
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
5-Ethylidene-2-norbornene (ENB) and 5-vinyl-2-norbornene (VNB) based alicyclic polyols for the synthesis of polyesters†
Brigita
Bratić
a,
Peter
Altenbuchner
b,
Thomas
Heuser
b and
Bernhard
Rieger
 *a
*a
aTechnical University of Munich, WACKER Chair of Macromolecular Chemistry, Department of Chemistry, D-85748 Garching bei München, Germany. E-mail: rieger@tum.de; Tel: +49 (0)89 289 54448
bEvonik Operations GmbH, Paul-Baumann-Straße 1, D-45772 Marl, Germany. E-mail: peter.altenbuchner@evonik.com; thomas.heuser@evonik.com
First published on 21st May 2025
Abstract
Amorphous polyesters derived from rigid alicyclic monomers with high glass transition temperatures are of great interest as potential substitutes for poly(carbonate)s. Therefore, 5-ethylidene-2-norbornene (ENB) and 5-vinyl-2-norbornene (VNB) were identified as interesting diol monomer precursors to enhance the thermal properties of polyesters. The regioselective synthesis of the corresponding diols from ENB and VNB was optimized and the first scale-up experiments gave promising results for a potential large-scale production of these polyester monomers. Moreover, for proof of concept, an alternative procedure was established to synthesize exclusively branched diols. Polymerization experiments were conducted with dimethyl terephthalate (DMT) as a model compound to compare the polyesters with commonly used poly(ethylene terephthalate)s. Therefore, a series of polyesters containing branched or linear regioisomers or a mixture of both regioisomers were produced, and structure–property relationships were investigated using GPC, TGA and DSC analyses. The Tg values ranged from 75 to 103 °C, depending on the branched and linear moieties in the polyester microstructure and their molecular weights. Moreover, isosorbide (IS) was introduced as a biobased comonomer, resulting in amorphous copolyesters with promising Tgs ranging from 81 to 97 °C.
Introduction
Polymers, with their versatile properties, have become indispensable in today's world since they fulfil a wide range of needs across industries, driving innovation and sustainability efforts. Their adaptability ensures that they will remain at the forefront of technological advancements for years to come.1,2 The development of new polymer materials is therefore of utmost importance due to the increasing requirements regarding performance and safety.1–4 One example of increasing safety standards is the impending worldwide ban of bisphenol-A-based polycarbonates (BPA-PC) due to their endocrine disrupting effects and toxicity.5–8 A potential candidate for the replacement of such widely used BPA-based polymers could be amorphous polyesters, ensuring that they exhibit comparable properties such as high heat resistance and clarity to fulfil market requirements. However, in this regard, most amorphous polyesters are often limited due to their low glass transition temperature (Tg), which is an important thermomechanical property for their practical application in industries such as food packaging materials and coil coatings.4,9,10 Amorphous polyesters with high Tgs are thermally stable, enabling them to endure higher temperatures without deformation or structure loss. This stability is required in food packaging, where products may undergo heating during processing or storage, as high Tg ensures the shape and barrier properties of packaging materials.10,11Moreover, high Tg polyesters exhibit superior barrier properties against gases, moisture and scents, crucial for preserving product freshness and quality.12 Additionally, their enhanced mechanical properties ensure better protection for packaged products during handling, transportation and storage.3 In applications such as coil coating, where the coating must withstand mechanical stresses and deformation, a high Tg guarantees the maintenance of structural integrity.13
An option to improve the thermal properties of polyesters is the incorporation of rigid cyclic structures into the polyester backbone, which has already been demonstrated in the literature. For example, tricyclodecanedimethanol (TCD-DM) was studied by the group of Fenouillot resulting in amorphous polyester materials with Tgs ranging from 116 to 163 °C.14 Among TCD-DM, isosorbide (IS) was studied extensively in the synthesis and modification of polyester materials with varying isosorbide content.10,14–24 Further interesting examples are 2,2,4,4-tetramethyl-1,3-cyclobutanediol (TMCD),12,25–27 which is known for the production of high-performance Tritan™ (Eastman), and spiroglycol (SPG),1 which is used for the production of Akestra™ (Perstorp AB). The substitution of terephthalic acid (TPA) or dimethyl terephthalate (DMT) has been a topic of research in food packaging as well, leading to many examples of dimethyl ester 2,5-furanedicarboxylate (DMFD)-based polyesters with remarkable thermal and barrier material properties.10,11,28–30 All these polymers exhibit high Tgs and can even be produced from biobased sources.
Nature provides a rich diversity of diene structures, which are potential precursors for the synthesis of diols, yet their availability for large-scale polymer production is often constrained by factors such as reactivity issues, extraction and high cost.31 With our search for new polyesters in mind, we identified 5-ethylidene-2-norbornene (ENB, 1) and 5-vinyl-2-norbornene (VNB, 2) as interesting monomer precursors, a readily available feedstock from the ethylene-propylene-diene monomer (EPDM) industry, which are produced on a multi-million metric ton per year scale.32 Their bicyclic core not only provides rigidity, but the corresponding diols (4) are also accessible via a hydroformylation–reduction sequence (Scheme 1). Our research aims to develop an efficient process that offers a new pathway to produce cycloaliphatic diols in a cost-effective and scalable manner. Moreover, this development could enable the production of additional downstream products, such as amines, carboxylic acids, and methyl methacrylates. Furthermore, the nature of the exocyclic double bonds in ENB (1) and VNB (2) allows control of the regioselectivity (linear vs. branched diols). Thus, the polyester backbone can be modified, and therefore, the properties of the polymer can be fine-tuned to its respective requirement. In the current study, we investigated the influence of the isomer composition of the aforementioned diols on the thermal properties of polyesters.
 | ||
| Scheme 1 Catalytic hydroformylation of ENB (1) or VNB (2) to a mixture of dialdehyde regioisomers (3) followed by hydrogenation to the corresponding diols (4). | ||
In this context, we conducted an optimization of the hydroformylation reaction and developed modified synthesis strategies for diols 4 (Scheme 1). Our goal was to scale up the production process of compounds 3 and 4 to a multigram scale, aiming to produce amorphous polyesters with promising thermal properties. These polyesters show potential applications in food packaging and coil coating.
Results and discussion
Optimizing monomer synthesis for industrial-scale production
While the synthesis of diols from ENB (1) and VNB (2) has been previously described in a patent, the reported conditions required high temperatures and pressures, which are not ideal for large-scale production.33 Furthermore, the available synthetic procedures and analytical data were limited. Given that diols 4 are excellent candidates for polyester monomers, the reaction sequence was re-evaluated to establish milder conditions that enhance scalability and reduce production cost.ENB (1) and VNB (2) are commercially available as a mixture of cis/trans and/or endo/exo diastereomers, and the reactivity of their endo- and exocyclic double bonds may influence the regioselectivity of their di-hydroformylation. Due to the inherent ring strain of the bicyclic structures, hydroformylation typically proceeds via an initial reaction at the endocyclic double bond (I), followed by the less favoured hydroformylation of the exocyclic double bond (II) (Scheme 2). Optimizing the reaction conditions, including pressure, temperature, reaction time, and phosphorus ligand selection, was therefore necessary to minimize undesired byproducts. The hydroformylation conditions were screened using bis(1,5-cyclooctadiene)rhodium(I) tetrafluoroborate [Rh(COD2)]BF4 as the precatalyst, in conjunction with the regio-directing phosphorus ligands Alkanox® and Oxophos®, which allow milder reaction conditions.31,34,35 Additionally, (Sax,S,S)-bobphos36,37 was tested, and a full screening of the reaction conditions did not improve conversion or regioselectivity. Consequently, due to these limitations and the scope of this study, this approach was not pursued further. For analysis, a combination of nuclear magnetic resonance (NMR) spectroscopy and gas chromatography mass spectrometry (GC MS) was employed due to the formation of multiple isomers, including diastereomers and regioisomers.
 | ||
| Scheme 2 Possible reaction sequences in di-hydroformylation of ENB (1) and VNB (2) using Alkanox® or Oxophos® as ligands. | ||
 | ||
| Fig. 1 Stacked 1H NMR spectra from hydroformylation reactions of ENB (1) at different temperatures after three hours using Alkanox® as the ligand; reaction conditions: 0.10 mol% [Rh(COD)2]BF4, 0.35 mol% Alkanox®, CO/H2 = 1/1, 50 bar, 40–140 °C, 3 h, (PhMe, 1 M). (A): characteristic resonances of aldehyde groups. (B): characteristic resonances of endocyclic double bonds. (C): characteristic resonances of cis/trans exocyclic double bonds. | ||
The information gathered from the proton NMR spectra gave important insights into the reaction sequence of ENB (1) hydroformylation. However, signal overlap impeded further analysis and understanding of the reaction. Consequently, the experiments were also examined with GC MS and additional information could be collected (Fig. 2, left). Hydroformylation of ENB (1) at 40 and 60 °C resulted in aldehydes 1′ with exclusive mono-hydroformylation (m/z = 150). Increasing the temperature to 80 and 100 °C led to the desired dialdehydes 3 (m/z = 180), but with aldehydes 1′ being not yet fully converted. Full conversion of aldehydes 1′ was achieved at 120 and 140 °C, but partial reduction of the desired product was observed as a side reaction (m/z = 182). Thus, for initial scale-up, the reaction was performed overnight at 100 °C to achieve full conversion of 1′ without the formation of undesired by-products.
 | ||
| Fig. 2 Stacked GC chromatograms from hydroformylation reactions of ENB (1) and VNB (2) at different temperatures after three hours; reaction conditions: (left) 24.96 mmol ENB (1), 0.10 mol% [Rh(COD)2]BF4, 0.31 mol% Alkanox®, CO/H2 = 1/1, 50 bar, 40–140 °C, (PhMe, 1 M). (middle) 24.96 mmol VNB (2), 0.10 mol% [Rh(COD)2]BF4, 0.11 mol% Oxophos®, CO/H2 = 1/1, 50 bar, 40–140 °C, (PhMe, 1 M). (right) 24.96 mmol VNB (2), 0.10 mol% [Rh(COD)2]BF4, 0.11 mol% Oxophos®, CO/H2 = 3/2, 50 bar, 40–140 °C, (PhMe, 1 M). Byproducts: aldehydes 1′ (m/z = 150) result from ENB (1) hydroformylation; aldehydes 2′ (m/z = 150) and 2′′ (m/z = 152) result from VNB (2) hydroformylation; hydrogenated compounds 5a (m/z = 122) and 5b (m/z = 124) emerge as byproducts from VNB (2) hydroformylation with a syngas ratio of CO/H2 = 1/1. | ||
Despite the use of Alkanox® to control regioselectivity in ENB (1) hydroformylation, selectivity challenges remained. To address this, Oxophos® was used in the hydroformylation of VNB (2) to preferentially generate linear dialdehydes 3c.31 This approach leveraged VNB's (2) less sterically hindered exocyclic double bond, in combination with the regio-directing effects of Oxophos®, facilitating the selective formation of linear dialdehydes 3c. Examination of the reaction between 40 and 140 °C using a syngas ratio of CO/H2 = 1/1 resulted in a mixture of products (Fig. 2, middle). Across this temperature range, byproducts 5a (m/z = 122) and 5b (m/z = 124) were detected along with aldehydes 2′ (m/z = 150) and 2′′ (m/z = 152). Compared to the reaction of ENB (1) with Alkanox® (Fig. 2, left) the number of signals for dialdehydes 3 decreased and could be assigned to linear dialdehydes 3c by 1H and 13C NMR spectroscopy. In an attempt to prevent reduction as a side reaction, the syngas ratio of CO/H2 was adjusted from 1/1 to 3/2 (Fig. 2, right).35,38 At 40 °C, aldehydes 2′ and byproduct 2′′ were observed along with the desired linear dialdehydes 3c, but the formation of 2′ and 2′′ was significantly decreased at higher temperatures (60–100 °C). At 120 and 140 °C, the signal intensities of 2′ and 2′′ increased again, presumably due to the decomposition of the ligand Oxophos®.39 Therefore, the best result was obtained at 100 °C and CO/H2 = 3/2, and these conditions were used for the initial scale-up.
To explore iso-selective hydroformylation of branched dialdehydes 3a + 3b, the chiral ligand (Sax,S,S)-bobphos was tested with ENB (1). Reactions were conducted at 40–80 °C (CO/H2 = 1/1) for 24 h and analysed by GC MS. At 40 °C, mainly aldehydes 1′ were formed. At 60 °C, a mixture of branched dialdehydes 3a + 3b and aldehydes 1′ was detected. At 80 °C, selectivity decreased due to the formation of a mixture of 3 and undesired reduction products. Further screening of catalyst and ligand loading at 60 °C did not improve conversion or regioselectivity, and consequently, this approach was not pursued further.
The hydroformylation reactions were optimized for scalability by adjusting the temperature and pressure in Rh-catalysed reactions of ENB (1) and VNB (2). Initial scale-up was performed in a 2 L batch reactor with a batch size of 416 mmol. Purification via vacuum distillation yielded colourless liquids with 76% yield for dialdehydes 3 and 40% yield for linear dialdehydes 3c. Despite high conversions, yield losses were observed during vacuum distillation, likely due to viscosity increases in the residue. Gradual temperature increases were applied to mitigate yield loss, but aldehyde side reactions (e.g. dehydrogenation or decomposition) may occur due to the low decomposition temperature of dialdehydes 3 (Td = 107 °C, see the ESI†). Moreover, stability studies of dialdehydes 3 revealed acetalization as a potential side reaction, which was identified using FT-IR and ESI-MS (see ESI Fig. S2 and Table S1†).
Initial hydrogenation attempts of dialdehydes 3 were conducted following strategy (a) on a 24.96 mmol scale, using 2 wt% RANEY® nickel and 10 wt% H2O. The addition of water could significantly improve the yield; thus, water was incorporated into the crude reaction mixture of 3 post-hydroformylation.40 The reactions were evaluated across a temperature range of 25 to 140 °C at 50 bar H2, and the desired products were subsequently isolated via vacuum distillation (Table 1). Reaction progress was meticulously monitored using GC MS (see ESI Fig. S1†). Alongside the formation of diols 4 (m/z = 184, C3; see ESI Fig. S1†), byproducts such as partially reduced compounds (m/z = 182; C2, Fig. S1†) and unreacted dialdehydes 3 were also detected (m/z = 180, C1; see ESI Fig. S1†). Additionally, light-boiling byproducts from the hydroformylation step remained in the reaction mixture (A and B; see ESI Fig. S1†). The desired products 4 were successfully isolated after full conversion of 3 with yields ranging from 45% to 67%, based on ENB (1), using vacuum distillation. For comparison, the patent procedure using dicyclopentadiene (DCPD) achieved a 71% yield of TCD-DM.40 Prolonged reaction times exceeding 14 hours were noted at temperatures below 140 °C. However, at 140 °C, hydrogenation was completed within five hours. Trace amounts of the Rh-catalyst could interfere with conversion rates and hinder product isolation during vacuum distillation. Consequently, strategy (b) was employed for the reduction of purified dialdehydes 3, leading to improved yields. The initial scale-up was conducted at a 171 mmol scale at 140 °C under optimized conditions using 4 wt% RANEY® nickel and 20 wt% H2O, yielding 82% diols 4 (Scheme 3).
 | ||
| Scheme 3 Optimized hydrogenation strategy for the scale-up synthesis of diols 4 and linear diols 4c. | ||
| # | T [°C] | H2 usage [bar] | Time [h] | Yield [%] | Full conversion of 3 |
|---|---|---|---|---|---|
| 1 | r.t. | 5.0 | o.n. | n.d | No |
| 2 | 40 | 6.0 | o.n. | 45 | Yes |
| 3 | 60 | 7.5 | o.n. | n.d. | No |
| 4 | 80 | 9.0 | o.n. | 66 | Yes |
| 5 | 100 | 10 | o.n. | 66 | Yes |
| 6 | 120 | 9.0 | 14 | n.d. | No |
| 7 | 140 | 10 | 4.5 | 67 | Yes |
Simultaneously, the hydrogenation of linear dialdehydes 3c was conducted on the crude product mixture at reaction temperatures of 80 °C and 100 °C. The resulting yields ranged from 16% to 31%, which are considerably lower compared to the hydrogenation of dialdehydes 3. In both cases, the catalyst concentration was maintained at 2 wt% RANEY® nickel with 10 wt% H2O. To achieve complete conversion of linear dialdehydes 3c while preventing undesired side reactions, temperatures of at least 80 °C were deemed sufficient. Based on the previously optimized hydrogenation conditions of 3 using 4 wt% RANEY® nickel and 20 wt% H2O, the hydrogenation process was successfully implemented on a 155 mmol scale. Hydrogenation of purified 3c at 90 °C resulted in a 60% yield of linear diols 4c (Scheme 3).
Proof-of-concept synthesis of branched diols 4b
The targeted synthesis of linear diols 4c suggests the possibility of selectively synthesizing branched dialdehydes. This approach may aid in screening structural dependencies on the thermal properties of polyesters. Building on the observed differences in the nature of the endo- and exocyclic double bonds of ENB (1) and VNB (2) under hydroformylation conditions, an alternative route towards branched diols 4b was investigated (Scheme 4).41 Therefore, the branched moiety in 3b was designed in two steps via the Wittig reaction, followed by hydrolysis. The substrate 5-acetyl-2-norbornene (AcNB, 6) was converted with in situ generated phosphorus ylide (methoxymethylene)triphenyl phosphane to cis/trans enolether 7 in 92% yield. The hydrolysis was performed with 20 mol% p-toluenesulfonic acid monohydrate, affording the corresponding branched aldehyde 8 in 84% yield. These two transformations laid the foundation for the branched moiety in 4b. Hence, the endocyclic double bond in 8 was implemented with the Rh-catalysed hydroformylation using Alkanox® as the ligand. The crude product 3b was initially reduced with NaBH4 and MeOH to the corresponding branched diols 4b. After purification with silica gel column chromatography, compound 4b was isolated in 64% yield. The alternative route towards branched diols 4b might not provide industrial scalability, but it allowed access to branched isomers. Hence, the three diols 4, 4b, and 4c possess structural differences and could show an intriguing influence on the thermal properties in polyester synthesis.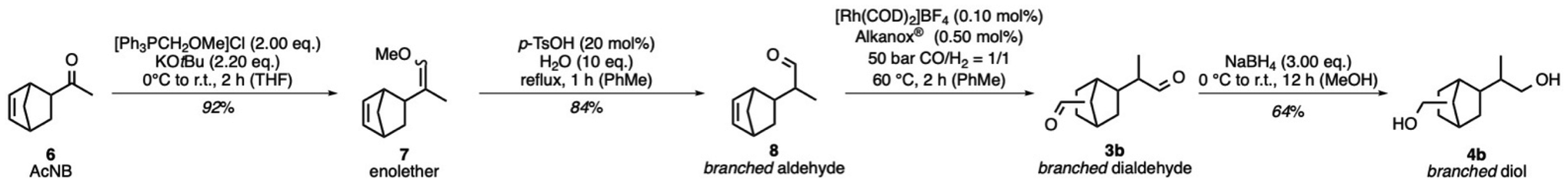 | ||
| Scheme 4 Synthesis of branched dialdehydes 3bvia Rh-catalysed hydroformylation with Alkanox® as the ligand, followed by reduction to the corresponding branched diols 4b. | ||
Synthesis of amorphous polyesters and thermal structure–property relationships
To investigate the impact of branched and linear moieties on the polyester microstructure, diols 4, 4b, and 4c were synthesized and incorporated into polyesters via a two-step melt polycondensation reaction using dimethyl terephthalate (DMT) as a model compound. Titanium tetra-n-butoxide [Ti(OnBu)4, 0.20 mol%] was employed as a catalyst (Scheme 5) to facilitate polymerization and enable a comparative study of the structural influence on material properties. DMT was selected for its well-characterized thermal properties, allowing direct comparison with commercially relevant poly(ethylene terephthalate)s. Additionally, this method accommodates bio-based diacids of diesters such as dimethyl furan dicarboxylate (DMFD). | ||
| Scheme 5 General polycondensation procedure for polyester synthesis (R1/R2: alkyl). | ||
While purified terephthalic acid (TPA) is the predominant monomer in PET production, its use in laboratory-scale synthesis poses challenges due to the presence of residual carboxylic acid groups, which can lead to side reactions, hinder chain growth, and potentially worsen alcoholysis. These issues are especially pronounced with thermally sensitive diols, such as 1,4-butanediol, which may undergo dehydration to tetrahydrofuran (THF) under esterification conditions.
In the transesterification step, a diol-to-diester mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) was heated to 160 °C under ambient pressure, initiating methanol evolution. The temperature was incrementally increased by 10 °C to 220 °C until methanol evolution ceased (∼5 to 6 hours), forming oligomeric precursor esters (step 1, Scheme 5). Subsequently, under reduced pressure (0.05 mbar), the oligomeric mixture was heated to 240 °C for 2–3 hours to remove volatilized diols via distillation (step 2, Scheme 5). The resulting amber-coloured polymer melt was dissolved in dichloromethane, precipitated, washed with cold methanol, and dried overnight at 70 °C under vacuum, yielding colourless to pale yellow polyesters (P4T, P4bT, and P4cT).
:1 molar ratio) was heated to 160 °C under ambient pressure, initiating methanol evolution. The temperature was incrementally increased by 10 °C to 220 °C until methanol evolution ceased (∼5 to 6 hours), forming oligomeric precursor esters (step 1, Scheme 5). Subsequently, under reduced pressure (0.05 mbar), the oligomeric mixture was heated to 240 °C for 2–3 hours to remove volatilized diols via distillation (step 2, Scheme 5). The resulting amber-coloured polymer melt was dissolved in dichloromethane, precipitated, washed with cold methanol, and dried overnight at 70 °C under vacuum, yielding colourless to pale yellow polyesters (P4T, P4bT, and P4cT).
Polymerization with diols 4 produced medium-to-high-molecular weight polymers (Mn,rel = 7.89–19.2 kg mol−1) with moderate to broad dispersities (Đ = 1.65–4.74) (P4T1–4; see ESI Table S2†). While reaction conditions such as temperature, catalyst loading, and time were kept constant, the observed differences in molecular weight and dispersity across the P4T1–4 series are attributed to variations in the reaction scale. Initial reactions were performed on a smaller scale to assess monomer reactivity, followed by larger-scale polymerizations to obtain sufficient material for characterization. These scale differences likely influenced mixing efficiency and substrate dispersion, affecting polymer chain growth. Consistently, diols 4b generated similar molecular weight distributions (Mn,rel = 9.39–23.8 kg mol−1) but with narrower dispersities (Đ = 2.10–2.70) (P4bT1–4; see ESI Table S2†). However, the highest molecular weights in the P4T and P4bT series were obtained with Mn,rel = 19.2 kg mol−1 and Mn,rel = 23.8 kg mol−1. The polymer containing linear diols 4c exhibited a high molecular weight (Mn,rel = 12.8 kg mol−1) with a broad dispersity (Đ = 4.48) (Table 2).
:1) at 220–240 °C yielding the respective polyesters P4T, P4bT and P4cT
| Polyester |
M
n,rela [kg mol−1] |
M
w,rela [kg mol−1] |
Đ [—] |
T
db [°C] |
T
gc [°C] |
|---|---|---|---|---|---|
| a Relative molecular weight and dispersity measured using GPC in THF at 30 °C (25 mmol L−1) relative to polystyrene. b Onset decomposition temperatures (Td) of the first decomposition step determined via TGA measurements. c T g determined via DSC measurements. | |||||
| P4T | 19.2 | 60.8 | 3.17 | 364 | 100 |
| P4bT | 23.8 | 63.3 | 2.70 | 379 | 103 |
| P4cT | 12.8 | 57.3 | 4.48 | 371 | 75 |
Achieving high-molecular-weight polymers requires precise end-group stoichiometry and efficient condensate removal to drive the reaction equilibrium toward polymer formation. The observed constraints in molecular weights and broad dispersities are characteristic of polycondensation due to inherent challenges, including:
• The necessity for exact stoichiometric balance between diester and diol, as minor deviations significantly reduce achievable molecular weights.
• Potential alcohol elimination during heating, leading to unreactive chain ends. Gas chromatography (GC) analysis detected volatile alcoholysis byproducts from excess diols removed during polymerization.
For industrial scalability, solid-state polymerization (SSP) can enhance molecular weight; however, its applicability is typically limited to semicrystalline polymers and may therefore be restricted for the predominantly amorphous polyesters described in this work.42–44
Understanding the thermal behaviour of these polyesters is essential for evaluating their potential high-temperature applications. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to assess decomposition temperatures (Td) and glass transition temperatures (Tg), revealing strong correlations with polymer chain lengths and structural differences.
TGA revealed Td values between 361 and 381 °C for the P4T series, while P4bT and P4cT demonstrated slightly higher Td values up to 387 °C (see ESI Table S2†). While this trend reflects general increases in molecular weight, it also highlights the influence of chain uniformity. Notably, the P4bT series displayed narrower molecular weight distributions and more consistent TGA profiles than P4T, suggesting that structural uniformity contributes to thermal stability. The broader variability observed in P4T may stem from the mixed regioisomer content of diol 4, which could lead to microstructural deviations or sequence blockiness, depending on differences in reactivity and volatility among the isomers. These factors warrant further investigations, particularly with purified diol isomers to assess their impact on polymer architecture and thermal behaviour.
DSC analysis further highlighted the relationship between molecular weight and Tg. The Tg of P4T increased progressively with molecular weight, reaching 100 °C at Mn,rel = 19.2 kg mol−1. The incorporation of branched moieties into P4bT resulted in higher Tg values reaching 103 °C at Mn,rel = 23.8 kg mol−1, whereas the incorporation of linear diols 4c into P4cT notably reduced Tg to 75 °C at Mn,rel = 12.8 kg mol−1. The lower Tg of P4cT is consistent with its lower molecular weight and the presence of flexible linear alkyl side chains, both of which contribute to increased segmental mobility. Although the linear structure could facilitate tighter chain packaging, DSC analysis did not reveal any evidence of crystallinity (see ESI Fig. S28†). Compared to previously reported biobased polyesters, none were found to combine Mn above 20.0 kg mol−1 and Tg above 100 °C, highlighting the favourable thermal properties of P4bT.12,20,28,45–51 Although direct comparison with commercial polyesters such as Tritan™ and PETG is premature due to differences in molecular weight and processing history, the reported Tg values (up to 103 °C) and thermal stability (Td up to 387 °C) of the synthesized polyesters indicate promising potential for future development toward high-performance applications.
Incorporation of isosorbide into copolyesters: synthesis, characterization and thermal properties
The incorporation of isosorbide (IS, 9; Fig. 3) as a renewable diol into polyesters has been widely studied due to its ability to significantly increase Tg while enhancing optical properties, including improved transmittance and reduced haze.14 Additionally, dimethyl furan dicarboxylate (DMFD, 10; Fig. 3) is emerging as a promising alternative to DMT in polyester production. Literature reports indicate that polyesters synthesized with DMFD exhibit thermal and barrier properties comparable to petroleum-based poly(ethylene terephthalate)s (PET), making them attractive for sustainable polymer development.11,45,52,53 | ||
| Fig. 3 Depicted diol and diester monomers for the synthesis of copolyesters P4coIST, P4ccoIST and P4coISF. | ||
In this study, a series of copolyesters – P4coIST, P4ccoIST, and P4coISF – were synthesized using a well-established two-step melt polycondensation process. These copolyesters incorporated diols 4, linear diols 4c, and isosorbide (IS), with DMT (T) and DMFD (F) serving as the corresponding diesters. The reaction was conducted using a diol-to-diacid molar ratio of 2:1. The chemical structures of the resulting copolyesters were characterized using 1H NMR and DOSY NMR spectroscopy, while their thermal properties were evaluated using TGA and DSC. These properties were then compared to those of reference polyesters P4T and P4cT. Although diol 4b demonstrated superior molecular control over molecular weight and dispersity, along with high Tg values, it was not included in the copolymerization due to the increased synthetic effort required for its preparation. The more scalable synthesis of diols 4 and 4c made them more suitable for evaluating industrially relevant systems.
The incorporation of IS into copolyesters P4coIST, P4ccoIST, and P4coISF presented specific challenges. Among these, P4coIST exhibited the lowest molecular weight with a narrow dispersity (Table 3). The 1H NMR analysis confirmed the presence of characteristic resonances for DMT and ester groups (–CH2O–) of diols 4 and 4c, similar to those observed in polyesters P4T and P4cT. The aliphatic proton resonances of IS were identified between δ1H = 5.51 and 4.00 ppm, with chain end groups appearing at δ1H = 5.00 and 3.60 ppm. Aliphatic protons of DMFD were detected at δ1H = 4.20 ppm and δ1H = 1.10 ppm, respectively. By comparing the intensity peaks of aromatic protons in DMT and DMFD to those of the ester groups (–CH2O–) of diols 4 and 4c, the final molar ratios of IS in the copolyesters were determined (Table 3). DOSY NMR spectroscopy further confirmed successful copolymerization, as the diffusion coefficients of proton signals from DMT and DMFD were identical to those of the respective diols (see ESI Fig. S14, S16 and S18†).
:1) at 220–240 °C yielding the respective copolyesters P4coIST, P4ccoIST and P4coISF
| Copolyester | Mole fraction of IS in the copolyestera [mol%] |
M
n,relb [kg mol−1] |
M
w,relb [kg mol−1] |
Đ [—] |
T
dc [°C] |
T
gd [°C] |
|---|---|---|---|---|---|---|
| a Mole fraction of IS in the copolyester, determined by integration in 1H NMR spectra. b Relative molecular weight and dispersity measured using GPC in THF at 30 °C (25 mmol L−1) relative to polystyrene. c Onset decomposition temperatures (Td) of the first decomposition step determined via TGA measurements. d T g determined via DSC measurements. | ||||||
| P4coIST | 5.00 | 7.4 | 12.9 | 1.76 | 373 | 97 |
| P4ccoIST | 1.50 | 17.3 | 38.5 | 2.07 | 363 | 81 |
| P4coISF | 2.50 | 13.4 | 31.9 | 1.88 | 340 | 89 |
Achieving high transesterification conversion, which facilitates polycondensation, requires efficient removal of alcohol byproducts to drive the reaction equilibrium. However, IS negatively impacted the polycondensation process, reducing the conversion rate and yielding shorter oligomers. Additionally, co-diols with lower boiling points than isosorbide reacted more quickly and evaporated more readily, further complicating the reaction.14,54
Despite these challenges, the transesterification step remained unaffected, with methanol evolution completing within 5–6 hours. However, the polycondensation step was more complex, relying on rapid alcoholysis and efficient elimination of terminal diols under high vacuum (<1 mbar). The boiling points of IS14 and diols 4 overlapped (105–120 °C vs. 120 °C, respectively), complicating selective removal. Linear diols 4c exhibited a slightly higher boiling point (130 °C), which influenced their behaviour in polycondensation. Our findings align with previous studies by Fenouillot et al., who investigated the reactivity of IS. They found that primary bicyclic diols, such as diols 4 and 4c, can induce steric hindrance, whereas the secondary bicyclic structure of IS allows for better accessibility to ester groups.14 Furthermore, the hydrophobic properties of diols 4 and 4c contrast with the hydrophilic properties of IS, making hydrophilic ester bond formation more challenging when diols 4b or 4c are present at both chain ends.14 Fenouillot's study also suggested that IS can act as a promoter, accelerating the polycondensation step.14
Comparing the molecular weights of P4coIST (Mn,rel = 7.37 kg mol−1) and P4ccoIST (Mn,rel = 17.3 kg mol−1), it was evident that copolymerization of IS with linear diols 4c resulted in higher molecular weights. This suggests that IS acted more effectively as a “chain linker” than linear diols 4c, leading to a lower IS content (1.50 mol%) in P4ccoIST. In contrast, the copolymerization of IS with diols 4 proved more complex, likely due to a mixture of sterically hindered isomers (branched vs. linear). The overlapping boiling points further complicated the removal process, leading to a higher IS content in P4coIST (5.00 mol%) compared to P4ccoIST (1.50 mol%). These observations suggest that future studies should explore separating regioisomers 4a, 4b, and 4c as model compounds to better understand reactivity differences between hydrophobic diols and hydrophilic IS.
Despite precipitation and methanol washing, P4coISF (2.50 mol% IS content) exhibited a pale-yellow colour, a common issue associated with high polymerization temperatures.11,45,53 Although DMFD was used to mitigate decarboxylation, prolonged reaction times (>16 hours) and high melt viscosity hindered heat and mass transfer. However, replacing DMT with DMFD provides significant advantages, including improved crystallization behaviour, ensuring amorphous morphologies suitable for industrial processing. Additionally, poly(ethylene 2,5-furanedicarboxylate) polyesters exhibit significantly lower gas permeabilities (O2, CO2, and H2O) due to reduced chain mobility, making them promising candidates for food packaging applications.11
Thermal analysis using TGA and DSC revealed distinct structure–property relationships in copolyesters incorporating IS (Table 3). P4coIST and P4ccoIST demonstrated high thermal stability, with Tds of 373 °C and 363 °C, comparable to Td = 363–379 °C (see ESI Table S2†) observed in reference polyesters P4T and P4cT. In contrast, P4coISF exhibited a lower Td of 340 °C. Despite this, all copolyesters displayed excellent thermal stability, making them suitable for high-temperature applications such as injection molding. DSC analysis highlighted the influence of molecular weight on Tg. The Tg of P4coIST (97 °C) was significantly higher than that of P4T1 (87 °C; see ESI Table S2†) despite similar molecular weights, aligning with previous studies that 6 mol% IS can increase Tg by 10 °C.18 Conversely, the incorporation of 2.50 mol% IS into P4coISF did not significantly affect Tg, which remained at 89 °C, compared to 92 °C for the polyester P4T2 (see ESI Table S2†). In contrast, in the copolyester P4ccoIST, 1.50 mol% IS content increased Tg to 81 °C, compared to 75 °C in P4cT. This suggests that even a low IS content can have a noticeable influence on the thermal properties, particularly in systems incorporating linear diols. Additionally, the variation in Tg between different copolyesters highlights the importance of precise monomer selection and polymerization conditions to achieve desired thermal characteristics.
Further investigation into the molecular dynamics of these copolyesters could provide deeper insights into the structural effects of IS incorporation. Future studies could explore the interplay between chain rigidity, segmental mobility, and hydrogen bonding interactions to fully understand the role of rigid cyclic diols in polyester performance. Moreover, optimizing reaction conditions to further increase molecular weight while maintaining favourable thermal properties will be crucial for advancing these materials in industrial applications such as sustainable packaging and coatings.
Conclusions
This study highlights the potential of utilizing alicyclic-diene-based polyols as key components for synthesizing amorphous, high Tg polyesters. The successful synthesis and scale-up of dialdehydes 3 and 3c were achieved through optimized hydroformylation reactions using two regio-directing ligands. The subsequent hydrogenation process to produce diols 4 and linear diols 4c was effectively carried out using RANEY® nickel, a widely used industrial heterogeneous catalyst. Despite these advancements, further improvements in purification methods are necessary to maximize the yields of reactive dialdehydes.Additionally, an alternative pathway for synthesizing branched diols 4b was established, allowing for an investigation into the influence of different regio- and diastereomers on polyester properties. The synthesized diols 4, 4b and 4c provided a platform for developing amorphous polyesters with medium-to-high molecular weights. While complete molecular weight optimization was not achieved, the resulting polyesters exhibit promising thermal properties, with Tg values ≥100 °C.
Although the synthesis of monomers 4 and 4c still relies on petroleum-based raw materials, this work advances the development of partially biobased polyesters by incorporating sustainable building blocks such as IS and DMFD. Notably, the incorporation of 5.00 mol% IS into the P4coIST-type copolyester significantly increased the glass transition temperature (Tg = 97 °C), despite its low molecular weight (Mn,rel = 7.37 kg mol−1). Similarly, in the P4ccoIST-type copolyester, IS incorporation increased Tg from 75 °C to 81 °C. The replacement of DMT with DMFD presents additional advantages in terms of mechanical and barrier properties. Although the P4coISF copolyester exhibited promising thermal properties, achieving higher molecular weight (Mn > 20 kg mol−1) will be essential for optimizing mechanical performance.
Cycloaliphatic structures offer significant advantages not only in polyesters but also in poly(amide)s (PA),54 poly(urethane)s (PU)55 or poly(methyl methacrylate)s (PMMA).56 Incorporating cycloaliphatic moieties into these polymers enhances their properties, making them more suitable for demanding applications that require enhanced UV stability, mechanical strength, and thermal resistance. These enhanced properties enable their use in a wide range of applications, including automotive parts, optical components, coatings, adhesives, and consumer goods.
The polymer precursors developed in this study open new avenues for synthesizing novel monomers, including amines, carboxylic acids, and methyl methacrylates. These advancements will facilitate the development of innovative copolymers tailored to meet the stringent requirements of high-performance applications, extending both the usability and lifespan of advanced materials.
Author contributions
Brigita Bratić: conceptualization, synthesis, funding acquisition, writing – original draft, visualization, data curation, and formal analysis; Peter Altenbuchner: writing – review & editing, visualization, data curation, and formal analysis; Thomas Heuser: writing – review & editing, visualization, data curation, and formal analysis; Bernhard Rieger: funding acquisition, supervision, project administration, resources, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank Dr Fabian Martin Hörmann and Stefanie Hörl for the great proofreading process. Additionally, the authors are grateful for the good discussion with the group of the WACKER Chair of Macromolecular Chemistry and Evonik Industries. The authors additionally want to thank Evonik Industries for support and funding.References
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2019, 38, 1–67 Search PubMed.
- R. Mori, RSC Sustainability, 2023, 1, 179–212 RSC.
- M. Zhang, G. M. Biesold, W. Choi, J. Yu, Y. Deng, C. Silvestre and Z. Lin, Mater. Today, 2022, 53, 134–161 CrossRef CAS.
- V. T. Weligama Thuppahige and M. A. Karim, Compr. Rev. Food Sci. Food Saf., 2022, 21, 689–718 Search PubMed.
- C. Lambré, J. M. Barat Baviera, C. Bolognesi, A. Chesson, P. S. Cocconcelli, R. Crebelli, D. M. Gott, K. Grob, E. Lampi, M. Mengelers, A. Mortensen, G. Rivière, V. Silano, I. L. Steffensen, C. Tlustos, L. Vernis, H. Zorn, M. Batke, M. Bignami, E. Corsini, R. Fitzgerald, U. Gundert-Remy, T. Halldorsson, A. Hart, E. Ntzani, E. Scanziani, H. Schroeder, B. Ulbrich, D. Waalkens-Berendsen, D. Woelfle, Z. Al Harraq, K. Baert, M. Carfì, A. F. Castoldi, C. Croera and H. Van Loveren, EFSA J., 2023, 21, e06857 Search PubMed.
- J. S. Siracusa, L. Yin, E. Measel, S. Liang and X. Yu, Reprod. Toxicol., 2018, 79, 96–123 Search PubMed.
- A. Abraham and P. Chakraborty, Rev. Environ. Health, 2019, 2, 201–210 Search PubMed.
- Y. B. Wetherill, B. T. Akingbemi, J. Kanno, J. A. McLachlan, A. Nadal, C. Sonnenschein, C. S. Watson, R. T. Zoeller and S. M. Belcher, Reprod. Toxicol., 2007, 24, 178–198 Search PubMed.
- S. Mangaraj, A. Yadav, L. M. Bal, S. K. Dash and N. K. Mahanti, J. Package Technol. Res., 2019, 3, 77–96 Search PubMed.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J.-P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 Search PubMed.
- D. Zhang and M. J. Dumont, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1478–1492 CrossRef CAS.
- J. Wang, X. Liu, Z. Jia, Y. Liu, L. Sun and J. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3298–3307 CrossRef CAS.
- M. N. S. Kumar, Z. Yaakob and H. Siddaramaiah, in Handbook of Bioplastics and Biocomposites Engineering Applications, ed. S. Pilla, 2011, pp. 121–160 Search PubMed.
- S. Legrand, N. Jacquel, H. Amedro, R. Saint-Loup, M. Colella, J.-P. Pascault, F. Fenouillot and A. Rousseau, ACS Sustainable Chem. Eng., 2020, 8, 15199–15208 Search PubMed.
- J. Thiem and H. Lüders, Polym. Bull., 1984, 11, 365–369 CrossRef CAS.
- R. Storbeck, M. Rehahn and M. Ballauff, Makromol. Chem., 1993, 194, 53–64 CrossRef CAS.
- R. Storbeck and M. Ballauff, J. Appl. Polym. Sci., 1996, 59, 1199–1202 CrossRef CAS.
- H. R. Kricheldorf, G. Behnken and M. Sell, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 679–684 CrossRef CAS.
- R. Storbeck and M. Ballauff, Polymer, 1993, 34, 5003–5006 Search PubMed.
- W. J. Yoon, S. Y. Hwang, J. M. Koo, Y. J. Lee, S. U. Lee and S. S. Im, Macromolecules, 2013, 46, 7219–7231 Search PubMed.
- T. Kim, J. M. Koo, M. H. Ryu, H. Jeon, S.-M. Kim, S.-A. Park, D. X. Oh, J. Park and S. Y. Hwang, Polymer, 2017, 132, 122–132 Search PubMed.
- S. Legrand, N. Jacquel, H. Amedro, R. Saint-Loup, J.-P. Pascault, A. Rousseau and F. Fenouillot, Eur. Polym. J., 2019, 115, 22–29 CrossRef CAS.
- S. R. Turner, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5847–5852 Search PubMed.
- R. Quintana, A. M. De Ilarduya, A. Alla and S. Muñoz-Guerra, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2252–2260 Search PubMed.
- G. W. Baell, C. E. Powell, J. Hancock, M. Kindinger, H. R. McKenzie, A. V. Bray and C. J. Booth, Appl. Clay Sci, 2007, 37, 295–306 Search PubMed.
- M. Zhang, R. B. Moore and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3710–3718 CrossRef CAS.
- J. Wang, S. Mahmud, X. Zhang, J. Zhu, Z. Shen and X. Liu, ACS Sustainable Chem. Eng., 2019, 7, 6401–6411 CrossRef CAS.
- J. Wang, X. Q. Liu, Y. J. Zhang, F. Liu and J. Zhu, Polymer, 2016, 103, 1–8 Search PubMed.
- S. Hong, K.-D. Min, B.-U. Nam and O. O. Park, Green Chem., 2016, 18, 5142–5150 Search PubMed.
- J. Wang, X. Q. Liu, Z. Jia, L. Sun, Y. J. Zhang and J. Zhu, Polymer, 2018, 137, 173–185 Search PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 Search PubMed.
- Y. G. Osokin, Pet. Chem., 2007, 47, 1–11 Search PubMed.
- D. Mijolovic, V. Wendel and A. Suckert, BASF SE, WO Pat., WO2011023540A2, 2011 Search PubMed.
- A. Bara-Estaún, C. L. Lyall, J. P. Lowe, P. G. Pringle, P. C. J. Kamer, R. Franke and U. Hintermair, Faraday Discuss., 2021, 229, 422–442 RSC.
- C. Kubis, R. Ludwig, M. Sawall, K. Neymeyr, A. Börner, K.-D. Wiese, D. Hess, R. Franke and D. Selent, ChemCatChem, 2010, 2, 287–295 CrossRef CAS.
- G. M. Noonan, J. A. Fuentes, C. J. Cobley and M. L. Clarke, Angew. Chem., 2012, 124, 2527–2530 Search PubMed.
- P. Dingwall, J. A. Fuentes, L. E. Crawford, A. M. Z. Slawin, M. Bühl and M. L. Clarke, J. Am. Chem. Soc., 2017, 139, 15921–15932 CrossRef CAS PubMed.
- D. Selent, R. Franke, C. Kubis, A. Spannenberg, W. Baumann, B. Kreidler and A. Börner, Organometallics, 2011, 30, 4509–4514 Search PubMed.
- B. Kreidler, D. Fridag, B. Schemmer, B. Wechsler, A. Christiansen and D. Neumann, Evonik Oxeno GmbH, EP Pat., EP2663573B2, 2011 Search PubMed.
- W. Dukat, E. Storm and K. Schmid, Oxea Deutschland GmbH, US Pat., US7301057B2, 2005 Search PubMed.
- P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670–6696 Search PubMed.
- S. N. Vouyiouka, E. K. Karakatsani and C. D. Papaspyrides, Prog. Polym. Sci., 2005, 30, 10–37 Search PubMed.
- F. C. Chen, R. G. Griskey and G. H. Beyer, AIChE J., 1969, 15, 680–685 Search PubMed.
- K. D. Samant and K. M. Ng, AIChE J., 1999, 45, 1808–1829 Search PubMed.
- R. J. I. Knoop, W. Vogelzang, J. Van Haveren and D. S. Van Es, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4191–4199 CrossRef CAS.
- J. G. Wang, X. Q. Liu, J. Zhu and Y. Jiang, Polymers, 2017, 9, 305 CrossRef PubMed.
- J. G. Van Berkel, N. Guigo, J. J. Kolstad, L. Sipos, B. Wang, M. A. Dam and N. Sbirrazzuoli, Macromol. Mater. Eng., 2015, 300, 466–474 CrossRef CAS.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1026–1036 CrossRef CAS.
- M. Vannini, P. Marchese, A. Celli and C. Lorenzetti, Green Chem., 2015, 17, 4162–4166 RSC.
- G. Z. Papageorgiou, D. G. Papageorgiou, Z. Terzopoulou and D. N. Bikiaris, Eur. Polym. J., 2016, 83, 202–229 CrossRef CAS.
- M. Garaleh, T. Yashiro, H. R. Kricheldorf, P. Simon and S. Chatti, Macromol. Chem. Phys., 2010, 211, 1206–1214 Search PubMed.
- J. C. Morales-Huerta, A. Martínez de Ilarduya and S. Muñoz-Guerra, Polymer, 2016, 87, 148–158 Search PubMed.
- W. H. Carothers, Trans. Faraday Soc., 1936, 32, 39–49 RSC.
- C.-W. Chen, C.-W. Lin, Y.-H. Chen, T.-F. Wei, S.-P. Rwei and R. Sasikumar, Polym. Bull., 2020, 77, 235–253 CrossRef CAS.
- A. Mouren and L. Avérous, Chem. Soc. Rev., 2023, 52, 277–317 Search PubMed.
- S. K. Asha, V. Deepthimol and M. Lekshmi, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5617–5626 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental (monomer synthesis and polymerisation); additional 1H, 13C and DOSY NMR spectra, thermal analysis (TGA and DSC), and GPC data. See DOI: https://doi.org/10.1039/d5py00247h |
| This journal is © The Royal Society of Chemistry 2025 |