
CO2-binding alcohols as potential candidates for poly(vinyl chloride) upcycling†
Juliette
Delcorps
,
Emna
Ben Ayed
and
Olivier
Coulembier
 *
*
Laboratory of Polymeric and Composite Materials, Centre of Innovation and Research in Materials and Polymers, University of Mons, Place du Parc 23, 7000 Mons, Belgium. E-mail: olivier.coulembier@umons.ac.be
First published on 2nd June 2025
Abstract
Despite the increasing global production of poly(vinyl chloride) (PVC), its recycling remains a major challenge, primarily due to its high chlorine content and limited compatibility with conventional recycling processes. This study explores the use of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)-based CO2-binding alcohols (CO2BALs) as nucleophiles for PVC functionalization, aiming to enhance its upcycling potential. The impact of solvent polarity, CO2BAL conversion, and reaction time on the substitution-to-elimination ratio was systematically investigated. Although the degree of substitution remained below 10 wt%, a promising SN2/E2 selectivity of 94/6 was achieved. The functionalized materials were characterized using 1H NMR, FT-IR, SEC, and TGA, confirming the successful grafting of carbonate moieties and highlighting thermal stability trends. While CO2BAL stabilization in polar solvents may limit reactivity, alternative approaches, such as flow chemistry, are currently under consideration to improve substitution efficiency. This work provides new insights into CO2-based strategies for PVC modification, bridging the gap between polymer upcycling and sustainable chemistry.
Introduction
With a global annual production of 45 million metric tons in 2022 and a projected increase to 65 million metric tons by 2030,1 poly(vinyl chloride) (PVC) ranks as the third most produced polymer worldwide, following polyethylene (PE) and polypropylene (PP).2 Its widespread use makes it a key target for the scientific community recycling efforts. Despite its reputation as a challenging material to recycle, Europe has seen significant progress in this area through initiatives such as Vinyl2010 and VinylPlus.3 These programs have driven an impressive increase in PVC recycling, from just 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons in 2004 to 264000 tons in 2010, and further to 731000 tons in 2020. Projections suggest this figure could reach 1 million tons per year by 2030.4–6 However, with an annual production of 6.5 million tons across the EU-27, Norway, Switzerland, and the United Kingdom,2 the current recycling rates leave room for considerable improvement.
000 tons in 2004 to 264000 tons in 2010, and further to 731000 tons in 2020. Projections suggest this figure could reach 1 million tons per year by 2030.4–6 However, with an annual production of 6.5 million tons across the EU-27, Norway, Switzerland, and the United Kingdom,2 the current recycling rates leave room for considerable improvement.
Two main approaches are currently employed for large-scale PVC recycling, i.e. mechanical recycling and feedstock (or chemical) recycling.4–7 Unfortunately, both methods face significant limitations. Mechanical recycling requires thoroughly sorted and homogeneous PVC waste to produce a material with properties comparable to the original polymer.5–7 This stringent requirement complicates waste management, and the process itself often degrades polymer chains, leading to a loss of physical and mechanical properties that restrict the applications of the recycled material.5 Feedstock recycling, on the other hand, is hindered by PVC's high chlorine content. This method typically involves thermal treatments, during which the easily broken C–Cl bond releases chlorinated derivatives (Fig. 1a). If not effectively captured, these emissions can cause equipment corrosion and air pollution. Moreover, dehydrochlorination generates conjugated polyene sequences that may undergo cyclization to form regular and chlorinated aromatic compounds, further increasing the environmental burden of feedstock recycling.7 The recovery of chlorine from PVC and other chlorinated polymers, such as polyvinylidene chloride (PVDC), is thus crucial to their recycling process.8–10
 | ||
| Fig. 1 Chlorine reactivity and selectivity in PVC modifications: (a) common reactivities of C–Cl bonds in PVC, (b) nucleophile-dependent substitution (SN2) and elimination (E2) ratios (I− in DMF at 60 °C, SCN− in DMF at 100 °C, HO− in DMF at 21 °C, N3− in DMF at 100 °C, phthalimide anion in DMF at 60 °C, ethylenediamine at 75 °C in bulk, 1-octanethiol/K2CO3 in DMF at 40 °C), and (c) enhanced selectivity using CO2BALs in our work (R = Decyl). | ||
To overcome the limitations of traditional recycling methods, innovative approaches such as biological and electrochemical recycling,8 as well as chemical upcycling,11 have been explored and warrant further research.5,12 Among these, chemical upcycling through nucleophilic substitution has garnered significant attention.13 However, the efficiency of these substitution reactions is influenced by a variety of factors, including the leaving group properties, the polymer structure, and the reaction environment.14–16 For instance, while chlorine is a modest leaving group that can undergo substitution and elimination reactions under various conditions,17 product selectivity remains a challenge due to the competition between these pathways. Factors such as solvent polarity, nucleophile identity, temperature, and nucleophile concentration play a critical role in directing the reaction.17 Polar aprotic solvents like dimethylformamide (DMF) have been shown to favor substitution over elimination, while higher temperatures tend to promote elimination.18,19 Similarly, less basic nucleophiles increase substitution selectivity,17 offering avenues for tailoring reaction conditions to improve outcomes.
Substitution reactions with nucleophiles offer the potential to convert PVC into new polymeric materials for specialized applications, including catalysis,20,21 separations,22,23 and antimicrobial polymers.24 Nevertheless, such transformations often face practical limitations due to incomplete dechlorination and competition with elimination, leading to residual chlorine atoms and alkenes that can alter polymer properties.25 These challenges underscore the need for innovative strategies to enhance both the efficiency and selectivity of substitution reactions, as demonstrated by recent studies.26
Optimizing these reaction parameters could enable the development of versatile and sustainable recycling methods for PVC. For instance, a variety of nucleophiles, including iodide anion, hydroxide, azide, and thiocyanate, have been explored for PVC substitution reactions (Fig. 1b).19 Thiols have also been employed, with Lu et al. achieving partial substitution of PVC chlorine atoms using potassium carbonate and N,N-diisopropylethylamine.27 Additionally, amination of PVC has enabled its upcycling into carbon dioxide absorbents, as demonstrated by Sneddon et al.28 However, while the use of nucleophiles for PVC modification has been extensively studied, research on employing carbonates as nucleophiles remains notably sparse. This gap is particularly surprising given the potential for carbonates to provide functionalized PVC materials with unique properties. Furthermore, although substitution reactions are often accompanied by elimination processes, the latter is either underreported or dominates in most studies,16 underscoring the need for approaches that favour selective substitution over elimination.
In this work, we aimed to functionalize PVC using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)-based CO2-binding alcohol-derived materials (CO2BALs), previously developed for the synthesis of “simple” dialkyl carbonates (Fig. 1c).29 The effects of temperature, solvent polarity, CO2BAL conversion, and reaction time were systematically studied to evaluate their impact on both the degree of substitution and the substitution-to-elimination ratio. Although substitution levels higher than 10 wt% were not achieved in optimal conditions, this result makes the CO2BAL approach particularly appealing, as the nature of the by-product is a TBD·HCl salt, which is harmless. However, for future investigations, it will be necessary to consider other functionalization strategies, such as flow chemistry.
Results and discussion
Carbon dioxide-binding organic liquids (CO2BOLs) are an innovative class of materials formed by combining alcohols with liquid organic bases, such as amidines or guanidines.30–33 These systems are capable of binding CO2 through reversible interactions, making them highly versatile for applications in CO2 capture and utilization.CO2BOLs form intermediates that can act as nucleophiles in substitution reactions,29,34–36 opening new avenues for polymer transformations and upcycling strategies. Among the organic bases used in CO2BOLs, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) has recently garnered particular attention.37 TBD-based CO2BOLs exhibit a unique ability to promote selective SN2 (bimolecular nucleophilic substitution) reactions with primary and secondary iodo-alkanes.29 This selectivity primarily arises from the formation of a TBD·H+ guanidinium cation during the reaction, which interacts with and stabilizes the iodine ion released as the leaving group in the SN2 process. By minimizing competing reactions such as elimination or base quaternization, this mechanism enables highly efficient and controlled transformations.
Building on these properties, this study extends the application of TBD-based CO2BOLs beyond small-molecule transformations to the upcycling of poly(vinyl chloride) (PVC), a widely used polymer with significant recycling challenges. To better highlight the improvements introduced in this work, several key aspects are compared in Table 1, showcasing the differences between TBD-based CO2BOLs used for small-molecule synthesis (cf. ref. 29) and their application in PVC recycling. As far as the environmental impact is concerned, it was hypothesized that these systems could limit the release of chloride ions during both dechlorination and dehydrochlorination (Fig. 2). By leveraging the chloride-trapping ability of TBD·H+, this approach seeks to reduce the formation of toxic by-products while enhancing substitution efficiency on the PVC backbone. Moreover, this work underscores the dual sustainability of the proposed method: integrating CO2 into the reaction process transforms this abundant greenhouse gas into a valuable resource for polymer modification. This innovative use of CO2 not only aligns with broader goals in carbon capture and utilization but also addresses critical challenges in PVC recycling.
 | ||
| Fig. 2 Trapping of chloride by guanidinium TBD·H+ during (a) dechlorination (SN2) and (b) dehydrochlorination (E2) reactions. | ||
| Criteria | Ref. 29 | This work |
|---|---|---|
| Main objective | Synthesis of dialkyl carbonates via SN2 with TBD-based CO2BOLs | Upcycling of PVC with CO2BOLs for sustainable valorization |
| Main substrates | Primary and secondary alkyl iodides | PVC secondary methine chlorides |
| Final product | Dialkyl carbonates | Carbonate-modified PVC through an almost exclusively SN2 process and a limited HCl-liberating E2 reaction |
| Reaction temperature | 21–65 °C | 21 °C (optimal conditions) |
| Solvents used | Acetonitrile or solvent-free condition | DMF (optimal conditions) |
| Environmental impact | Green alternative to toxic metal-based methods | Plastic waste valorization, reduction of HCl waste by-product by an exclusive TBD·HCl benign salt production |
| Major innovation | Guanidinium-assisted SN2 ion-pair process | Extension to polymers, contribution to PVC recycling by CO2 valorization |
To better reflect the versatility of the systems explored in this study, the term CO2BALs (“CO2-binding alcohols”) is proposed to describe alcohol-based systems that extend beyond the liquid-phase CO2BOLs typically described in the literature. This broader definition emphasizes the adaptability of CO2BALs in enabling sustainable and efficient polymer transformations.
Thermal properties of CO2BALs: melting points and degradation
In developing a method to functionalize PVC using CO2BALs, a solvent-free approach was prioritized to enhance the environmental sustainability of the process and minimize waste generation. Such an approach requires both reactants to be in a compatible physical state to ensure effective interaction. For PVC, this means operating above its Vicat softening temperature (∼92 °C)38 but below its thermal degradation threshold (∼250 °C).39 The CO2BALs, on the other hand, must remain liquid under these conditions, as this enhances their ability to mix with the polymer and facilitates reactivity.To evaluate the feasibility of these conditions, the melting points and thermal stability of CO2BALs were studied. A series of TBD alkylcarbonate salts were prepared by mixing equimolar amounts of alcohol and TBD in bulk, as previously detailed by us.29 Mixtures were then heated to a constant temperature and exposed to CO2 (pCO2 = 0.2 bar) under vigorous stirring (500 rpm). The temperature was maintained at 50 °C for methanol, ethanol, and 1-propanol, while 90 °C was used for 1-butanol, 1-hexanol, 1-octanol, and 1-decanol (Fig. S1, S3, S5, S7, S9, and S11†). The thermal properties of the obtained CO2BALs were evaluated by determining their melting points, which are influenced by the length of the alkyl chain. As shown in Fig. 3, with the exception of the TBD butylcarbonate salt (which remains a viscous liquid with a melting temperature of 13.5 °C), all other CO2BALs are solid at room temperature. The melting points follow a clear trend: they decrease with the length of the alkyl chain down to a minimum for the 1-butanol-based salt, after which they slightly increase for longer chains. These results were compared to those of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)- and 1,1,3,3-tetramethylguanidine (TMG)-based CO2BALs,33 with the latter exhibiting negative melting points across all alkyl chain lengths. Notably, the melting points of the TBD-based CO2BALs are generally higher than those of DBU-based salts, particularly for shorter alkyl chains such as methanol, ethanol, and 1-propanol. This suggests that the TBD salts form more stable crystalline structures compared to DBU and TMG salts. This thermal behavior is significant when considering the conditions required for the upcycling of PVC, as the melting point influences both the stability and reactivity of the CO2BALs during polymer functionalization.
 | ||
| Fig. 3 Melting points of TBD-based CO2BALs as a function of the alkyl chain length, compared with the melting points of DBU- and TMG-based CO2BALs. Thermal data for DBU (in orange) and TMG (in purple) are adapted from ref. 33. | ||
Initially encouraged by the relatively high melting points of CO2BALs derived from methanol, ethanol, and 1-propanol, we faced a critical limitation when examining their thermal stability. Thermogravimetric analyses (TGA) revealed that the degradation onset temperatures (T5%) of these salts are consistently close to their melting points (ca. 80–85 °C, Fig. S2, S4, and S6†), excluding their use in liquid form at temperatures compatible with PVC's Vicat softening point. Furthermore, the TGA data showed that the degradation temperatures of CO2BALs are independent of alkyl chain length, falling within a narrow range of 80–90 °C (Fig. S2, S4, S6, S8, S10, S12 and S14†).
To gain deeper insight into the thermal behavior of CO2BALs, we sought to determine the activation energy (Ea) and pre-exponential factor (A) by applying the Arrhenius equation across a broad temperature range (40–110 °C). This required selecting a CO2BAL with a melting point sufficiently distinct from its T5% to ensure reliable thermal measurements. The 1-decanol-based CO2BAL emerged as a good candidate, with a melting point of 31.5 °C and a degradation onset temperature of 86.4 °C. These properties provided a stable and practical platform for assessing thermal stability and reactivity. It should also be noted that, as previously reported,29 the longer alkyl chain of 1-decanol poses a barrier to achieving quantitative carbonate conversion. The synthesis of the corresponding CO2BAL used here was indeed limited to ∼80% conversion, as determined by 1H NMR analysis (Fig. S13c†). Despite this slight limitation, the thermal properties of this compound made it a good choice for the subsequent experiments.
In the simplest approach, the decarboxylation reaction rate constant for the 1-decanol-based CO2BAL, k, can be calculated according to the eqn (1):
 | (1) |
 | (2) |
 | ||
| Fig. 4 (a) Decarboxylation kinetics of 1-decanol-based CO2BAL at different temperatures and (b) corresponding Arrhenius plot. | ||
The activation energy (Ea) and pre-exponential factor (A) obtained for the 1-decanol-based CO2BAL (62 kJ mol−1 and 2.4 × 107 min−1) fall within the typical range observed for first-order reactions involving complex organic transformations. For instance, Moreno et al. reported a Ea values of 58.7–70.5 kJ mol−1 for cannabinoid decarboxylation,40 illustrating that the thermal behavior of the 1-decanol-based CO2BAL is comparable to other thermally activated systems. While this moderate Ea reflects a certain degree of thermal stability, it also indicates that degradation becomes non-negligible at elevated temperatures. Combined with the relatively low degradation onset temperature (T5% = 86.4 °C), this limits the practical use of the CO2BAL in bulk conditions, particularly for PVC functionalization near its Vicat softening temperature (∼92 °C). These findings further justify transitioning to solution-phase reactions, where thermal constraints are minimized, allowing for more controlled and efficient reactivity.
Reaction between PVC and CO2BALs in solution
Unlike high-temperature bulk processes, solution-phase methods offer greater flexibility in selecting both the reaction temperature and the polarity of the medium. For PVC, only tetrahydrofuran (THF, dipole moment = 1.75 D), N,N-dimethylformamide (DMF, dipole moment = 3.82 D), and methyl ethyl ketone (MEK, dipole moment = 2.81 D) have been considered to investigate the ability of CO2BAL to react with PVC at room temperature.41–44 While THF is known to dissolve PVC completely, it was directly discarded due to its inability to solubilize CO2BAL of 1-decanol (conv. ∼80%) whatever its concentration. DMF and MEK, which exhibit satisfactory PVC dissolution performances and known to perfectly solubilize CO2BALs, have then been compared.Before initiating any reaction, the absolute number-average molar mass of PVC was determined. The theoretical number-average molar mass (Mnth) provided by Sigma is estimated at 48000 g mol−1. Experimentally, Size Exclusion Chromatography (SEC) was performed using a polystyrene (PS) standard calibration in THF at 25 °C, with the Mark–Houwink relationship applied for both PS and PVC ([η] = KMa, where KPS = 1.1 × 10−4 dL g−1 and aPS = 0.725 for PS; KPVC = 1.658 × 10−4 dL g−1 and aPVC = 0.76 for PVC).45 This analysis yielded an experimental number-average molar mass (MnSEC) of 41400 g mol−1 and a dispersity value (ĐM = Mw/Mn) of 2.2. Prior to correction, the relative MnSEC was 63500 g mol−1.
To limit the significance of elimination reactions, which typically play a major role in the SN2–E2 competition with PVC,19,27,28 reactions were performed at 21 °C by reacting a CO2BAL of 1-decanol (conv. ∼80%) in a PVC solution ([PVC]0 = 0.7 mmol L−1) for an initial [Cl]0-to-[CO2BAL]0 molar ratio of 1. Even though MEK is capable of perfectly dissolving both PVC and the CO2BAL of 1-decanol individually, their combination in this solvent and under the applied concentration conditions renders the CO2BAL of 1-decanol insoluble. Under vigorous stirring, and after several hours, the insolubilization of CO2BAL gradually diminishes, eventually disappearing and leaving behind an insoluble black precipitate that does not dissolve in any of the organic solvents used. In our opinion, such a black insoluble precipitate is likely due to the formation of a polyacetylene-like structure, possibly resulting from the degradation of CO2BAL, which releases 1-decanol and TBD into the solution. The free TBD, or the alkoxide formed by deprotonation of 1-decanol by the guanidine, could then act as a base, promoting E2 elimination reactions on the PVC. Interestingly, the rate of insolubilization has been observed variable with the degree of CO2BAL conversion, i.e., CO2BALs of varying degrees of carbonate conversion. Two TBD decanol-based carbonate salts with different degrees of carbonate conversion were then prepared under CO2 by mixing equimolar amounts of 1-decanol and TBD at 90 °C for different reaction times. This approach allowed us to prepare new CO2BALs with carbonate conversions of 50 and 75 mol% (Fig. S13a and b†). Note that a conversion of 75% corresponds to a salt composed of 75 mol% carbonate and 25 mol% TBD alkoxide. A TBD alkoxide (referred to as the “0% conversion”) also served as a reference for a CO2BAL system entirely devoid of CO2. These CO2BALs were then reacted with PVC previously dissolved in MEK ([PVC]0 = 0.7 mmol L−1) for a reaction time limited to 1 hour and a [Cl]0-to-[1-decanol]0 = 1. 1H NMR analyses of the three reactions reveal no apparent reaction with the CO2BAL but a substantial increase in vinyl protons showing up at δ = 5.5–6 ppm (Fig. S15 and S16†). By integrating these protons relative to the CH-Cl signals of PVC (δ = 4.35–4.65 ppm), the proportion of E2 elimination per chain (= 100 × (I5.5–6/2)/[(I5.5–6/2) + I4.35–4.65]) rises with decreasing CO2BAL conversion. Moreover, the relative MnSEC values also decrease accordingly, in agreement with the literature.46 These trends are summarized in Fig. 5, which compares the proportion of E2 elimination with the corresponding MnSEC values. It should be noted that for the sample obtained with CO2BAL 0% (i.e., an equimolar mixture of 1-decanol and TBD), only the soluble fraction was analyzed by SEC, while the majority remained insoluble in the SEC eluent, likely due to a high unsaturation content.
 | ||
| Fig. 5 Influence of CO2BAL carbonate conversion on the reaction outcome with PVC in MEK at 21 °C ([PVC]0 = 0.7 mmol L−1, [Cl]0-to-[1-decanol]0 = 1, reaction time = 1 h). The relative MnSEC values (in orange) and the proportion of E2 elimination (in blue) are plotted as a function of the CO2BAL carbonate conversion. | ||
To overcome the limitations observed in MEK, reactions were finally carried out at 21 °C in DMF, a solvent ensuring complete solubilization of both PVC and the CO2BAL of 1-decanol. Five functionalization reactions were performed in parallel for reaction times of 30 minutes, 1 hour, 2 hours, 18 hours, and 72 hours. Each reaction medium was subsequently precipitated, washed, and dried to ensure complete removal of decanol derivatives. 1H NMR analyses revealed signals associated with the α-methine proton of a decyl carbonate, which typically appears at δ = 4.87 ppm for alkyl carbonates and δ = 4.97 ppm for acyl carbonates.29,47 In the present case, a signal was detected at approximately δ = 4.76 ppm, overlapping with the PVC methine protons, suggesting the formation of a decyl-based carbonate. Additionally, a triplet at δ = 0.89 ppm, corresponding to terminal methyl protons of decyl chains, confirmed the successful reaction of CO2BAL with PVC (Fig. 6a). Notably, vinyl proton signals (δ = 5.5–6 ppm) first appeared at the 2-hour mark, corresponding to an SN2/E2 ratio of 94/6. Beyond this point, the selectivity shifted in favour of elimination, with SN2/E2 ratios decreasing to 54/46 after 18 hours and 38/62 after 72 hours. This shift was accompanied by a decrease in relative MnSEC values, which initially increased from 63500 g mol−1 for PVC to over 89000 g mol−1 after 2 hours, before declining for longer reaction times (Fig. S17 and S18†). These observations suggest that prolonged reaction times lead to kinetic instability and the gradual release of free TBD into the reaction medium, promoting elimination over substitution.
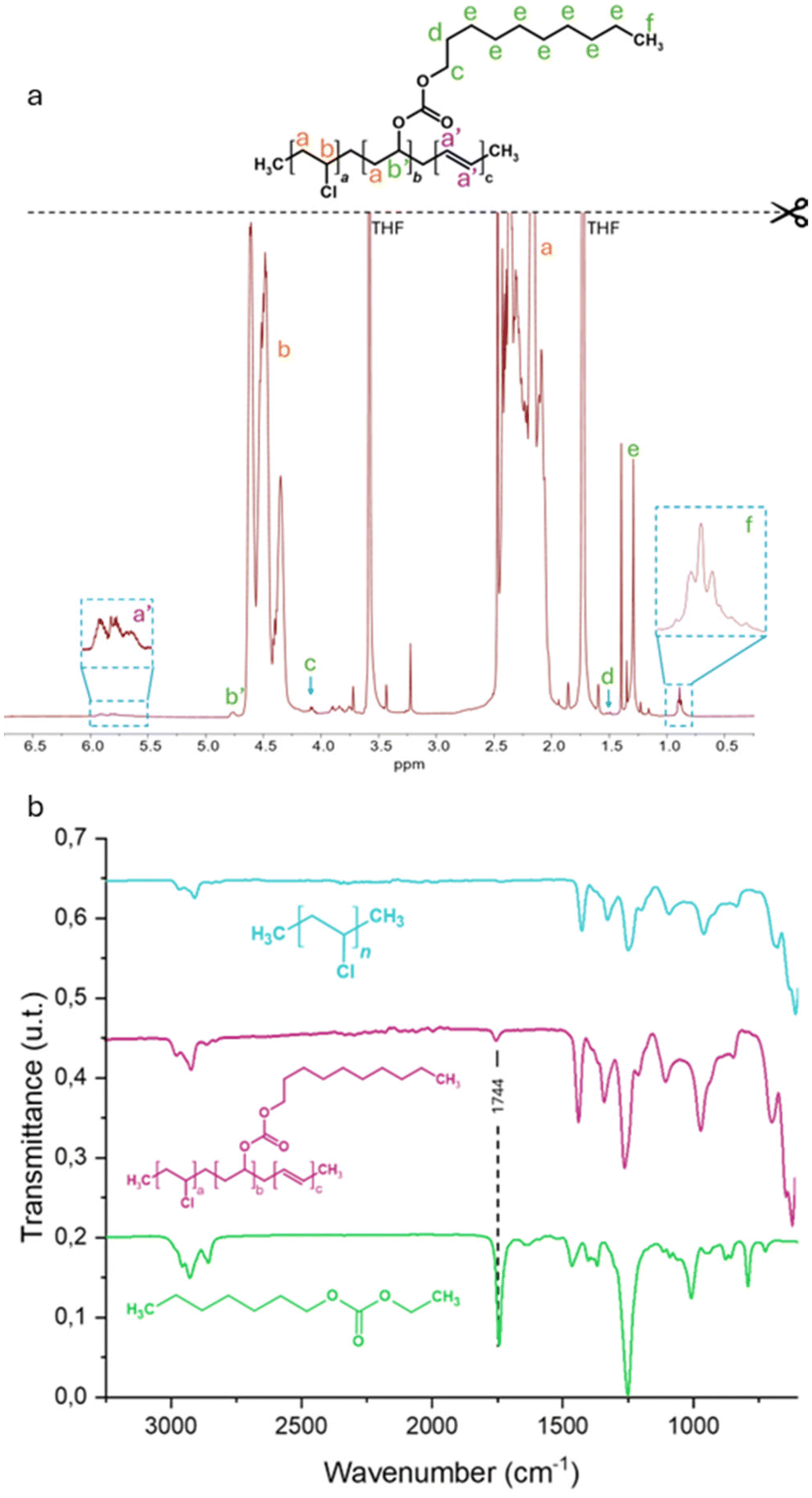 | ||
| Fig. 6 (a) 1H NMR (THF-d8, 500 MHz) of modified PVC (b) FT-IR of PVC (blue), modified PVC (purple), and a heptyl-ethyl carbonate (green) used in comparison and prepared as reported in ref. 29. | ||
The success of the reaction was further confirmed by Fourier Transform Infrared (FTIR) spectroscopy. The spectrum of the sample obtained after 2 hours of reaction exhibited a new absorption band at 1744 cm−1, attributed to the asymmetric stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. This frequency matches that observed for a model small molecule in our previous work (Fig. 6b).29 Notably, no significant absorption bands corresponding to CC stretching vibrations were detected, indicating that vinyl unsaturations formed via elimination remain below the FT-IR detection limit.
O bond. This frequency matches that observed for a model small molecule in our previous work (Fig. 6b).29 Notably, no significant absorption bands corresponding to CC stretching vibrations were detected, indicating that vinyl unsaturations formed via elimination remain below the FT-IR detection limit.
The sample obtained after 2 hours of reaction was further analyzed by thermogravimetric analysis (TGA) to accurately determine the mass fraction of grafted carbonate moieties and compared to the pristine PVC (Fig. 7). As expected, the thermogram of pristine PVC exhibits thermal stability up to 230 °C, beyond which rapid dehydrochlorination occurs, spanning the temperature range of 230 to 370 °C.48 Between 375 and 525 °C, the degraded polyene structures undergo transformation into condensed aromatic hydrocarbons, eventually yielding infusible coke with a residual carbon content of approximately 6.8% at 650 °C. Three key differences were observed in the thermogram of modified PVC. First, an additional mass loss step appears at ca. 125 °C, attributed to the degradation of the grafted carbonate groups. Second, the chlorinated backbone of PVC is destabilized upon functionalization, as evidenced by a shift in the dehydrochlorination onset temperature from 230 °C (pristine PVC) to approximately 180 °C. This phenomenon is well-documented in the literature.27,28,49,50 Finally, the coke residue at 650 °C is slightly higher for modified PVC compared to pristine PVC, likely reflecting the small proportion of elimination reactions detected by 1H NMR spectroscopy.
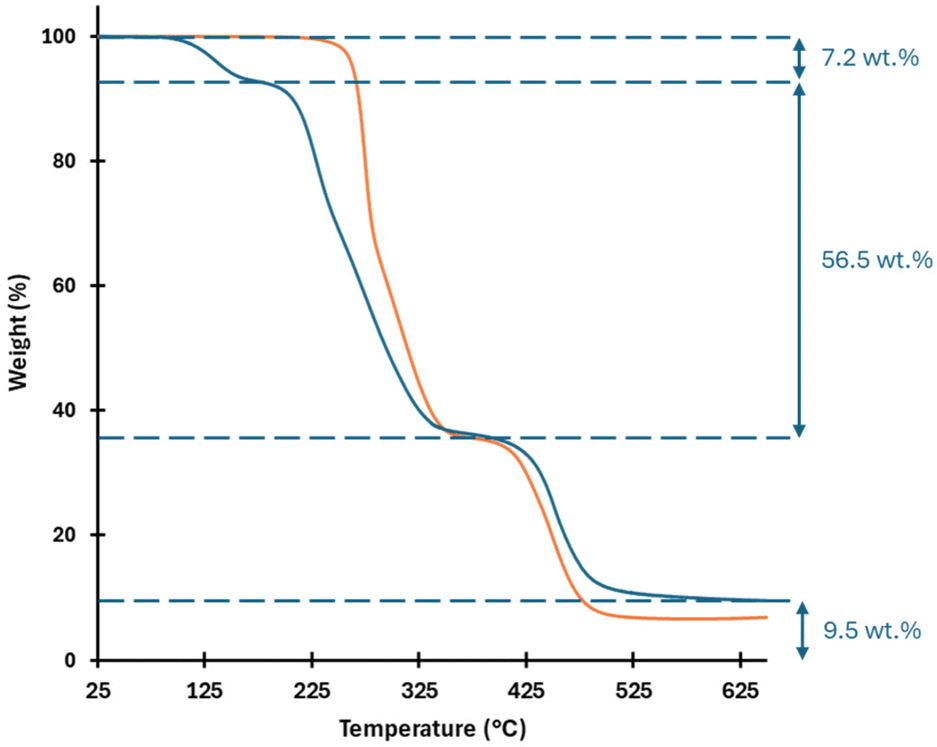 | ||
| Fig. 7 Thermal stability of pristine PVC (orange) and PVC modified (blue) with 1-decanol-based CO2BAL (conv. 80%) in DMF for 2 hours. | ||
The limited grafting efficiency observed in DMF can be attributed to a combination of steric and electronic factors. CO2BALs are known to be significantly stabilized in highly polar solvents like DMF due to dipolar interactions,29 which reduces their overall reactivity and may hinder efficient nucleophilic attack. In SN2 reactions, the substitution of the halogenated carbon is governed by both the dynamics and reactivity of the system.51 Typically, SN2 reactions proceed via a backside attack, resulting in Walden inversion. However, the introduction of bulky substituents can sterically hinder this pathway, potentially favoring an SN2 frontside mechanism despite its slightly higher energetic cost. In the case of PVC, a statistical coil polymer with randomly distributed CH-Cl electrophilic sites, steric hindrance plays a crucial role in dictating the reaction pathway. The steric environment likely promotes frontside SN2 substitution, especially when assisted by a guanidinium ion-pair mechanism (Scheme 1).29 The combination of CO2BAL stabilization in DMF and steric constraints on the PVC backbone could thus explain the observed limitation in grafting efficiency. Further investigations, such as a flow chemistry-driven reaction, are currently underway to explore alternative strategies aimed at improving reaction yields.
 | ||
| Scheme 1 Mechanism proposed for PVC substitution from CO2BAL by a frontside approach. | ||
Conclusions
Bulk-phase reactions revealed that CO2BALs undergo degradation under these conditions, necessitating the use of solution-phase functionalization for efficient PVC modification at 21 °C. Despite a moderate degree of substitution (<10 wt%) compared to some already existing and present in the introduction, this approach achieved a remarkable SN2 selectivity (94/6) while generating a single, benign by-product (TBD·HCl). Spectroscopic and thermal analyses confirmed the successful grafting of carbonate functionalities, though solvent effects influenced reaction efficiency. Specifically, the strong stabilization of CO2BALs in polar solvents like DMF reduced their nucleophilicity at room temperature, highlighting the need for alternative strategies to enhance substitution yields. Beyond PVC functionalization, this strategy holds broader environmental benefits. The use of CO2BALs introduces a novel approach for integrating CO2 into polymer modifications, aligning with circular economy principles. A potential concern for large-scale implementation may be the cost of TBD; however, this limitation can be mitigated, as demonstrated by Fritz-Langhals, who developed a straightforward and cost-effective one-pot synthesis of a liquid TBD formulation.37 Moreover, the generation of a single, benign by-product (TBD·HCl) enhances the process sustainability. As a guanidinium chloride, TBD·HCl has well-established applications in biochemistry, notably as a protein denaturant and nucleic acid stabilizer,52,53 which supports its potential for recovery and reuse. To overcome current limitations in substitution efficiency, future work will explore process intensification strategies, such as flow chemistry, to improve both reaction kinetics and scalability. By bridging CO2 utilization with PVC upcycling, this work contributes to the development of greener methodologies for the valorization of chlorine-rich polymers.Experimental
Materials
Carbon dioxide N50 (≥99.999%) was supplied by Air Liquide. 1-Propanol (99+%), 1-butanol (≥99.4%, ACS reagent), 1-hexanol (anhydrous, ≥99%), deuterated chloroform (0.03 vol% TMS, deuteration degree min. 99.8% for NMR spectroscopy, stabilized with silver), 1-decanol (for synthesis), poly(vinyl chloride) (low molecular weight), as well as N,N-dimethylformamide (anhydrous, 99.8%) were purchased from Sigma-Aldrich. Methanol (GPR Rectapur) and octan-1-ol (purified) were purchased from VWR. 2-Butanone (or methyl ethyl ketone (MEK), ACS Reagent) was acquired from Acros Organics. Ethanol (denaturated with 3% diethylether, Desinfectol) was purchased from Chemlab. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) was supplied by Apollo Scientific. Deuterated tetrahydrofuran (99.5% D) was supplied by CortecNet. All reagents were used as received.Synthetic procedures
δ
H(60 MHz; CDCl3; MeOCO2−,TBD·H+) 3.59 (3H, s, MeOC(O)O−), 3.29 (8H, dt, NCH2), 1.98 (4H, quint, CH2).
δ
H(60 MHz; CDCl3; EtOCO2−,TBD·H+) 4.03 (2H, q, CH2OC(O)O−), 3.29 (8H, dt, NCH2), 1.98 (4H, quint, CH2), 1.23 (3H, t, CH3).
δ
H(60 MHz; CDCl3; PrOCO2−,TBD·H+) 3.87 (2H, q, CH2OC(O)O−), 3.29 (8H, dt, NCH2), 1.98 (4H, quint, CH2), 1.57 (2H, quint, CH2), 0.94 (3H, t, Me).
δ
H(500 MHz; CDCl3; ButOCO2−,TBD·H+) 3.91 (2H, q, CH2OC(O)O−), 3.29 (8H, dt, NCH2), 1.98 (4H, quint, CH2), 1.59 (2H, quint, CH2), 1.40 (2H, m, CH2), 0.91 (3H, t, Me).
δ
H(500 MHz; CDCl3; HexOCO2−,TBD·H+) 3.91 (2H, q, CH2OC(O)O−), 3.29 (8H, dt, NCH2), 1.98 (4H, quint, CH2), 1.61 (2H, quint, CH2), 1.38 (2H, m, CH2), 1.28 (4H, m, (CH2)2CH3), 0.87 (3H, t, Me).
δ
H(500 MHz; CDCl3; OctOCO2−,TBD·H+) 3.91 (2H, q, CH2OC(O)O−), 3.30 (8H, dt, NCH2), 1.99 (4H, quint, CH2), 1.61 (2H, quint, CH2), 1.37 (2H, m, CH2), 1.27 (8H, m, (CH2)4CH3), 0.87 (3H, t, Me).
δ
H(500 MHz; CDCl3; DecOCO2−,TBD·H+) 3.90 (2H, q, CH2OC(O)O−), 3.30 (8H, dt, NCH2), 1.99 (4H, quint, CH2), 1.60 (2H, quint, CH2), 1.38 (2H, m, CH2), 1.25 (12H, m, (CH2)6CH3), 0.87 (3H, t, Me).
ν
max/cm−1 3232 (NH), 3152 (NH), 1655 (CO), 1570 (CN), 1388 (CO).
δ
H(500 MHz; THF-d8; modified PVC) 5.79 (2H, br t, CHCH), 4.75 (1H, br t, CH2CHOC(O)OC10H21), 4.49 (1H, br t, CH2CHCl), 4.09 (1H, br t, CH2CHOC(O)OCH2C9H19), 2.24 (2H, br m, CH2CHCl, CH2CHOC(O)OC10H21), 1.46 (2H, t, CH2CHOC(O)OCH2CH2C8H17), 1.29 (14H, br m, CH2CHOC(O)OCH2CH2(CH2)7CH3), 0.89 (3H, t, CH2CHOC(O)O(CH2)9CH3).
ν
max/cm−1 1744 (CO), 1429 (PVC), 1330 (PVC), 1250 (PVC), 1117 (C–O), 964 (PVC).
Methods
Products of the reaction between CO2BAL and PVC were subjected to TGA as well. This allowed for thermal stability evaluation as well as the determination of the substitution rate. Measurements were performed under nitrogen flow at a rate of 2.5 °C per minute using a TA Q500 TGA.
In order to evaluate the CO2BALs’ thermal stability, degradation kinetics analysis was performed. Samples were therefore subjected to a nitrogen flow (p(N2) = 0.2 bar) at different temperatures, using the same experimental setup as for their synthesis. Degradation was monitored regularly through 1H NMR spectroscopy. The neperian logarithm of the degradation percentage was plotted against time. After linear regression, the slope obtained gave the kinetic constant of the degradation reaction (k) at each temperature. Consequently, the plot of ln(k) against 1/T (K−1) allowed to work out the frequency factor (A) and the activation energy (Ea) parameters of the Arrhenius equation for the degradation reaction.
Final CO2BAL and substitution products were all characterized by 1H NMR using a Bruker AVANCEII 500 MHz apparatus at room temperature in chloroform-d1 (CDCl3) and THF-d8, respectively. All spectra were acquired according to the following parameters: aq = 6.8 s, d1 = 10 s, sw = 12, o1p = 5, ns = 32 for small molecules and 512 for PVC-based compounds.
Author contributions
Conceptualization: O. C.; methodology: J. D.; validation: J. D. and E. B. A.; investigation: J. D. and E. B. A.; writing – original draft: J. D.; writing – review & editing: O. C., J. D., and E. B. A.; visualization: J. D., E. B. A., and O. C.; supervision: O. C.Data availability
The data supporting this study, including spectroscopic, thermal, and chromatographic analyses, are available in the ESI† of this article. No additional datasets were generated or analyzed beyond those included in the main text and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. and E. B. A. acknowledge the support of the AXA Research Fund for the funding of this project. O. C. acknowledges support for his position as a Senior Research Associate for the F.R.S.-FNRS of Belgium and AXA Professor in Chemistry.References
- ChemAnalyst, Poly Vinyl Chloride (PVC) Market Analysis: Industry Market Size, Plant Capacity, Production, Operating Efficiency, Demand & Supply, End-User Industries, Sales Channel, Regional Demand, Foreign Trade, Company Share, Manufacturing Process, 2015–2030, ChemAnalyst, 2023 Search PubMed.
- The Circular economy for Plastics - A European Analysis, Plastics Europe, 2024.
- A. Buekens and A. Sevenster, J. Mater. Cycles Waste Manag., 2010, 12, 184–192 Search PubMed.
- K. Lewandowski and K. Skórczewska, Polymers, 2022, 14, 3035 Search PubMed.
- Z. Ait-Touchente, M. Khellaf, G. Raffin, N. Lebaz and A. Elaissari, Polym. Adv. Technol., 2024, 35, e6228 CrossRef CAS.
- M. Sadat-Shojai and G.-R. Bakhshandeh, Polym. Degrad. Stab., 2011, 96, 404–415 Search PubMed.
- X. Jiang, B. Zhu and M. Zhu, Green Chem., 2023, 25, 6971–7025 RSC.
- D. E. Fagnani, D. Kim, S. I. Camarero, J. F. Alfaro and A. J. McNeil, Nat. Chem., 2023, 15, 222–229 Search PubMed.
- M. Liu, X. Wu and P. J. Dyson, Nat. Chem., 2024, 16, 700–708 CrossRef CAS PubMed.
- S. Mishra, S. Kar, R. Rangappa, P. Patil, V. Kadam, S. H. Chikkali and R. C. Samanta, Chem. – Eur. J., 2025, 31, e202403980 CrossRef CAS PubMed.
- J. L. S. Zackasee, V. Srivardhan, B. L. Truesdell, E. J. Vrana and C. S. Sevov, Chem, 2025, 11, 102298 CAS.
- S. Zhang, H. Han, M. Cao, Y. Xie and J. Chen, Interdiscip. Mater., 2025, 4, 5–23 CAS.
- T. Yoshioka and G. Grause, in Topical Themes in Energy and Resources, ed. Y. Tanaka, M. Norton and Y.-Y. Li, Springer Japan, Tokyo, 2015, pp. 195–214 Search PubMed.
- S. Moulay, J. Res. Updates Polym. Sci., 2015, 4, 79–122 Search PubMed.
- S. Moulay, Prog. Polym. Sci., 2010, 35, 303–331 CrossRef CAS.
- R. K. Jha, B. J. Neyhouse, M. S. Young, D. E. Fagnani and A. J. McNeil, Chem. Sci., 2024, 15, 5802–5813 RSC.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 Search PubMed.
- G. Martinez, N. Guarrotxena, J. M. Gomez-Elvira and J. Millan, Polym. Bull., 1992, 28, 427–433 CrossRef CAS.
- T. Kameda, M. Ono, G. Grause, T. Mizoguchi and T. Yoshioka, Polym. Degrad. Stab., 2009, 94, 107–112 CrossRef CAS.
- L. Shao, C. Qi and X.-M. Zhang, RSC Adv., 2014, 4, 53105–53108 RSC.
- M. Bakherad, A. Keivanloo and S. Samangooei, Chin. J. Catal., 2014, 35, 324–328 CrossRef CAS.
- J. Zhu, Y. Su, X. Zhao, Y. Li, J. Zhao, X. Fan and Z. Jiang, Ind. Eng. Chem. Res., 2014, 53, 14046–14055 CrossRef CAS.
- A. Siekierka, J. Wolska, W. Kujawski and M. Bryjak, Sep. Sci. Technol., 2018, 53, 1191–1197 CrossRef CAS.
- S. Rabie, D. Ali, N. Khaireldin, M. El-Hashash, M. Elsaidi, A. El-Sayed and N. Wahed, Egypt. J. Chem., 2020, 63, 4833–4849 Search PubMed.
- E. J. Park, B. C. Park, Y. J. Kim, A. Canlier and T. S. Hwang, Macromol. Res., 2018, 26, 913–923 CrossRef CAS.
- N. G. Bush, M. K. Assefa, S. Bac, S. Mallikarjun Sharada and M. E. Fieser, Mater. Horiz., 2023, 10, 2047–2052 RSC.
- L. Lu, S. Kumagai, T. Kameda, L. Luo and T. Yoshioka, RSC Adv., 2019, 9, 28870–28875 RSC.
- G. Sneddon, J. C. McGlynn, M. S. Neumann, H. M. Aydin, H. H. P. Yiu and A. Y. Ganin, J. Mater. Chem. A, 2017, 5, 11864–11872 RSC.
- J. Delcorps, K. S. Rawat, M. Wells, E. Ben Ayed, B. Grignard, C. Detrembleur, B. Blankert, P. Gerbaux, V. Van Speybroeck and O. Coulembier, Cell Rep. Phys. Sci., 2024, 5, 102057 CrossRef CAS.
- D. J. Heldebrant, C. R. Yonker, P. G. Jessop and L. Phan, Energy Procedia, 2009, 1, 1187–1195 CrossRef CAS.
- D. J. Heldebrant, C. R. Yonker, P. G. Jessop and L. Phan, Energy Environ. Sci., 2008, 1, 487–493 RSC.
- D. J. Heldebrant, P. K. Koech, M. T. C. Ang, C. Liang, J. E. Rainbolt, C. R. Yonker and P. G. Jessop, Green Chem., 2010, 12, 713–721 RSC.
- L. Phan, D. Chiu, D. J. Heldebrant, H. Huttenhower, E. John, X. Li, P. Pollet, R. Wang, C. A. Eckert, C. L. Liotta and P. G. Jessop, Ind. Eng. Chem. Res., 2008, 47, 539–545 CrossRef CAS.
- M. Verdecchia, M. Feroci, L. Palombi and L. Rossi, J. Org. Chem., 2002, 67, 8287–8289 CrossRef CAS PubMed.
- Á. Mesías-Salazar, R. S. Rojas, F. Carrillo-Hermosilla, J. Martínez, A. Antiñolo, O. S. Trofymchuk, F. M. Nachtigall, L. S. Santos and C. G. Daniliuc, New J. Chem., 2024, 48, 105–111 RSC.
- S. G. Khokarale and J.-P. Mikkola, RSC Adv., 2019, 9, 34023–34031 RSC.
- E. Fritz-Langhals, Org. Process Res. Dev., 2022, 26, 3015–3023 CrossRef CAS.
- Heat distortion temperature, https://pvc.org/about-pvc/pvcs-physical-properties/heat-distortion-temperature/, (accessed 10 August 2024).
- J. Yu, L. Sun, C. Ma, Y. Qiao and H. Yao, Waste Manage., 2016, 48, 300–314 CrossRef CAS PubMed.
- T. Moreno, P. Dyer and S. Tallon, Ind. Eng. Chem. Res., 2020, 59, 20307–20315 CrossRef CAS.
- G. Grause, A. Buekens, Y. Sakata, A. Okuwaki and T. Yoshioka, J. Mater. Cycles Waste Manag., 2011, 13, 265–282 CrossRef.
- Z. Hou and R. M. Broughton, J. Coated Fabr., 1998, 28, 37–55 CrossRef.
- S. Adanur, Z. Hou and R. M. Broughton, J. Coated Fabr., 1998, 28, 145–168 CrossRef CAS.
- G. Grause, S. Hirahashi, H. Toyoda, T. Kameda and T. Yoshioka, J. Mater. Cycles Waste Manag., 2017, 19, 612–622 CrossRef CAS.
- A. J. De Vries, C. Bonnebat and M. Carrega, Pure Appl. Chem., 1971, 26, 209–240 CrossRef CAS.
- A. Wirsén and P. Flodin, J. Appl. Polym. Sci., 1978, 22, 3039–3056 Search PubMed.
- S. Y. Lee, J. M. Murphy, A. Ukai and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 15149–15153 CrossRef CAS PubMed.
- N. Choudhury, A. Kim, M. Kim and B.-S. Kim, Adv. Mater., 2023, 35, 2304113 CrossRef CAS PubMed.
- A. Salazar Avalos, M. Hakkarainen and K. Odelius, Polymers, 2017, 9, 84 CrossRef PubMed.
- P. Jia, M. Zhang, L. Hu, R. Wang, C. Sun and Y. Zhou, Polymers, 2017, 9, 621 CrossRef PubMed.
- E. Carrascosa, J. Meyer, J. Zhang, M. Stei, T. Michaelsen, W. L. Hase, L. Yang and R. Wester, Nat. Commun., 2017, 8, 25 CrossRef PubMed.
- K. C. Aune and C. Tanford, Biochemistry, 1969, 8, 4579–4585 CrossRef CAS PubMed.
- L. M. Mayr and F. X. Schmid, Biochemistry, 1993, 32, 7994–7998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00350d |
| This journal is © The Royal Society of Chemistry 2025 |