
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DNA extraction from bacteria using a gravity-driven microcapillary siphon†
Crescenzo
Ianniello
ab,
Julia
Sero
bc,
David
Gough
d,
Barbara
Kasprzyk-Hordern
ef and
Nuno M.
Reis
 *abfg
*abfg
aDepartment of Chemical Engineering, Claverton Down, University of Bath, Bath BA2 7AY, UK. E-mail: n.m.reis@bath.ac.uk
bCentre for Bioengineering & Biomedical Technologies (CBio), University of Bath, Bath BA2 7AY, UK
cDepartment of Life Sciences, University of Bath, Bath BA2 7AY, UK
dLamina Dielectrics Ltd, Billingshurst RH14 9SJ, UK
eDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK
fCentre of Excellence in Water-Based Early Warning Systems for Health Protection (CWBE), University of Bath, Bath BA2 7AY, UK
gCentre of Biological Engineering (CEB), University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal
First published on 14th May 2025
Abstract
Nucleic acid amplification tests (NAATs) are used in many applications ranging from human diagnostics to environmental monitoring. However, currently NAATs rely on access to bulky equipment and laboratory facilities. In particular, DNA extraction remains a key limiting step, with current methods depending on large devices and extensive manual pipetting. The design of portable NAAT devices and wider access to molecular testing is therefore of critical scientific and medical importance. Here we report the first application of a gravity-driven microcapillary siphon for the extraction of bacterial DNA. Making use of magnetic DNA-binding beads and an external magnet, capturing, washing and DNA elution were performed in 10-bored, 200 μm internal diameter siphons using passive flow, without the need for external pumps nor other specialised equipment. The method was successfully evaluated using real-time qPCR, from where we observed a linear relationship between colony forming units (CFUs) and threshold cycle (Ct) for E. coli spiked in several matrices relevant for pathogen analysis: PBS buffer, sheep blood, and river water. Importantly, this method was not only cost-effective but also outperformed the standard manual protocol, returning higher recovery efficiency (>90% vs. 52%) and better removal of assay inhibitors, which we believe linked to the higher shear rates and short diffusion distances obtained in the flow-through, microcapillary siphons. In addition, we have shown microcapillary siphons are reusable without detectable cross-contamination between consecutive DNA extractions. These findings demonstrate that high quality DNA can be isolated without access to expensive equipment or complex fluid handling. Future work will explore integration with isothermal amplification methods for development of a fully integrated, passive-flow devices for portable NAATs.
1. Introduction
Nucleic acid amplification tests (NAATs) are widely used for the detection of pathogens,1–3 also finding relevant applications in ecological studies4,5 and environmental monitoring, including antimicrobial-resistance genes detection.6–8 NAATs have low limits of detection,9–12 therefore are suitable for the detection of a vast number of microorganisms and viruses. However, reagents for DNA extraction and amplification are expensive and often limited to well-equipped laboratory facilities.13–17 Current purification protocols require several manual pipetting steps and access to bulky equipment, such as benchtop centrifuges, which has so far limited the application of NAATs to field or point-of-care testing. It is therefore desirable to develop technical capabilities for efficient DNA extraction enabling near-the-source environmental surveillance or point-of-care clinical testing of key pathogens.Although the purification step is not always necessary, as shown for SARS-CoV2 in saliva samples,18,19 nucleic acid testing from complex matrices such as blood or wastewater often requires a DNA extraction step to remove enzymatic inhibitors.20 For example, amplification protocols from blood samples may be affected by the presence of heparin, IgG, haemoglobin and lactoferrin.21,22 Wastewater and river water, used in wastewater-based epidemiology8,14,16,23–27 and environmental monitoring, can contain fats, proteins, polyphenols, heavy metals, polysaccharides, metal ions and RNases.28–33
In essence, DNA purification is a multi-step process, where reagents are sequentially added to remove contaminants. Several studies have previously proposed microfluidic devices to overcome current limitations and decentralising NAATs.34–36 Microfluidic devices can use smaller sample volumes, reducing the cost of consumables and reagents. In addition, operation can be automated reducing the number of manual steps, therefore minimising the number of technical errors resulting from the user. Most previous reports of DNA extraction in microfluidic devices used solid-phase extraction (SPE), with a surface that enabled DNA binding, followed by washing and elution. Examples of SPE include Chelex-100,37 silica,38–40 carboxylic-group-modified pillars41 and amine-modified surfaces.42 Most of these devices require reagents to be loaded through syringes or pumps, although centrifugal disks were also reported for liquids' transfer.37,43 Jagannath et al.39 controlled the reagents' flow from a microfluidic cassette using a combination of on–off valves and check valves, while Thang et al.44 used a pre-loaded cartridge with plungers and an automated rotation system for reagents delivery.
Magnetic beads are the most common method for SPE in microfluidic devices, mostly using centrifugal disks, where washing and elution steps are carried out with the help of a magnet.43 Centrifugal forces enable the sequential control of liquid reagents retained in a set of chambers through the magnetic beads trapped with a magnet.45–51 Other variants proposed moving the magnetic beads across the device rather than the reagents, with the assistance of an external magnet.52–56 Alternative microfluidic designs used droplet handling based on electrowetting,57 cartridges with integrated switching valves58 and paper-based microfluidic devices.59 Paper devices allow power-free transfers of liquid, however still requiring manual loading of reagents.
We report for the first time a microfluidic device for DNA extraction that uses a gravity-driven passive flow microcapillary siphon. Gravity-driven siphon was previously reported by Reis et al.60 for rapid immunoassays proving to be an effective power-free multi-step ELISA solution. In this work we have tailored the microfluidic siphon concept to power-free DNA extraction, washing and elution. The microfluidic siphons consisted of 10-bored, ∼200 μm-diameter hydrophilic microcapillaries allowing power-free liquid transfer between two volumes placed at different heights, with the liquid being transferred from the higher to the lower point by gravity and molecular cohesion. Capturing of magnetic beads in microcapillaries revealed feasible and the microcapillary siphons revealed reusable, with no cross-contamination observed after three independent extractions. Despite its simplicity, microcapillary siphons revealed reliable and reproducible results on a variety of matrices spiked with DNA from E. coli, thus representing a valid and robust solution, a prototype for the design of portable DNA testing devices.
2. Materials and method
2.1. Media and assay reagents
LB agar (BP1425-500), LB broth (BP1426-500), and Sheep Blood in Alsever Solution (SR0053) were sourced from Fisher Scientific (Loughborough, UK). Ten-bored FEP-Teflon® microcapillary films (MCF), with ∼200 μm internal diameter (Fig. 1A) were supplied by Lamina Dielectrics Ltd (Billingshurst, West Sussex, UK). For bacterial DNA extraction, the MagaZorb® DNA Mini-Prep kit (MB1004, Promega) was used. Thermo Scientific™ PCR Master Mix (K0171), Thermo Scientific™ GeneRuler 100 bp DNA Ladder (SM0241), DNA oligos (Thermo Fisher Scientific), GelRed® Nucleic Acid Stain 10000X Water (STC123, Merck) and Applied Biosystems™ PowerUp™ SYBR™ Green Master Mix for qPCR (A25779) were used for PCR, gel electrophoresis, and real-time qPCR. Phosphate buffered saline powder (P5368-10PAK), 0.05% Tween 20 PBS tablets (524653) and polyvinyl alcohol powder (363065-25G) were from Sigma Aldrich (Dorset, UK). River water was collected downstream of a local wastewater treatment plant (pH 7.96). | ||
| Fig. 1 Design of the microcapillary siphon. (A) Front view of a FEP-Teflon® MCF. Each strip is made of 10 individual microcapillaries with mean internal diameter ∼200 μm. (B) Microphotographic and (C) schematic side views of microcapillary siphons. An 85 mm long strip of hydrophilic MCF was bent and held in place using a plastic holder. (D) Capturing of magnetic beads in the microcapillary siphon (isolation step). Using an external magnet at the back of the microcapillary strip (shown in the background, enclosed in a white case), beads were isolated in each of the 10 microcapillaries. (E) Magnetic beads in microcapillary siphon at end of enrichment's step. | ||
2.2. Bacterial cultures
E. coli K12 M1655 cells were grown on LB agar plates overnight at 37 °C. A colony was picked from the LB agar plate and incubated overnight in 10.0 ml of sterile LB broth at 37 °C on shaker at 300 rpm. The bacteria were washed three times in PBS buffer through centrifugation at 4000 rpm for 10 min, re-suspended in PBS and kept at room temperature in the dark.61 Before each experiment, colony-counting on LB agar plate was performed to estimate the colony forming units (CFU) per ml.2.3. Experimental design and preparation steps for microfluidic siphon assay
As melt-extruded FEP-Teflon® MCF is hydrophobic, we first coated the inner surface of the microcapillary by adsorption of polyvinyl alcohol (PVOH) using the protocol previously reported by Reis et al.60,62 To achieve this, a 5.0 mg ml−1 solution of high-molecular weight PVOH was loaded into short MCF fragments (200 mm in length) for two hours and then washed with 0.05% Tween 20 in PBS. The coated MCF strips were then emptied by injecting air with a clean plastic syringe and then trimmed into 85 mm long strips and stored between 2–8 °C for at least one week before use and up to 1 month. To evaluate the efficacy of the hydrophilic coating, a 10 mm-long MCF strip was immersed in a cuvette containing 1 ml of distilled water and the liquid rise in the MCF strip was recorded (Fig. S1†).DNA was extracted from E. coli cells using an adaptation of the manufacturer's MagaZorb® DNA Mini-Prep kit manual. All reagents from the kit were stored at room temperature, therefore most reagents were pre-aliquoted and stored in plastic tubes. In preparation for the experiments, 5.0 μl of proteinase K were added to a fresh 0.2 ml PCR tube, 200.0 μl of washing buffer were added in two 0.2 ml PCR tube (for a total of 400.0 μl) and 200.0 μl of elution buffer were added in two 0.2 ml PCR tube (for a total of 400.0 μl). Furthermore, 20.0 μl of washing buffer were added into three clean wells of a 96-well Nunc plate, and 20.0 μl of elution buffer was added into two clean wells of the same plate. Alternatively, the 96-well plate could be replaced by individual plastic tubes, provide these contained enough volume to immerse the bottom-end of the microcapillary siphon.
A good control of the discharge rate, Q of the microcapillary siphons is crucial to deliver the correct volumes of each reagent, and is determined by gravity, g; dynamic fluid viscosity, μ; fluid density, ρ; and the inner diameter of the microcapillary, dc:60,63
| Qμdc−4N−1 = πρg/128·H/L | (1) |
To establish the duration of each step, Q values were calculated by weighting the volume of the fluid using a laboratory scale. For each reagent, three PCR tubes containing 200.0 μl of the reagent were weighed to work out the density and initial mass, m1. Using hydrophilic microcapillaries pre-filled with magnetic beads, liquid was transferred from the tubes to a 96-well Nunc plate, and the tubes re-weighted to work out final mass m2. Values of Q were calculated based on the density of the respective fluid, ρ and change in mass transferred and time interval, t as follows:
| Q = (m1 − m2)/ρ × t | (2) |
2.4. DNA extraction in microcapillary siphons
Firstly, 50.0 μl of the sample were added to the PCR tube containing pre-aliquoted 5.0 μl of proteinase K. After a brief hand-swirling, 50.0 μl of lysis buffer was added to the tube. The sample was gently vortexed and then incubated for 10 minutes at 56 °C using a tight-fitting heating block. Afterwards, 125.0 μl of binding buffer was added to the tube, followed by 5.0 μl of MagaZorb reagent containing the magnetic beads. The sample was then incubated for 10 minutes at room temperature (20 °C) to allow adsorption of DNA on the beads' surface, inverting the tube 5 times every 2 minutes to avoid particles precipitation.Subsequently, a PVOH coated 85 mm long microcapillary strip was placed into a plastic holder and shaped with a neck and a ‘swan beak’, yielding the geometry required for a siphon (Fig. 1B and C), with the inlet at a higher liquid hydraulic height than the outlet, as it works at the driving force for operation of the microcapillary siphon. Priming, isolation and enrichment were illustrated in Fig. 2. The top end of the microcapillary siphon was dipped in the binding-beads mix tube and 28.5 μl of the sample were passively loaded into the siphon through microcapillary action and gravity (priming step) (Fig. 2-I). Using a magnet (MagneSphere® Technology Magnetic Separation Stands, Z5331, Promega, UK), the beads were pelleted on one side around the centre of the siphon (isolation step, Fig. 2-II and 1D). At this stage, two different regions of beads' accumulation were visible. The beads at the lower region were collected by moving the magnet from the lower end of the siphon to its final position (12 mm above the lower end). This was done to avoid loss of magnetic beads from the lower part. Once the siphon was fully loaded with liquid, it was used to transfer the sample from a “source” to a “destination” point, with the liquid transfer entirely driven by gravity due to the height difference between the source and the destination, as predicted by eqn (1). The siphon remained filled with liquid throughout the whole extraction process. Following this step, an additional binding-beads mix was run through the siphon to capture more beads and DNA (enrichment step, Fig. 2-III). During the enrichment step, beads only accumulated in the upper accumulation region (Fig. 1E), immediately captured by the external magnet. A pre-loaded Nunc-plate well with 20.0 μl of washing buffer was used as the “destination” point. The incubation time for the enrichment step, described as the time between addition and removal of the siphon's “beak” from the source volume, was set specifically as 2.0 minutes, allowing the flow of 60.0 μl of binding-beads mix through the microcapillaries. We have estimated the inner volume of the 85 mm long, 10-bored ∼200 μm-diameter microcapillaries as 28.5 μl, which is around 3 times lower than the total volume of binding-beads mix flowing through the microcapillary siphons, calculated as 88.5 μl (i.e. 28.5 μl during the priming and 60.0 μl during the enrichment steps). A ratio between these volumes around 3 is important to yield a good replacement of sequential reagents in the flow-through, microcapillaries due to levels of axial dispersion.
 | ||
| Fig. 2 Diagrammatic workflow of microcapillary siphon for DNA extraction. The microcapillary siphons were first self-primed with binding mix including magnetic beads, identified as step (I). From that moment, the siphon remained filled with liquid throughout all steps, with reagents' flow exclusively driven by gravity and cohesion forces. Subsequently, beads were isolated using a magnet (step II) before more beads were flown and captured through the microcapillaries (enrichment, step III). The microcapillary siphon was then washed twice with washing buffer (step IV). Elution buffer was loaded, pushing out the washing buffer; after 5–10 minutes off the magnet, the DNA was released from the beads (step V). Finally, the magnet was replaced and one additional elution buffer step carried out to allow full recovery of the eluted DNA into a fresh well (step VI). | ||
Lately, a washing step (Fig. 2-IV) was performed to discard contaminants that could affect downstream DNA amplification testing. A PCR tube with pre-aliquoted washing buffer was used as the source, and a 96-well Nunc-plate well pre-loaded with 20.0 μl of washing buffer was used as destination. The incubation time was set at 4 minutes and repeated twice to ensure a proper removal of contaminants. During the entire step, a magnet was kept on the microcapillary siphon to ensure the beads remained isolated during the process and that were not washed away by the reagents' flow.
Finally, an elution step was carried out which consisted of two parts. In step 1, 35.0 μl of elution buffer was run through the microcapillary siphon using a pre-loaded tube as source and a 96-well plate well preloaded with 20.0 μl of elution buffer as destination for 70 seconds (Fig. 2-V). This step was performed to replace the washing buffer inside the microcapillaries with elution buffer. Following this first incubation, the magnet was removed and the microcapillary siphon left for 10 minutes at room temperature, keeping the bottom end of the siphon immersed in liquid in order to avoid evaporation. Incubation without the magnet allowed the release of the DNA from the magnetic beads to the elution buffer. In subsequent step, the magnet was replaced for 1 minute, and then the second part of the elution step performed to allow the recovery of DNA in elution buffer (Fig. 2-VI). A total of 28.0 μl of elution buffer was run through the siphon using a pre-loaded tube as source and a 96-well plate well preloaded with 20.0 μl of elution buffer as destination for 23 seconds. The eluted DNA sample (measured as 48.0 μl) was then transferred to a fresh 0.2 ml PCR tube in preparation for further testing.
2.5. Analysis of captured beads in microcapillaries via ImageJ
Images of magnetic beads in microcapillaries were captured with a Canon PowerShot S120 digital camera and evaluated via ImageJ analysis (NIH, USA). The binding-beads mix solution was allowed to flow through the microcapillary siphons for 125 seconds and magnetic beads isolated throughout the process using an external magnet. An absorbance plot was then obtained for all RGB photographs following split into red, green and blue channels using ImageJ. Absorbance was calculated as log10(Io/I), where Io is the intensity of the grey scale light intensity entering the microcapillaries and I is the grey scale light intensity leaving the microcapillaries.2.6. Manual DNA extraction
As benchmark, full manual extraction was performed using the MagaZorb® DNA Mini-Prep kit (Promega, UK) following the user manual protocol for the isolation of DNA from cultured cells. Briefly, 20.0 μl of proteinase K was added to a 1.5 ml tube containing 200.0 μl of the sample. This was followed by 200.0 μl of lysis buffer added to the sample and gently vortexed and incubated for 10 minutes at 56 °C using a tight-fitting heating block. Afterwards, 500.0 μl of binding buffer was added to the tube, followed by 20.0 μl of MagaZorb reagent containing the magnetic beads for DNA adsorption. The sample was then incubated for further 10 minutes at room temperature to allow adsorption of DNA. Magnetic beads were isolated using a magnetic stand, and the supernatant was discarded prior to the addition of 1.0 ml of washing buffer. The PCR tube was quickly inverted, the beads re-isolated and the supernatant discarded. This process was repeated twice, for a total of two washing. Finally, 200.0 μl of elution buffer was added to the tube, and the sample was incubated for 10 minutes to allow the release of the DNA from the magnetic beads. Finally, the beads were re-isolated, and the supernatant was collected in a fresh 200.0 μl PCR tube and stored between −15 °C and −25 °C.2.7. DNA quantification and polymerase chain reaction (PCR)
For an initial evaluation of DNA extraction in the microcapillary siphons, DNA was extracted from 2.3 × 109 CFU ml−1E. coli K12 MG1655 in PBS. Sterile PBS was used as negative control and all experiments repeated at least in triplicate for both positive and negative control samples. Normalised DNA concentration was calculated by subtracting the background noise observed in the negative controls from the value obtained for the bacterial sample. The same process was repeated through manual extraction. The extracted DNA was measured using a NanoDrop™ 2000 spectrophotometer and a PCR protocol was run to evaluate the integrity of the DNA. The recovery efficiency for each method was calculated, and the calculations are shown in our ESI.† The primers (5′-AGCAACAGGCAGCAGAGGCG-3′ forward, 5′-GACGTTCGCGCTGTTTCGGC-3′ reverse) were selected from a previous study64 and added to the PCR mix at a final concentration of 500 nM. The PCR products were run on 1% agarose electrophoresis gel.2.8. DNA extraction from E. coli in PBS, sheep blood and river water
DNA extraction in the microcapillary siphons was tested for PBS, blood and river water spiked with E. coli K12 M1655, and the outcome evaluated through real-time qPCR, using a StepOnePlus Real-Time PCR (Applied Biosystems). For the reaction, 3.0 μl of the samples was added to 5.0 μl of master mix and 1.0 μl of each primer. Because only 3.0 μl from the 48.0 μl of extracted DNA were used, the total CFU of each sample was diluted 16 times which was reflected in the results' plots. The primers, described in section 2.7, were used to a final concentration of 500 nM. Each sample was tested in triplicate biological and technical replicates. Any outliers, likely resulting from technical errors, were identified and removed according to the Tukey's fences and k-distance methods.2.9. Evaluation of inhibitors removal via real-time qPCR
The removal of PCR inhibitors in the microcapillary siphons was evaluated via real-time qPCR and compared with the manual protocol. A sterile sample of PBS was run through the microcapillary siphons, while another sample was run through the manual method. The same process was repeated for river water. Afterwards, 250 pg of DNA was added to 21.5 μl of each sample, and five replicates were added to an optical plate for real-time qPCR. Each reaction consisted of 5.0 μl of master mix, 1.0 μl of each primer and 3.0 μl of the sample.2.10. Reusability of microcapillary siphon for DNA extraction
We have tested reusability of microcapillary siphons via real-time qPCR. Three different siphons were used for this series of experiments to assess potential cross-contaminations that may occur from multiple uses. This was done by carrying out DNA purification in a set of three samples and three siphons loaded with different sequences. One microcapillary siphon was first used with 104.8 CFUs, subsequently loaded with the negative control (PBS only) and finally with 102.8 CFUs. A second microcapillary siphon was first used with the negative control, and then with 102.8 CFUs and 104.8 CFUs. A third microcapillary siphon was used first with 102.8 CFUs, followed by 104.8 CFUs and negative control. Between each use, microcapillary siphons were washed for 1 minute with PBS without the magnet to displace the magnetic beads from the microcapillaries. Using a manual syringe and a tubing connector, the microcapillary siphon was fully emptied and allowed to dry for 24 hours at room temperature to ensure fully dried microcapillaries.3. Results
3.1. Isolation of magnetic beads and protocol design
Magnetic beads from the MagaZorb® DNA Mini-Prep kit were successfully isolated in microcapillary siphons. Fig. 3A shows the accumulation of the beads over time in the presence of an external magnet using a fully passive gravity-driven flow. Despite the small diameter (∼200 μm), the presence of the beads did not prevent further liquid passing through the microcapillaries and successful multi-step DNA extraction protocol. However, we have noticed an excessive accumulation of magnetic beads interfered with the flow rates (Fig. S2†), so we limited the protocol to a 2.0-minute enrichment step for loading magnetic beads. The accumulation of beads was quantified via ImageJ analysis to observe the light absorbed by the particles, with the results showing a plateau after 100 seconds, suggesting there is not further capability to accumulate magnetic beads in the microcapillaries (Fig. 3B). The size of the beads was estimated to be between 130–260 nm when tested in distilled water on a Zetasizer instrument (Malvern Panalytical) (Fig. S3†).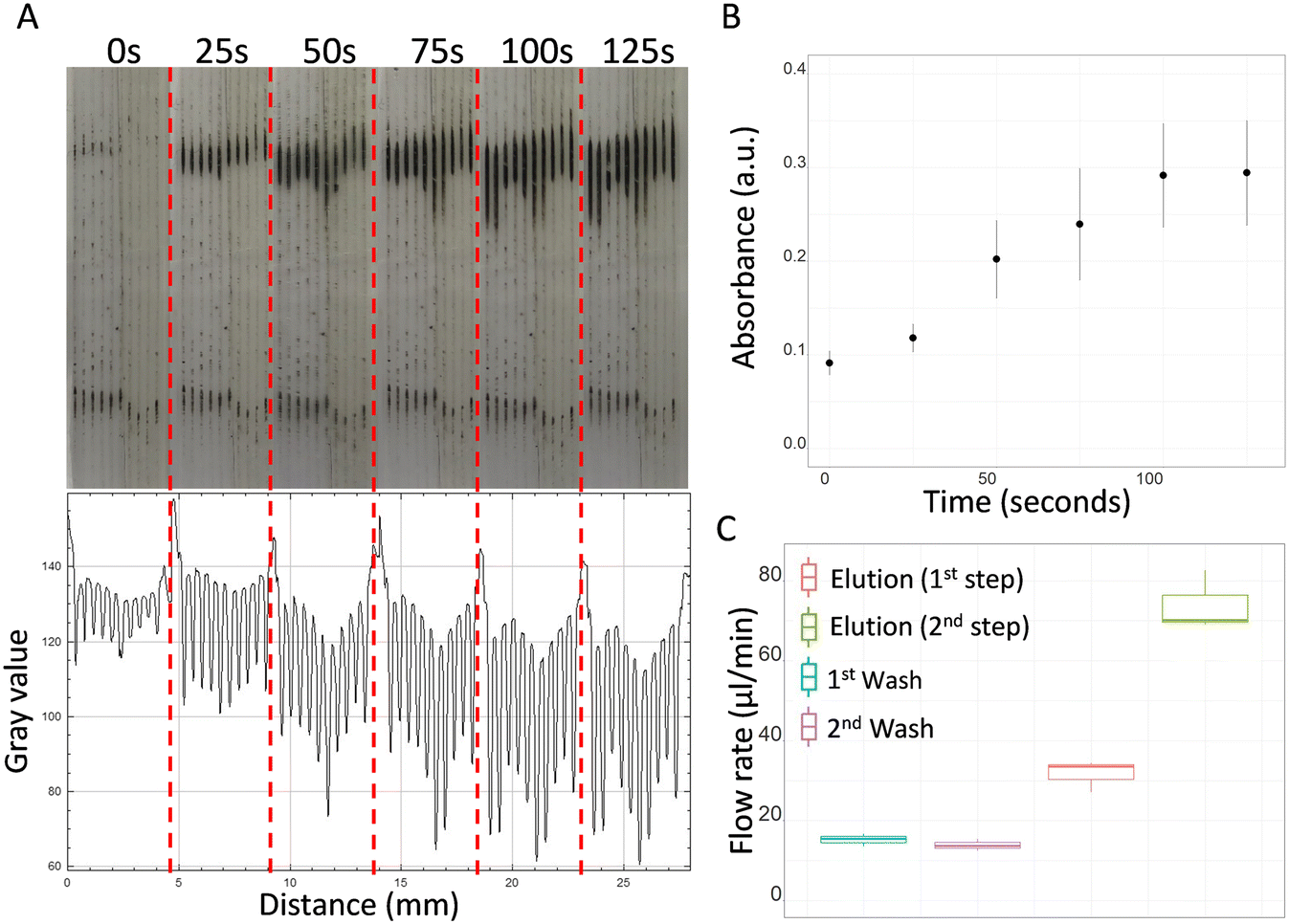 | ||
| Fig. 3 Isolation of magnetic beads in the microcapillary siphons. (A) Isolation from the priming step (0 seconds) up to 125 seconds of enrichment, forming clear lines near the upper part of the external magnet, and ImageJ absorbance profile obtained using RGB channels. (B) Mean absorbance (± SD of n = 3 technical replicates) of magnetic beads in the microcapillary siphons over time measured from ImageJ absorbance profiles. (C) Flow rates calculated for washing and elution steps. No substantial difference was observed between the first and second washing steps. The flow rate for the elution buffer significantly improved in the second step due to reduction in viscosity of the fluid within the microcapillaries. | ||
The flow rate of the washing buffer remained constant between the first and second washes (Fig. 3C). Since an average flow rate of 15.2 μl min−1 was observed for the first wash and 13.9 μl min−1 for the second wash, both steps were fixed to 4.0 minutes to allow the transfer of at least 50.0 μl of buffer per wash. It was observed that Q of elution buffer was significantly affected by the viscosity of the reagents previously loaded in the microcapillaries. Note all reagents used for DNA purification exhibited very distinct viscosities. In the first part of the elution step, where the microcapillaries were pre-loaded with washing buffer (i.e. with higher viscosity), the flow rate was found 2.1 times slower than the second part of the elution step, where the microcapillaries were already filled with elution buffer (i.e. lower viscosity fluid). For this reason, different incubation times were assigned to the two parts of the elution step: 70 seconds for the first part, to allow collection of 35.0 μl, and 23 seconds for the second, to allow collection of 28.0 μl.
3.2. Microfluidic vs. manual extraction
Both microcapillary siphon and manual protocols showed a successful DNA extraction, although absorbance values were in general found higher with the manual extraction (Fig. 4A and B). However, the normalised DNA concentration, obtained subtracting the negative control value from the DNA concentration of the bacterial sample, was 10% higher with the microcapillary siphons (Fig. 4C). The recovery efficiency for the microcapillary siphons was higher than 90% against the median value of 51.47 obtained with manual extraction (Fig. 4D). The ratio between the absorbance at 260 nm and 280 nm was 1.84 and 1.97, so within the expected range of 1.8–2.0 (Fig. 4E), for microfluidic and manual protocol respectively, although the ratios between 260 nm and 230 nm absorbances, also expected to be between 1.8–2.0 or higher, were 0.74 and 0.71 (Fig. 4F).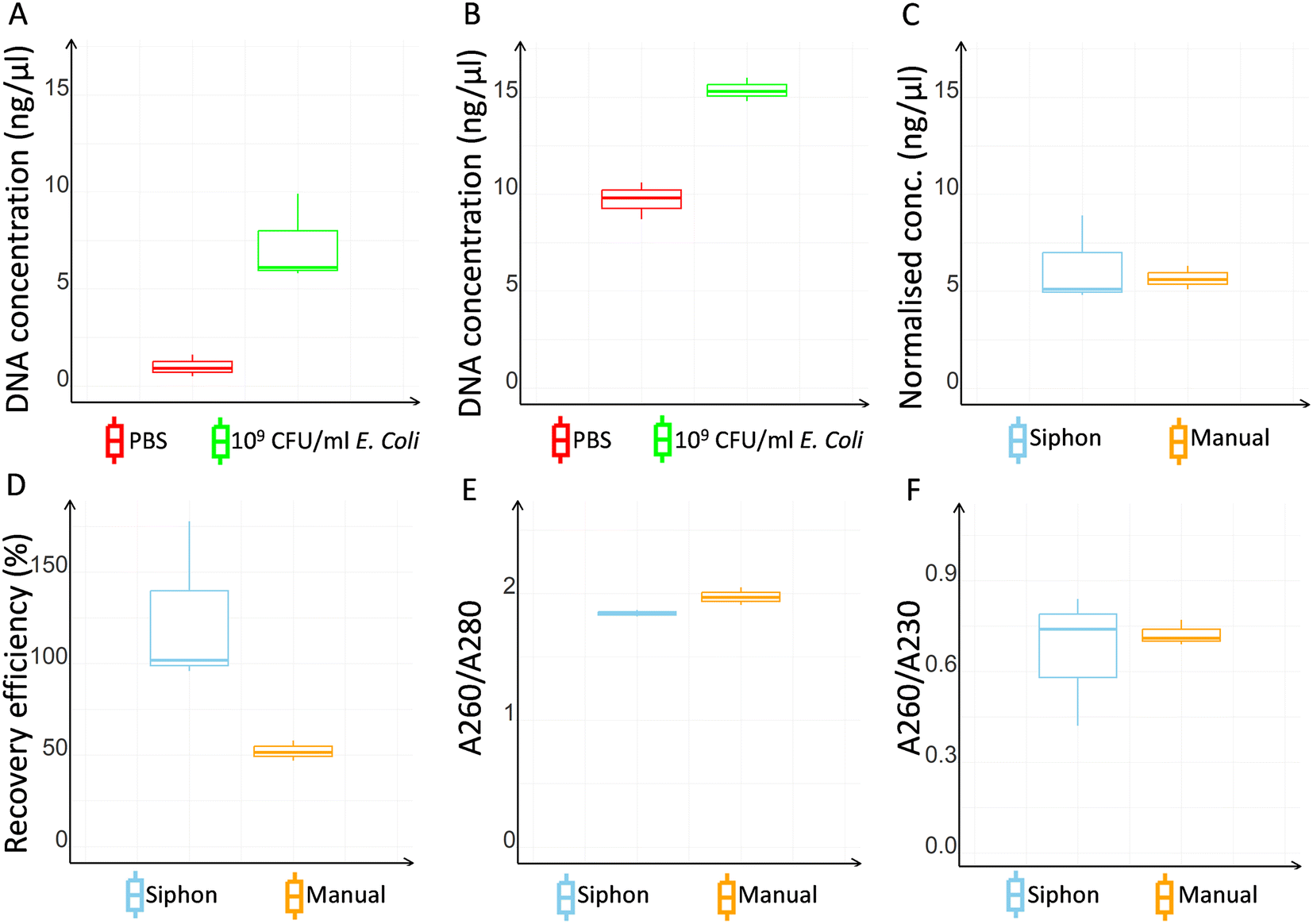 | ||
| Fig. 4 Microfluidic vs. manual DNA extraction. (A) and (B) show DNA quantification in PBS only (negative control) and PBS spiked with E. coli in the microcapillary siphon (A) or manual extraction (B). (C) DNA concentrations normalised to negative controls for microcapillary and manual extraction. (D) Recovery efficiency for microcapillary siphon and manual extraction. (E) 260 nm/280 nm absorbance for microcapillary siphon and manual extraction. (F) 260 nm/230 nm absorbance for microcapillary siphon and manual extraction. All data shown for at least N = 3 biological replicates. | ||
As the manual extraction yielded consistently higher blank readings with the NanoDrop, with an average value of 9.8 ng μl−1, we tested how extra wash steps affected absorbance in the spectrophotometer. The results showed strong noise for the manual extraction even when additional washes were performed, with average value of 8.0 ng μl−1 with one extra wash and 6.5 ng μl−1 with two extra washes (Fig. 5A), suggesting the presence of other molecules that interfere with the DNA absorbance at 260 nm, while the background signal remained very low for the microcapillary siphons (1.3 ng μl−1). The high absorbance in PBS extracts using the standard kit protocol suggested that contaminants, such as salts or washing agents, remained in the buffer even after extra washes. To evaluate the potential impact of the presence of contaminants in the efficiency of PCR amplification, real-time qPCR was performed to compare microcapillary siphon versus manual extraction (Fig. 5B). The microcapillary siphons yielded significantly lower threshold cycle (Ct), 28.8 against 35.4 for the manual extraction, for the same amount of starting material, indicating a more efficient amplification. River water, which contains more possible contaminants than PBS, yielded similar results for microcapillary siphons and manual extraction (average Ct was 28.0 and 33.3 respectively).
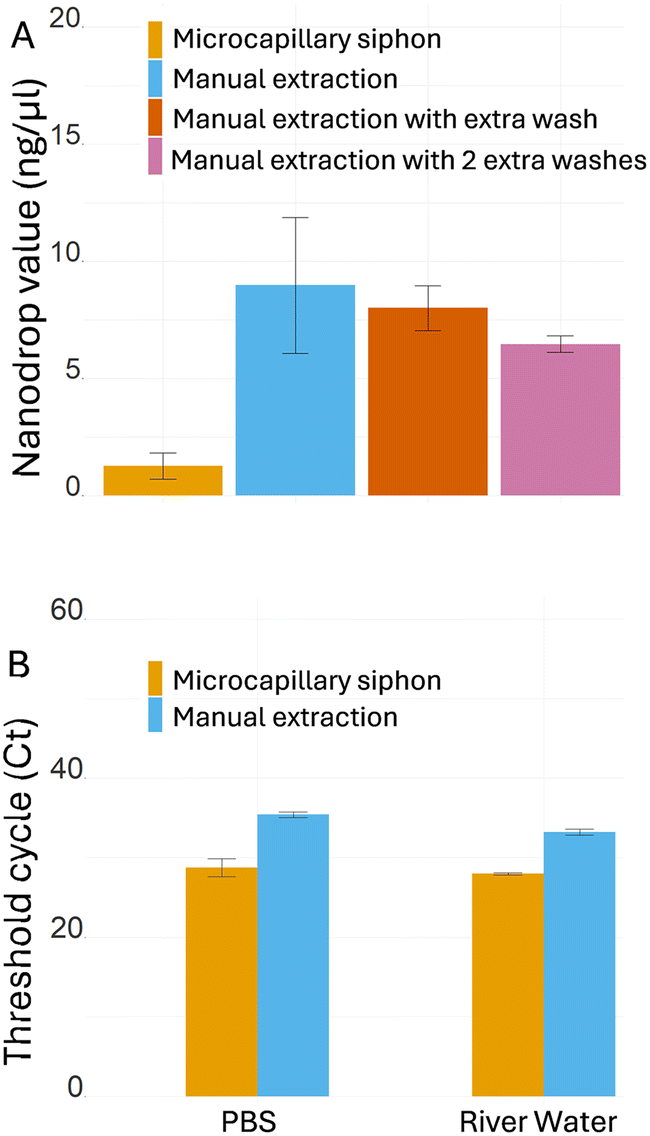 | ||
| Fig. 5 Washing efficiency and removal of PCR inhibitors. (A) DNA quantification of negative controls (PBS only) from microcapillary siphons, standard manual extraction and manual extraction with one or two extra washes (N = 3 biological replicates). (B) Real-time qPCR after microcapillary siphon and manual extraction from PBS and river water (n = 5 technical replicates). For both carrier liquids, the spiked DNA was detected much earlier when using the microcapillary siphon, leading to a significant reduction of the Ct. The graphs show means ± SD. | ||
3.3. Real-time qPCR following microfluidic extraction
To test the ability of the microcapillary siphons to extract bacterial DNA from various liquid matrices and to probe the limit of detection by RT-qPCR, E. coli was spiked into PBS, river water, and sheep's blood and subsequently extraction and amplification steps performed as explained previously. The median Ct values plotted against spiked-in CFUs showed a decrease with increasing numbers of bacteria, with a linear relationship between 102 and 105 CFUs for all three carrier liquids (Fig. 6A–C). Late amplification events were seen in negative control and in samples with low concentrations after 34 PCR cycles. | ||
| Fig. 6 Bacteria DNA detection extracted in microfluidic siphons using qPCR. Real-time qPCR for E. coli detection in PBS (A) sheep blood (B) and river water (C). The plots show the median threshold cycle (Ct) values by CFUs of spiked E. coli ± median absolute deviation of N = 3 biological replicates and n = 3 technical replicates. | ||
3.4. Reusability of microcapillary siphon for multiple extractions
Although clinical in vitro diagnostics is dominated by a disposable approach, in order to avoid cross-contamination of samples, near the source environmental monitoring can benefit from reduced costs in sample preparation, therefore we have tested the potential of reusing the microcapillary siphons in multiple extractions. Three microcapillary siphons were each used three times to extract DNA from three different concentrations of E. coli in PBS in varying sequences, with DNA quantified by real-time qPCR (Fig. 7A). Reusing microcapillary siphons that had previously handled high bacterial concentration did not affect subsequent experiments where the same was used for lower or null bacterial concentrations. Negative controls showed average Ct values of 33.54 and 35.44 when tested after high bacterial concentration (104.8 CFUs), compared to 35.15 when tested in unused siphon. Multiple operations of the same microcapillary siphon did not affect the PCR reactions, with no significant difference observed between first, second and third iterations with a median value of 24.51 for high bacterial concentration (104.8 CFUs), with a maximum and minimum of 25.35 and 24.32, and 31.65 for low bacterial concentration (102.8 CFUs), with a maximum and minimum of 32.61 and 31.17 (Fig. 7B). | ||
| Fig. 7 Reusability shown for three microcapillary siphons used for DNA purification from three samples in distinct order. (A) Average Ct values (± SD of n = 3 technical replicates) of real-time qPCR following DNA extraction from reused microcapillary siphons. The first microcapillary siphon (I) was used for 104.8 CFUs first, for negative control and finally for 102.8 CFUs. The second microcapillary siphon (II) was used for negative control first, and then for 102.8 CFUs and 104.8 CFUs. Siphon III was used for 102.8 CFUs first, followed by 104.8 CFUs and negative control. (B) Summary of the experiments with reused microcapillary siphons (N = 3 biological replicates and n = 3 technical replicates) showing Ct value remained similar independent on the order or sequence of CFUs processed in the microcapillary siphons. | ||
4. Discussion
The microcapillary siphons designed and fully characterised in this experimental work were intended to provide a portable solution for DNA extraction from complex matrices. For this reason, in addition to PBS buffer, whole blood and environmental river water were tested. Although technological solutions have already been reported in literature, some problems were yet to be addressed. For instance, some of the technologies previously proposed require the support of syringe pumps to drive samples from an inlet to an outlet, or centrifuges to operate pre-loaded disks, or even larger equipment to move liquid through a sophisticated system of valves. For in-field testing, reagents' storage is also an important factor to take into consideration, particularly when reagents need freezing or refrigeration. In addition, reagents like magnetic particles can precipitate very quickly and may require vigorous shaking before use, therefore not suitable for long on-chip storage. Finally, most of the devices were designed for testing single samples and may not be suitable for multiple testing, creating bulkiness if a large number of samples needs to be tested. Our microcapillary siphon overcomes all those limitations. No instruments are needed to drive liquid through the microcapillaries, and all reagents stored between 15 °C and 30 °C off-chip in 200 μl tubes, minimising refrigeration and facilitating manual mixing before loading. Reusing the same siphon multiple times has the potential to contribute to further reduction in waste and simplify storage and transportation for in-field testing. Microcapillary siphons can also be adapted for multiplex testing (Fig. S4†) while maintaining miniaturisation and portability.The microcapillary siphon has a simpler design in comparison to devices with multiple chambers, where magnetic beads are moved across with an external magnet, and is more suitable for scalable testing. Similar key performances were described in literature for pressure-driven preloaded cartridges,44 with comparable CFUs detected in downstream real-time PCR experiments. While the pressure-driven cartridges ensure a fully automated process, the siphon require reagents' tubes to be replaced manually for each step. However, the siphon showed increased miniaturisation, making it more suitable when a large number of samples is to be tested, and is reusable for at least three times, without carrying risk of cross-contamination or affecting test performance. Although reusable testing device may not be feasible in in vitro diagnostics, it could be a useful resource in other settings, such as environmental water monitoring or food testing, where results are less critical.
DNA extraction in microcapillary siphon devices was compared to the manufacturer's protocol to verify whether the reduced volumes and the small diameter of the microcapillaries had any impacts on the extraction. The microcapillary siphons outperformed the manual protocol on various aspects, providing a recovery efficiency of 90% or above versus 52% obtained with the manual extraction and matching the efficiency of centrifugal arrays and solid phase extraction previously reported for food and cell-free DNA testing.36,37,39,50Fig. 4D shows that some replicates yielded a recovery efficiency higher than 100%, but the median value showed an expected value. Recoveries higher than 100% were previously reported in literature also for other microfluidic systems.50 Note we have used a volume of eluent of 28.0 μl which matched the internal volume of the microcapillary siphon, so a recovery higher than 100% should not be linked to a potential concentration effect of the elution step. The high variability of DNA concentration and 260/230 nm ratio shown by the NanoDrop™ at high CFU ml−1 was not observed in real-time qPCR, where the data were highly consistent (Fig. 6). The ratio between the 260 nm and 280 nm absorbance, used to identify potential contaminants, was within the expected range (Fig. 4E), while the ratio between the 260 nm and 230 nm absorbance (Fig. 4F) was lower than expected for both microcapillary siphons and manual extraction, presumably due to the low DNA concentration used. Nevertheless, bacterial samples were successfully amplified via PCR, as shown by the gel electrophoresis in Fig. S5,† proving that the reaction was not inhibited.
Negative control from the manual extraction showed an average DNA concentration of 9.7 ng μl−1 even when no DNA was present, agreeing with standards already reported in literature.34 This higher noise in the control can be partially explained by the larger volume used by the manual protocol, that could lead to a higher accumulation of unwanted contaminants. However, the larger volume used to recover the DNA, four times larger than the volume used for the siphon, should reduce the gap by diluting the sample. Instead, we believe that the lower noise of the microcapillary siphons demonstrate the higher washing efficiency, and this could be explained by the siphon design that minimises the presence of dead volumes in the sample and the co-purification of undesired molecules but also due to viscous effect including higher shear in shorter diffusion distances in the microcapillaries. High efficiency of washing using a gravity-driven siphon was already reported by Reis et al.,60 showing how the concentration of dyes in microcapillaries sharply drops after washing with 0.05% Tween 20 in PBS. On the other hand, a full removal of liquid using a pipette tip is more challenging, leading to less efficient washing when performing the manual protocol. The higher washing efficiency was also confirmed by the real-time qPCR results, where the samples extracted with microcapillary siphon yielding a lower Ct (19% for PBS and 16% for river water) than samples extracted with the manual method. The lower Ct obtained with the microcapillary siphons is a clear demonstration of a more efficient removal of PCR inhibitors, that caused a strong delay in the DNA amplification when using the manual extraction. The higher washing efficiency enabled by a flow-through configuration, large surface area, and short diffusion distances were very likely the main reasons behind the higher recovery efficiency obtained with the microcapillary siphon, with the increased purity of the elution buffer improving the DNA recovery from the beads' surface. In the manual procedure, some beads can be accidentally aspirated and discarded by the pipette tips during manual washes, causing loss of DNA and drop in recovery efficiency.
The efficacy of the microcapillary siphon was tested on PBS, sheep blood and river water collected downstream of a wastewater treatment. The real-time qPCR results suggested that it is a viable method for bacterial detection from these matrices. As expected, a linear relationship was observed for Ct and CFUs between 102 and 105 CFUs, similar range compared to literature.42,44 The experiments showed a late increase of fluorescence for low CFUs and negative controls potentially due to events of non-specific amplification, as suggested by the melt region curves in Fig. S6–S8.† Non-specific amplification products are common during the last cycles of real-time qPCR, particularly when using DNA intercalant such as SYBR green rather than more specific solutions such as TaqMan probes; in particular, these events are usually observed in sample at low concentration.
The microcapillary siphon proved to be a versatile solution for nucleic acid extraction. We opted for melt-extruded FEP-Teflon® MCF due to its optical transparency, with a refractive index of 1.34–1.35,65,66 hoping in future to combine DNA extraction and optical detection in MCF. The efficient removal of inhibitors and the suitability for both blood and environmental water suggests its hypothetical application in various settings, ranging from pathogen detection for human and animal diagnostics67,68 to analysis of cell-free DNA,69,70 eDNA for metagenomics studies71,72 and detection of pathogens and antimicrobial-resistance genes detection in water.73,74 DNA testing is often challenged by the presence of reaction inhibitors. For instance, immunoglobulins G in blood bind single-strand DNA, leading to delays in DNA amplification in real-time approaches, while haemoglobin inhibits the DNA polymerase and interact with fluorescent dyes.75 For environmental water, the inhibition is strictly dependent on the sample's nature, with humic acid76,77 being one of the main factors of reaction's delay.
The microcapillary siphon concept has the potential to pave the way for future design of integrated portable devices for NAATs. We have demonstrated that the use of microcapillaries in our device is compatible with magnetic beads, that currently represent the most suitable solution for portable devices since minimal equipment is required. The use of small bore microcapillaries reduces volumes and costs, without compromising assay performance, and our model does not require liquid pumps or complex designs, being fully compatible with near-the-source or in-field testing. Our system can be easily coupled with portable nucleic acid detection for a fully integrated device; loop-mediated isothermal amplification (LAMP) represents the most promising solution, potentially reaching very low limits of detection in short testing time.
5. Conclusions
Gravity-driven microcapillary siphons were tested for the first time for DNA extraction, representing an innovative solution for the design of portable and integrated DNA testing without prior sample cleanup and concentration. The flow of reagents is driven solely by gravity and molecular cohesion forces, therefore not requiring any syringe pumps, electric patterns nor articulated systems of valves. Miniaturisation of the sample and reagent volumes was found not to compromise assay's performance. DNA purification in microcapillary siphon devices outperformed manual extraction, with a high recovery efficiency (>90%), a better DNA quality and a higher removal of PCR inhibitors. The method tested on PBS and more complex liquids such as whole sheep blood and river water demonstrated the methodology is suitable for both complex matrices with potential interferences and for low bacterial concentrations. Furthermore, each microcapillary siphon was demonstrated to be reusable for at least three times without affecting the test performance. The sensitivity could be improved by combining this method with pre-concentration steps or increasing the number of beads captured; furthermore, given the simplicity of its design, it could be coupled with isothermal DNA amplification to obtain a fully integrated device. Portable DNA testing has a wide spectrum of applications, varying from diagnostics in human tissues, such as blood and urines, to environmental matrices, including sea and freshwater, but also wastewater. The use of inexpensive, power-free, microfluidic siphon devices brings new bioanalytical capabilities, cutting down the costs of magnetic bead purification, and freeing routine DNA testing from the need of trained personnel, laboratory facilities and expensive equipment. Future work will explore the potential of integration of DNA extraction with in-field nucleic acid amplification.Data availability
The data supporting the findings of this study are included within the manuscript and the ESI† provided. All data can be shared by the authors upon request.Author contributions
Crescenzo Ianniello: conceptualization, methodology, formal analysis, investigation, writing – original draft, visualization. Julia Sero: resources, writing – review & editing. David Gough: conceptualization, methodology. Barbara Kasprzyk-Hordern: conceptualization, resources, writing – review & editing, funding acquisition. Nuno M. Reis: conceptualization, methodology, resources, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to University of Bath for financial support through funding a Ph.D. scholarship and funding by the NATO Science for Peace and Security Programme under grant reference MYP G6305. NMR's contribution has also benefited from funding from the European Research Executive Agency (REA), under ERA Chair project REACTORS 5.0, grant agreement 101186592.References
- M. Chen, X. Lan, L. Zhu, P. Ru, W. Xu and H. Liu, Foods, 2022, 11, 2675, DOI:10.3390/foods11172675.
- M. W. McCarthy and T. J. Walsh, Expert Rev. Mol. Diagn., 2016, 16, 1025–1036 CrossRef CAS PubMed.
- K. Sachse, Applied Biochemistry and Biotechnology - Part B Molecular Biotechnology, 2004, 26, 61–79 CAS.
- B. Cindy, J. Simon Neil, S. Tiffany, M. Haylea, S. Todd, K. John Kenneth and B. Oliver, Commun. Biol., 2021, 4, 236, DOI:10.1038/s42003-021-01760-8.
- G.-J. Jeunen, U. von Ammon, H. Cross, S. Ferreira, M. Lamare, R. Day, J. Treece, X. Pochon, A. Zaiko, N. J. Gemmell, N. J. Gemmell and J.-A. L. Stanton, Environ. DNA, 2022, 4, 1420–1433, DOI:10.1002/edn3.356.
- N. Sims, A. Kannan, E. Holton, K. Jagadeesan, L. Mageiros, R. Standerwick, T. Craft, R. Barden, E. J. Feil and B. Kasprzyk-Hordern, Environ. Pollut., 2023, 333, 122020, DOI:10.1016/j.envpol.2023.122020.
- M. R. Williams, R. D. Stedtfeld, X. Guo and S. A. Hashsham, Water Environ. Res., 2016, 88, 1951–1967 CrossRef CAS PubMed.
- L. Xu, J. Zang, W. Cong, E. Holton, L. Jiang, S. K. Sheppard, Y. Wang, N. Wang, J. Weeks, C. Fu, H. Lambert and B. Kasprzyk-Hordern, Water Res., 2022, 222, 118942, DOI:10.1016/j.watres.2022.118942.
- X. Hua, E. Yang, W. Yang, R. Yuan and W. Xu, Chem. Commun., 2019, 55, 12463–12466 RSC.
- G. Cao, Y. Qiu, K. Long, Y. Xiong, M. Shi, J. Yang, Y. Li, F. Nie, D. Huo and C. Hou, Microchim. Acta, 2022, 189, 342, DOI:10.1007/s00604-022-05390-7.
- M. G. Mills, K. B. Juergens, J. P. Gov, C. J. McCormick, R. Sampoleo, A. Kachikis, J. K. Amory, F. C. Fang, A. C. Pérez-Osorio, N. A. P. Lieberman, N. A. P. Lieberman and A. L. Greninger, J. Clin. Virol., 2023, 159, 105373, DOI:10.1016/j.jcv.2022.105373.
- K.-A. Hwang, J. H. Ahn and J.-H. Nam, J. Microbiol., 2018, 56, 593–599 CrossRef CAS PubMed.
- S. Jarju, R. D. Wenlock, M. Danso, D. Jobe, Y. J. Jagne, A. Darboe, M. Kumado, Y. Jallow, M. Touray, E. A. Ceesay, B. Kampmann and T. I. de Silva, Nat. Commun., 2024, 15, 3814, DOI:10.1038/s41467-024-48098-3.
- H. Schenk, P. Heidinger, H. Insam, N. Kreuzinger, R. Markt, F. Nägele, H. Oberacher, C. Scheffknecht, M. Steinlechner, G. Vogl, A. O. Wagner and W. Rauch, Sci. Total Environ., 2023, 873, 162149, DOI:10.1016/j.scitotenv.2023.162149.
- L. J. Reynolds, G. Gonzalez, L. Sala-Comorera, N. A. Martin, A. Byrne, S. Fennema, N. Holohan, S. R. Kuntamukkula, N. Sarwar, T. M. Nolan, C. De Gascun and W. G. Meijer, Sci. Total Environ., 2022, 838, 155828, DOI:10.1016/j.scitotenv.2022.155828.
- B. Kasprzyk-Hordern, B. Adams, I. D. Adewale, F. O. Agunbiade, M. I. Akinyemi, E. Archer, F. A. Badru, J. Barnett, I. J. Bishop, M. Di Lorenzo, G. Wolfaardt and C. O. Yinka-Banjo, Environ. Int., 2022, 161, 107143, DOI:10.1016/j.envint.2022.107143.
- B. Na, J. Park, S. Park, E. Park, J. Jang, Y.-H. Kim, J. Lee and H.-S. Chung, Sci. Rep., 2025, 15, 4167, DOI:10.1038/s41598-025-87225-y.
- A. J. Colbert, D. H. Lee, K. N. Clayton, S. T. Wereley, J. C. Linnes and T. L. Kinzer-Ursem, Anal. Chim. Acta, 2022, 1203, 339702, DOI:10.1016/j.aca.2022.339702.
- S. Papamatthaiou, J. Boxall-Clasby, E. J. A. Douglas, P. Jajesniak, H. Peyret, J. Mercer-Chalmers, V. K. S. Kumar, G. P. Lomonossoff, J. Reboud, M. Laabei, B. Kasprzyk-Hordern and D. Moschou, Lab Chip, 2023, 23, 4400–4412 RSC.
- C. Schrader, A. Schielke, L. Ellerbroek and R. Johne, J. Appl. Microbiol., 2012, 113, 1014–1026 CrossRef CAS PubMed.
- W. A. Al-Soud and P. Rådström, J. Clin. Microbiol., 2001, 39, 485–493 CrossRef CAS PubMed.
- M. E. García, J. L. Blanco, J. Caballero and D. Gargallo-Viola, J. Clin. Microbiol., 2002, 40, 1567–1568 CrossRef PubMed.
- C. Fuschi, H. Pu, M. Negri, R. Colwell and J. Chen, ACS ES&T Water, 2021, 1, 1352–1362 Search PubMed.
- J. Saththasivam, S. S. El-Malah, T. A. Gomez, K. A. Jabbar, R. Remanan, A. K. Krishnankutty, O. Ogunbiyi, K. Rasool, S. Ashhab, S. Rashkeev, J. Lawler and K. A. Mahmoud, Sci. Total Environ., 2021, 774, 145608, DOI:10.1016/j.scitotenv.2021.145608.
- B. Joseph-Duran, A. Serra-Compte, M. Sàrrias, S. Gonzalez, D. López, C. Prats, M. Català, E. Alvarez-Lacalle, S. Alonso and M. Arnaldos, Sci. Rep., 2022, 12, 15073, DOI:10.1038/s41598-022-18518-9.
- V. Kisand, P. Laas, K. Palmik-Das, K. Panksep, H. Tammert, L. Albreht, H. Allemann, L. Liepkalns, K. Vooro, C. Ritz, V. Hauryliuk and T. Tenson, Water Res., 2023, 231, 119617, DOI:10.1016/j.watres.2023.119617.
- N. Z. Masri, K. G. Card, E. A. Caws, A. Babcock, R. Powell, C. J. Lowe, S. Donovan, S. Norum, S. Lyons, S. De Pol, T. M. Fyles and H. L. Buckley, Environ. Adv., 2022, 9, 100310, DOI:10.1016/j.envadv.2022.100310.
- C. Rock, A. Alum and M. Abbaszadegan, Appl. Environ. Microbiol., 2010, 76, 8102–8109 CrossRef CAS PubMed.
- Y.-S. C. Shieh, D. Wait, L. Tai and M. D. Sobsey, J. Virol. Methods, 1995, 54, 51–66 CrossRef CAS PubMed.
- T. Demeke and R. P. Adams, BioTechniques, 1992, 12, 332–334 CAS.
- L. Rossen, P. Nørskov, K. Holmstrøm and O. F. Rasmussen, Int. J. Food Microbiol., 1992, 17, 37–45 CrossRef CAS PubMed.
- H. A. Erlich, PCR technology: Principles and applications for DNA amplification, 2015 Search PubMed.
- A. Kuffel, A. Gray and N. N. Daeid, Int. J. Leg. Med., 2021, 135, 63–72 CrossRef PubMed.
- S. M. Azimi, G. Nixon, J. Ahern and W. Balachandran, Microfluid. Nanofluid., 2011, 11, 157–165 CrossRef CAS.
- R. N. Deraney, L. Schneider and A. Tripathi, Analyst, 2020, 145, 2412–2419 RSC.
- K. Perez-Toralla, I. Pereiro, S. Garrigou, F. Di Federico, C. Proudhon, F.-C. Bidard, J.-L. Viovy, V. Taly and S. Descroix, Sens. Actuators, B, 2019, 286, 533–539 CrossRef CAS.
- X. Xiang, J. Lu, M. Tao, X. Xu, Y. Wu, Y. Sun, S. Zhang, H. Niu, Y. Ding and Y. Shang, Food Chem., 2024, 443, 138507, DOI:10.1016/j.foodchem.2024.138507.
- S. Petralia, E. L. Sciuto and S. Conoci, Analyst, 2017, 142, 140–146 RSC.
- A. Jagannath, Y. Li, H. Cong, J. Hassan, G. Gonzalez, W. Wang, N. Zhang and M. D. Gilchrist, ACS Appl. Mater. Interfaces, 2023, 15, 31159–31172 CrossRef CAS PubMed.
- B. Lin, Z. Geng, Y. Chen, W. Zeng, B. Li, Y. Zhang and P. Liu, Lab Chip, 2023, 23, 1794–1803 RSC.
- C. D. M. Campos, S. S. T. Gamage, J. M. Jackson, M. A. Witek, D. S. Park, M. C. Murphy, A. K. Godwin and S. A. Soper, Lab Chip, 2018, 18, 3459–3470 RSC.
- C. E. Jin, T. Y. Lee, B. Koo, K.-C. Choi, S. Chang, S. Y. Park, J. Y. Kim, S.-H. Kim and Y. Shin, Anal. Chem., 2017, 89, 7502–7510 CrossRef CAS PubMed.
- Y. Xiao, M. Zhou, C. Liu, S. Gao, C. Wan, S. Li, C. Dai, W. Du, X. Feng, Y. Li, P. Chen and B.-F. Liu, Biosens. Bioelectron., 2024, 255, 116240, DOI:10.1016/j.bios.2024.116240.
- L. T. H. Thang, W. Han, J. Shin and J. H. Shin, Sens. Actuators, B, 2023, 375, 132948, DOI:10.1016/j.snb.2022.132948.
- A. Hatami, M. Saadatmand and M. Garshasbi, Talanta, 2024, 267, 125245, DOI:10.1016/j.talanta.2023.125245.
- E. L. Sciuto, S. Petralia, G. Calabrese and S. Conoci, Biotechnol. Bioeng., 2020, 117, 1554–1561 CrossRef CAS PubMed.
- H. V. Nguyen, V. D. Nguyen, E. Y. Lee and T. S. Seo, Biosens. Bioelectron., 2019, 136, 132–139 CrossRef CAS PubMed.
- B. H. Park, S. J. Oh, J. H. Jung, G. Choi, J. H. Seo, D. H. Kim, E. Y. Lee and T. S. Seo, Biosens. Bioelectron., 2017, 91, 334–340 CrossRef CAS PubMed.
- G. Czilwik, T. Messinger, O. Strohmeier, S. Wadle, F. Von Stetten, N. Paust, G. Roth, R. Zengerle, P. Saarinen, J. Niittymäki, J. O'Leary and D. Mark, Lab Chip, 2015, 15, 3749–3759 RSC.
- F. Schlenker, P. Juelg, J. Lüddecke, N. Paust, R. Zengerle and T. Hutzenlaub, Analyst, 2023, 148, 932–941 RSC.
- L. M. Dignan, M. S. Woolf, C. J. Tomley, A. Q. Nauman and J. P. Landers, Anal. Chem., 2021, 93, 7300–7309 CrossRef CAS PubMed.
- R. Wimbles, L. M. Melling, B. Cain, N. Davies, J. Doherty, B. Johnson and K. J. Shaw, Ecol. Evol., 2021, 11, 1535–1543 CrossRef PubMed.
- J. Yin, Z. Zou, Z. Hu, S. Zhang, F. Zhang, B. Wang, S. Lv and Y. Mu, Lab Chip, 2020, 20, 979–986 RSC.
- S. Sharma, E. Thomas, M. Caputi and W. Asghar, Biosensors, 2022, 12, 298, DOI:10.3390/bios12050298.
- J. Yin, J. Hu, J. Sun, B. Wang and Y. Mu, Analyst, 2019, 144, 7032–7040 RSC.
- A. Sun, P. Vopařilová, X. Liu, B. Kou, T. Řezníček, T. Lednický, S. Ni, J. Kudr, O. Zítka, Z. Fohlerová, H. Zhang and P. Neužil, Microsyst. Nanoeng., 2024, 10, 66, DOI:10.1038/s41378-024-00677-6.
- S. Hu, Y. Jie, K. Jin, Y. Zhang, T. Guo, Q. Huang, Q. Mei, F. Ma and H. Ma, Biosensors, 2022, 12, 324, DOI:10.3390/bios12050324.
- K. Yang, J. Zhou, J. Zhao, L. Liu, C. Hua, C. Hong, M. Wang, A. Hu, W. Zhang, J. Cui, Y. Liu and L. Zhu, Sens. Actuators, B, 2024, 419, 136413, DOI:10.1016/j.snb.2024.136413.
- J. Reboud, G. Xu, A. Garrett, M. Adriko, Z. Yang, E. M. Tukahebwa, C. Rowell and J. M. Cooper, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4834–4842 CrossRef CAS PubMed.
- N. M. Reis, S. H. Needs, S. M. Jegouic, K. K. Gill, S. Sirivisoot, S. Howard, J. Kempe, S. Bola, K. Al-Hakeem, I. M. Jones, C. Puttikhunt and A. D. Edwards, ACS Sens., 2021, 6, 4338–4348 CrossRef CAS PubMed.
- C.-H. Liao and L. M. Shollenberger, Lett. Appl. Microbiol., 2003, 37, 45–50 CrossRef PubMed.
- N. M. Reis, J. Pivetal, A. L. Loo-Zazueta, J. M. S. Barros and A. D. Edwards, Lab Chip, 2016, 16, 2891–2899 RSC.
- N. Akhras, G. Singh, K. K. Gill, S. Bola, K. Al-Hakeem and N. M. Reis, Front. Chem. Eng., 2024, 6, 1443949, DOI:10.3389/fceng.2024.1443949.
- A. Salman, H. Carney, S. Bateson and Z. Ali, Talanta, 2020, 207, 120303, DOI:10.1016/j.talanta.2019.120303.
- N. M. Reis, J. Pivetal, A. L. Loo-Zazueta, J. M. S. Barros and A. D. Edwards, Lab Chip, 2016, 16, 2891–2899 RSC.
- A. I. Barbosa, A. P. Castanheira, A. D. Edwards and N. M. Reis, Lab Chip, 2014, 14, 2918–2928 RSC.
- H. Y. A. H. Mahmoud, T. Tanaka, A. O. Ali and W. F. A. Emeish, BMC Vet. Res., 2024, 20, 260, DOI:10.1186/s12917-024-04109-5.
- T. Bunse, A. Ziel, P. Hagen, G. Rigopoulos, U. Yasar, H. Inan, G. Köse, U. Eigner, R. Kaiser, N. Bardeck, U. Protzer and J. M. Wettengel, Med. Microbiol. Immunol., 2024, 213, 18, DOI:10.1007/s00430-024-00800-4.
- X.-L. Cui, J. Nie, H. Zhu, K. Kowitwanich, A. V. Beadell, D. C. West-Szymanski, Z. Zhang, U. Dougherty, A. Kwesi, Z. Deng, M. Chen and C. He, Genome Biol., 2024, 25, 157, DOI:10.1186/s13059-024-03262-2.
- R. Stokowski, E. Wang, K. White, A. Batey, B. Jacobsson, H. Brar, M. Balanarasimha, D. Hollemon, A. Sparks, K. Nicolaides, K. Nicolaides and T. J. Musci, Prenatal Diagn., 2015, 35, 1243–1246 CrossRef CAS PubMed.
- M. Osathanunkul and C. Suwannapoom, Sci. Rep., 2024, 14, 8885, DOI:10.1038/s41598-024-58752-x.
- C. O. Carvalho, W. Gromstad, M. Dunthorn, H. E. Karlsen, A. Schrøder-Nielsen, J. S. Ready, T. Haugaasen, G. Sørnes, H. de Boer and Q. Mauvisseau, Sci. Rep., 2024, 14, 10154, DOI:10.1038/s41598-024-60762-8.
- J. Cheng, P. Wang, J.-F. Ghiglione, L. Liu, Z. Cai, J. Zhou and X. Zhu, J. Hazard. Mater., 2024, 462, 132741, DOI:10.1016/j.jhazmat.2023.132741.
- T. D. Bedane, B. Megersa, F. Abunna, H. Waktole, F. T. Woldemariyam, M. Tekle, E. Shimelis and F. D. Gutema, Sci. Rep., 2024, 14, 12461, DOI:10.1038/s41598-024-61810-z.
- M. Sidstedt, J. Hedman, E. L. Romsos, L. Waitara, L. Wadsö, C. R. Steffen, P. M. Vallone and P. Rådström, Anal. Bioanal. Chem., 2018, 410, 2569–2583 CrossRef CAS PubMed.
- C. N. Albers, A. Jensen, J. Bælum and C. S. Jacobsen, Geomicrobiol. J., 2013, 30, 675–681 CrossRef CAS.
- K. Uchii, H. Doi, T. Okahashi, I. Katano, H. Yamanaka, M. K. Sakata and T. Minamoto, Environ. DNA, 2019, 1, 359–367 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00735b |
| This journal is © The Royal Society of Chemistry 2025 |