
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and depolymerization studies of biohybrid polycarbonates derived from terpenes†
Thirusangumurugan
Senthamarai
a,
Enrico
Lanaro
 ab,
Jack
Tinker
c,
Antoine
Buchard
c and
Arjan W.
Kleij
*ad
ab,
Jack
Tinker
c,
Antoine
Buchard
c and
Arjan W.
Kleij
*ad
aInstitute of Chemical Research of Catalonia (ICIQ), the Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 – Tarragona, Spain. E-mail: akleij@iciq.es
bDepartament de Química Física i Inorgànica/Universitat Rovira i Virgili, Marcel·lí Domingo s/n, 43007 – Tarragona, Spain
cGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, YO10 5DD York, UK
dCatalan Institute of Research and Advanced Studies (ICREA), Pg. Lluís Companys 23, 08010 – Barcelona, Spain
First published on 23rd May 2025
Abstract
We here report the catalytic ring-opening copolymerization of 2-menthene oxide (MO), a terpene-based monomer derived from L-menthol, and CO2 to provide poly(menthene carbonate), PMC, with a maximum molecular weight (Mn) of 10.2 kg mol−1. The terpene monomer MO can also be combined with both limonene oxide (LO) and CO2 in a formal terpolymerization process providing, depending on the monomer feed ratio, different types of biohybrid polycarbonates (PLMC) with different degrees of functionality. These terpolymerizations could be extended to the use of an acyclic terpene oxide and either MO/CO2 or LO/CO2, and a previously reported xylose-derived bicyclic oxetane. A selection of MO/LO based biohybrid PLMCs were conveniently depolymerized under TBD catalysis to regenerate the original mixture (>95%) of terpene oxides thereby providing a suitable starting point for the circular use of these biohybrid macromolecules.
Biobased polymers are receiving increasing attention from the chemical community as a way to answer to a growing need for materials with an improved sustainability footprint and recyclability while replacing fossil fuel derived analogues.1–5 Commercial polycarbonate (PC) produced from bis-phenol A (BPA) is a thermoplastic polymer whose mechanical, thermal and optical properties have enabled a wide range of applications in for instance the automotive and electronics industries.6,7 The safe disposal and reuse of commercial PC is, however, complicated by toxicity concerns of BPA metabolites.8 Furthermore, the commercial PC production process makes use of diphenylcarbonate (DPC) as a carbonylating agent.9 DPC is very toxic for aquatic life while its conversion releases corrosive and toxic phenol as a byproduct. The development of ring-opening copolymerization (ROCOP) of epoxides and CO2 is intrinsically a much safer alternative to prepare polycarbonates and at present represents the most versatile approach to produce polycarbonates.10–12
Over the years a plethora of epoxide monomers have been shown to effectively participate in catalytic ROCOP providing different kinds of (functionalized) homo and hetero polycarbonates with control over the microscopic and macroscopic properties.13–20 Building on these seminal achievements, a more recent trend has illustrated that bioderived epoxy/oxetane monomers are also suitable precursors to develop new types of polycarbonate macromolecules with a controlled proportion of biocontent.21–27 Apart from the trend to devise polycarbonates based on bio-epoxy monomers, there has been interesting progress in the reuse of polycarbonates through catalytic depolymerization, allowing to recycle the polycarbonates into polymerizable monomers and thus getting a step closer to circular materials.28–30
We have been interested in the use of terpene oxides (with a main focus on limonene oxide, a cyclic and rigid precursor) as monomers for both polycarbonate and polyesters produced via catalytic ROP.31,32 As far as we are aware, the Greiner group reported the only example of the ROCOP of sterically demanding 2-menthene oxide (MO) and CO2 to provide poly(menthene carbonate), PMC.33 Menthene has a rigid and structurally related backbone compared to limonene, and inspired by this work and the proven potential of Al(aminotriphenolate) complexes in coupling reactions involving sterically challenging epoxy compounds/monomers, we set out to prepare a series of high Tg biohybrid polycarbonates derived from different combinations of terpene oxide monomers. Such a process should facilitate the control over the ratio between more or less rigid monomers and functionalized ones, thereby creating opportunities to further tailor various macromolecular properties through catalytic control with the known poly(limonene) and poly(menthene) carbonates functioning as biopolycarbonate reference materials.
Herein, biohybrid macromolecules are presented that incorporate different combinations of cyclic and acyclic terpene oxides, and a bicyclic sugar-based oxetane.27,34 A pre-selected biohybrid polycarbonate containing both menthene and limonene fragments was further decorated with long chain alkyl thiols thereby reducing its glass transition as a way to post-synthetically adjust its processability. In addition, various biohybrid polycarbonates were selectively depolymerized (>95%) to mixtures of epoxy monomers, paving the way for their repolymerization into the pristine biohybrid structures.
Results and discussion
Given the previous success31,32,35 we attained with the copolymerization of limonene oxide (LO) and CO2 by binary catalysts comprising Al(III) aminotriphenolate complexes in combination with halide-derived initiators affording poly(limonene carbonate), PLC, we started our research by identifying suitable ROCOP conditions for the coupling between limonene oxide (LO), 2-menthene oxide (MO) and CO2 (Table 1) promoted by the Al(III) complex AlMe and PPNCl (Table 1; PPN = bis(triphenyl)iminium).|
|
||||||
|---|---|---|---|---|---|---|
| Entry |
LO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MO :MO |
Solv. | T/tb | Conv.c (%) |
m:nd |
M n/Đe |
|
a Reaction conditions: MO:LO (6.5 mmol) ratio as indicated, AlMe (1 mol%) and PPNCl (0.5 mol%), toluene (0.5 M), 15 bar CO2, 45 °C, 72 h.
b
T denotes the reaction temperature in °C and t is the reaction time in h.
c Values correspond to the overall epoxide conversion as determined by 1H NMR (CDCl3).
d Determined by 1H NMR (CDCl3) using the signal integrals of the methine hydrogens of the LO and MO based repeat units.
e
M
n values are given in kg mol−1 and Đ = Mw/Mn, all values determined by GPC in THF calibrated with polystyrene standards.
f Carried out at 30 bar CO2 pressure.
g Using 0.5 mol% AlMe and 0.25 mol% PPNCl. For the majority of the experiments done, the theoretical mass of the polycarbonates using 1 mol% of AlMe is approximately 19.7 kg mol−1 with MO and LO having nearly the same molar mass (154.25 vs. 152.24 g mol−1). The carbonate selectivity for all polymers was >95% as evidenced by 1H NMR.
|
||||||
| 1 | 1:0 |
Tol | 45, 72 | 75 | 100:0 |
6.84, 1.26 |
| 2 | 0:1 |
Tol | 45, 72 | 51 | 0:100 |
5.21, 1.19 |
| 3 | 1:1 |
Tol | 45, 72 | 75 | 64:36 |
4.89, 1.25 |
| 4 | 1:1 |
Tol | 60, 72 | 53 | 56:44 |
4.15, 1.12 |
| 5f | 1:1 |
Tol | 45, 72 | 38 | 69:31 |
7.12, 1.21 |
| 6 | 1:1 |
Tol | 45, 48 | 71 | 67:33 |
4.74, 1.25 |
| 7 | 1:1 |
Tol | 45, 24 | 57 | 58:42 |
4.59, 1.15 |
| 8g | 1:1 |
Tol | 45, 72 | 32 | 78:22 |
3.76, 1.23 |
| 9 | 3:1 |
Tol | 45, 72 | 67 | 79:21 |
7.09, 1.28 |
| 10 | 1:3 |
Tol | 45, 72 | 68 | 37:63 |
6.25, 1.18 |
| 11 | 1:0 |
— | 45, 72 | 56 | 100:0 |
6.65, 1.37 |
| 12 | 0:1 |
— | 45, 72 | 60 | 0:100 |
7.00, 1.22 |
| 13 |
1:1 |
— | 45, 72 | 75 |
65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :35 :35
|
10.2, 1.29 |
| 14 | 3:1 |
— | 45, 72 | 39 | 85:15 |
3.36, 1.19 |
| 15 | 1:3 |
— | 45, 72 | 43 | 57:43 |
7.92, 1.21 |
| 16 | 1:1 |
— | 55, 72 | 73 | 64:36 |
7.92, 1.25 |
| 17f | 1:1 |
— | 45, 72 | 71 | 64:36 |
8.87, 1.25 |
| 18g | 1:1 |
— | 45, 72 | 39 | 80:20 |
4.01, 1.24 |
| 19 | 1:1 |
— | 45, 48 | 67 | 64:36 |
4.06, 1.32 |
| 20 | 1:1 |
— | 45, 24 | 42 | 68:32 |
2.27, 1.31 |

The copolymerization of both LO and MO with CO2 and promoted by AlMe/PPNCl proceeds similarly (entries 1 and 2), though the overall conversion of MO (versusLO) was substantially lower (51 versus 75%). This resulted in a slightly lower molecular weight PMC (Mn = 5.21 kg mol−1) compared to PLC derived from LO and CO2 (Mn = 6.84 kg mol−1). From these data it can be inferred that MO seems to be less reactive than LO.
The next step was to combine both monomers at an equimolar ratio (1:1) and evaluate this formal terpolymerization process (entry 3) in solution phase. A total epoxide (LO + MO) conversion of 75% was observed, and the isolated terpolymer (abbreviated as PLMC) had an Mn of 4.89 kg mol−1 with a LO:MO incorporation ratio of 64:36. The higher incorporation of LO can be expected on the basis of the relative reactivity displayed in entries 1 and 2. We then set out to further improve the formation of this hybrid biopolycarbonate PLMC (entries 5–10). Increasing the CO2 pressure in this terpolymerization reaction (cf., entries 3 and 5) improved the molecular weight of PLMC (Mn = 7.12 kg mol−1), which is likely the result of an improved relative propagation-to-chain transfer rate. However, in this latter case also a lower epoxide conversion is noted and the CO2-enriched reaction medium possibly causes some degree of phase separation that can affect the efficient mixing of the reaction components, and thus the overall epoxide conversion. Time-dependent terpolymerization (entries 3, 6 and 7) showed that after 48 h there is little improvement in total epoxide conversion in line with the eventual viscous nature of the reaction mixture blocking further propagation of the macromolecule.
A lower amount of catalyst and initiator (entry 8) led to lower molecular weight PLMC likely due the fact that under these conditions the propagation rate would be lower due to a higher dilution of both catalyst components leading to lower epoxide conversion.36 Finally, experiments were conducted with either a three-fold excess of LO (entry 9; 3:1) or MO (entry 10; 1:3), which resulted in terpolymers with a higher (Mn = 7.09 kg mol−1, m:n = 79:21) or lower amount (Mn = 6.25 kg mol−1, m:n = 37:63) of incorporated LO. This is a useful observation as it demonstrates that the monomer feed ratio can be used to dictate the amount of incorporated functionalized monomer (i.e., LO; C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond) while retaining an essentially fully biosourced origin.
C bond) while retaining an essentially fully biosourced origin.
Encouraged by the solution phase results for the terpolymerization of LO, MO and CO2, next we examined a solvent-free approach (Table 1, entries 11–20). Similar trends in terms of Mn and LO-to-MO incorporation ratios for the produced PLMC were seen and with similar order of magnitude conversion levels. Under the best conditions, this biohybrid polycarbonate was isolated with an improved Mn of 10.2 kg mol−1 (entry 13; Đ = 1.29; see Fig. 1 for an 1H NMR comparison), and the more concentrated nature of the monomers is apparently productive towards higher molecular weight polycarbonate. In this latter case, compared to solution-phase results, a comparable epoxide conversion (entries 13 vs. 3; 75%) was noted together with a similar LO:MO incorporation ratio (entry 13 vs. 3, m:n = 65:35 vs. 64:36). Performing the neat terpolymerization process at 30 bar CO2 pressure was also successful (entry 17) though the PLMC product had a slightly lower Mn of 8.87 kg mol−1. In this latter case, much higher monomer conversion (71%) was comparatively noted (cf., entries 3 and 5) thus compensating for the dilution caused by the CO2-expanded liquid phase. The ability to perform the polymerization without solvent adds value to the practical nature of the process and its sustainability in terms of atom-efficiency.
 | ||
| Fig. 1 1H NMR spectral comparison between PLC, PMC and the PLMC from entry 13 in Table 1. A selected zoom of the region where the methine and olefinic H are resonating is shown. For more detailed peak assignments, see the ESI, Fig. S53.† | ||
In order to assess the potential recycling of PLMC, we carried out depolymerization studies (Table 2) using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as an organocatalyst under various conditions (see the ESI for further details, Fig. S1–S3†).28,29 First, toluene was probed as a reaction medium and under reflux, the presence of 4 mol% TBD (entry 1) resulted in a modest conversion (27%) of preselected P1 (see for details entry 13, Table 1) after 24 h. Under these conditions, the “epoxide” selectivity, i.e. the relative amount of formed LO and MO, was 44% while the remainder of the converted polycarbonate were the respective cyclic carbonates LC (limonene carbonate) and MC (2-menthene carbonate). By reducing the reaction time to 15 h but increasing the amount of TBD (8–16 mol%; entries 2–5), a much higher epoxide selectivity and quantitative conversion of P1 could be achieved. We finally identified optimized conditions, reported in entry 6, by using acetonitrile instead of toluene as solvent in the presence of 11 mol% TBD, providing a total epoxide selectivity of >98% at full conversion of P1 (>98%). Furthermore, the LO:MO ratio in the crude reaction mixture (59:41) was only slightly lower than originally determined in P1 (66:34), thus reasonably in line with its terpene constitution. To further evaluate the depolymerization, two other samples (P2: entry 14, and P3: entry 15, see Table 1) were also treated under similar reaction conditions (results in entries 7 and 8, Table 2). From the obtained data it can be concluded that quantitative polymer conversion takes place producing epoxide mixtures close to those originally determined for P2 and P3 by 1H NMR spectroscopy (see ESI for details, Fig. S1–S3†). The high epoxide selectivity for the depolymerization of P1–P3 holds great promise to recycle these monomers into PLMC using AlMe/PPNCl, providing a catalysis-enabled circular process.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | P | t, TBD |
m:nb |
Conv.c (%) | Sel.d (%) |
m:nc |
| a Reaction conditions: polycarbonate (100 mg), toluene (0.5 mL), 15 h, 111 °C. b Values for the starting polymer as determined by 1H NMR (CDCl3). c Determined by 1H NMR (CDCl3) of the depolymerized polymer sample. d Total epoxide selectivity, the remaining products were the cyclic carbonates. e The solvent was CH3CN. f The solvent was CH3CN (0.5 mL), at reflux (82 °C). P stands for the polymer sample used, t is the reaction time in hours and TBD is the organocatalyst with the amount indicated in mol%. LC = cyclic limonene carbonate, MC = cyclic 2-menthene carbonate. | ||||||
| 1 | P1 | 24, 4 | 65:35 |
27 | 44 | 61:39 |
| 2 | P1 | 15, 8 | 65:35 |
87 | 79 | 87:13 |
| 3 | P1 | 15, 12 | 65:35 |
93 | 70 | 83:17 |
| 4 | P1 | 15, 16 | 65:35 |
>98 | 98 | 66:34 |
| 5e | P1 | 15, 12 | 65:35 |
>98 | 93 | 66:34 |
| 6f | P1 | 15, 11 | 65:35 |
>98 | >98 | 59:41 |
| 7f | P2 | 15, 11 | 85:15 |
>98 | >98 | 83:17 |
| 8f | P3 | 15, 11 | 57:43 |
>98 | >98 | 55:45 |

The biohybrid PLMCs have typically high glass transitions (Tg's) comparable to parent PLC (Table 3) due to the presence of the more bulky menthene fragments, and some signs of semi-crystalline behavior was noted for these hybrid PCs (see the ESI, see section 5 and Fig. S7–S21†). We wondered whether we could use the CC double bonds present in the LO-based repeat units to manipulate their processability features. Thereto, we used a thiol–ene approach to functionalize the olefinic fragments present in both PLC and PLMC (entry 13, Table 1 and ESI, Fig. S12, S15 and S21†). Radical initiated functionalization by a C12-derived thiol proved to both efficient and easy allowing to convert >95% of the CC bonds (supported by 1H NMR analysis and a substantial increase in Mn as measured by GPC) in both samples to their respective poly-thioether derivatives. While both PLC and PLMC are white solids, the appearance of their functionalized counterparts (PLCs and PLMCs) was quite different being highly viscous and oily in nature. This indicated that the functionalization process significantly altered their initial thermal behavior. Indeed, upon analysis by DSC, clear melting and crystallization temperatures (Tm and Tc) were observed. For both PLCs (Tm = −25 °C, with Tc = −39 °C) and PLMCs (Tm = +13 °C, with a Tc of −47 °C, see Fig. 2 and the ESI, Fig. S12 and S21†) these temperatures are significantly below ambient conditions, and clearly show that the introduction of the long tail alkyl groups increases the molecular ordering in these macromolecules most likely being the result of substantial intertwining. TGA analyses showed that the functionalization process resulted in materials with higher decomposition temperatures (T10d) at least 30 °C higher than the non-functionalized polymers.
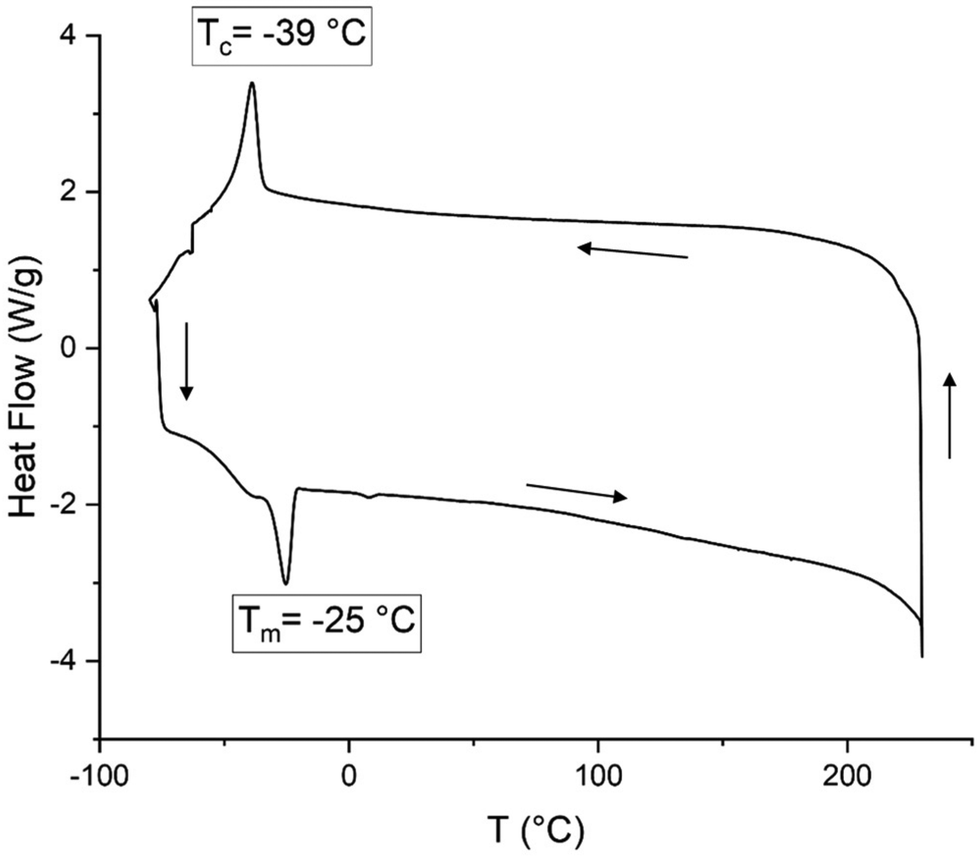 | ||
| Fig. 2 DSC analysis of PLCs, note that the data refer to the second heating/cooling. | ||
C bonds in PLC and a hybrid polycarbonatea
|
|
||||
|---|---|---|---|---|
| Data ↓ | PLC | PLCS | PLMC | PLMCS |
|
a Reaction conditions: thiol-to-double bond ratio was 2:1, 70 °C, 18 h.
b
M
n and Đ values were determined by GPC in THF calibrated with polystyrene standards, a previously prepared sample of PLC was utilized.
c As determined by 1H NMR spectroscopy (CDCl3) using the signal integrals of the remaining CC hydrogens versus the methine–H; data refer to the CC bonds present in the entire polymer product.
d Carbonate absorbance.
e Taken from ref. 31.
f Data collected by differential scanning calorimetry (DSC), the data refer to the second heating.
g The value represents a Tm (melting point).
h Measured by thermogravimetric analysis (TGA) using the Td value at 5% weight loss.
i Oil here refers to a transparent, highly viscous material.
|
||||
| M n (kg mol−1)b | 7.8 | 13.2 | 10.2 | 16.2 |
| Đ | 1.38 | 1.32 | 1.28 | 1.21 |
| % CCc |
>99 | <5 | 65 | <4 |
| IR (cm−1)d | 1742e | 1744 | 1744 | 1746 |
| T g (°C)f | 104 | −25g | 109g | 13g |
| T 10d (°C)h | 217 | 265 | 237 | 269 |
| Appearancei | Solid | Oil | Solid | Oil |

We then sought to extend the types of monomers that can be used to create bio-hybrid polycarbonates (Table 4). The originally used monomers MO and LO were combined with either an acyclic terpene oxide derived from O-protected citronellol (abbreviated as CO) or a sugar-derived oxetane (OX).26,27,34 First the copolymerization of CO with CO2 was attempted but the low conversion of CO (entry 1, 20%; P4, Mn = 1.34 kg mol−1) after three days was a testament to the more difficult nature of the coupling between acyclic trisubstituted terpene oxides and CO2.37 In the presence of LO or MO, terpolymers P5 (entry 2; Mn = 4.62 kg mol−1) and P6 (entry 3; Mn = 2.26 kg mol−1) were formed, respectively, with low CO-incorporation levels of around 10%. This reconfirms the sluggish nature of the monomer CO. We then shifted our focus to bicyclic xylose-based OX (entries 4–8). At 70 °C, the copolymerization of OX with CO2 under AlMe/PPNCl catalysis affords only oligomeric P7 (entry 4; Mn = 1.51 kg mol−1) at a low OX conversion (10%). Raising the reaction temperature to 100 °C (entry 5) allowed to increase both the OX conversion (to 85%) and the molecular weight of the oligomer P7 formed (Mn = 4.28 kg mol−1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Monomers | Conv.b (%) | P |
m:nc |
M n/Đd |
|
a Reaction conditions: monomer ratio 1:1 (where applicable), neat, 65 °C, 15 bar CO2, 72 h.
b Values correspond to the overall epoxide conversion as determined by 1H NMR spectroscopy (CDCl3).
c Determined by 1H NMR (CDCl3) using the signal integrals of the methine hydrogens of the LO and MO based repeat units.
d
M
n values are given in kg mol−1 and Đ = Mw/Mn, all values determined by GPC in THF calibrated with polystyrene standards. Note that crude samples from entries 1, 3, 4 and 6–8 were measured, whereas the data related to entries 2 and 5 are from the isolated oligo/polymers.
e Reaction was performed at 50 °C.
f Reaction was performed at 70 °C for 48 h using 2.9 mmol of OX in toluene as a medium.
g Reaction was performed at 100° and 20 bar CO2 pressure for 72 h using 2.9 mmol of OX in toluene (0.25 M) as a medium.
h Conversion refers to OX, the conversion of MO was <5%.
|
|||||
| 1 | CO | 25 | P4 | — | 1.34, 1.00 |
| 2e | LO + CO | 41 | P5 | 9:1 |
4.62, 1.24 |
| 3e | MO + CO | 36 | P6 | 9:1 |
2.26, 1.10 |
| 4f | OX | 10 | P7 | — | 1.51, 1.41 |
| 5g | OX | 85 | P7 | — | 4.28, 1.09 |
| 6e | LO + OX | 14 | P8 | >99:1 |
1.81, 1.10 |
| 7 | MO + OX | 20 | P9 | >99:1 |
2.12, 1.13 |
| 8g | MO + OX | 96h | P9 | <1:99 |
1.62, 1.07 |
Then, monomers LO and OX were combined under the optimized conditions of Table 1 (entry 13; with T = 50 °C)38 showing only a very modest monomer conversion of 14% (Table 4, entry 6). The oligomeric carbonate that had formed (P8) only had the LO monomer incorporated representing thus a co-instead of the targeted terpolymer (m:n > 99:1). Under these conditions the OX monomer remained unaffected and could not be activated.
Next, we examined a ternary combination of MO, OX and CO2 (entries 7 and 8) to give access to P9. At 65 °C and 15 bar CO2 pressure (entry 7), the presence of binary system AlMe/PPNCl again only afforded an oligomeric carbonate P9 at low MO conversion (20%) with no observable incorporation of OX. When increasing the reaction temperature to 100 °C, effective activation of OX takes place (96% conversion as measured by 1H NMR with low Mn for the oligomer P9 formed; Mn = 1.62 kg mol−1) though with virtually no incorporation of MO. The combined data collected for samples P4–P9 in Table 4 demonstrate that matching the relative reactivities of somewhat related monomers is not an easy task as multiple parameters such as sterics, electronics and stability need to be simultaneously controlled.
Conclusions
In summary, we here describe a catalytic process that allows to combine different monomers derived from terpenes (LO and MO) thereby creating access to nearly fully biobased polycarbonates with different degrees of double bond densities. The thermal properties can be significantly altered by introducing different amounts of long-chain alkyl groups via classic radical-mediated thiol–ene chemistry providing brush-type highly viscous, oily materials that show an increased macromolecular ordering expressed by their respective melting temperatures. Selected biohybrid PLMC samples were subjected to catalytic and controlled degradation allowing to selectively generate the initial monomers (LO and MO), which in principle should allow for their repolymerization to either PLC, PMC or PLMC. Furthermore, the depolymerization-based NMR data help to confirm the initially assessed terpene monomer incorporation ratios. Preliminary studies with other types of monomers (acyclic CO and xylose-derived OX) illustrate the need for combining biobased monomers with similar reactivities to be able to create a wider range of polycarbonates with modular thermal, mechanical and structural (cf., functionality) properties. Future work focuses on a wider use of terpene-based monomers to forge new types of biobased polymers with control over their macromolecular architectures and functionality.Experimental section
Typical procedure for the terpolymerization of MO, LO and CO2
Inside a glove box, a 50 mL autoclave was charged with complex AlMe (33.0 mg, 65.7 μmol, 0.01 equiv.) and PPNCl (18.3 mg, 32.8 μmol, 0.005 equiv.) dissolved in limonene oxide (LO, 3.88 mmol, 1 equiv.) and 2-menthene oxide (MO, 3.28 mmol, 1 equiv.). The reactor was then closed and pressurized to 15 bar of CO2. The mixture was allowed to stir at 45 °C for 72 h. Hereafter the reactor was first cooled to r.t. and carefully depressurized. An aliquot was collected to determine the epoxide conversion by 1H NMR (CDCl3) analysis. The mixture was dissolved into a minimum amount of dichloromethane and then poured into acidified methanol (1 M) under stirring causing precipitation of the polymer product. The precipitate was washed twice with cold methanol and dried for 24 h under vacuum at 50 °C. All terpolymerization reactions were carried out in similar fashion, and the collected analytical data for selected PLMC products P1–P3 (31–57% isolated yield, white solids) can be found in the ESI.†Typical procedure for the depolymerization of the terpolymers
In a typical procedure, in a glovebox a Schlenk tube equipped with a stirring bar was charged with a preselected PLMC (P1–P3; 0.10 g, 0.654 mmol carbonate repeat units) and TBD (16.5 mg, 0.118 mmol, 0.18 equiv.). The reaction mixture was dissolved in acetonitrile (0.5 M) and kept at gentle reflux (82 °C) for 15 h. The reaction was monitored by 1H NMR spectroscopy (CDCl3), taking small aliquots from the reactive mixture. At the end of the reaction, the crude product was analyzed by 1H NMR to determine the percentage of each component (primarily LO and MO) and their relative ratio. An example of how this was done can be found in the ESI.†Author contributions
TS, EL and JT carried out all syntheses regarding the monomer and carbonate polymers. AWK and AB assisted with continuous feedback during the project and scientific input. TS, EL and AWK designed the project, while AWK and AB wrote the manuscript with feedback from all authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Cerca program/Generalitat de Catalonia, ICREA, MICINN (PID2020-112684GB-100, PID2023-149295NB-I00 and Severo Ochoa Excellence Accreditation 2020–2023 CEX2019-000925-S) and AGAUR (2021-SGR-00853) for financial support. TS is thankful to European Union's Horizon 2020 research and innovation program for a Marie Skłodowska-Curie grant (101110356), and EL acknowledges financial support from the European Union (Marie Skłodowska-Curie Grant Agreement No. 101073223, D-Carbonize project). Research funding from the Royal Society (URF\R\221027: fellowship to AB and studentship to JT) is also acknowledged.References
- J.-G. Rosenboom, R. Langer and G. Traverso, Bioplastics for a circular economy, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- C. Veith, F. Diot-Néant and S. A. Miller, Synthesis and polymerization of bio-based acrylates: a review, Polym. Chem., 2020, 11, 7452–7470 RSC.
- T. O. Machado, C. J. Stubbs, V. Chiaradia, M. A. Alraddadi, A. Brandolese, J. C. Worch and A. P. Dove, A renewably sourced, circular photopolymer resin for additive manufacturing, Nature, 2024, 629, 1069–1074 CrossRef CAS PubMed.
- C. J. Stubbs, J. C. Worch, H. Prydderch, Z. Wang, R. T. Mathers, A. V. Dobrynin, M. L. Becker and A. P. Dove, Sugar-Based Polymers with Stereochemistry-Dependent Degradability and Mechanical Properties, J. Am. Chem. Soc., 2022, 144, 1243–1250 CrossRef CAS PubMed.
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y.-X. Chen, Bio-Based Polymers with Performance-Advantaged Properties, Nat. Rev. Mater., 2022, 7, 83–103 CrossRef CAS.
- M. Karhankova, A. Mizera, M. Adamek, V. Mach, P. Stoklasek and M. Gracla, Mechanical resistance of safety elements in transportation, Transp. Res. Proc., 2023, 74, 732–739 Search PubMed.
- A. L. De la Colina Martínez, G. Martínez Barrera, C. E. Barrera Díaz, L. I. Ávila Córdoba, F. Ureña Núñez and D. J. Delgado Hernández, Recycled polycarbonate from electronic waste and its use in concrete: Effect of irradiation, Constr. Build. Mater., 2019, 201, 778–785 CrossRef.
- Y. Ma, H. Liu, J. Wu, L. Yuan, Y. Wang, X. Du, R. Wang, M. Marwa, P. Petlulu, X. Chen and H. Zhang, The adverse health effects of bisphenol A and related toxicity mechanisms, Environ. Res., 2019, 176, 108575 CrossRef CAS PubMed.
- For details on the use of DPC consult: https://cdn.intratec.us/docs/reports/previews/pc-e21a-b.pdf.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Sainia and C. K. Williams, Ring-opening copolymerization (ROCOP): synthesis and properties of polyesters and polycarbonates, Chem. Commun., 2015, 51, 6459–6479 RSC.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, Multifunctional Catalysts for Ring-Opening Copolymerizations, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- X. Xie, Z. Huo, E. Jang and R. Tong, Recent advances in enantioselective ring-opening polymerization and copolymerization, Commun. Chem., 2023, 6, 202 CrossRef CAS PubMed.
- G.-P. Wu, P.-X. Xu, X.-B. Lu, Y.-P. Zu, S.-H. Wei, W.-M. Ren and D. J. Darensbourg, Crystalline CO2 Copolymer from Epichlorohydrin via Co(III)-Complex-Mediated Stereospecific Polymerization, Macromolecules, 2013, 46, 2128–2133 CrossRef CAS.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, B. Li, Y.-P. Zu and D. J. Darensbourg, Alternating Copolymerization of CO2 and Styrene Oxide with Co(III)-Based Catalyst Systems: Differences Between Styrene Oxide and Propylene Oxide in Reactivity, Polymer Selectivity, and Regioselective Ring-Opening, Energy Environ. Sci., 2011, 4, 5084–5092 RSC.
- D. J. Darensbourg and S. J. Wilson, The Synthesis of Poly(indene carbonate) from Indene Oxide and Carbon Dioxide—A Polycarbonate with a Rigid Backbone, J. Am. Chem. Soc., 2011, 133, 18610–18613 CrossRef CAS PubMed.
- W. C. Ellis, Y. Jung, M. Mulzer, R. Di Girolamo, E. B. Lobkovsky and G. W. Coates, Copolymerization of CO2 and meso Epoxides Using Enantioselective β-Diiminate Catalysts: A Route to Highly Isotactic Polycarbonates, Chem. Sci., 2014, 5, 4004–4011 RSC.
- J. G. Kim and G. W. Coates, Synthesis and Polymerization of Norbornenyl-Terminated Multiblock Poly(cyclohexene carbonate)s: A Consecutive Ring-Opening Polymerization Route to Multisegmented Graft Polycarbonates, Macromolecules, 2012, 45, 7878–7883 CrossRef CAS.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Renewable Polycarbonates and Polyesters from 1,4-Cyclohexadiene, Green Chem., 2015, 17, 300–306 RSC.
- T. M. McGuire, A. C. Deacy, A. Buchard and C. K. Williams, Solid-State Chemical Recycling of Polycarbonates to Epoxides and Carbon Dioxide Using a Heterodinuclear Mg(II)Co(II) Catalyst, J. Am. Chem. Soc., 2022, 144, 18444–18449 CrossRef CAS PubMed.
- C. Martín and A. W. Kleij, Terpolymers derived from Limonene Oxide and Carbon Dioxide: Access to Cross-Linked Polycarbonates with Improved Thermal Properties, Macromolecules, 2016, 49, 6285–6295 CrossRef.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Bio-Based Polycarbonate from Limonene Oxide and CO2 with High Molecular Weight, Excellent Thermal Resistance, Hardness and Transparency, Green Chem., 2016, 18, 760–770 RSC.
- V. Bonamigo Moreira, J. Rintjema, F. Bravo, A. W. Kleij, L. Franco, J. Puiggalí, C. Alemán and E. Armelin, Novel Biobased Epoxy Thermosets and Coatings from Poly(Limonene Carbonate) Oxide and Synthetic Hardeners, ACS Sustainable Chem. Eng., 2022, 10, 2708–2719 CrossRef CAS PubMed.
- O. Hauenstein, S. Agarwal and A. Greiner, Bio-Based Polycarbonate as Synthetic Toolbox, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. E. Koning, Bio-derived Polymers for Coating Applications: Comparing Poly(Limonene Carbonate) and Poly(Cyclohexadiene Carbonate), Polym. Chem., 2017, 8, 6099–6105 RSC.
- S. Reiter, A. Vagin, C. Kronast, B. Jandl and B. Rieger, Lewis Acid β-Diiminato-Zinc-Complex as All-rounder for Co- and Terpolymerisation of various Epoxides with Carbon Dioxide, Chem. Sci., 2017, 8, 1876–1882 RSC.
- T. M. McGuire and A. Buchard, Polymers from Sugars and CS2: Ring Opening Copolymerisation of a D-xylose Anhydrosugar Oxetane, Polym. Chem., 2021, 12, 4253 RSC.
- D. K. Tran, A. Z. Rashad, D. J. Darensbourg and K. L. Wooley, Sustainable Synthesis of CO2-derived Polycarbonates from D-xylose, Polym. Chem., 2021, 12, 5271 RSC.
- C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, Unique Base-Initiated Depolymerization of Limonene-Derived Polycarbonates, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS PubMed.
- D. H. Lamparelli, A. Villar-Yanez, L. Dittrich, J. Rintjema, F. Bravo, C. Bo and A. W. Kleij, Bicyclic Guanidine Promoted Mechanistically Divergent Depolymerization and Recycling of a Biobased Polycarbonate, Angew. Chem., Int. Ed., 2023, 62, e202314659 CrossRef CAS PubMed.
- F. N. Singer, A. C. Deacy, T. M. McGuire, C. K. Williams and A. Buchard, Chemical Recycling of Poly(Cyclohexene Carbonate) Using a Di-MgII Catalyst, Angew. Chem., Int. Ed., 2022, 61, e202201785 CrossRef CAS PubMed.
- L. Peña Carrodeguas, J. González-Fabra, F. Castro-Gómez, C. Bo and A. W. Kleij, AlIII-Catalysed Formation of Poly(limonene)-carbonate: DFT Analysis of the Origin of Stereoregularity, Chem. – Eur. J., 2015, 21, 6115–6122 CrossRef PubMed.
- N. Kindermann, À. Cristòfol and A. W. Kleij, Access to Biorenewable Polycarbonates with Unusual Glass-Transition Temperature (Tg) Modulation, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS.
- A. Wambach, S. Agarwal and A. Greiner, Synthesis of Biobased Polycarbonate by Copolymerization of Menth-2-ene Oxide and CO2 with Exceptional Thermal Stability, ACS Sustainable Chem. Eng., 2020, 8, 14690–11469 CrossRef CAS.
- T. M. McGuire, J. Bowles, E. Deane, E. H. E. Farrar, M. N. Grayson and A. Buchard, Control of Crystallinity and Stereocomplexation of Synthetic Carbohydrate Colymers from d- and l-xylose, Angew. Chem., Int. Ed., 2021, 60, 4524–4528 CrossRef CAS PubMed.
- A. Brandolese and A. W. Kleij, Catalyst Engineering Empowers the Creation of Biomass-Derived Polyesters and Polycarbonates, Acc. Chem. Res., 2022, 55, 1634–1645 CrossRef CAS PubMed.
- The observation of lower molecular weight polycarbonates with similar types of binary catalysts using biobased epoxide monomer has been observed in other cases, see: A. Brandolese, D. H. Lamparelli, I. Grimaldi, S. Impemba, P. Baglioni and A. W. Kleij, Access to Functionalized Polycarbonates derived from Fatty Acid Esters via Catalytic ROCOP and their Potential in Gel Formulations, Macromolecules, 2024, 57, 3816–3823 CrossRef CAS , See also ref. 31 and 32.
- G. Fiorani, M. Stuck, C. Martín, M. Martínez-Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Catalytic Coupling of Carbon Dioxide with Terpene Scaffolds: Access to Challenging Bio-Based Organic Carbonates, ChemSusChem, 2016, 9, 1304–1311 CrossRef CAS PubMed.
- Note that typically the LO/CO2 copolymerization using AlMe/PPNCl can be carried out at around 45 °C justifying the temperature conditions for entry 7 in Table 4, see ref. 31 and 32.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data for all monomers and polycarbonates, and copies of relevant spectra and chromatograms. See DOI: https://doi.org/10.1039/d5py00285k |
| This journal is © The Royal Society of Chemistry 2025 |