EPOC effect can be quantitatively expressed by the use of two dimensionless parameters,40,389Λ = Δr/(I/nF) and ρ = r/r0, where F is Faraday's constant, ∼96485 C mol−1, n is the charge of the transported ion, r is the catalytic reaction rate obtained under closed circuit conditions, r0 is the open-circuit catalytic rate (i.e., at I = 0), Δr = r − r0 and I/nF is the ionic flux through the electrolyte based on the Faraday law. If Λ = 1, the effect is faradaic, where the reaction rate equals the rate of ion transport rate. In many cases, a strong non-faradaic effect was observed with Λ values ≫1, even as high as 3 × 105, and ρ values as high as 150 (Table 4). The observed change in the catalytic rate is non-faradaic since the products formed surpass considerably the amount of ions supplied to the reaction.
| Reaction | Reactor | Λ (times) | ρ | References |
|---|---|---|---|---|
| C2H4 oxidation | Ag/ZrO2(Y2O3)/Ag | — | 400 | 387 |
| H2O + CH4 for H2 | Pt-YSZ/YSZ/Au, single chamber | 3–4 | — | 359 |
| CH4 oxidation | Pd–Co3O4/YSZ | 80 | — | 395 |
| RWGS | Pt/YSZ/Pt | 10 | — | 396 |
| CO2 hydrogenation | Ni–Ri on carbon fiber/YSZ/Au | 1600 | 3 | 397 |
| Tubular Pt/YSZ/Ru | — | 2 | 116 | |
| Ru–Co3O4/BZY/Au | 10 | 2 | 398 | |
| Pt, Pd, Ag/YSZ/Pt, Pd, Ag | 6000 | 100–3 × 105 | 393 | |
| Pt/YSZ/Au | 74000 |
26 | 388 | |
| Ru–Co/BZY/Au | 60 | — | 391 | |
| Ru/YSZ/Au | 5 | 1.4 | 392 | |
| Ru/BZY/Au | 500 | — | 390 | |
| Ammonia synthesis | Fe/CaIn0.1Zr0.9O3−α/Ag | 13 | 6 | 399 |
| C2H4 + NOx to N2 | Pt/β′′-alumina/Au | — | 15 | 400 |
| NO + CO/C3H6 | Pt/β′′-alumina/Au | — | — | 401 |
| NO + CO | Pd/YSZ/Au | 700 | 1.5 | 402 |
| NO reduction | Ni-YSZ/YSZ/LSMC-GDC/Pd-GDC | — | 100–1000 | 165 |
| NOx reduction | La0.8Sr0.2Co0.9Ru0.1O3−δ/YSZ/Pt | 30 | — | 403 |
| Ethylene oxidation | Pt | 105 | 74000 |
404 |
EPOC effect is universally observed in both single-chamber and two-chamber configurations, which are well suitable for SEMR operation, either in fuel cell mode or electrolysis mode. For example, Caravaca et al.359 experimentally found that the H2 formation rate in single-chamber steam electrolysis cells (Pt-YSZ/YSZ/Au) varied significantly under methane partial oxidation, steam electrolysis, and methane auto-thermal reforming conditions, respectively. The highest H2 production rates were achieved under the auto-thermal reforming conditions and with an application of negative currents over the Pt catalyst-working electrode at 600 °C. It was demonstrated that the application of the negative currents modified the work function of the Pt catalyst and altered its catalytic properties, which allowed an additional H2 production based on the intrinsic EPOC effect, therefore, overcoming the limitation by Faraday's law and reducing the cost of Zagoraios et al.395 reported the electrochemical promotion of methane oxidation effect on monodispersed Pd/Co3O4 catalysts with YSZ solid-state electrolytes. It was found that the catalytic rate increased by 2.5-fold and faradaic efficiency values increased by 8000%. In addition, they confirmed that the EPOC effects enhance the reaction rate at temperatures lower than those observed in previous studies. Another interesting case is its application in reverse water gas shift reaction (RWGS) based on SEMRs with proton conductors.396 Under such H2/CO2 conditions, hydrogen was electrochemically supplied as H+ other than as molecular H2. Under closed circuit operation, Λ value up to 10 for CO formation was achieved, which should be ascribed to direct electrocatalysis together with the enhancement of the open circuit reaction rate. More attention to the EPOC effect is given to CO2 hydrogenation with SEMR both on oxygen-ion conducting electrolytes116,397 and on proton-conducting electrolytes.116,390–392,398 In the thermochemical catalytic process, the rate-determining process is generally ascribed to either the dissociation of the C–O bond or the hydrogenation of the intermediates, while in the O-SEMRs, the CO2 electrolysis was suggested to go through a CO32− intermediate, which is promoted by the applied anodic bias.81,82 For example, Chatzilias intentionally adopted a low temperature (340–400 °C) O-SOFC to the electrochemical promoting reactor for CO2 hydrogenation (Fig. 25a).116 At the first glance, O2− conducting from the cathode moving to H2/CO2 may be detrimental to hydrogenation. Even with the EPOC effect, the low temperature operation can only result in quite low fuel cell performance. Indeed, the peak power density could only reach 0.45 mW cm−2 at 400 °C (Fig. 25b), while rate enhancement ratios of up to 3 (at 440 °C) and faradaic efficiency values of up to 1600 were found at lower temperatures (<360 °C) with H2 and CO2 partial pressure of 7 kPa and 1 kPa, and the selectivity of CH4 reduced with the increased temperature (Fig. 25c). In contrast, the faradaic efficiency values observed for CO formation were between −20 and −10, with the corresponding rate enhancement ratios being close to 0.5 (Fig. 25c). If sufficient power (as a fuel cell) could be produced for the intensification of the EPOC effect, it can be applied for the valorization of CO2 to value products.
 | ||
| Fig. 25 (a) Schematic representation of the tubular SOFC reactor with reactions at each electrode under OCV and short-circuit conditions, (b) power generation characteristics, and (c) temperature dependence under steady-state conditions on the Λ and ρ. PH2 = 7 kPa, PCO2 = 1 kPa, FT = 200 cm3 min−1. Reproduced with permission from ref. 116. | ||
In PCECs, a CO32− intermediate was observed in the fuel electrode,318via in situ Raman and diffuse reflectance infrared Fourier transform spectroscopy characterization. To further reduce CO2, the continuously replenished protons play a critical role in CO2 conversion and CH4/CO selectivity.390,398 Kalaitzidou prepared a Ru-deposited BZY proton conductor as the model catalyst for the hydrogenation of CO2 at temperatures of 300–450 °C and atmospheric pressure.390 It was observed that the rate and the selectivity to CH4 are enormously enhanced by the removal of a proton from the catalyst surface to the proton-conducting support. The EPOC effect parameter Λ for CH4 reached 500 and depended strongly on the thickness of the porous Ru catalyst film. Such an enormous improvement in the CH4 yield may suggest the practical application of such a reaction of both environmental and industrial importance. Moreover, it has been found that the catalytic activity of ammonia synthesis and NOx reduction can be promoted by the EPOC effect.399 The catalytic activity of industrially commercialized ammonia synthesis catalysts (BASF S6-10RED) was enhanced by up to 1300%, or higher, by depositing on a CaIn0.1Zr0.9O3−α proton conductor with electrochemical supply of protons to the catalyst surface at 400 °C and 50 atmospheric pressures according to Yiokai's report.399 Furthermore, the NH3 production rate is 6 times larger than the proton pumping rate, which indicates the EPOC effect. This work also showed the first demonstration of the scale-up of an SEMR with 24 electrically connected catalyst pellets, which highlights the important role of the EPOC effect in promoting the wide application of SEMRs for electrochemical NH3 synthesis. Besides, the literature reveals EPOC effects in the reduction of nitric oxide (NOx) to N2 under lean conditions168 with or without reducing agents using β′′-alumina400–402 or zirconia165,402,403,405 based SEMRs. The selectivity and electrochemical promotion reduction of NOx significantly reduce the system complexity without compromising the conversion efficiency, suggesting the wide application in SEMR technology.
As can be seen from the above discussion, EPOC could have a significant impact on catalytic research in energy and environmental fields with multiple benefits. (1) Unlike gas phase or solid-state promotors, the modification in EPOC is electrochemical, which is much more easily controlled, in particular, when one of the reactants in the reactor could act both as a power generator for the EPOC effect and the reactant for the catalytic reaction, resulting in much-reduced system complexity. Moreover, the energy consumption for the EPOC effect is much lower than the stoichiometry required.39,116 From a practical viewpoint, SEMRs consume very little electrical energy to operate and product generation is not limited by Faraday's law. (2) With the combined faradaic and EPOC effects, the overall efficiency of SEMRs could be significantly increased, which could facilitate the temperature reduction to the range of interest.359 Taking the methanation reaction as a typical example, the current H2O/CO2 electrolysis studies are performed at a temperature above 600 °C to ensure sufficient kinetics, while the methanation reaction is preferred at 300–500 °C. If the EPOC effect is coupled, the possibility of dynamic modification of the catalyst activity could favor such a temperature reduction, which could lead to a on-site electrolysis and methanation process. In addition, sharing the same benefits of the current temperature reduction in SOFCs, the stability, lifespan as well as system cost will be guaranteed because of the absence of thermal stresses. Under the reduced temperature, the economic potential of cogeneration to a large number of reactions could be expected, promoting the practical application of those cogeneration processes. (3) EPOC allows for the controlled enhancement of the catalytic activity/selectivity by potential or small current application between a catalyst (working electrode) supported on a solid electrolyte and a counter electrode.57,308,390 A typical example with a very high NEMCA effect is the production of CO2 from ethylene and O2 on the Pt catalyst using the YSZ electrolyte and temperatures in the range of 260–450 °C where a potentiostat–galvanostatic operation was employed.27 Kalaitzidou et al.390 also observed threefold enhancement in the catalytic rate of CH4 formation with concomitant 50% suppression of the CO formation rate when protons were removed from the catalysts to the electrolyte supporter, which suggests that the EPOC effect parallelly regulated the methanation reaction and CO yield reaction (water–gas shift reaction) are parallel route (Fig. 26b). Such ionic-mediated electrochemical processes could potentially be harnessed to promote the activity of other reactions, such as the electrochemical ammonia synthesis where hydrogen evolution reaction occurs in parallel.
 | ||
| Fig. 26 (A) Schematic of the basic principle of EPOC when ions (oxide ions or cations) are moved to the catalyst of the working electrode408 and (B) CH4 and CO production rates when CO2 hydrogenation is carried out over a range of negative and positive polarization.390 Reproduced with permission from ref. 390 and 408. | ||
Different mechanisms have been proposed to explain the EPOC effect in SEMRs: (i) With the applied current or direct potential, ion migration from the solid electrolyte (spillover) to the catalyst surface or removal from the catalyst surface changes the work function or electronic structure of the electrode materials, which leads to the dramatic change of catalytic properties in a reversible manner.359,398,404,406 In the context of SOECs, it is believed that catalytic activity is enhanced due to the promotion of the work function of catalytic surfaces generated by oxygen ion pumping to/from the electrolyte onto the catalyst surface.407 Unlike structural promoters that improve the dispersion and the chemical stability of the active catalyst phase, electronic promoters directly modify the catalytic activity of the catalyst itself (Fig. 26a). By controlling the surface work function, one can control the adsorption, and surface diffusion properties for intermediates and partially discharged promoters. Such an EPOC effect has been well demonstrated in the initial discovery of oxygen ions on an Ag catalyst for the ethylene oxidation reaction.117,387
(ii) Reactant activation includes improved reactant chemical adsorption and/or charge transfer process.401,405 Under the specific electrical field conditions, the ion migrated to the triple phase boundary, which formed an effective electrochemical double layer,394 altering the chemisorptive properties of the catalyst. The reactant may be polarized and induced with the improved chemical adsorption and then is activated subsequently resulting in improved charge transfer between the electrode and reactant.
(iii) The ionic species from the fuel/oxidant for power generation also serves as a sacrificial promoter. In other words, these ionic species/promoters can be destroyed as easily as they are generated, just by varying the polarization of the catalytic surface.395 Hence, the EPOC phenomenon allows in situ control and optimizes the number of promoter species supplied to the catalyst/electrode, which has very important implications for practical purposes over other controlled technologies.
(iv) Intensified metal–support interaction.395 By carefully comparing the additional effect of Co3O4 to Pd supported on YSZ (over Pd/YSZ film), a 2.5-fold increase of the catalytic rate and a faradaic efficiency of 80 was obtained. Therefore, metal–support interactions significantly improve the EPOC effect, in which Co3O4 behaved as a “bridge” to allow for the Oδ− promoter ions to reach the metal nanoparticles, showing a much improved EPOC effect.
Finally, the maturity of the EPOC effect which has resulted from more than 40 years of intensive and extensive research because of the multiple benefits should offer the opportunity for many attractive applications. However, application of the EPOC effect in SEMRs to produce targeted chemicals requires further R&D activities. Future work may focus on a clear perception of the governing reactions, and reaction kinetics thereof, choice of electrode–electrolyte–catalyst combination, and finally, optimization of the operating conditions.
7. Summary and perspectives
Solid electrolyte membrane reactors (SEMRs) provide a promising alternative to conventional thermal catalysis because of the well-controlled reaction rate, avoidance of chemical balance, catalytic selectivity as well as possible EPOC effect. This review summarizes the applications of high temperature solid membranes for energy (among thermal, electrical, and chemical) conversion and storage, chemical generation, and environmental protection, with a focus on (reversible) conversion of small chemical molecules such as water, H2, CO, CO2, CH4, NH3, and NOx through (de)oxygenates, (de)hydrogenation in form of oxidative or non-oxidative processes. Currently, the application of oxygen ion conductors for SEMRs is still the most important field of research and applications, even though proton conducting membranes offer new possibilities for the implementation of electrochemical reactors. In both cases, a combined effect such as cogeneration of electricity and chemicals, detoxification, and electrical energy generation, “achieving two things through a single action”, could be well designed and deployed. Detailed case studies of the application of versatile ionic conductors, like O2−, H+, CO32−, Na+ and their hybrid conductors, their multi-functional application in SEMRs as well as the corresponding key assembly units (e.g., electrode materials and microstructure membranes and catalysts), and electrochemical performances like conversion, selectivity, and yields were presented. High temperature SEMRs showed remarkable applicability for different targeted reactions because of the coupled thermal and electrochemical process, as well as diverse cell configurations, such as two chamber-based (panel and tubular), single chamber-based, porous electrolyte-based, non-symmetric and symmetric SEMRs. Among these, electrolysis and co-electrolysis of H2O in the form of steam in SEMRs and CO2 are considered a cornerstone for the future chemical industry and sustainable energy systems, enabling efficient conversion and long-term storage of electrical energy and production of sustainable fuels and chemicals from renewable sources. Electrochemical selective oxidation or coupling of an alkane, their conversion to an alkene or arene/aromatics, and NH3 synthesis in both O- and H-SEMRs are of interest for both fundamental research regarding elementary catalytic steps and practical applications for improved conversion efficiency and large-scale deployment. In addition, concepts/strategies of process intensification/integration of SEMRs with other effective and renewable energy forms such as photo, thermoelectric, and plasma energy also showed improvement in energy efficiency, reaction selectivity and accelerated targeting chemical activation and conversion. Another benefit in SEMRs is the non-faradaic process/effect on the existing reactions, i.e., EPOC effect, which dramatically alters the conversion rate and selectivity of expected reactions over the typical faradaic reaction, which has been widely used in both studies and influence the rates of catalytic reactions. With the EPOC effect, it is expected to extend the measurements to lower temperatures of general interest and help elucidate the reaction mechanism that could not be obtained otherwise. In other words, SEMRs provide a vast opportunity to address important challenges beyond other technologies' capabilities and have therefore become an emergent technology/device to promote electrification of the current chemical/catalytic industry.However, the progress of SEMRs has not been sufficient to promote these processes on a larger scale, i.e., very limited cases with stack-level or large-scale cell applications.15,117,399 It should be noted that the massive commercialization of SEMRs relies on their capital and operating costs, lifespan, and reliability, while the operating cost is partly dependent on the system's output performance and operational stability. For example, the electrochemical output of a chemical and power co-generation reactor is much lower than that of the state-of-the-art SOFC technology, which is also integral to other functions of SEMRs. The conversion rate, selectivity and yield of CO2 reduction to hydrocarbons is still not well controlled and understood.308 Increasing reactor durability and decreasing capital costs are the main challenges to achieving market penetration. These essential technologies are currently at very low technology readiness levels (TRLs), among which the high-temperature CO2 electrolyzers using solid oxide electrolytes is approaching a mature level (TRL 8) and have been demonstrated with over 1000 h of stability.409 The durability and capital cost reduction of SEMRs highly rely on developing reliable reactor materials and robust devices. Leaning to experience from SOFCs, for example, we can build electrolyte-supported instead of anode-supported cell configuration based on the state-of-the-art cell material which remarkably improves the cell/reactor durability. With more industrial exploration or demonstration, combining the intrinsic high energy efficiency, and high reaction kinetics, it is expected that the capital cost will be much reduced, making them compete with other commercial processes and promoting the SEMR process on a larger scale.
There are also a variety of fundamental issues for the performance and degradation of SEMRs, including SEMR itself (key components' chemical, thermal, and mechanical, properties and their associated compatibility), the interaction between the reactant and impurity, and lifespan requirement of at least 10–20 years.62 Finding the degradation factors and developing means for reviving degraded performance would offer great value in improving SEMRs' commercial viability. Progress in materials science and solid-state ionics could be used for SEMRs with multiple purposes with minor modifications. There is also a significant gap between laboratory-scale research and industrial processes. In general, the industry has been reluctant to adopt the SEMR process because of limited ionic fluxes.117 In addition, the SEMRs should be scaled up into larger flat cells or stacks and, even pilot scale, to validate stable and high-efficiency operation. Moreover, the operation under application-sound conditions is necessary, for instance, by testing at higher total reactor pressure or pulsed current/voltage conditions which is matched with the nature of intermittent renewable energy, as well as grid-scale application. Another challenge is that there is a lack of techno-economic studies and bottom-up cost analysis of SEMR systems to replace the current chemical industrial plants. The techno-economic aspects should be carefully evaluated to meet further industrial needs.
Regarding the EPOC or NEMCA effect in SEMRs, many classic reactions including CO2 hydrogenation and hydrocarbon fuel oxidation have been demonstrated with an impressively improved effect. However, no consensus mechanism for EPOC has been reached. Detailed primitive reaction step investigation with advanced electrochemical and photo-spectroscopy characterization techniques is preferred. The good news is that the increasing number of publications/literature dealing with practical aspects of NEMCA application address questions related to pilot plant scale tests, and reactor designs but are also relevant to catalyst and electrolyte manufacturing technology.410
Finally, SEMRs generally provide one or more reactants for continuous chemical synthesis, therefore, a combination of SEMRs with catalysts of targeted catalytic reactions in appropriate electrode–electrolyte–catalyst structure design/geometry,15,47 and a clear perception of the governing reactions and reaction kinetics is required. High energy efficiency,15 reaction conversion rate/yield, and system stability20 can be expected through optimization of the working conditions. Learning from commercial chloralkali processes, electrolysis of alumina, and organic chemical synthesis like acrylonitrile dimerization and fluorination of ethers will be important for the scale-up of electrochemically driven energy conversion and storage, chemical synthesis and environmental treatment in SEMRs.
Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this review.Author contributions
L. F.: Conceptualization, Writing – original draft, Resources, Funding acquisition, Writing – review & editing, Supervision; W. L., Q. F., Q. H., and Y. F.: Writing – review & editing; T.-W. C. and P. D. L.: Resources, Writing – review & editing, Supervision; all authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Abbreviations
| 6Sc1CeSZ | Sc0.06Ce0.01Zr0.93O2 |
| ASR | Area-specific resistance |
| BSCF | (Ba,Sr)(Co,Fe)O3−δ |
| BZCFY | BaCo0.4Fe0.4Zr0.1Y0.1O3−δ |
| BZCY | BaZr0.7Ce0.2Y0.1O3−d |
| BZCYYb | BaZr0.1Ce0.7Y0.1Yb0.1O3−δ |
| BZY | BaZr0.8Y0.2O3−δ |
| C2 | Hydrocarbon with two carbon atoms |
| DCFC | Direct carbon fuel cell |
| De-NOx | Removal of nitric oxide |
| DF-SOFCs | Direct flame solid oxide fuel cells |
| DRIFTS | In situ diffuse reflectance infrared spectroscopy |
| EPOC | Electrochemical promotion of catalysis |
| FAE | Fuel-assisted electrolysis |
| F–T | Fischer–Tropsch process |
| GDC or CGO | Gd doped ceria, normally Gd0.1Ce0.9O2−δ |
| H-SEMRs | Hydrogen ionic conductive SEMRs |
| j 0 | Exchange current density |
| LSCF | (La,Sr)(Co,Fe)O3−δ |
| LSGM | (La,Sr)(Ga,Mg2)O3−δ |
| LSM | (La,Sr)MnO3−δ |
| LSMC | La0.8Sr0.2Mn0.9Cu0.1O3 |
| NASA | National aeronautics and space administration |
| NEMCA | Non-faradaic electrochemical modification of catalytic activity |
| NOx | NO or NO2 |
| OCM | Oxidative coupling of methane |
| OCV | Open circuit voltage |
| O-SEMRs | Oxygen ionic conductive SEMRs |
| O-SOEC | Oxygen ionic conductive SOEC |
| O-SOFCs | Oxygen ionic conductive SOFCs |
| PBM | PrBaMn2O5+δ |
| PCEC | Proton ceramic electrolysis cell |
| PCFCs | Proton ceramic fuel cell |
| PEM | Proton exchange membrane |
| PENs | Photo-electrochemical cells |
| RSOCs | Reversible solid oxide cells |
| RWSR | Reverse water shift reaction |
| SC-SOFCs | Single-chamber solid oxide fuel cells |
| ScSZ | Scandia stabilized zirconia |
| SDC | Sm doped ceria, normally Sm0.2Ce0.8O2−δ |
| SEMRs | Solid electrolyte membrane reactors |
| SFM | Sr2Fe1.5Mo0.5O6−δ |
| SOCs | Solid oxide cells |
| SOECs | Solid oxide electrolysis cells |
| SOFCs | Solid oxide fuel cells |
| STEM | Scanning transmission electron microscope |
| TPO-CO2 | Temperature-programmed oxidation using CO2 |
| TRL | Technology readiness levels |
| XPS | X-ray photoelectron spectroscopy |
| YSZ | Yttrium-stabilized zirconia |
Acknowledgements
L. Fan is thankful for the financial support from the National Natural Science Foundation of China (22378268), the Guangdong Basic and Applied Basic Research Foundation (2024A1515012212), and the Shenzhen University-National Taipei University of Technology Joint Research Program (2023011).References
- V. Thyssen, V. Vilela, D. de Florio, A. Ferlauto and F. Fonseca, Direct Conversion of Methane to C2 Hydrocarbons in Solid-State Membrane Reactors at High Temperatures, Chem. Rev., 2022, 122, 3966–3995 CrossRef CAS PubMed.
- X. Hou, Y. Jiang, K. Wei, C. Jiang, T. Jen, Y. Yao, X. Liu, J. Ma and J. Irvine, Syngas Production from CO2 and H2O via Solid-Oxide Electrolyzer Cells: Fundamentals, Materials, Degradation, Operating Conditions, and Applications, Chem. Rev., 2024, 124, 5119–5166 CrossRef CAS PubMed.
- T. Zheng, M. Ou, S. Xu, X. Mao, S. Wang and Q. He, Recent Progress of Bifunctional Electrocatalysts for Oxygen Electrodes in Unitized Regenerative Fuel Cells, J. Electrochem., 2023, 29, 2205301 Search PubMed.
- S. McIntosh and R. J. Gorte, Direct hydrocarbon solid oxide fuel cells, Chem. Rev., 2004, 104, 4845–4865 CAS.
- Z. J. Schiffer and K. Manthiram, Electrification and Decarbonization of the Chemical Industry, Joule, 2017, 1, 10–14 Search PubMed.
- J. L. Barton, Electrification of the chemical industry, Science, 2020, 368, 1181–1182 CAS.
- I. Jang, J. S. A. Carneiro, J. O. Crawford, Y. J. Cho, S. Parvin, D. A. Gonzalez-Casamachin, J. Baltrusaitis, R. P. Lively and E. Nikolla, Electrocatalysis in Solid Oxide Fuel Cells and Electrolyzers, Chem. Rev., 2024, 124, 8233–8306 CAS.
- A. Hauch, R. Küngas, P. Blennow, A. Hansen, J. Hansen, B. Mathiesen and M. Mogensen, Recent advances in solid oxide cell technology for electrolysis, Science, 2020, 370, eaba6118 CAS.
- S. Foit, I. Vinke, L. de Haart and R. Eichel, Power-to-Syngas: An Enabling Technology for the Transition of the Energy System?, Angew. Chem., Int. Ed., 2017, 56, 5402–5411 CAS.
- R. Daiyan, I. MacGill and R. Amal, Opportunities and Challenges for Renewable Power-to-X, ACS Energy Lett., 2020, 5, 3843–3847 CrossRef CAS.
- L. Zheng, M. Niu, T. Zeng, X. Ge, Y. Wang, C. X. Guo, W. Yuan, D. Cao, L. Y. Zhang and C. M. Li, Assembling molybdenum-doped platinum clusters into a coral-like nanostructure for highly enhanced oxygen reduction, eScience, 2024, 4, 100187 Search PubMed.
- S. Shangqing, M. Yalan, L. Fang, Z. Shukang, S. Yidan, G. Qiang and L. Xiaojing, Recent advances in nanoscale engineering of Pd-based electrocatalysts for selective CO2 electroreduction to formic acid/formate, Energy Mater., 2024, 4, 400027 Search PubMed.
- R. D. Farr and C. G. Vayenas, Ammonia High Temperature Solid Electrolyte Fuel Cell, J. Electrochem. Soc., 1980, 127, 1478–1483 CAS.
- A. I. Stankiewicz and H. Nigar, Beyond electrolysis: old challenges and new concepts of electricity-driven chemical reactors, React. Chem. Eng., 2020, 5, 1005–1016 CAS.
- D. Clark, H. Malerød-Fjeld, M. Budd, I. Yuste-Tirados, D. Beeaff, S. Aamodt, K. Nguyen, L. Ansaloni, T. Peters, K. Vestre Per, K. Pappas Dimitrios, I. Valls María, S. Remiro-Buenamañana, T. Norby, S. Bjørheim Tor, M. Serra Jose and C. Kjølseth, Single-step hydrogen production from NH3, CH4, and biogas in stacked proton ceramic reactors, Science, 2022, 376, 390–393 CAS.
- P. Boldrin, E. RuizTrejo, J. Mermelstein, B. M. José Miguel, R. r. Tomás and N. Brandon, Strategies for Carbon and Sulfur Tolerant Solid Oxide Fuel Cell Materials, Incorporating Lessons from Heterogeneous Catalysis, Chem. Rev., 2016, 116, 13633–13684 CrossRef CAS.
- W. Wang, C. Su, Y. Wu, R. Ran and Z. Shao, Progress in Solid Oxide Fuel Cells with Nickel-Based Anodes Operating on Methane and Related Fuels, Chem. Rev., 2013, 113, 8104–8151 CrossRef CAS PubMed.
- D. Brett, A. Atkinson, N. Brandon and S. Skinner, Intermediate temperature solid oxide fuel cells, Chem. Soc. Rev., 2008, 37, 1568–1578 CAS.
- K. Sundmacher, L. Rihko-Struckmann and V. Galvita, Solid electrolyte membrane reactors: Status and trends, Catal. Today, 2005, 104, 185–199 CAS.
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O'Hayre, Highly efficient reversible protonic ceramic electrochemical cells for power generation and fuel production, Nat. Energy, 2019, 4, 230–240 CAS.
- C. Duan, R. Kee, H. Zhu, C. Karakaya, Y. Chen, S. Ricote, A. Jarry, E. Crumlin, D. Hook, R. Braun, N. Sullivan and R. O'Hayre, Highly durable, coking and sulfur tolerant, fuel-flexible protonic ceramic fuel cells, Nature, 2018, 557, 217–222 CAS.
- L. Dittrich, M. Nohl, E. E. Jaekel, S. Foit, L. G. J. de Haart and R.-A. Eichel, High-Temperature Co-Electrolysis: A Versatile Method to Sustainably Produce Tailored Syngas Compositions, J. Electrochem. Soc., 2019, 166, F971–F975 CAS.
- J. M. Serra, Electrifying chemistry with protonic cells, Nat. Energy, 2019, 4, 178–179 Search PubMed.
- E. Wachsman, C. Marlowe and K. Lee, Role of solid oxide fuel cells in a balanced energy strategy, Energy Environ. Sci., 2012, 5, 5498–5509 RSC.
- X. Yan, Y. Yang, Y. Zeng, B. Shalchi Amirkhiz, J. Luo and N. Yan, Generating C4 Alkenes in Solid Oxide Fuel Cells via Cofeeding H2 and n-Butane Using a Selective Anode Electrocatalyst, ACS Appl. Mater. Interfaces, 2020, 12, 16209–16215 CrossRef CAS.
- M. Li, B. Hua and J. Luo, Alternative Fuel Cell Technologies for Cogenerating Electrical Power and Syngas from Greenhouse Gases, ACS Energy Lett., 2017, 2, 1789–1796 CrossRef CAS.
- F. Alcaide, P. Cabot and E. Brillas, Fuel cells for chemicals and energy cogeneration, J. Power Sources, 2006, 153, 47–60 CAS.
- H. Spacil and J. Tedmon, Electrochemical dissociation of water vapor in solid oxide electrolyte cells, J. Electrochem. Soc., 1969, 116, 1618–1626 CAS.
- R. W. Coughlin and M. Farooque, Hydrogen production from coal, water and electrons, Nature, 1979, 279, 301–303 CAS.
- H. Iwahara, High temperature proton conducting oxides and their applications to solid electrolyte fuel cells and steam electrolyzer for hydrogen production, Solid State Ionics, 1988, 28–30, 573–578 Search PubMed.
- M. Stoukides, Applications of solid electrolytes in heterogeneous catalysis, Ind. Eng. Chem. Res., 1988, 27, 1745–1750 CAS.
- I. Metcalfe, Stabilised-zirconia solid electrolyte membranes in catalysis, Catal. Today, 1994, 20, 283–293 CAS.
- M. P. Heddrich, S. Gupta and S. Santhanam, Electrochemical Ceramic Membrane Reactors in Future Energy and Chemical Process Engineering, Chem. Ing. Tech., 2019, 91, 809–820 CAS.
- W. Li and J. Luo, High-Temperature Electrochemical Devices Based on Dense Ceramic Membranes for CO2 Conversion and Utilization, Electrochem. Energy Rev., 2021, 4, 518–544 CAS.
- X. Zhang, Y. Song, G. Wang and X. Bao, Co-electrolysis of CO2 and H2O in high-temperature solid oxide electrolysis cells: Recent advance in cathodes, J. Energy Chem., 2017, 26, 839–853 CrossRef.
- L. Zhang, S. Hu, X. Zhu and W. Yang, Electrochemical reduction of CO2 in solid oxide electrolysis cells, J. Energy Chem., 2017, 26, 593–601 CrossRef.
- J. C. Fornaciari, D. Primc, K. Kawashima, B. R. Wygant, S. Verma, L. Spanu, C. B. Mullins, A. T. Bell and A. Z. Weber, A Perspective on the Electrochemical Oxidation of Methane to Methanol in Membrane Electrode Assemblies, ACS Energy Lett., 2020, 5, 2954–2963 CAS.
- L. Bi, S. Boulfrad and E. Traversa, Steam electrolysis by solid oxide electrolysis cells (SOECs) with proton-conducting oxides, Chem. Soc. Rev., 2014, 43, 8255–8270 CAS.
- P. Vernoux, L. Lizarraga, M. N. Tsampas, F. M. Sapountzi, A. De Lucas-Consuegra, J.-L. Valverde, S. Souentie, C. G. Vayenas, D. Tsiplakides, S. Balomenou and E. A. Baranova, Ionically Conducting Ceramics as Active Catalyst Supports, Chem. Rev., 2013, 113, 8192–8260 CAS.
- G. Marnellos and M. Stoukides, Catalytic studies in electrochemical membrane reactors, Solid State Ionics, 2004, 175, 597–603 CAS.
- T. Tagawa, K. Moe, M. Ito and S. Goto, Fuel cell type reactor for Chemicals-energy co-generation, Chem. Eng. Sci., 1999, 54, 1553–1557 CAS.
- S. Ebbesen, S. Jensen, A. Hauch and M. Mogensen, High temperature electrolysis in alkaline cells, solid proton conducting cells, and solid oxide cells, Chem. Rev., 2014, 114, 10697–10734 CAS.
- M. Stoukides, Solid-Electrolyte Membrane Reactors: Current Experience and Future Outlook, Catal. Rev., 2000, 42, 1–70 CAS.
- I. Garagounis, V. Kyriakou, C. Anagnostou, V. Bourganis, I. Papachristou and M. Stoukides, Solid Electrolytes: Applications in Heterogeneous Catalysis and Chemical Cogeneration, Ind. Eng. Chem. Res., 2011, 50, 431–472 CAS.
- C. Kokkofitis, M. Ouzounidou, A. Skodra and M. Stoukides, High temperature proton conductors: Applications in catalytic processes, Solid State Ionics, 2007, 178, 507–513 CAS.
- S. Monika, P. Sara, S. Akhilesh Kumar, S. Ranjan, S. Aradhana and S. Manish, Recent advancement of solid oxide fuel cells towards semiconductor membrane fuel cells, Energy Mater., 2024, 4, 400012 Search PubMed.
- S. Morejudo, R. Zanón, S. Escolástico, I. Yuste-Tirados, H. Malerød-Fjeld, P. Vestre, W. Coors, A. Martínez, T. Norby, J. Serra and C. Kjølseth, Direct conversion of methane to aromatics in a catalytic co-ionic membrane reactor, Science, 2016, 353, 563–566 CrossRef CAS.
- Y. Guo, M. Bessaa, S. Aguado, M. Steil, D. Rembelski, M. Rieu, J. Viricelle, N. Benameur, C. Guizard, C. Tardivat, P. Vernoux and D. Farrusseng, An all porous solid oxide fuel cell (SOFC): a bridging technology between dual and single chamber SOFCs, Energy Environ. Sci., 2013, 6, 2119–2123 RSC.
- Z. Shao, S. Haile, J. Ahn, P. Ronney, Z. Zhan and S. Barnett, A thermally self-sustained micro solid-oxide fuel-cell stack with high power density, Nature, 2005, 435, 795–798 CrossRef CAS PubMed.
- Z. Shao, C. Zhang, W. Wang, C. Su, W. Zhou, Z. Zhu, H. J. Park and C. Kwak, Electric power and synthesis gas co-generation from methane with zero waste gas emission, Angew. Chem., Int. Ed., 2011, 50, 1792–1797 CrossRef CAS PubMed.
- Q. Van Overmeere, K. Kerman and S. Ramanathan, Energy Storage in Ultrathin Solid Oxide Fuel Cells, Nano Lett., 2012, 12, 3756–3760 CAS.
- T. Defferriere, D. Klotz, J. Gonzalez-Rosillo, J. Rupp and H. Tuller, Photo-enhanced ionic conductivity across grain boundaries in polycrystalline ceramics, Nat. Mater., 2022, 21, 438–444 CAS.
- D. Gu, G. Zhang and J. Zou, High temperature thermo-photocatalysis driven carbon removal in direct biogas fueled solid oxide fuel cells, Chin. Chem. Lett., 2021, 32, 3548–3552 CAS.
- X. Zhang, L. Ye, H. Li, F. Chen and K. Xie, Electrochemical Dehydrogenation of Ethane to Ethylene in a Solid Oxide Electrolyzer, ACS Catal., 2020, 10, 3505–3513 CAS.
- I. Zvonareva, X. Fu, D. Medvedev and Z. Shao, Electrochemistry and energy conversion features of protonic ceramic cells with mixed ionic-electronic electrolytes, Energy Environ. Sci., 2022, 15, 439–465 RSC.
- J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T. Lim and G. Kim, Hybrid-solid oxide electrolysis cell: A new strategy for efficient hydrogen production, Nano Energy, 2018, 44, 121–126 CrossRef CAS.
- S. Gunduz, D. Dogu, D. Deka, K. Meyer, A. Fuller, A. Co and U. Ozkan, Application of solid electrolyte cells in ion pump and electrolyzer modes to promote catalytic reactions: An overview, Catal. Today, 2019, 323, 3–13 CAS.
- Y. Tao, M. Wu, M. Hu, X. Xu, M. I. Abdullah, J. Shao and H. Wang, High-performance porous transport layers for proton exchange membrane water electrolyzers, SusMat, 2024, 4, e230 CAS.
- Y. Li, N. Mushtaq, Y. Chen, W. Ye, Z. Zhuang, M. Singh, Y. Jing and L. Fan, Revisiting Mo-Doped SrFeO3-δ Perovskite: The Origination of Cathodic Activity and Longevity for Intermediate-Temperature Solid Oxide Fuel Cells, Adv. Funct. Mater., 2025, 35, 2411025 CAS.
- L. Fan, B. Zhu, P. Su and C. He, Nanomaterials and technologies for low temperature solid oxide fuel cells: Recent advances, challenges and opportunities, Nano Energy, 2018, 45, 148–176 CAS.
- Z. Pan, Q. Liu, Z. Yan, Z. Jiao, L. Bi, S. Chan and Z. Zhong, On the delamination of air electrodes of solid oxide electrolysis cells: A mini-review, Electrochem. Commun., 2022, 137, 107267 CAS.
- J. Schefold, A. Brisse and H. Poepke, 23,000 h steam electrolysis with an electrolyte supported solid oxide cell, Int. J. Hydrogen Energy, 2017, 42, 13415–13426 CrossRef CAS.
- L. Blum, L. G. J. de Haart, J. Malzbender, N. Margaritis and N. H. Menzler, Anode-supported solid oxide fuels cell achieves 70000 hours of continuous operation, Energy Technol., 2016, 4, 939–942 CAS.
- Q. Fang, L. Blum and D. Stolten, Electrochemical Performance and Degradation Analysis of an SOFC Short Stack Following Operation of More than 100,000 Hours, J. Electrochem. Soc., 2019, 166, F1320–F1325 CAS.
- C. Athanassiou, G. Pekridis, N. Kaklidis, K. Kalimeri, S. Vartzoka and G. Marnellos, Hydrogen production in solid electrolyte membrane reactors (SEMRs), Int. J. Hydrogen Energy, 2007, 32, 38–54 CAS.
- L. Lei, J. Zhang, Z. Yuan, J. Liu, M. Ni and F. Chen, Progress Report on Proton Conducting Solid Oxide Electrolysis Cells, Adv. Funct. Mater., 2019, 29, 1903805 Search PubMed.
- M. Mogensen, Materials for reversible solid oxide cells, Curr. Opin. Electrochem., 2020, 21, 265–273 CAS.
- Y. Zheng, Z. W. Chen and J. J. Zhang, Solid Oxide Electrolysis of H2O and CO2 to Produce Hydrogen and Low-Carbon Fuels, Electrochem. Energy Rev., 2021, 4, 508–517 CAS.
- Y. Tian, N. Abhishek, C. Yang, R. Yang, S. Choi, B. Chi, J. Pu, Y. Ling, J. Irvine and G. Kim, Progress and potential for symmetrical solid oxide electrolysis cells, Matter, 2022, 5, 482–514 CAS.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, A review of high temperature co-electrolysis of H2O and CO2 to produce sustainable fuels using solid oxide electrolysis cells (SOECs): advanced materials and technology, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- C. Graves, S. Ebbesen, S. Jensen, S. Simonsen and M. Mogensen, Eliminating degradation in solid oxide electrochemical cells by reversible operation, Nat. Mater., 2015, 14, 239–244 CrossRef CAS.
- A. Jun, J. Kim, J. Shin and G. Kim, Achieving High Efficiency and Eliminating Degradation in Solid Oxide Electrochemical Cells Using High Oxygen-Capacity Perovskite, Angew. Chem., Int. Ed., 2016, 55, 12512–12515 CAS.
- M. Li, B. Hua, J. Chen, Y. Zhong and J. Luo, Charge transfer dynamics in RuO2/perovskite nanohybrid for enhanced electrocatalysis in solid oxide electrolyzers, Nano Energy, 2019, 57, 186–194 CAS.
- J. Cao, Y. Li, Y. Zheng, S. Wang, W. Zhang, X. Qin, G. Geng and B. Yu, A Novel Solid Oxide Electrolysis Cell with Micro-/Nano Channel Anode for Electrolysis at Ultra-High Current Density over 5 A cm−2, Adv. Energy Mater., 2022, 12, 2200899 CrossRef CAS.
- T. Li, T. Wang, T. Wei, X. Hu, Z. Ye, Z. Wang, D. Dong, B. Chen, H. Wang and Z. Shao, Robust Anode-Supported Cells with Fast Oxygen Release Channels for Efficient and Stable CO2 Electrolysis at Ultrahigh Current Densities, Small, 2021, 17, 2007211 CAS.
- Y. Luo, T. Liu, Y. Wang and M. Ding, High-temperature CO2 electrolysis in solid oxide electrolysis cells cathode: Advances and perspective, Chem Catal., 2023, 3, 100815 CAS.
- M. Hecht, J. Hoffman, D. Rapp, J. McClean, J. SooHoo, R. Schaefer, A. Aboobaker, J. Mellstrom, J. Hartvigsen, F. Meyen, E. Hinterman, G. Voecks, A. Liu, M. Nasr, J. Lewis, J. Johnson, C. Guernsey, J. Swoboda, C. Eckert, C. Alcalde, M. Poirier, P. Khopkar, S. Elangovan, M. Madsen, P. Smith, C. Graves, G. Sanders, K. Araghi, M. de la Torre Juarez, D. Larsen, J. Agui, A. Burns, K. Lackner, R. Nielsen, T. Pike, B. Tata, K. Wilson, T. Brown, T. Disarro, R. Morris, R. Schaefer, R. Steinkraus, R. Surampudi, T. Werne and A. Ponce, Mars Oxygen ISRU Experiment (MOXIE), Space Sci. Rev., 2021, 217, 9 Search PubMed.
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, High-temperature CO2 electrolysis in solid oxide electrolysis cells: Developments, challenges, and prospects, Adv. Mater., 2019, 31, e1902033 Search PubMed.
- X. Chen, J. Zhao, G. Li, D. Zhang and H. Li, Recent advances in photocatalytic renewable energy production, Energy Mater., 2022, 2, 200001 CAS.
- Y. Yang, Y. Li, Y. Jiang, M. Zheng, T. Hong, X. Wu and C. Xia, The electrochemical performance and CO2 reduction mechanism on strontium doped lanthanum ferrite fuel electrode in solid oxide electrolysis cell, Electrochim. Acta, 2018, 284, 159–167 CAS.
- A. Opitz, A. Nenning, C. Rameshan, M. Kubicek, T. Götsch, R. Blume, M. Hävecker, K. Axel, G. Rupprechter, B. Klötzer and J. Fleig, Surface chemistry of perovskite-type electrodes during high temperature CO2 electrolysis investigated by operando photoelectron spectroscopy, ACS Appl. Mater. Interfaces, 2017, 9, 35847–35860 CrossRef CAS.
- Y. Yu, B. Mao, A. Geller, R. Chang, K. Gaskell, Z. Liu and B. W. Eichhorn, CO2 activation and carbonate intermediates: an operando AP-XPS study of CO2 electrolysis reactions on solid oxide electrochemical cells, Phys. Chem. Chem. Phys., 2014, 16, 11633–11639 RSC.
- W. Lin, Y. Li, M. Singh, H. Zhao, R. Yang, P.-C. Su and L. Fan, Electronic engineering and oxygen vacancy modification of La0.6Sr0.4FeO3-δ perovskite oxide by low-electronegativity sodium substitution for efficient CO2/CO fueled reversible solid oxide cells, Green Chem., 2024, 26, 3202–3210 RSC.
- W. Lin, W. Su, Y. Li, T. Chiu, M. Singh, Z. Pan and L. Fan, Enhancing Electrochemical CO2 Reduction on Perovskite Oxide for Solid Oxide Electrolysis Cells through In Situ A-Site Deficiencies and Surface Carbonate Deposition Induced by Lithium Cation Doping and Exsolution, Small, 2023, 19, 2303305 CrossRef CAS PubMed.
- A. Shaur, M. Drazkowski, S. Zhu, B. Boukamp and H. Bouwmeester, Single-phase gadolinium-doped ceria cathode for highly efficient CO2 electrolysis, J. Mater. Chem. A, 2023, 11, 25020–25030 RSC.
- Y. Ye, W. Lee, J. Pan, X. Sun, M. Zhou, J. Li, N. Zhang, J. Han and Y. Chen, Tuning the product selectivity of CO2/H2O co-electrolysis using CeO2-modified proton-conducting electrolysis cells, Energy Environ. Sci., 2023, 16, 3137–3145 RSC.
- S. Wang, H. Tsuruta, M. Asanuma and T. Ishihara, Ni-Fe-La(Sr)Fe(Mn)O3 as a new active cermet cathode for intermediate-temperature CO2 electrolysis using a LaGaO3-based electrolyte, Adv. Energy Mater., 2015, 5, 1401003 Search PubMed.
- Y. Shen, T. Liu, R. Li, H. Lv, N. Ta, X. Zhang, Y. Song, Q. Liu, W. Feng, G. Wang and X. Bao, In situ electrochemical reconstruction of Sr2Fe1.45Ir0.05Mo0.5O6-δ perovskite cathode for CO2 electrolysis in solid oxide electrolysis cells, Natl. Sci. Rev., 2023, 10, nwad078 CAS.
- Y. Song, J. Min, Y. Guo, R. Li, G. Zou, M. Li, Y. Zang, W. Feng, X. Yao, T. Liu, X. Zhang, J. Yu, Q. Liu, P. Zhang, R. Yu, X. Cao, J. Zhu, K. Dong, G. Wang and X. Bao, Surface Activation by Single Ru Atoms for Enhanced High-Temperature CO2 Electrolysis, Angew Chem. Int. Ed. Engl., 2024, 63, e202313361 CAS.
- X. Xi, J. Liu, Y. Fan, L. Wang, J. Li, M. Li, J. Luo and X. Fu, Reducing d-p band coupling to enhance CO2 electrocatalytic activity by Mg-doping in Sr2FeMoO6-δ double perovskite for high performance solid oxide electrolysis cells, Nano Energy, 2021, 82, 105707 CAS.
- X. Xi, J. Liu, W. Luo, Y. Fan, J. Zhang, J. L. Luo and X. Z. Fu, Unraveling the Enhanced Kinetics of Sr2Fe1+xMo1-xO6-δ Electrocatalysts for High-Performance Solid Oxide Cells, Adv. Energy Mater., 2021, 11, 2102845 CAS.
- Y. Li, Y. Li, Y. Wan, Y. Xie, J. Zhu, H. Pan, X. Zheng and C. Xia, Perovskite oxyfluoride electrode enabling direct electrolyzing carbon dioxide with excellent electrochemical performances, Adv. Energy Mater., 2019, 9, 1803156 CrossRef.
- Y. Zhou, L. Lin, Y. Song, X. Zhang, H. Lv, Q. Liu, Z. Zhou, N. Ta, G. Wang and X. Bao, Pd single site-anchored perovskite cathode for CO2 electrolysis in solid oxide electrolysis cells, Nano Energy, 2020, 71, 104598 CrossRef CAS.
- S. Lee, M. Kim, K. Lee, J. Irvine and T. Shin, Enhancing electrochemical CO2 reduction using Ce(Mn,Fe)O2 with La(Sr)Cr(Mn)O3 cathode for high-temperature solid oxide electrolysis cells, Adv. Energy Mater., 2021, 11, 2100339 CAS.
- X. Yang, K. Sun, M. Ma, C. Xu, R. Ren, J. Qiao, Z. Wang, S. Zhen, R. Hou and W. Sun, Achieving strong chemical adsorption ability for efficient carbon dioxide electrolysis, Appl. Catal., B, 2020, 272, 118968 CAS.
- Y. Tian, Y. Liu, A. Naden, L. Jia, M. Xu, W. Cui, B. Chi, J. Pu, J. T. S. Irvine and J. Li, Boosting CO2 electrolysis performance via calcium-oxide-looping combined with in situ exsolved Ni–Fe nanoparticles in a symmetrical solid oxide electrolysis cell, J. Mater. Chem. A, 2020, 8, 14895–14899 CAS.
- H. Lv, L. Lin, X. Zhang, Y. Song, H. Matsumoto, C. Zeng, N. Ta, W. Liu, D. Gao, G. Wang and X. Bao, In Situ Investigation of Reversible Exsolution/Dissolution of CoFe Alloy Nanoparticles in a Co-Doped Sr2Fe1.5Mo0.5O6-δ Cathode for CO2 Electrolysis, Adv. Mater., 2020, 32, e1906193 Search PubMed.
- H. Lv, T. Liu, X. Zhang, Y. Song, H. Matsumoto, N. Ta, C. Zeng, G. Wang and X. Bao, Atomic-Scale Insight into Exsolution of CoFe Alloy Nanoparticles in La0.4Sr0.6Co0.2Fe0.7Mo0.1O3−δ with Efficient CO2 Electrolysis, Angew. Chem., Int. Ed., 2020, 59, 15968–15973 CAS.
- Y. Li, Y. Li, S. Zhang, C. Ren, Y. Jing, F. Cheng, Q. Wu, P. Lund and L. Fan, Mutual conversion of CO-CO2 on a perovskite fuel electrode with endogenous alloy nanoparticles for reversible solid oxide cells, ACS Appl. Mater. Interfaces, 2022, 14, 9138–9150 CAS.
- Y. Li, M. Singh, Z. Zhuang, Y. Jing, F. Li, K. Maliutina, C. He and L. Fan, Efficient reversible CO/CO2 conversion in solid oxide cells with a phase-transformed fuel electrode, Sci. China Mater., 2021, 64, 1114–1126 CAS.
- S. Liu, Q. Liu and J. Luo, Highly stable and efficient catalyst with in situ exsolved Fe–Ni alloy nanospheres socketed on an oxygen deficient perovskite for direct CO2 electrolysis, ACS Catal., 2016, 6, 6219–6228 CAS.
- Y. Li, Y. Li, L. Yu, Q. Hu, Q. Wang, K. Maliutina and L. Fan, Achieving excellent and durable CO2 electrolysis performance on a dual-phase fuel electrode in solid oxide electrolysis cells, J. Power Sources, 2021, 491, 229599 CrossRef CAS.
- W. Wang, L. Gan, J. P. Lemmon, F. Chen, J. T. S. Irvine and K. Xie, Enhanced carbon dioxide electrolysis at redox manipulated interfaces, Nat. Commun., 2019, 10, 1550 CrossRef.
- L. Ye, M. Zhang, P. Huang, G. Guo, M. Hong, C. Li, J. Irvine and K. Xie, Enhancing CO2 electrolysis through synergistic control of non-stoichiometry and doping to tune cathode surface structures, Nat. Commun., 2017, 8, 14785 CrossRef CAS.
- H. Lv, L. Lin, X. Zhang, R. Li, Y. Song, H. Matsumoto, N. Ta, C. Zeng, Q. Fu, G. Wang and X. Bao, Promoting exsolution of RuFe alloy nanoparticles on Sr2Fe1.4Ru0.1Mo0.5O6−δ via repeated redox manipulations for CO2 electrolysis, Nat. Commun., 2021, 12, 5665 CrossRef CAS PubMed.
- M. Wang, N. Li, Q. Shen, Z. Zhan and C. Chen, A highly efficient and stable perovskite cathode with in situ exsolved NiFe alloy nanoparticles for CO2 electrolysis, Sustainable Energy Fuels, 2022, 6, 2038–2044 CAS.
- Y. Li, P. Li, B. Hu and C. Xia, A nanostructured ceramic fuel electrode for efficient CO2/H2O electrolysis without safe gas, J. Mater. Chem. A, 2016, 4, 9236–9243 CAS.
- L. Yu, J. Wang, X. Hu, Z. Ye, C. Buckley and D. Dong, A nanocatalyst network for electrochemical reduction of CO2 over microchanneled solid oxide electrolysis cells, Electrochem. Commun., 2018, 86, 72–75 CAS.
- J. Tong, N. Ni, B. Zhou, C. Yang, K. M. Reddy, H. Tu, Y. Liu, Z. Tan, L. Xiang, H. Li, X. Zhou, Y. Zhang, Y. Li, H. Zhang, L. Zhu and Z. Huang, Toward High CO Selectivity and Oxidation Resistance Solid Oxide Electrolysis Cell with High-Entropy Alloy, ACS Catal., 2024, 14, 2897–2907 CAS.
- M. Torrell, S. Garcia-Rodriguez, A. Morata, G. Penelas and A. Tarancon, Co-electrolysis of steam and CO2 in full-ceramic symmetrical SOECs: a strategy for avoiding the use of hydrogen as a safe gas, Faraday Discuss., 2015, 182, 241–255 CAS.
- Q. Li, Y. Zheng, Y. Sun, T. Li, C. Xu, W. Wang and S. H. Chan, Understanding the occurrence of the individual CO2 electrolysis during H2O-CO2 co-electrolysis in classic planar Ni-YSZ/YSZ/LSM-YSZ solid oxide cells, Electrochim. Acta, 2019, 318, 440–448 CAS.
- D. Deka, J. Kim, S. Gunduz, M. Ferree, A. Co and U. Ozkan, Temperature-induced changes in the synthesis gas composition in a high-temperature H2O and CO2 co-electrolysis system, Appl. Catal., A, 2020, 602, 117697 CAS.
- M. Zheng, S. Wang, Y. Yang and C. Xia, Barium carbonate as a synergistic catalyst for the H2O/CO2 reduction reaction at Ni–yttria stabilized zirconia cathodes for solid oxide electrolysis cells, J. Mater. Chem. A, 2018, 6, 2721–2729 RSC.
- D. Chen, D. Niakolas, V. Papaefthimiou, E. Ioannidou, S. Neophytides and S. Zafeiratos, J. Catal., 2021, 404, 518–528 CrossRef CAS.
- B. Chen, H. Xu, L. Chen, Y. Li, C. Xia and M. Ni, Modelling of One-Step Methanation Process Combining SOECs and Fischer-Tropsch-like Reactor, J. Electrochem. Soc., 2016, 163, F3001–F3008 CAS.
- C. Chatzilias, E. Martino, C. G. Vayenas, G. Kyriakou and A. Katsaounis, A low temperature SOFC as a self-promoted reactor for CO2 catalytic hydrogenation, Appl. Catal., B, 2022, 317, 121778 CAS.
- Z. Pan, C. Duan, T. Pritchard, A. Thatte, E. White, R. Braun, R. O'Hayre and N. P. Sullivan, High-yield electrochemical upgrading of CO2 into CH4 using large-area protonic ceramic electrolysis cells, Appl. Catal., B, 2022, 307, 121196 CAS.
- D. Bierschenk, J. Wilson and S. Barnett, High efficiency electrical energy storage using a methane-oxygen solid oxide cell, Energy Environ. Sci., 2011, 4, 944–951 CAS.
- Q. Fu, C. Mabilat, M. Zahid, A. Brisse and L. Gautier, Syngas production via high-temperature steam/CO2 co-electrolysis: an economic assessment, Energy Environ. Sci., 2010, 3, 1382–1397 CAS.
- S. Jensen, C. Graves, M. Mogensen, C. Wendel, R. Braun, G. Hughes, Z. Gao and S. Barnett, Large-scale electricity storage utilizing reversible solid oxide cells combined with underground storage of CO2 and CH4, Energy Environ. Sci., 2015, 8, 2471–2479 CAS.
- L. Chen, F. Chen and C. Xia, Direct synthesis of methane from CO2-H2O co-electrolysis in tubular solid oxide electrolysis cells, Energy Environ. Sci., 2014, 7, 4018–4022 RSC.
- S. Bebelis, H. Karasali and C. Vayenas, Electrochemical promotion of CO2 hydrogenation on Rh/YSZ electrodes, J. Appl. Electrochem., 2008, 38, 1127–1133 CrossRef CAS.
- E. Papaioannou, S. Souentie, A. Hammad and C. Vayenas, Electrochemical promotion of the CO2 hydrogenation reaction using thin Rh, Pt and Cu films in a monolithic reactor at atmospheric pressure, Catal. Today, 2009, 146, 336–344 CAS.
- K. Xie, Y. Zhang, G. Meng and J. T. S. Irvine, Direct synthesis of methane from CO2/H2O in an oxygen-ion conducting solid oxide electrolyser, Energy Environ. Sci., 2011, 4, 2218–2222 RSC.
- L. Lei, T. Liu, S. Fang, J. P. Lemmon and F. Chen, The co-electrolysis of CO2–H2O to methane via a novel micro-tubular electrochemical reactor, J. Mater. Chem. A, 2017, 5, 2904–2910 RSC.
- Y. Luo, W. Li, Y. Shi, T. Cao, X. Ye, S. Wang and N. Cai, Experimental Characterization and Theoretical Modeling of Methane Production by H2O/CO2 Co-Electrolysis in a Tubular Solid Oxide Electrolysis Cell, J. Electrochem. Soc., 2015, 162, F1129–F1134 CAS.
- Y. Luo, Y. Shi, W. Li and N. Cai, Synchronous enhancement of H2O/CO2 co-electrolysis and methanation for efficient one-step power-to-methane, Energy Convers. Manage., 2018, 165, 127–136 CAS.
- B. Ewan and O. Adeniyi, A Demonstration of Carbon-Assisted Water Electrolysis, Energies, 2013, 6, 1657–1668 CAS.
- B. Alexander, R. Mitchell and T. Gur, Steam-Carbon Fuel Cell Concept for Cogeneration of Hydrogen and Electrical Power, J. Electrochem. Soc., 2011, 158, B505–B513 CAS.
- S. Gopalan, G. Ye and U. B. Pal, Regenerative, coal-based solid oxide fuel cell-electrolyzers, J. Power Sources, 2006, 162, 74–80 CAS.
- Y. Luo, Y. Shi, W. Li, M. Ni and N. Cai, Elementary reaction modeling and experimental characterization of solid oxide fuel-assisted steam electrolysis cells, Int. J. Hydrogen Energy, 2014, 39, 10359–10373 CAS.
- J. Martinez-Frias, A. Pham and S. M. Aceves, A natural gas-assisted steam electrolyzer for high-efficiency production of hydrogen, Int. J. Hydrogen Energy, 2003, 28, 483–490 CrossRef CAS.
- W. Wang, R. Gorte and J. Vohs, Analysis of the performance of the electrodes in a natural gas assisted steam electrolysis cell, Chem. Eng. Sci., 2008, 63, 765–769 CrossRef CAS.
- F. Liu, T. Wang, J. Li, T. Wei, Z. Ye, D. Dong, B. Chen, Y. Ling and Z. Shao, Elevated-temperature bio-ethanol-assisted water electrolysis for efficient hydrogen production, Chem. Eng. J., 2022, 434, 134699 CrossRef CAS.
- V. Kyriakou, D. Neagu, G. Zafeiropoulos, R. Sharma, C. Tang, K. Kousi, L. Metcalfe, M. van de Sanden and M. Tsampas, Symmetrical Exsolution of Rh Nanoparticles in Solid Oxide Cells for Efficient Syngas Production from Greenhouse Gases, ACS Catal., 2020, 10, 1278–1288 CAS.
- S. Giddey, A. Kulkarni and S. Badwal, Low emission hydrogen generation through carbon assisted electrolysis, Int. J. Hydrogen Energy, 2015, 40, 70–74 CAS.
- A. Lee, R. Mitchell and T. Gür, Feasibility of hydrogen production in a steam-carbon electrochemical cell, Solid State Ionics, 2011, 192, 607–610 CAS.
- L. Lei, Y. Wang, S. Fang, C. Ren, T. Liu and F. Chen, Efficient syngas generation for electricity storage through carbon gasification assisted solid oxide co-electrolysis, Appl. Energy, 2016, 173, 52–58 CAS.
- A. de Lucas-Consuegra, N. Gutiérrez-Guerra, A. Caravaca, J. Serrano-Ruiz and J. Valverde, Coupling catalysis and electrocatalysis for hydrogen production in a solid electrolyte membrane reactor, Appl. Catal., A, 2014, 483, 25–30 CAS.
- A. Pham, P. Wallman and R. Glass, Natural gas-assisted steam electrolyzer, US Pat., 6051125, 2000 Search PubMed.
- W. Kiatkittipong, T. Tagawa, S. Goto, S. Assabumrungrat and P. Praserthdam, Oxygen transport through LSM/YSZ/LaAlO system for use of fuel cell type reactor, Chem. Eng. J., 2005, 106, 35–42 CAS.
- H. Jiang, H. Wang, S. Werth, T. Schiestel and J. Caro, Simultaneous production of hydrogen and synthesis gas by combining water splitting with partial oxidation of methane in a hollow-fiber membrane reactor, Angew. Chem., Int. Ed., 2008, 47, 9341–9344 CAS.
- Y.-F. Sun, Y.-Y. Wu, Y.-Q. Zhang, J.-H. Li, Y. Luo, Y.-X. Shi, B. Hua and J. Luo, A bifunctional solid oxide electrolysis cell for simultaneous CO2 utilization and synthesis gas production, Chem. Commun., 2016, 52, 13687–13690 CAS.
- Y. Wang, T. Liu, S. Fang, G. Xiao, H. Wang and F. Chen, A novel clean and effective syngas production system based on partial oxidation of methane assisted solid oxide co-electrolysis process, J. Power Sources, 2015, 277, 261–267 CAS.
- T. Liu, H. Liu, X. Zhang, L. Lei, Y. Zhang, Z. Yuan, F. Chen and Y. Wang, A robust solid oxide electrolyzer for highly efficient electrochemical reforming of methane and steam, J. Mater. Chem. A, 2019, 7, 13550–13558 RSC.
- J. Lu, C. Zhu, C. Pan, W. Lin, J. Lemmon, F. Chen, C. Li and K. Xie, Highly efficient electrochemical reforming of CH4/CO2 in a solid oxide electrolyser, Sci. Adv., 2018, 4, eaar5100 CrossRef PubMed.
- G. Keller and M. Bhasin, Synthesis of ethylene via oxidative coupling of methane: I. Determination of active catalysts, J. Catal., 1982, 73, 9–19 CAS.
- M. Stoukides, Methane conversion to C2 hydrocarbons in solid electrolyte membrane reactors, Res. Chem. Intermed., 2006, 32, 187–204 CAS.
- T. Tagawa, K. Kuroyanagi, S. Goto, S. Assabumrungrat and P. Praserthdam, Selective oxidation of methane in an SOFC-type reactor: Effect of applied potential, Chem. Eng. J., 2003, 93, 3–9 CAS.
- H. Nagamoto, K. Hayashi and H. Inoue, Methane oxidation by oxygen transported through solid electrolyte, J. Catal., 1990, 126, 671–673 CAS.
- L. Ye, Z. Shang and K. Xie, Selective Oxidative Coupling of Methane to Ethylene in a Solid Oxide Electrolyser Based on Porous Single-Crystalline CeO2 Monoliths, Angew. Chem., Int. Ed., 2022, 61, e202207211 CAS.
- A. Caravaca, A. de Lucas-Consuegra, J. González-Cobos, J. Valverde and F. Dorado, Simultaneous production of H2 and C2 hydrocarbons by gas phase electrocatalysis, Appl. Catal., B, 2012, 113–114, 192–200 CrossRef CAS.
- W. Kiatkittipong, T. Tagawa, S. Goto, S. Assabumrungrat, K. Silpasup and P. Praserthdam, Comparative study of oxidative coupling of methane modeling in various types of reactor, Chem. Eng. J., 2005, 115, 63–71 CrossRef CAS.
- S. Kyun Kim, Y. Kwon, Y. BeomKim, J. Jung, S. Kang and J. HoonJoo, Novel approach to integrate CO2 utilization coupled with direct methane conversion to C2 products using solid oxide electrolysis cell, Chem. Eng. J., 2022, 444, 136619 CAS.
- N. Lapeña-Rey and P. Middleton, The selective oxidation of methane to ethane and ethylene in a solid oxide electrolyte reactor, Appl. Catal., A, 2003, 240, 207–222 CrossRef.
- Y. Song, L. Lin, W. Feng, X. Zhang, Q. Dong, X. Li, H. Lv, Q. Liu, F. Yang, Z. Liu, G. Wang and X. Bao, Interfacial Enhancement by γ-Al2O3 of Electrochemical Oxidative Dehydrogenation of Ethane to Ethylene in Solid Oxide Electrolysis Cells, Angew. Chem., Int. Ed., 2019, 58, 16043–16046 CrossRef CAS PubMed.
- L. Ye, X. Duan and K. Xie, Electrochemical Oxidative Dehydrogenation of Ethane to Ethylene in a Solid Oxide Electrolyzer, Angew. Chem., Int. Ed., 2021, 60, 21746–21750 CrossRef CAS PubMed.
- C. Zhu, S. Hou, X. Hu, J. Lu, F. Chen and K. Xie, Electrochemical conversion of methane to ethylene in a solid oxide electrolyzer, Nat. Commun., 2019, 10, 1173 Search PubMed.
- T. Li, T. Wang, C. Shi, S. Chang, M. Zhang, Y. Zhao, C. E. Buckley, Y. Dong and D. Dong, Efficient Reduction of Low-Concentration NO via Dendritically Channeled Solid Oxide Cells, ACS Appl. Energy Mater., 2021, 4, 6968–6974 CAS.
- Y. Kwon, S. Kim, Y. Kim, S. Son, G. Nam, H. Park, W. Cho, H. Yoon and J. Joo, Nitric oxide utilization for ammonia production using solid electrolysis cell at atmospheric pressure, ACS Energy Lett., 2021, 6, 4165–4172 CAS.
- Q. Hu, Y. Qin, X. Wang, Z. Wang, X. Huang, H. Zheng, K. Gao, H. Yang, P. Zhang, M. Shao and C. He, Reaction intermediate-mediated electrocatalyst synthesis favors specified facet and defect exposure for efficient nitrate–ammonia conversion, Energy Environ. Sci., 2021, 14, 4989–4997 CAS.
- H. Patel, R. Sharma, V. Kyriakou, A. Pandiyan, S. Welzel, M. Sanden and M. Tsampas, Plasma-Activated Electrolysis for Cogeneration of Nitric Oxide and Hydrogen from Water and Nitrogen, ACS Energy Lett., 2019, 4, 2091–2095 CAS.
- J. Wang, L. Ma, W. Tan, S. Wang, J. Wen, Z. Zhang, H. Yu and W. Li, NiO and Co3O4 nanoparticles decorated La0.8Sr0.2MnO3-based electrodes for electrochemical NOx removal in solid electrolyte cells, Chem. Eng. J., 2023, 466, 143248 CAS.
- T. Huang, S. Hsu and C. Wu, Simultaneous NOx and Hydrocarbon Emissions Control for Lean-Burn Engines Using Low-Temperature Solid Oxide Fuel Cell at Open Circuit, Environ. Sci. Technol., 2012, 46, 2324–2329 CrossRef CAS.
- T. Huang, C. Wu, S. Hsu and C. Wu, Complete emissions control for highly fuel-efficient automobiles via a simulated stack of electrochemical-catalytic cells, Energy Environ. Sci., 2011, 4, 4061–4067 RSC.
- J. Shao, Y. Tao and K. K. Hansen, Highly selective NOx reduction for diesel engine exhaust via an electrochemical system, Electrochem. Commun., 2016, 72, 36–40 CAS.
- T. Huang and C. Chou, Electrochemical NOx Reduction with Power Generation in Solid Oxide Fuel Cells with Cu-Added (LaSr)(CoFe)O3–(Ce,Gd)O2−x Cathode, J. Electrochem. Soc., 2010, 157, P28 CAS.
- K. Kammer, Electrochemical DeNOx in solid electrolyte cells—an overview, Appl. Catal., B, 2005, 58, 33–39 CAS.
- L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu, Z. Cheng and M. Liu, Enhanced sulfur and coking tolerance of a mixed ion conductor for SOFCs: BaZr0.1Ce0.7Y0.2-xYbxO3-δ, Science, 2009, 326, 126–129 CAS.
- Z. Shao and S. Haile, A high-performance cathode for the next generation of solid-oxide fuel cells, Nature, 2004, 431, 170–173 CAS.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O'Hayre, Readily processed protonic ceramic fuel cells with high performance at low temperatures, Science, 2015, 349, 1321–1326 CAS.
- Z. Zhuang, Y. Li, R. Yu, L. Xia, J. Yang, Z. Lang, J. Zhu, J. Huang, J. Wang, Y. Wang, L. Fan, J. Wu, Y. Zhao, D. Wang and Y. Li, Reversely trapping atoms from a perovskite surface for high-performance and durable fuel cell cathodes, Nat. Catal., 2022, 5, 300–310 CAS.
- W. Bian, W. Wu, B. Wang, W. Tang, M. Zhou, C. Jin, H. Ding, W. Fan, Y. Dong, J. Li and D. Ding, Revitalizing interface in protonic ceramic cells by acid etch, Nature, 2022, 604, 479–485 CrossRef CAS.
- H. Zhang, K. Xu, Y. Xu, F. He, F. Zhu, K. Sasaki, Y. Choi and Y. Chen, In situ formed catalysts for active, durable, and thermally stable ammonia protonic ceramic fuel cells at 550 °C, Energy Environ. Sci., 2024, 17, 3433–3442 RSC.
- S. S. Shin, J. H. Kim, K. T. Bae, K.-T. Lee, S. M. Kim, J.-W. Son, M. Choi and H. Kim, Multiscale structured low-temperature solid oxide fuel cells with 13 W power at 500 °C, Energy Environ. Sci., 2020, 13, 3459–3468 CAS.
- E. Wachsman and K. Lee, Lowering the temperature of solid oxide fuel cells, Science, 2011, 334, 935–939 CAS.
- Y. Zhang, R. Knibbe, J. Sunarso, Y. Zhong, W. Zhou, Z. Shao and Z. Zhu, Recent Progress on Advanced Materials for Solid-Oxide Fuel Cells Operating Below 500 °C, Adv. Mater., 2017, 29, 1700132 Search PubMed.
- S. Park, J. Vohs and R. Gorte, Direct oxidation of hydrocarbons in a solid-oxide fuel cell, Nature, 2000, 404, 265–267 CAS.
- X. Ge, S. Chan, Q. Liu and Q. Sun, Solid oxide fuel cell anode materials for direct hydrocarbon utilization, Adv. Energy Mater., 2012, 2, 1156–1181 CAS.
- Y. Chen, B. deGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. Kim, Y. Ding, P. Hu, F. Tao and M. Liu, A robust fuel cell operated on nearly dry methane at 500 °C enabled by synergistic thermal catalysis and electrocatalysis, Nat. Energy, 2018, 3, 1042–1050 CAS.
- K. Xu, H. Zhang, Y. Xu, F. Zhu, F. He, Y. Liu, K. Sasaki, Y. Choi and Y. Chen, An efficient construction of nano-interfaces for excellent coking tolerance of cermet anodes, Mater. Today, 2024, 79, 28–35 CAS.
- K. Xu, H. Zhang, Y. Xu, F. Zhu, F. He, K. Sasaki, Y. Choi and Y. Chen, Realizing efficient operations of Ni-cermet-based fuel cells on hydrocarbons via an in situ self-assembled metal/oxide nano-heterostructured catalyst, Appl. Catal., B, 2024, 355, 124208 CAS.
- S. Kim, C. Kim, J. H. Lee, J. Shin, T.-H. Lim and G. Kim, Tailoring Ni-based catalyst by alloying with transition metals (M = Ni, Co, Cu, and Fe) for direct hydrocarbon utilization of energy conversion devices, Electrochim. Acta, 2017, 225, 399–406 CAS.
- L. Yang, Y. Choi, W. Qin, H. Chen, K. Blinn, M. Liu, P. Liu, J. Bai, T. A. Tyson and M. Liu, Promotion of water-mediated carbon removal by nanostructured barium oxide/nickel interfaces in solid oxide fuel cells, Nat. Commun., 2011, 2, 357 CrossRef.
- Z. Zhan and S. Barnett, An octane-fueled solid oxide fuel cell, Science, 2005, 308, 844–847 CrossRef CAS PubMed.
- T. Hibino, A. Hashimoto, T. Inoue, J. Tokuno, S. Yoshida and M. Sano, A low-operating-temperature solid oxide fuel cell in hydrocarbon-air mixtures, Science, 2000, 288, 2031–2033 CrossRef CAS.
- E. Perry Murray, T. Tsai and S. A. Barnett, A direct-methane fuel cell with a ceria-based anode, Nature, 1999, 400, 649–651 CrossRef.
- S. Tao and J. Irvine, A redox-stable efficient anode for solid-oxide fuel cells, Nat. Mater., 2003, 2, 320–323 CAS.
- Y. Huang, R. Dass, Z. Xing and J. Goodenough, Double perovskites as anode materials for solid-oxide fuel cells, Science, 2006, 312, 254–257 CAS.
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. S. Irvine and G. Kim, Layered oxygen-deficient double perovskite as an efficient and stable anode for direct hydrocarbon solid oxide fuel cells, Nat. Mater., 2015, 14, 205–209 CAS.
- C. Yang, Z. Yang, C. Jin, G. Xiao, F. Chen and M. Han, Sulfur-tolerant redox-reversible anode material for direct hydrocarbon solid oxide fuel cells, Adv. Mater., 2012, 24, 1439–1443 CAS.
- M. Qin, Y. Xiao, H. Yang, T. Tan, Z. Wang, X. Fan and C. Yang, Ru/Nb co-doped perovskite anode: Achieving good coking resistance in hydrocarbon fuels via core-shell nanocatalysts exsolution, Appl. Catal., B, 2021, 299, 120613 CAS.
- B. Li, S. He, J. Li, X. Yue, J. T. S. Irvine, D. Xie, J. Ni and C. Ni, A Ce/Ru Codoped SrFeO3−δ Perovskite for a Coke-Resistant Anode of a Symmetrical Solid Oxide Fuel Cell, ACS Catal., 2020, 10, 14398–14409 CAS.
- S. Liu, Q. Liu, X. Fu and J. Luo, Cogeneration of ethylene and energy in protonic fuel cell with an efficient and stable anode anchored with in-situ exsolved functional metal nanoparticles, Appl. Catal., B, 2018, 220, 283–289 CAS.
- M. Pillai, I. Kim, D. Bierschenk and S. Barnett, Fuel-flexible operation of a solid oxide fuel cell with Sr0.8La0.2TiO3 support, J. Power Sources, 2008, 185, 1086–1093 CAS.
- W. Rosensteel, S. Babiniec, D. Storjohann, J. Persky and N. Sullivan, Use of anode barrier layers in tubular solid-oxide fuel cells for robust operation on hydrocarbon fuels, J. Power Sources, 2012, 205, 108–113 CrossRef CAS.
- H. Shi, C. Su, G. Yang, R. Ran, Y. Hao, M. O. Tade and Z. Shao, Fabrication and operation of flow-through tubular SOFCs for electric power and synthesis gas cogeneration from methane, AlChE J., 2014, 60, 1036–1044 CrossRef CAS.
- D. Fan, F. Liu, J. Li, T. Wei, Z. Ye, Z. Wang, X. Hu, D. Dong, H. Wang and Z. Shao, A microchannel reactor-integrated ceramic fuel cell with dual-coupling effect for efficient power and syngas co-generation from methane, Appl. Catal., B, 2021, 297, 120443 CrossRef CAS.
- T. Suzuki, Z. Hasan, Y. Funahashi, T. Yamaguchi, Y. Fujishiro and M. Awano, Impact of anode microstructure on solid oxide fuel cells, Science, 2009, 325, 852–855 CrossRef CAS PubMed.
- J. Wang, D. Fan, L. Yu, T. Wei, X. Hu, Z. Ye, Z. Wang, Y. Wang, C. Li, J. Yao and D. Dong, Efficient conversion of methane into power via microchanneled solid oxide fuel cells, J. Power Sources, 2020, 453, 227848 CrossRef CAS.
- D. Fan, Y. Gao, F. Liu, T. Wei, Z. Ye, Y. Ling, B. Chen, Y. Zhang, M. Ni and D. Dong, Autothermal reforming of methane over an integrated solid oxide fuel cell reactor for power and syngas co-generation, J. Power Sources, 2021, 513, 230536 CrossRef CAS.
- S. Najari, S. Saeidi, P. Concepcion, D. Dionysiou, S. Bhargava, A. Lee and K. Wilson, Oxidative dehydrogenation of ethane: catalytic and mechanistic aspects and future trends, Chem. Soc. Rev., 2021, 50, 4564–4605 CAS.
- T. Tan, Z. Wang, K. Huang and C. Yang, High-Performance Co-production of Electricity and Light Olefins Enabled by Exsolved NiFe Alloy Nanoparticles from a Double-Perovskite Oxide Anode in Solid Oxide-Ion-Conducting Fuel Cells, ACS Nano, 2023, 17, 13985–13996 CAS.
- F. Akin and Y. Lin, Selective oxidation of ethane to ethylene in a dense tubular membrane reactor, J. Membr. Sci., 2002, 209, 457–467 CAS.
- J. Coronas, M. Menendez and J. Santamaria, Use of a Ceramic Membrane Reactor for the Oxidative Dehydrogenation of Ethane to Ethylene and Higher Hydrocarbons, Ind. Eng. Chem. Res., 1995, 34, 4229–4234 CAS.
- O. Czuprat, S. Werth, S. Schirrmeister, T. Schiestel and J. Caro, Olefin Production by a Multistep Oxidative Dehydrogenation in a Perovskite Hollow-Fiber Membrane Reactor, ChemCatChem, 2009, 1, 401–405 CAS.
- Y. Gao, F. Haeri, F. He and F. Li, Alkali Metal-Promoted LaxSr2–xFeO4−δ Redox Catalysts for Chemical Looping Oxidative Dehydrogenation of Ethane, ACS Catal., 2018, 8, 1757–1766 CAS.
- D. Dogu, K. Meyer, A. Fuller, S. Gunduz, D. Deka, N. Kramer, A. Co and U. Ozkan, Effect of lanthanum and chlorine doping on strontium titanates for the electrocatalytically-assisted oxidative dehydrogenation of ethane, Appl. Catal., B, 2018, 227, 90–101 CAS.
- L. Zhang, C. Yang, A. I. Frenkel, S. Wang, G. Xiao, K. Brinkman and F. Chen, Co-generation of electricity and chemicals from propane fuel in solid oxide fuel cells with anode containing nano-bimetallic catalyst, J. Power Sources, 2014, 262, 421–428 CAS.
- S. Lei, A. Wang, J. Xue and H. Wang, Catalytic ceramic oxygen ionic conducting membrane reactors for ethylene production, React. Chem. Eng., 2021, 6, 1327–1341 CAS.
- I. Yentekakis, Y. Jiang, M. Makri and C. Vayenas, Ethylene production from methane in a gas recycle electrocatalytic reactor separator, Ionics, 1995, 1, 286–291 CAS.
- B. L. Farrell, V. O. Igenegbai and S. Linic, A Viewpoint on Direct Methane Conversion to Ethane and Ethylene Using Oxidative Coupling on Solid Catalysts, ACS Catal., 2016, 6, 4340–4346 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. Jaffer, T. Jaramillo and E. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506 CrossRef CAS.
- J. Kim, Y. Kim, M. Ferree, S. Gunduz, A. Co, M. Kim and U. Ozkan, In-situ exsolution of bimetallic CoFe nanoparticles on (La,Sr)FeO3 perovskite: Its effect on electrocatalytic oxidative coupling of methane, Appl. Catal., B, 2023, 321, 122026 CrossRef CAS.
- T. Tagawa, K. Kyaw Moe, T. Hiramatsu and S. Goto, Design of electrode for solid oxide fuel cells reactor, Solid State Ionics, 1998, 106, 227–235 CrossRef CAS.
- K. Liu, J. Zhao, D. Zhu, F. Meng, F. Kong and Y. Tang, Oxidative coupling of methane in solid oxide fuel cell tubular membrane reactor with high ethylene yield, Catal. Commun., 2017, 96, 23–27 CAS.
- Y. Jiang, I. Yentekakis and C. Vayenas, Methane to Ethylene with 85 Percent Yield in a Gas Recycle Electrocatalytic Reactor-Separator, Science, 1994, 264, 1563–1566 CAS.
- W. Appamana, S. Charojrochkul, S. Assabumrungrat and W. Wiyaratn, Synthesis of Na2WO4-Mn supported YSZ as a potential anode catalyst for oxidative coupling of methane in SOFC reactor, Eng. J., 2015, 19, 13–20 Search PubMed.
- A. Cruellas, J. J. Bakker, M. van Sint Annaland, J. A. Medrano and F. Gallucci, Techno-economic analysis of oxidative coupling of methane: Current state of the art and future perspectives, Energy Convers. Manage., 2019, 198, 111789 CAS.
- A. Cruellas, J. Heezius, V. Spallina, M. van Sint Annaland, J. A. Medrano and F. Gallucci, Oxidative Coupling of Methane in Membrane Reactors; A Techno-Economic Assessment, Processes, 2020, 8, 274 CAS.
- A. Torabi, J. Barton, C. Willman, H. Ghezel-Ayagh, N. Li, A. Poozhikunnath, R. Maric and O. A. Marina, Developing Low-Intermediate Temperature Fuel Cells for Direct Conversion of Methane to Methanol Fuel, ECS Trans., 2016, 72, 193–199 CAS.
- S. Neophytides and C. Vayenas, Chemical Cogeneration in Solid Electrolyte Cells: The Oxidation of CH3OH to H2CO, J. Electrochem. Soc., 1990, 137, 839–845 CAS.
- S. McIntosh, J. Vohs and R. Gorte, An examination of lanthanide additives on the performance of Cu-YSZ cermet anodes, Electrochim. Acta, 2002, 47, 3815–3821 CAS.
- B. Ji, J. Wang, W. Chu, W. Yang and L. Lin, Acrylic acid and electric power cogeneration in an SOFC reactor, Chem. Commun., 2009, 2038–2040 CAS.
- C. Vayenas and R. Farr, Cogeneration of electric energy and nitric oxide, Science, 1980, 208, 593–594 CAS.
- N. Kiratzis and M. Stoukides, The Synthesis of Hydrogen Cyanide in a Solid Electrolyte Fuel Cell, J. Electrochem. Soc., 1987, 134, 1925–1929 CAS.
- N. Kiratzis and M. Stoukides, The synthesis of HCN in a solid electrolyte cell, J. Catal., 1991, 132, 257–262 CAS.
- A. Raj, R. Rudkin and A. Atkinson, Cogeneration of HCN in a Solid Oxide Fuel Cell, J. Electrochem. Soc., 2010, 157, B719–B725 CAS.
- T. Li, M. F. Rabuni, L. Kleiminger, B. Wang, G. H. Kelsall, U. W. Hartley and K. Li, A highly-robust solid oxide fuel cell (SOFC): simultaneous greenhouse gas treatment and clean energy generation, Energy Environ. Sci., 2016, 9, 3682–3686 RSC.
- I. Yentekakis and C. Vayenas, Chemical Cogeneration in Solid Electrolyte Cells: The Oxidation of H2S to SO2, J. Electrochem. Soc., 1989, 136, 996–1002 CrossRef CAS.
- M. Liu, G. Wei, J. Luo, A. Sanger and K. Chuang, Use of metal sulfides as anode catalysts in H2S-Air SOFCs, J. Electrochem. Soc., 2003, 150, A1025–A1029 CrossRef CAS.
- G. Wei, J. Luo, A. Sanger and K. Chuang, High-performance anode for H2S-Air SOFCs, J. Electrochem. Soc., 2004, 151, A232–A237 CrossRef CAS.
- X. Yan, M. Zhou, Y. Zhang, Q. Qiu, Q. Chen, W. Cai, Y. Tang and J. Liu, An all-solid-state carbon-air battery reaching an output power over 10 W and a specific energy of 3600 Wh kg−1, Chem. Eng. J., 2021, 404, 127057 CrossRef CAS.
- C. Jiang, J. Ma, G. Corre, S. L. Jain and J. T. S. Irvine, Challenges in developing direct carbon fuel cells, Chem. Soc. Rev., 2017, 46, 2889–2912 CAS.
- Y. Xie, W. Cai, J. Xiao, Y. Tang, J. Liu and M. Liu, Electrochemical gas–electricity cogeneration through direct carbon solid oxide fuel cells, J. Power Sources, 2015, 277, 1–8 CrossRef CAS.
- Q. Chen, Q. Qiu, X. Yan, M. Zhou, Y. Zhang, Z. Liu, W. Cai, W. Wang and J. Liu, A compact and seal-less direct carbon solid oxide fuel cell stack stepping into practical application, Appl. Energy, 2020, 278, 115657 CAS.
- H. Xu, B. Chen, J. Liu and M. Ni, Modeling of direct carbon solid oxide fuel cell for CO and electricity cogeneration, Appl. Energy, 2016, 178, 353–362 CAS.
- B. Yang, R. Ran, Y. Zhong, C. Su, M. O. Tade and Z. Shao, A carbon-air battery for high power generation, Angew. Chem., Int. Ed., 2015, 54, 3722–3725 CAS.
- Y. Zhong, C. Su, R. Cai, M. O. Tadé and Z. Shao, Process Investigation of a Solid Carbon-Fueled Solid Oxide Fuel Cell Integrated with a CO2-Permeating Membrane and a Sintering-Resistant Reverse Boudouard Reaction Catalyst, Energy Fuels, 2016, 30, 1841–1848 CAS.
- H. Xie, S. Zhai, T. Liu, H. Liao, Y. Zhang, W. Zhou, Z. Shao, M. Ni and B. Chen, Cu-modified Ni foams as three-dimensional outer anodes for high-performance hybrid direct coal fuel cells, Chem. Eng. J., 2021, 410, 128239 CAS.
- W. J. Bian, W. Wu, C. J. Orme, H. P. Ding, M. Zhou and D. Ding, Dual 3D Ceramic Textile Electrodes: Fast Kinetics for Carbon Oxidation Reaction and Oxygen Reduction Reaction in Direct Carbon Fuel Cells at Reduced Temperatures, Adv. Funct. Mater., 2020, 30, 1910096 CAS.
- W. Wu, Y. Zhang, D. Ding and T. He, A High-Performing Direct Carbon Fuel Cell with a 3D Architectured Anode Operated Below 600 °C, Adv. Mater., 2018, 30, 1704745 Search PubMed.
- M. Ma, X. Yang, R. Ren, C. Xu, J. Qiao, W. Sun, K. Sun and Z. Wang, Honeycombed Porous, Size-Matching Architecture for High-Performance Hybrid Direct Carbon Fuel Cell Anode, ACS Appl. Mater. Interfaces, 2020, 12, 30411–30419 CAS.
- J. Liu, H. Yuan, J. Qiao, J. Feng, C. Xu, Z. Wang, W. Sun and K. Sun, Hierarchical hollow nanofiber networks for high-performance hybrid direct carbon fuel cells, J. Mater. Chem. A, 2017, 5, 17216–17220 CAS.
- A. Jayakumar, R. Kungas, S. Roy, A. Javadekar, D. Buttrey, J. Vohs and R. Gorte, A direct carbon fuel cell with a molten antimony anode, Energy Environ. Sci., 2011, 4, 4133–4137 CAS.
- C. Jiang, J. Ma, A. D. Bonaccorso and J. T. S. Irvine, Demonstration of high power, direct conversion of waste-derived carbon in a hybrid direct carbon fuel cell, Energy Environ. Sci., 2012, 5, 6973–6980 CAS.
- N. Xu, X. Li, X. Zhao, J. B. Goodenough and K. Huang, A novel solid oxide redox flow battery for grid energy storage, Energy Environ. Sci., 2011, 4, 4942–4946 CAS.
- A. Inoishi, S. Ida, S. Uratani, T. Okano and T. Ishihara, High capacity of an Fe-air rechargeable battery using LaGaO3-based oxide ion conductor as an electrolyte, Phys. Chem. Chem. Phys., 2012, 14, 12818–12822 CAS.
- X. Zhao, X. Li, Y. Gong and K. Huang, Enhanced reversibility and durability of a solid oxide Fe-air redox battery by carbothermic reaction derived energy storage materials, Chem. Commun., 2014, 50, 623–625 CAS.
- A. Inoishi, J. Hyodo, H. Kim, T. Sakai, S. Ida and T. Ishihara, Low temperature operation of a solid-oxide Fe-air rechargeable battery using a La0.9Sr0.1Ga0.8Mg0.2O3 oxide ion conductor, J. Mater. Chem. A, 2015, 3, 8260–8264 CAS.
- Q. Tang, C. Morey, Y. Zhang, N. Xu, S. Sun and K. Huang, Proton-Mediated and Ir-Catalyzed Iron/Iron-Oxide Redox Kinetics for Enhanced Rechargeability and Durability of Solid Oxide Iron-Air Battery, Adv. Sci., 2022, 9, e2203768 Search PubMed.
- X. Zhao, Y. Gong, X. Li, N. Xu and K. Huang, A new solid oxide molybdenum-air redox battery, J. Mater. Chem. A, 2013, 1, 14858–14861 RSC.
- A. Inoishi, T. Sakai, Y.-W. Ju, S. Ida and T. Ishihara, A rechargeable Si-air solid state oxygen shuttle battery incorporating an oxide ion conductor, J. Mater. Chem. A, 2013, 1, 15212–15215 RSC.
- X. Zhao, X. Li, Y. Gong, N. Xu, K. Romito and K. Huang, A high energy density all solid-state tungsten-air battery, Chem. Commun., 2013, 49, 5357–5359 RSC.
- A. Inoishi, Y.-W. Ju, S. Ida and T. Ishihara, Mg-air oxygen shuttle batteries using a ZrO2-based oxide ion-conducting electrolyte, Chem. Commun., 2013, 49, 4691–4693 RSC.
- A. Inoishi, M. Matsuka, T. Sakai, Y.-W. Ju, S. Ida and T. Ishihara, Lithium–Air Oxygen Shuttle Battery with a ZrO2-Based Ion-Conducting Oxide Electrolyte, ChemPlusChem, 2015, 80, 359–362 CrossRef CAS.
- A. Javadekar, A. Jayakumar, R. J. Gorte, J. M. Vohs and D. J. Buttrey, Energy Storage in Electrochemical Cells with Molten Sb Electrodes, J. Electrochem. Soc., 2012, 159, A386–A389 CrossRef CAS.
- Q. Tang, Y. Zhang, N. Xu, X. Lei and K. Huang, Demonstration of 10+ hour energy storage with ϕ1′′ laboratory size solid oxide iron–air batteries, Energy Environ. Sci., 2022, 15, 4659–4671 RSC.
- C. Zhang and K. Huang, An Intermediate-Temperature Solid Oxide Iron–Air Redox Battery Operated on O2–-Chemistry and Loaded with Pd-Catalyzed Iron-Based Energy Storage Material, ACS Energy Lett., 2016, 1, 1206–1211 CrossRef CAS.
- J. Irvine, D. Neagu, M. Verbraeken, C. Chatzichristodoulou, C. Graves and M. Mogensen, Evolution of the electrochemical interface in high-temperature fuel cells and electrolysers, Nat. Energy, 2016, 1, 15014 CAS.
- G. Glenk and S. Reichelstein, Reversible Power-to-Gas systems for energy conversion and storage, Nat. Commun., 2022, 13, 2010 CrossRef CAS PubMed.
- H. Iwahara, T. Esaka, H. Uchida and N. Maeda, Proton conduction in sintered oxides and its application to steam electrolysis for hydrogen production, Solid State Ionics, 1981, 3–4, 359–363 CAS.
- H. Matsumoto, M. Okubo, S. Hamajima, K. Katahira and H. Iwahara, Extraction and production of hydrogen using high-temperature proton conductor, Solid State Ionics, 2002, 152–153, 715–720 CAS.
- T. Norby, The promise of protonics, Nature, 2001, 410, 877–878 CAS.
- C. Duan, J. Huang, N. Sullivan and R. O'Hayre, Proton-conducting oxides for energy conversion and storage, Appl. Phys. Rev., 2020, 7, 011314 CAS.
- G. Marnellos and M. Stoukides, Ammonia Synthesis at Atmospheric Pressure, Science, 1998, 282, 98–100 CAS.
- M. Li, B. Hua, L.-C. Wang, J. D. Sugar, W. Wu, Y. Ding, J. Li and D. Ding, Switching of metal–oxygen hybridization for selective CO2 electrohydrogenation under mild temperature and pressure, Nat. Catal., 2021, 4, 274–283 CrossRef CAS.
- Y. Zhou, X. Guan, H. Zhou, K. Ramadoss, S. Adam, H. Liu, S. Lee, J. Shi, M. Tsuchiya, D. Fong and S. Ramanathan, Strongly correlated perovskite fuel cells, Nature, 2016, 534, 231–234 CAS.
- Z. Wang, Y. Wang, J. Wang, Y. Song, M. J. Robson, A. Seong, M. Yang, Z. Zhang, A. Belotti, J. Liu, G. Kim, J. Lim, Z. Shao and F. Ciucci, Rational design of perovskite ferrites as high-performance proton-conducting fuel cell cathodes, Nat. Catal., 2022, 5, 777–787 CAS.
- F. Liu, H. Deng, D. Diercks, P. Kumar, M. Jabbar, C. Gumeci, Y. Furuya, N. Dale, T. Oku, M. Usuda, P. Kazempoor, L. Fang, D. Chen, B. Liu and C. Duan, Lowering the operating temperature of protonic ceramic electrochemical cells to <450 °C, Nat. Energy, 2023, 8, 1145–1157 CAS.
- M. Papac, V. Stevanovic, A. Zakutayev and R. O'Hayre, Triple ionic-electronic conducting oxides for next-generation electrochemical devices, Nat. Mater., 2021, 20, 301–313 CAS.
- S. Choi, C. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H. Ji and S. Haile, Exceptional power density and stability at intermediate temperatures in protonic ceramic fuel cells, Nat. Energy, 2018, 3, 202–210 CAS.
- H. An, H.-W. Lee, B.-K. Kim, J.-W. Son, K. J. Yoon, H. Kim, D. Shin, H.-I. Ji and J.-H. Lee, A 5 × 5 cm2 protonic ceramic fuel cell with a power density of 1.3 W cm−2 at 600 °C, Nat. Energy, 2018, 3, 870–875 CAS.
- H. Malerød-Fjeld, D. Clark, I. Yuste-Tirados, R. Zanón, D. Catalán-Martinez, D. Beeaff, S. H. Morejudo, P. K. Vestre, T. Norby, R. Haugsrud, J. M. Serra and C. Kjølseth, Thermo-electrochemical production of compressed hydrogen from methane with near-zero energy loss, Nat. Energy, 2017, 2, 923–931 Search PubMed.
- Z. Luo, Y. Zhou, X. Hu, N. Kane, T. Li, W. Zhang, Z. Liu, Y. Ding, Y. Liu and M. Liu, Critical role of acceptor dopants in designing highly stable and compatible proton-conducting electrolytes for reversible solid oxide cells, Energy Environ. Sci., 2022, 15, 2992–3003 RSC.
- E. Vøllestad, R. Strandbakke, M. Tarach, D. Catalán-Martínez, M.-L. Fontaine, D. Beeaff, D. R. Clark, J. M. Serra and T. Norby, Mixed proton and electron conducting double perovskite anodes for stable and efficient tubular proton ceramic electrolysers, Nat. Mater., 2019, 18, 752–759 CrossRef PubMed.
- C. Zuo, S. Zha, M. Liu, M. Hatano and M. Uchiyama, BaZr0.1Ce0.7Y0.2O3-δ as an electrolyte for low-temperature solid-oxide fuel cells, Adv. Mater., 2006, 18, 3318–3320 CrossRef CAS.
- H. Ding, W. Wu, C. Jiang, Y. Ding, W. Bian, B. Hu, P. Singh, C. J. Orme, L. Wang, Y. Zhang and D. Ding, Self-sustainable protonic ceramic electrochemical cells using a triple conducting electrode for hydrogen and power production, Nat. Commun., 2020, 11, 1907 CrossRef CAS PubMed.
- H. Iwahara, Y. Asakura, K. Katahira and M. Tanaka, Prospect of hydrogen technology using proton-conducting ceramics, Solid State Ionics, 2004, 168, 299–310 CrossRef CAS.
- E. Antolini, Low molecular weight alkane-fed solid oxide fuel cells for power and chemicals cogeneration, J. Energy Chem., 2023, 80, 711–735 CrossRef CAS.
- Y. Feng, J. Luo and K. T. Chuang, Conversion of propane to propylene in a proton-conducting solid oxide fuel cell, Fuel, 2007, 86, 123–128 CrossRef CAS.
- X. Fu, X. Luo, J. Luo, K. Chuang, A. Sanger and A. Krzywicki, Ethane dehydrogenation over nano-Cr2O3 anode catalyst in proton ceramic fuel cell reactors to co-produce ethylene and electricity, J. Power Sources, 2011, 196, 1036–1041 CrossRef CAS.
- J.-H. Li, X.-Z. Fu, J. Luo, K. T. Chuang and A. R. Sanger, Evaluation of molybdenum carbide as anode catalyst for proton-conducting hydrogen and ethane solid oxide fuel cells, Electrochem. Commun., 2012, 15, 81–84 CrossRef CAS.
- X. Fu, J. Lin, S. Xu, J. Luo, K. Chuang, A. Sanger and A. Krzywicki, CO2 emission free co-generation of energy and ethylene in hydrocarbon SOFC reactors with a dehydrogenation anode, Phys. Chem. Chem. Phys., 2011, 13, 19615–19623 RSC.
- S. Cui, J. Li, J. Luo, K. Chuang and L. Qiao, Co-generation of energy and ethylene in hydrocarbon fueled SOFCs with Cr3C2 and WC anode catalysts, Ceram. Int., 2014, 40, 11781–11786 CAS.
- J.-Y. Lin, L. Shao, F.-Z. Si, S.-B. Liu, X.-Z. Fu and J. Luo, Co2CrO4 Nanopowders as an Anode Catalyst for Simultaneous Conversion of Ethane to Ethylene and Power in Proton-Conducting Fuel Cell Reactors, J. Phys. Chem. C, 2018, 122, 4165–4171 CrossRef CAS.
- J. Li, J. Hou, X. Xi, Y. Lu, M. Li, Y. Fan, L. Wang, L. Wang, X.-Z. Fu and J. Luo, Cogeneration of ethylene and electricity in symmetrical protonic solid oxide fuel cells based on a La0.6Sr0.4Fe0.8Nb0.1Cu0.1O3−δ electrode, J. Mater. Chem. A, 2020, 8, 25978–25985 RSC.
- S. Liu, K. T. Chuang and J. Luo, Double-layered perovskite anode with in situ exsolution of a Co–Fe alloy to cogenerate ethylene and electricity in a proton-conducting ethane fuel cell, ACS Catal., 2016, 6, 760–768 CrossRef CAS.
- J. Luo, Y. Liu, J. Zhu, C. Wang, Y. Zhao, D. Yan, J. Li and L. Jia, A proton-conducting solid oxide fuel cell for co-production of ethylene and power via ethane conversion, Chin. Chem. Lett., 2024, 110171, DOI:10.1016/j.cclet.2024.110171.
- J.-Y. Lin, L. Shao, F.-Z. Si, X.-Z. Fu and J. Luo, Multiple-doped barium cerate proton-conducting electrolytes for chemical-energy cogeneration in solid oxide fuel cells, Int. J. Hydrogen Energy, 2018, 43, 19704–19710 CrossRef CAS.
- S. Lei, A. Wang, G. Weng, Y. Wu, J. Xue and H. Wang, Simultaneous generation of electricity, ethylene and decomposition of nitrous oxide via protonic ceramic fuel cell membrane reactor, J. Energy Chem., 2023, 77, 359–368 CAS.
- M. Wang, L.-C. Wang, H. Li, W. Wu, S. W. Snyder, G. Gao, F. Chen, Y. Yang and D. Ding, Nanostructured carbon as highly efficient and stable anodes for ethylene production and power generation in protonic ceramic electrochemical cells, Carbon, 2022, 199, 379–386 CrossRef CAS.
- R. Zhang, Y. Meng, L.-C. Wang, M. Wang, W. Wu, W. Wu and D. Ding, Boosting the performances of protonic solid oxide fuel cells for co-production of propylene and electricity from propane by integrating thermo- and electro- catalysis, Fuel, 2024, 357, 129685 CAS.
- N. Shi, S. Xue, Y. Xie, Y. Yang, D. Huan, Y. Pan, R. Peng, C. Xia, Z. Zhan and Y. Lu, Co-generation of electricity and olefin via proton conducting fuel cells using (Pr0.3Sr0.7)0.9Ni0.1Ti0.9O3 catalyst layers, Appl. Catal., B, 2020, 272, 118973 CAS.
- Y. Feng, J. Luo and K. Chuang, Carbon deposition during propane dehydrogenation in a fuel cell, J. Power Sources, 2007, 167, 486–490 CAS.
- X. Fu, J. Luo, A. Sanger, N. Danilovic and K. Chuang, An integral proton conducting SOFC for simultaneous production of ethylene and power from ethane, Chem. Commun., 2010, 46, 2052–2054 CAS.
- X. Fu, J. Luo, A. Sanger, N. Luo and K. Chuang, Y-doped BaCeO3-δ nanopowders as proton-conducting electrolyte materials for ethane fuel cells to co-generate ethylene and electricity, J. Power Sources, 2010, 195, 2659–2663 CAS.
- J. H. Lunsford, Catalytic conversion of methane to more useful chemicals and fuels: a challenge for the 21st century, Catal. Today, 2000, 63, 165–174 CAS.
- T. Hibino, S. Hamakawa and H. Iwahara, Electrochemical Methane Activation to C2-Hydrocarbons Using Protonic Conductor, Chem. Lett., 1992, 21, 1715–1716 Search PubMed.
- S. Hamakawa, T. Hibino and H. Iwahara, Electrochemical Methane Coupling Using Protonic Conductors, J. Electrochem. Soc., 1993, 140, 459–462 CAS.
- V. Kyriakou, C. Athanasiou, I. Garagounis, A. Skodra and M. Stoukides, Production of H2 and C2 hydrocarbons from methane in a proton conducting solid electrolyte cell using a Au–5Ce–5Na2WO4/SiO2 anode, Int. J. Hydrogen Energy, 2012, 37, 16636–16641 CAS.
- M. Sakbodin, Y. Wu, S. C. Oh, E. D. Wachsman and D. Liu, Hydrogen-Permeable Tubular Membrane Reactor: Promoting Conversion and Product Selectivity for Non-Oxidative Activation of Methane over an Fe©SiO2 Catalyst, Angew. Chem., Int. Ed., 2016, 128, 16383–16386 Search PubMed.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen, Science, 2014, 344, 616–619 CAS.
- M. Sakbodin, E. Schulman, S. Cheng, Y.-L. Huang, Y. Pan, P. Albertus, E. D. Wachsman and D. Liu, Direct Nonoxidative Methane Conversion in an Autothermal Hydrogen-Permeable Membrane Reactor, Adv. Energy Mater., 2021, 11, 2102782 CAS.
- B. Hua, N. Yan, M. Li, Y.-q. Zhang, Y.-f. Sun, J. Li, T. Etsell, P. Sarkar, K. Chuang and J. Luo, Novel layered solid oxide fuel cells with multiple-twinned Ni0.8Co0.2 nanoparticles: the key to thermally independent CO2 utilization and power-chemical cogeneration, Energy Environ. Sci., 2016, 9, 207–215 CAS.
- B. Hua, N. Yan, M. Li, Y. Sun, Y. Zhang, J. Li, T. Etsell, P. Sarkar and J. Luo, Anode-Engineered Protonic Ceramic Fuel Cell with Excellent Performance and Fuel Compatibility, Adv. Mater., 2016, 28, 8922–8926 CAS.
- F. Liu, H. Deng, H. Ding, P. Kazempoor, B. Liu and C. Duan, Process-intensified protonic ceramic fuel cells for power generation, chemical production, and greenhouse gas mitigation, Joule, 2023, 7, 1308–1332 CAS.
- F. Liu, L. Fang, D. Diercks, P. Kazempoor and C. Duan, Rationally designed negative electrode for selective CO2-to-CO conversion in protonic ceramic electrochemical cells, Nano Energy, 2022, 102, 107722 CrossRef CAS.
- F. Liu, H. Deng, Z. Wang, A. M. Hussain, N. Dale, Y. Furuya, Y. Miura, Y. Fukuyama, H. Ding, B. Liu and C. Duan, Synergistic Effects of In-Situ Exsolved Ni–Ru Bimetallic Catalyst on High-Performance and Durable Direct-Methane Solid Oxide Fuel Cells, J. Am. Chem. Soc., 2024, 146, 4704–4715 CrossRef CAS.
- D. Ding, Y. Zhang, W. Wu, D. Chen, M. Liu and T. He, A novel low-thermal-budget approach for the co-production of ethylene and hydrogen via the electrochemical non-oxidative deprotonation of ethane, Energy Environ. Sci., 2018, 11, 1710–1716 RSC.
- W. Wu, L. Wang, H. Hu, W. Bian, J. Gomez, C. Orme, H. Ding, Y. Dong, T. He, J. Li and D. Ding, Electrochemically Engineered, Highly Energy-Efficient Conversion of Ethane to Ethylene and Hydrogen below 550 °C in a Protonic Ceramic Electrochemical Cell, ACS Catal., 2021, 11, 12194–12202 CrossRef CAS.
- E. Rebollo, C. Mortalo, S. Escolastico, S. Boldrini, S. Barison, J. M. Serra and M. Fabrizio, Exceptional hydrogen permeation of all-ceramic composite robust membranes based on BaCe0.65Zr0.20Y0.15O3-δ and Y- or Gd-doped ceria, Energy Environ. Sci., 2015, 8, 3675–3686 RSC.
- Y. Tong, X. Meng, T. Luo, C. Cui, Y. Wang, S. Wang, R. Peng, B. Xie, C. Chen and Z. Zhan, Protonic Ceramic Electrochemical Cell for Efficient Separation of Hydrogen, ACS Appl. Mater. Interfaces, 2020, 12, 25809–25817 CrossRef CAS PubMed.
- S. Choi, T. C. Davenport and S. M. Haile, Protonic ceramic electrochemical cells for hydrogen production and electricity generation: exceptional reversibility, stability, and demonstrated faradaic efficiency, Energy Environ. Sci., 2019, 12, 206–215 CAS.
- I. Quina, L. Almar, D. Catalán-Martínez, A. M. Dayaghi, A. Martínez, T. Norby, S. Escolástico and J. M. Serra, Direct electrocatalytic CO2 reduction in a pressurized tubular protonic membrane reactor, Chem Catal., 2023, 3, 100766 CAS.
- X. Miao, J. Feng, Z. Dai, X. Zhu, J. Wen, L. Zhang, X. Ye, Y. Zhou and Z. Wen, A Regenerative Coking-resistant CO2 Hydrogenation Reactor using a Protonic Ceramic Electrolysis Cell with Thin and Robust Fuel Electrode, Adv. Energy Mater., 2024, 14, 2402208 CAS.
- K. Xie, Y. Zhang, G. Meng and J. T. S. Irvine, Electrochemical reduction of CO2 in a proton conducting solid oxide electrolyser, J. Mater. Chem., 2011, 21, 195–198 CAS.
- N. Shi, Y. Xie, D. Huan, Y. Yang, S. Xue, Z. Qi, Y. Pan, R. Peng, C. Xia and Y. Lu, Controllable CO2 conversion in high performance proton conducting solid oxide electrolysis cells and the possible mechanisms, J. Mater. Chem. A, 2019, 7, 4855–4864 CAS.
- G. Weng, S. Lei, R. Wang, K. Ouyang, J. Dong, X. Lin, J. Xue, L.-X. Ding and H. Wang, A high-efficiency electrochemical proton-conducting membrane reactor for ammonia production at intermediate temperatures, Joule, 2023, 7, 1333–1346 CAS.
- S. Giddey, S. Badwal and A. Kulkarni, Review of electrochemical ammonia production technologies and materials, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Recent Advances and Challenges of Electrocatalytic N2 Reduction to Ammonia, Chem. Rev., 2020, 120, 5437–5516 CAS.
- M. Ouzounidou, A. Skodra, C. Kokkofitis and M. Stoukides, Catalytic and electrocatalytic synthesis of NH3 in a H+ conducting cell by using an industrial Fe catalyst, Solid State Ionics, 2007, 178, 153–159 CAS.
- G. Marnellos, S. Zisekas and M. Stoukides, Synthesis of Ammonia at Atmospheric Pressure with the Use of Solid State Proton Conductors, J. Catal., 2000, 193, 80–87 CAS.
- C. Fernandez, N. Hortance, Y. Liu, J. Lim, K. Hatzell and M. Hatzell, Opportunities for intermediate temperature renewable ammonia electrosynthesis, J. Mater. Chem. A, 2020, 8, 15591–15606 CAS.
- W. B. Wang, X. B. Cao, W. J. Gao, F. Zhang, H. T. Wang and G. L. Ma, Ammonia synthesis at atmospheric pressure using a reactor with thin solid electrolyte BaCe0.85Y0.15O3−α membrane, J. Membr. Sci., 2010, 360, 397–403 CAS.
- E. Vasileiou, V. Kyriakou, I. Garagounis, A. Vourros and M. Stoukides, Ammonia synthesis at atmospheric pressure in a BaCe0.2Zr0.7Y0.1O2.9 solid electrolyte cell, Solid State Ionics, 2015, 275, 110–116 CAS.
- I. Amar, R. Lan, C. Petit and S. Tao, Solid-state electrochemical synthesis of ammonia: a review, J. Solid State Electrochem., 2011, 15, 1845–1860 CAS.
- B. Wang, J. Wang, R. Liu, Y. Xie and Z. Li, Synthesis of ammonia from natural gas at atmospheric pressure with doped ceria–Ca3(PO4)2–K3PO4 composite electrolyte and its proton conductivity at intermediate temperature, J. Solid State Electrochem., 2007, 11, 27–31 Search PubMed.
- I. Amar, R. Lan, C. T. Petit, V. Arrighi and S. Tao, Electrochemical synthesis of ammonia based on a carbonate-oxide composite electrolyte, Solid State Ionics, 2011, 182, 133–138 CAS.
- I. A. Amar, C. T. G. Petit, L. Zhang, R. Lan, P. J. Skabara and S. Tao, Electrochemical synthesis of ammonia based on doped-ceria-carbonate composite electrolyte and perovskite cathode, Solid State Ionics, 2011, 201, 94–100 CAS.
- I. A. Amar, C. T. G. Petit, G. Mann, R. Lan, P. J. Skabara and S. Tao, Electrochemical synthesis of ammonia from N2 and H2O based on (Li,Na,K)2CO3–Ce0.8Gd0.18Ca0.02O2−δ composite electrolyte and CoFe2O4 cathode, Int. J. Hydrogen Energy, 2014, 39, 4322–4330 CAS.
- R. Lan, K. A. Alkhazmi, I. A. Amar and S. W. Tao, Synthesis of ammonia directly from wet air at intermediate temperature, Appl. Catal., B, 2014, 152, 212–217 CrossRef.
- I. Amar, R. Lan and S. Tao, Electrochemical synthesis of ammonia directly from wet N2 using La0.6Sr0.4Fe0.8Cu0.2O3-δ-Ce0.8Gd0.18Ca0.02O2-δ composite catalyst, J. Electrochem. Soc., 2014, 161, H350–H354 CrossRef CAS.
- F. Kosaka, N. Noda, T. Nakamura and J. Otomo, In situ formation of Ru nanoparticles on La1-xSrxTiO3-based mixed conducting electrodes and their application in electrochemical synthesis of ammonia using a proton-conducting solid electrolyte, J. Mater. Sci., 2017, 52, 2825–2835 Search PubMed.
- L. Zhu, C. Cadigan, C. Duan, J. Huang, L. Bian, L. Le, C. H. Hernandez, V. Avance, R. O'Hayre and N. P. Sullivan, Ammonia-fed reversible protonic ceramic fuel cells with Ru-based catalyst, Commun. Chem., 2021, 4, 121 CAS.
- I. Amar, R. Lan, C. Petit and S. Tao, Electrochemical Synthesis of Ammonia Based on Co3Mo3N Catalyst and LiAlO2–(Li,Na,K)2CO3 Composite Electrolyte, Electrocatalysis, 2014, 6, 286–294 Search PubMed.
- X. Wang, J. Yin, J. Xu, H. Wang and G. Ma, Chemical Stability, Ionic Conductivity of BaCe0.9−xZrxSm0.10O3−α and Its Application to Ammonia Synthesis at Atmospheric Pressure, Chin. J. Chem., 2011, 29, 1114–1118 CAS.
- M. Li, B. Hua, W. Wu, L. Wang, Y. Ding, M. Welander, R. Walker and D. Ding, Activating nano-bulk interplays for sustainable ammonia electrosynthesis, Mater. Today, 2022, 60, 31–40 CAS.
- D. Yun, J. Joo, J. Yu, H. Yoon, J. Kim and C. Yoo, Electrochemical ammonia synthesis from steam and nitrogen using proton conducting yttrium doped barium zirconate electrolyte with silver, platinum, and lanthanum strontium cobalt ferrite electrocatalyst, J. Power Sources, 2015, 284, 245–251 CAS.
- F. Kosaka, T. Nakamura, A. Oikawa and J. Otomo, Electrochemical Acceleration of Ammonia Synthesis on Fe-Based Alkali-Promoted Electrocatalyst with Proton Conducting Solid Electrolyte, ACS Sustain. Chem. Eng., 2017, 5, 10439–10446 CAS.
- F. Wang, Y. Wang, L. Li, Z. Li, W. Zhang, Z. Xue, D. Liu, X. Meng, C. Li, J. Sunarso, S. Liu and N. Yang, Electrocatalytic ammonia synthesis on Fe@MXene catalyst as cathode of intermediate-temperature proton-conducting solid oxide cell, Int. J. Hydrogen Energy, 2023, 48, 17677–17688 CAS.
- R. Li, T. Li, X. Liu, C. Xie, Q. Zhen, S. Bashir and J. L. Liu, Green synthesis of ammonia from steam and air using solid oxide electrolysis cells composed of ruthenium-modified perovskite catalyst, Energy Sci. Eng., 2023, 11, 2293–2301 CAS.
- L. Ye, H. Li and K. Xie, Sustainable ammonia production enabled by membrane reactor, Nat Sustainability, 2022, 5, 787–794 Search PubMed.
- C. Zhou, J. Sunarso, Y. Song, J. Dai, J. Zhang, B. Gu, W. Zhou and Z. Shao, New reduced-temperature ceramic fuel cells with dual-ion conducting electrolyte and triple-conducting double perovskite cathode, J. Mater. Chem. A, 2019, 7, 13265–13274 CAS.
- J. Cao, C. Su, Y. Ji, G. Yang and Z. Shao, Recent advances and perspectives of fluorite and perovskite-based dual-ion conducting solid oxide fuel cells, J. Energy Chem., 2021, 57, 406–427 CrossRef CAS.
- S. Fop, K. McCombie, E. Wildman, J. Skakle, J. Irvine, P. Connor, C. Savaniu, C. Ritter and A. McLaughlin, High oxide ion and proton conductivity in a disordered hexagonal perovskite, Nat. Mater., 2020, 19, 752–757 CAS.
- F. He, Z. Teng, G. Yang, C. Zhou, D. Guan, S. Chen, R. Ran, W. Wang, W. Zhou and Z. Shao, Manipulating cation nonstoichiometry towards developing better electrolyte for self-humidified dual-ion solid oxide fuel cells, J. Power Sources, 2020, 460, 228105 CrossRef CAS.
- W. Ye, Q. Hu, H. Zhao, Y. Jing, M. Singh and L. Fan, In situ reconstruction of proton conductive electrolyte from self-assembled perovskite oxide-based nanocomposite for low temperature ceramic fuel cells, Chem. Eng. J., 2024, 497, 154977 CrossRef CAS.
- C. Xia, Y. Li, Y. Tian, Q. Liu, Z. Wang, L. Jia, Y. Zhao and Y. Li, Intermediate temperature fuel cell with a doped ceria-carbonate composite electrolyte, J. Power Sources, 2010, 195, 3149–3154 CrossRef CAS.
- C. Xia, Y. Li, Y. Tian, Q. Liu, Y. Zhao, L. Jia and Y. Li, A high performance composite ionic conducting electrolyte for intermediate temperature fuel cell and evidence for ternary ionic conduction, J. Power Sources, 2009, 188, 156–162 CrossRef CAS.
- R. Lan, K. A. Alkhazmi, I. A. Amar and S. Tao, Synthesis of ammonia directly from wet air using new perovskite oxide La0.8Cs0.2Fe0.8Ni0.2O3-δ as catalyst, Electrochim. Acta, 2014, 123, 582–587 CrossRef CAS.
- L. Zhang, N. Xu, X. Li, S. Wang, K. Huang, W. H. Harris and W. K. S. Chiu, High CO2 permeation flux enabled by highly interconnected three-dimensional ionic channels in selective CO2 separation membranes, Energy Environ. Sci., 2012, 5, 8310–8317 RSC.
- W. Xing, T. Peters, M. Fontaine, A. Evans, P. Henriksen, T. Norby and R. Bredesen, Steam-promoted CO2 flux in dual-phase CO2 separation membranes, J. Membr. Sci., 2015, 482, 115–119 CrossRef CAS.
- J. Wade, C. Lee, A. West and K. Lackner, Composite electrolyte membranes for high temperature CO2 separation, J. Membr. Sci., 2011, 369, 20–29 CrossRef CAS.
- J. Wade, K. Lackner and A. West, Transport model for a high temperature, mixed conducting CO2 separation membrane, Solid State Ionics, 2007, 178, 1530–1540 CrossRef CAS.
- Y. Li, Z. Rui, C. Xia, M. Anderson and Y. S. Lin, Performance of ionic-conducting ceramic/carbonate composite material as solid oxide fuel cell electrolyte and CO2 permeation membrane, Catal. Today, 2009, 148, 303–309 CAS.
- X. Dong and Y. Lin, Catalyst-free ceramic-carbonate dual phase membrane reactor for hydrogen production from gasifier syngas, J. Membr. Sci., 2016, 520, 907–913 CrossRef CAS.
- H. Wu, Z. Rui and J. Lin, Hydrogen production with carbon dioxide capture by dual-phase ceramic-carbonate membrane reactor via steam reforming of methane, J. Membr. Sci., 2020, 598, 117780 CAS.
- A. Caravaca, A. de Lucas-Consuegra, C. Molina-Mora, J. Valverde and F. Dorado, Enhanced H2 formation by electrochemical promotion in a single chamber steam electrolysis cell, Appl. Catal., B, 2011, 106, 54–62 CAS.
- Y. M. Guo, G. Largiller, C. Guizard, C. Tardivat and D. Farrusseng, Coke-free operation of an all porous solid oxide fuel cell (AP-SOFC) used as an O2 supply device, J. Mater. Chem. A, 2015, 3, 2684–2689 CAS.
- M. Horiuchi, S. Suganuma and M. Watanabe, Electrochemical Power Generation Directly from Combustion Flame of Gases, Liquids, and Solids, J. Electrochem. Soc., 2004, 151, A1402 CAS.
- L. Sun, Y. Hao, C. Zhang, R. Ran and Z. Shao, Coking-free direct-methanol-flame fuel cell with traditional nickel–cermet anode, Int. J. Hydrogen Energy, 2010, 35, 7971–7981 CAS.
- X. Zhu, Z. Lü, B. Wei, X. Huang, Z. Wang and W. Su, Direct flame SOFCs with La0.75Sr0.25Cr0.5Mn0.5O3-δ/Ni coimpregnated yttria-stabilized zirconia anodes operated on liquefied petroleum gas flame, J. Electrochem. Soc., 2010, 157, B1838–B1843 CAS.
- T. Li, X. Lu, M. F. Rabuni, B. Wang, N. M. Farandos, G. H. Kelsall, D. J. L. Brett, P. R. Shearing, M. Ouyang, N. P. Brandon and K. Li, High-performance fuel cell designed for coking-resistance and efficient conversion of waste methane to electrical energy, Energy Environ. Sci., 2020, 13, 1879–1887 CAS.
- Y. Wu, L. Liu, X. Yu, J. Zhang, L. Li, C. Yan and B. Zhu, Natural hematite ore composited with ZnO nanoneedles for energy applications, Composites, Part B, 2018, 137, 178–183 CAS.
- Y. Wu, B. Dong, J. Zhang, H. Song and C. Yan, The synthesis of ZnO/SrTiO3 composite for high-efficiency photocatalytic hydrogen and electricity conversion, Int. J. Hydrogen Energy, 2018, 43, 12627–12636 CrossRef CAS.
- M. Morgenbesser, A. Schmid, A. Viernstein, J. de Dios Sirvent, F. Chiabrera, N. Bodenmüller, S. Taibl, M. Kubicek, F. Baiutti, A. Tarancon and J. Fleig, SrTiO3 based high temperature solid oxide solar cells: Photovoltages, photocurrents and mechanistic insight, Solid State Ionics, 2021, 368, 115700 CrossRef CAS.
- B. Xie, D. Hu, P. Kumar, V. V. Ordomsky, A. Y. Khodakov and R. Amal, Heterogeneous catalysis via light-heat dual activation: A path to the breakthrough in C1 chemistry, Joule, 2024, 8, 312–333 CrossRef CAS.
- H. Jiang, L. Wang, H. Kaneko, R. Gu, G. Su, L. Li, J. Zhang, H. Song, F. Zhu, A. Yamaguchi, J. Xu, F. Liu, M. Miyauchi, W. Ding and M. Zhong, Light-driven CO2 methanation over Au-grafted Ce0.95Ru0.05O2 solid-solution catalysts with activities approaching the thermodynamic limit, Nat. Catal., 2023, 6, 519–530 CrossRef CAS.
- X. Ye, J. Melas-Kyriazi, Z. A. Feng, N. A. Melosh and W. C. Chueh, A semiconductor/mixed ion and electron conductor heterojunction for elevated-temperature water splitting, Phys. Chem. Chem. Phys., 2013, 15, 15459–15469 RSC.
- J. Fleig, G. Walch, A. K. Opitz, M. Kubicek, G. C. Brunauer, B. Rotter and K. Ponweiser, Solid Oxide Photo-electrochemistry with Oxides “Breathing” upon UV Light and Solar Cells Operating at 400°C, ECS Meeting Abstracts, 2016, vol. MA2016-02, p. 4184 Search PubMed.
- G. Walch, B. Rotter, G. C. Brunauer, E. Esmaeili, A. K. Opitz, M. Kubicek, J. Summhammer, K. Ponweiser and J. Fleig, A solid oxide photoelectrochemical cell with UV light-driven oxygen storage in mixed conducting electrodes, J. Mater. Chem. A, 2017, 5, 1637–1649 RSC.
- Y. Shi, L. Wang, Z. Wang, G. Vinai, L. Braglia, P. Torelli, C. Aruta, E. Traversa, W. Liu and N. Yang, Defect Engineering for Tuning the Photoresponse of Ceria-Based Solid Oxide Photoelectrochemical Cells, ACS Appl. Mater. Interfaces, 2021, 13, 541–551 CrossRef CAS PubMed.
- T. Wei, Y. Huang, Q. Zhang, L. Yuan, J. Yang, Y. Sun, X. Hu, W. Zhang and J. Goodenough, Thermoelectric Solid-Oxide Fuel Cells with Extra Power Conversion from Waste Heat, Chem. Mater., 2012, 24, 1401–1403 CrossRef CAS.
- R. Sharma, H. Patel, U. Mushtaq, V. Kyriakou, G. Zafeiropoulos, F. Peeters, S. Welzel, M. van de Sanden and M. Tsampas, Plasma Activated Electrochemical Ammonia Synthesis from Nitrogen and Water, ACS Energy Lett., 2020, 6, 313–319 CrossRef.
- M. Siebenhofer, A. Viernstein, M. Morgenbesser, J. Fleig and M. Kubicek, Photoinduced electronic and ionic effects in strontium titanate, Mater. Adv., 2021, 2, 7583–7619 CAS.
- A. Viernstein, M. Kubicek, M. Morgenbesser, T. M. Huber, E. Ellmeyer, M. Siebenhofer, C. A. F. Vaz and J. Fleig, Mechanism of photo-ionic stoichiometry changes in SrTiO3, Solid State Ionics, 2022, 383, 115992 CrossRef CAS.
- G. C. Brunauer, B. Rotter, G. Walch, E. Esmaeili, A. K. Opitz, K. Ponweiser, J. Summhammer and J. Fleig, UV-Light-Driven Oxygen Pumping in a High-Temperature Solid Oxide Photoelectrochemical Cell, Adv. Funct. Mater., 2016, 26, 120–128 CAS.
- A. Senocrate, E. Kotomin and J. Maier, On the Way to Optoionics, Helv. Chim. Acta, 2020, 103, e2000073 CrossRef CAS.
- H. Su and Y. H. Hu, Thermo-photo catalytic anode process for carbonate-superstructured solid fuel cells, Proc. Natl. Acad. Sci. U.S.A., 2024, 121, e2314996121 CAS.
- N. Ito, M. Iijima, K. Kimura and S. Iguchi, New intermediate temperature fuel cell with ultra-thin proton conductor electrolyte, J. Power Sources, 2005, 152, 200–203 CAS.
- S. Jeong, T. Yamaguchi, M. Okamoto, C. Zhu, H. Habazaki, M. Nagayama and Y. Aoki, Proton Pumping Boosts Energy Conversion in Hydrogen-Permeable Metal-Supported Protonic Fuel Cells, ACS Appl. Energy Mater., 2020, 3, 1222–1234 CAS.
- S. Jeong, N. Wang, S. Kitano, H. Habazaki and Y. Aoki, Metal/oxide heterojunction boosts fuel cell cathode reaction at low temperatures, Adv. Energy Mater., 2021, 11, 2102025 CAS.
- T. Wei, Y. Huang, R. Zeng, L. Yuan, X. Hu, W. Zhang, L. Jiang, J. Yang and Z. Zhang, Evaluation of Ca3Co2O6 as cathode material for high-performance solid-oxide fuel cell, Sci. Rep., 2013, 3, 1125 Search PubMed.
- H. Yoon, M. Choi, T. Lim, H. Kwon, K. Ihm, J. Kim, S. Choi and J. Son, Reversible phase modulation and hydrogen storage in multivalent VO2 epitaxial thin films, Nat. Mater., 2016, 15, 1113–1119 CAS.
- X. Guan, J. Jiang, J. Lattimer, M. Tsuchiya, C. M. Friend and S. Ramanathan, Hydride-Based Solid Oxide Fuel Cell-Battery Hybrid Electrochemical System, Energy Technol., 2017, 5, 616–622 CrossRef CAS.
- M. Stoukides and C. Vayenas, The effect of electrochemical oxygen pumping on the rate and selectivity of ethylene oxidation on polycrystalline silver, J. Catal., 1981, 70, 137–146 CrossRef CAS.
- C. Vayenas and S. Bebelis, Electrochemical promotion of heterogeneous catalysis, Catal. Today, 1999, 51, 581–594 CrossRef CAS.
- R. Imbihl, Electrochemical promotion of catalytic reactions, Prog. Surf. Sci., 2010, 85, 241–278 CrossRef CAS.
- I. Kalaitzidou, A. Katsaounis, T. Norby and C. Vayenas, Electrochemical promotion of the hydrogenation of CO2 on Ru deposited on a BZY proton conductor, J. Catal., 2015, 331, 98–109 CrossRef CAS.
- A. Kotsiras, I. Kalaitzidou, D. Grigoriou, A. Symillidis, M. Makri, A. Katsaounis and C. Vayenas, Electrochemical promotion of nanodispersed Ru-Co catalysts for the hydrogenation of CO2, Appl. Catal., B, 2018, 232, 60–68 CrossRef CAS.
- C. Chatzilias, E. Martino, A. Katsaounis and C. Vayenas, Electrochemical promotion of CO2 hydrogenation in a monolithic electrochemically promoted reactor (MEPR), Appl. Catal., B, 2021, 284, 119695 CrossRef CAS.
- C. Vayenas, S. Bebelis, S. Neophytides and I. Yentekakis, Non-faradaic electrochemical modification of catalytic activity in solid electrolyte cells, Appl. Phys. A, 1989, 49, 95–103 CrossRef.
- C. G. Vayenas, S. Bebelis, C. Pliangos, S. Brosda and D. Tsiplakides, Electrochemical Activation of Catalysis: Promotion, Electrochemical Promotion, and Metal-Support Interactions, Springer, 2001 Search PubMed.
- D. Zagoraios, A. Athanasiadi, I. Kalaitzidou, S. Ntais, A. Katsaounis, A. Caravaca, P. Vernoux and C. G. Vayenas, Electrochemical promotion of methane oxidation over nanodispersed Pd/Co3O4 catalysts, Catal. Today, 2020, 355, 910–920 CrossRef CAS.
- G. Pekridis, K. Kalimeri, N. Kaklidis, E. Vakouftsi, E. F. Iliopoulou, C. Athanasiou and G. E. Marnellos, Study of the reverse water gas shift (RWGS) reaction over Pt in a solid oxide fuel cell (SOFC) operating under open and closed-circuit conditions, Catal. Today, 2007, 127, 337–346 CrossRef CAS.
- V. Jiménez, C. Jiménez-Borja, P. Sánchez, A. Romero, E. I. Papaioannou, D. Theleritis, S. Souentie, S. Brosda and J. L. Valverde, Electrochemical promotion of the CO2 hydrogenation reaction on composite Ni or Ru impregnated carbon nanofiber catalyst-electrodes deposited on YSZ, Appl. Catal., B, 2011, 107, 210–220 CrossRef.
- D. Zagoraios, C. Panaritis, A. Krassakopoulou, E. A. Baranova, A. Katsaounis and C. G. Vayenas, Electrochemical promotion of Ru nanoparticles deposited on a proton conductor electrolyte during CO2 hydrogenation, Appl. Catal., B, 2020, 276, 119148 CAS.
- C. Yiokari, G. Pitselis, D. Polydoros, A. Katsaounis and C. Vayenas, High-Pressure Electrochemical Promotion of Ammonia Synthesis over an Industrial Iron Catalyst, J. Phys. Chem. A, 2000, 104, 10600–10602 CrossRef CAS.
- I. Harkness and R. Lambert, Electrochemical Promotion of the NO + Ethylene Reaction over Platinum, J. Catal., 1995, 152, 211–214 CrossRef CAS.
- A. Palermo, M. Tikhov, N. Filkin, R. Lambert, I. Yentekakis and C. Vayenas, Electrochemical promotion of NO reduction by CO and by propene, Stud. Surf. Sci. Catal., 1996, 101, 513–522 CrossRef CAS.
- M. Marwood and C. G. Vayenas, Electrochemical Promotion of the Catalytic Reduction of NO by CO on Palladium, J. Catal., 1997, 170, 275–285 CrossRef CAS.
- E. Wachsman, P. Jayaweera, G. Krishnan and A. Sanjurjo, Electrocatalytic reduction of NOx on La1−xAxB1−yB′yO3−δ: evidence of electrically enhanced activity, Solid State Ionics, 2000, 136–137, 775–782 CrossRef CAS.
- S. Bebelis and C. G. Vayenas, Non-faradaic electrochemical modification of catalytic activity: 1. The case of ethylene oxidation on Pt, J. Catal., 1989, 118, 125–146 CrossRef CAS.
- T. Huang, C. Wu, S. Hsu and C. Wu, Electrochemical-catalytic conversion for simultaneous NOx and hydrocarbons emissions control of lean-burn gasoline engine, Appl. Catal., B, 2011, 110, 164–170 CrossRef CAS.
- S. Biswas, A. P. Kulkarni, S. Giddey and S. Bhattacharya, A Review on Synthesis of Methane as a Pathway for Renewable Energy Storage With a Focus on Solid Oxide Electrolytic Cell-Based Processes, Front. Energy Res., 2020, 8, 570112 CrossRef.
- A. Nakos, S. Souentie and A. Katsaounis, Electrochemical promotion of methane oxidation on Rh/YSZ, Appl. Catal., B, 2010, 101, 31–37 CrossRef CAS.
- E. López, F. Dorado and A. de Lucas-Consuegra, Electrochemical promotion for hydrogen production via ethanol steam reforming reaction, Appl. Catal., B, 2019, 243, 355–364 CrossRef.
- R. Küngas, Review—Electrochemical CO2 Reduction for CO Production: Comparison of Low- and High-Temperature Electrolysis Technologies, J. Electrochem. Soc., 2020, 167, 044508 Search PubMed.
- N. Anastasijevic, NEMCA—From discovery to technology, Catal. Today, 2009, 146, 308–311 CAS.
| This journal is © The Royal Society of Chemistry 2025 |