
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInnovations in non-flammable and flame-retardant electrolytes for safer lithium-ion batteries
Won-Jang
Cho
a,
Yoo Jeong
Huh
a,
Soyeon
Choi
b,
Uddhav
Kulkarni
a,
Kiran P.
Shejale
a and
Gi-Ra
Yi
 *ab
*ab
aDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Republic of Korea. E-mail: yigira@postech.ac.kr
bDepartment of Battery Engineering, Graduate Institute of Ferros & Eco Materials Technology (GIFT), Pohang University of Science and Technology (POSTECH), Pohang, Republic of Korea
First published on 16th July 2025
Abstract
Lithium-ion batteries (LIBs) are essential energy storage solutions that support advancements in modern electronic applications. However, the inherent flammability of liquid electrolytes significantly increases fire safety risks, posing a significant barrier to their wider adoption. Although substantial research efforts have focused on developing non-flammable or flame-retardant electrolytes, the simultaneous attainment of optimal cell-level safety and excellent electrochemical performance remains a complex challenge that requires further exploration. A deep and comprehensive understanding of safer electrolyte designs is essential for the development of more effective strategies aimed at improving LIB safety. This review critically examines the latest advancements in non-flammable and flame-retardant electrolytes, covering areas such as molecular engineering, functional additive incorporation, and complex modifications to solvation structures. Furthermore, we provide valuable insights into future research directions, emphasizing the urgent need for the creation of multi-functional electrolyte solutions that seamlessly integrate safety, stability, and superior electrochemical performance.
1. Introduction
Lithium-ion batteries (LIBs) have become the cornerstone of modern energy storage technologies, playing a crucial role in portable electronics, electric vehicles, and grid-scale energy storage systems.1–5 Since their commercialization in the 1990s, LIBs have undergone significant advancements driven by their high energy density, long cycle life, and lightweight design. These attributes have positioned LIBs as the preferred choice for efficient and reliable energy storage applications.6 However, the widespread adoption of LIBs has been accompanied by critical safety concerns, particularly related to thermal runaway, battery fires, and explosions.7–13 Thermal runaway is a self-accelerating, exothermic process that occurs when the heat generation rate within a battery exceeds its ability to dissipate heat. This rapid and uncontrolled temperature rise can initiate a series of decomposition reactions, leading to gas evolution, electrode degradation, and electrolyte combustion. The severity of thermal runaway depends on multiple factors, including the battery chemistry, state of charge, and external operating conditions. The primary triggers of this failure mode include mechanical abuse, such as external forces that lead to internal short circuits and localized overheating; electrical abuse, including overcharging or deep discharging, which destabilizes the electrode–electrolyte interface and induces lithium plating or dendrite formation; and thermal abuse, where exposure to elevated temperatures accelerates electrolyte degradation and initiates exothermic side reactions that further propagate heat generation. The occurrence of thermal runaway not only threatens device and user safety but also poses significant challenges for large-scale LIB deployment, particularly in electric vehicles and energy storage systems. This issue has contributed to the so-called “battery chasm,” a term describing the widespread concerns and hesitations regarding LIB safety that hinder their broader adoption despite their potential to drive sustainability. Despite extensive efforts, achieving a complete understanding and prevention of thermal runaway remains highly complex. These events occur within milliseconds to seconds, making real-time monitoring and analysis challenging.14–17 Furthermore, post-incident investigations are hindered by material consumption, phase changes, and irreversible reactions, which obscure the precise sequence of chemical and physical events leading to failure.Among the various components of LIBs, the electrolyte plays a central role in determining battery safety.18–24 As an ion-conducting medium, the electrolyte facilitates charge transport between the anode and cathode. However, conventional liquid electrolytes, primarily based on carbonate solvents such as ethylene carbonate (EC), propylene carbonate (PC), diethyl carbonate (DEC), and dimethyl carbonate (DMC), are inherently flammable due to their low flash points and high vapor pressures. Under thermal abuse conditions, these electrolytes undergo exothermic decomposition, generating highly reactive radicals and flammable gases such as hydrogen, methane, and carbon monoxide, which exacerbate fire hazards. To mitigate these risks, research has focused on developing safer electrolyte formulations. Solid-state electrolytes, including ceramic, polymer, and hybrid systems, eliminate liquid components and thereby reduce flammability risks.25,26 However, these systems suffer from low room-temperature ionic conductivity, poor electrode–electrolyte interfacial contact, and mechanical brittleness, which limit their commercial viability. In contrast, non-flammable and flame-retardant liquid electrolytes have been widely investigated as an alternative approach, with strategies such as the incorporation of high flash point solvents, ionic liquids, and flame-retardant additives. Phosphate-based co-solvents and additives have shown significant promise in enhancing electrolyte non-flammability. However, these formulations often lead to the formation of a solid electrolyte interphase layer, which increases ionic resistance and may hinder long-term battery performance. In addition to solvent engineering, recent efforts have explored modifications to the solvation structure, fluorinated solvents, and radical scavengers as potential pathways to improve electrochemical stability while simultaneously addressing safety concerns.
This review provides a comprehensive analysis of recent advancements in non-flammable and flame-retardant electrolytes, emphasizing their role in enhancing LIB safety. The discussion begins with an in-depth examination of thermal runaway mechanisms, outlining their sequential stages, triggering factors, and analytical techniques used to study these high-speed failure events. The review then explores the latest developments in non-flammable electrolyte formulations, including the design principles of flame-retardant solvents, functional additives, and solvation structure modifications. Finally, it presents a forward-looking perspective on the role of electrolyte engineering in preventing thermal runaway, highlighting the key challenges, opportunities, and future research directions required to develop safer, high-performance lithium-ion batteries. By addressing the intricate interplay between electrolyte chemistry and thermal stability, this review contributes to the ongoing efforts toward safer, more reliable energy storage systems. These advancements will ultimately support the large-scale adoption of electric vehicles, renewable energy integration, and next-generation power solutions.
2. Thermal runaway
2.1 Basic mechanism of thermal runaway
Fig. 1 comprehensively illustrates the sequential process known as thermal runaway (TR). This phenomenon is characterized by an uncontrollable and exponential increase in temperature, primarily resulting from a series of exothermic chemical reactions occurring within the electrochemical cell.7–17 The onset of thermal runaway is a critical safety concern, particularly in high-energy-density systems such as lithium-ion batteries. Typically, the thermal runaway process can be delineated into three distinct and significant stages, each with unique thermodynamic properties and implications for system stability.7 Understanding these stages is essential for the development of effective prevention and mitigation strategies to ensure the safe operation of energy storage systems. | ||
| Fig. 1 Schematic illustration of the sequence of events during thermal runaway. | ||
As the temperature approaches ∼100 °C, the solid electrolyte interphase (SEI) layer continuously decomposes and regenerates, observed in DSC as an exothermic reaction peak.29,30 While inorganic components (e.g., LiF, Li2O, and Li2CO3) remain thermally stable, organic components (e.g., alkyl carbonates, RCO3) decompose at elevated temperatures, generating gases such as CxHy, H2, and CO2, which further accelerate thermal runaway. Among these, H2 and CxHy are particularly hazardous due to their low ignition temperatures, high diffusivity, and high thermal potentials. The accumulation of these gases within the cell leads to internal pressures building up and creates a flammable atmosphere that can be easily ignited in the next stages. Furthermore, these gaseous intermediates can react with the cathode surface through redox pathways, accelerating thermal feedback and propagation. The simultaneous regeneration and decomposition of the organic SEI layer reinforces a self-amplifying reaction loop. As a result, the system rapidly transitions into Stage II.31–33 Recently, Jo et al.33 found that the evolution of ethylene (C2H4) gas during SEI decomposition is a critical initiator of TR in its early stages. The breakdown of lithium alkyl carbonates within the SEI proceeds via the following equation 1.
 | (1) |
As schematically illustrated in Fig. 2a, this C2H4 generation triggers a sequence of crosstalk reactions between the anode and cathode that occur within the temperature range of 120–200 °C. In single-electrode systems, C2H4 evolves primarily from the anode through SEI decomposition, while oxygen release from the cathode remains negligible. However, in full-cell configurations, the C2H4 diffuses toward the cathode, where it is oxidized by lattice oxygen, thereby accelerating O2 release. This released O2 then migrates back to the anode and further promotes the formation of C2H4. This bidirectional chemical exchange forms a dynamic redox feedback loop, intensifying as the temperature exceeds 150 °C. Such gas-phase interplay between the electrodes amplifies structural degradation, accelerates cathode phase transitions, and significantly enhances heat generation. Compared to cathode-only or anode-only systems, this synergistic crosstalk leads to an earlier onset of exothermic events in full cells.
 | ||
| Fig. 2 (a) Gas evolution on the graphite anode during thermal runaway (adapted from ref. 33, under the terms of the Creative Commons Attribution 4.0 License, CC BY 4.0, https://creativecommons.org/licenses/by/4.0/, copyright 2024, Wiley). (b) DSC results for exothermic heat reaction of lithiated graphite anion (reproduced from ref. 36 with permission from Elsevier, copyright 2013). (c) Comparing total heat release and its reaction rate between LFP and NCM811 cathode (adapted from ref. 47, under the terms of the Creative Commons Attribution 4.0 License, CC BY 4.0, https://creativecommons.org/licenses/by/4.0/, copyright 2024, Elsevier). | ||
A key event at this stage is the thermal decomposition of the liquid electrolyte, including carbonate solvents such as EC, DMC, or DEC. These solvents degrade into flammable gases, heat, and reactive intermediates such as free radicals, further fuelling the exothermic reaction cascade. Simultaneously, the collapsed SEI layer exposes the pristine anode surface, which comes into direct contact with the electrolyte. These interfacial reactions are highly exothermic and play a critical role in accelerating TR propagation. Another major factor is separator shrinkage. Conventional polyolefin separators begin to shrink or melt at temperatures exceeding ∼130–150 °C. This mechanical failure may result in short circuits. These combined effects—electrolyte decomposition, anode–electrolyte interfacial reactions, and separator collapse—collectively lead to an uncontrollable escalation of thermal runaway.34,35
Salt decomposition, such as that of the bis(fluorosulfonyl)imide (FSI) anion, which is generally considered thermally stable, can undergo thermal reduction under extreme conditions, such as high temperatures and strong electric fields.36 This process results in the formation of lithium–FSI complex materials, which exhibit high thermal potential and contribute to heat generation. Moreover, a lithiated anode (charged state) undergoes intense exothermic reactions with the electrolyte.33,36–38 As shown in Fig. 2b, the DSC profiles of lithiated and delithiated graphite (LiC6 and Li0C6) paired with different anions reveal distinct thermal behaviors that influence the early stages of TR. In particular, delithiated graphite (Li0C6) shows negligible exothermic heat release compared to lithiated graphite (LiC6), due to its lower thermal reactivity and thermodynamic stability in the uncharged state. When LiC6 is combined with a 1 M LiPF6 EC/DMC electrolyte, two pronounced exothermic peaks appear between 130 and 250 °C. These events are attributed to the decomposition of the SEI layer followed by reactions between the freshly exposed graphite and the electrolyte. In contrast, when LiC6 is heated with a 1 M LiFSI electrolyte, a single sharp exothermic peak is observed near 200 °C, preceded by a minor broad exotherm starting from ∼70 °C. The total heat generated in the LiFSI system (∼1300 J g−1) is nearly double that of the LiPF6 system (∼725 J g−1), indicating that while LiFSI is more thermally stable, it may induce a more abrupt heat release upon reaching decomposition onset. This difference is critical in the context of TR mitigation. Although LiFSI based electrolytes delay the onset of thermal decomposition to higher temperatures, their interaction with lithiated graphite can lead to more concentrated and intense exothermic reactions. This behavior is consistent with prior findings that LiFSI tends to form more thermally stable SEI layers on delithiated graphite (Li0C6), as evidenced by the absence of exothermic features in that configuration. Therefore, the thermal characteristics presented in Fig. 2b demonstrate not only the anion-dependent stability of the SEI but also the potential energy release profiles that contribute to TR initiation under abuse conditions.
Notably, Si anodes exhibit higher exothermic intensity than graphite, and larger Si particles generate even greater heat release.39 In addition, the liquid electrolyte decomposes, and the layered cathode undergoes thermal collapse, releasing O2 gas inside the cell.40–42
On the other hand, the olivine structure of LFP, recognized for its superior thermal stability, offers a potential alternative to mitigate TR.42–48 The strong P–O bonding within LFP inhibits lattice oxygen release, resulting in greater structural integrity and reduced exothermic reactivity under abusive conditions. Schoberl et al.47 compared the TR behaviour of NMC811 and LFP cathodes and reported that LFP exhibits a slower heat release rate, despite a higher total energy output, due to its lower reactivity. As shown in Fig. 2c, the DSC profiles reveal that while LFP cells release more total thermal energy, the heat is dissipated gradually over time, delaying the onset of TR propagation. In contrast, NMC811 cells exhibit a more abrupt energy release, facilitating faster propagation even with a lower total energy output. This difference is primarily attributed to the disparity in venting behavior and thermal mass between the two chemistries. NMC811 cells undergo greater material ejection (∼45.8%), allowing hot gases to escape and transfer heat rapidly, whereas LFP cells retain a larger fraction of thermal energy internally due to lower mass loss (∼21.1%), resulting in a more localized and delayed heat transfer. The normalized remaining thermal energy in both systems (approximately 10–11 kJ Ah−1) is comparable; however, the rate of energy release and the thermal stability of the active materials are more critical factors in TR propagation dynamics. In stage II of thermal runaway, crosstalk reactions between the anode, electrolyte, and cathode become increasingly significant. Gaseous intermediates produced from SEI decomposition at the anode can react with oxygen evolving from the cathode, amplifying exothermic reactions and further accelerating the thermal runaway process.17,33,40,41,49,50 These findings collectively underscore the importance of considering both the thermal stability of materials and the kinetics of heat release when evaluating the TR risk of different cell chemistries.
2.2 Thermal analysis
Various thermal analysis techniques are employed to assess the safety behavior of lithium-ion battery electrolytes under abusive conditions (Fig. 3). These tools offer essential supporting data for evaluating non-flammable and flame-retardant electrolytes. Therefore, this section briefly introduces three representative methods that are critical for understanding electrolyte-related safety.53 In Fig. 3a, differential scanning calorimetry (DSC) provides insights into the thermal decomposition behavior of individual battery components such as SEI layers, electrolytes, and electrodes. It identifies onset temperatures and exothermic transitions at the material level, aiding the understanding of thermal risks during initial TR stages.54–58 However, the conventional sample chamber used in DSC (typically referred to as PAN) prohibits the testing of liquid or wet samples due to the evaporation caused by the liquid-to-gas phase transition during temperature increases. As a result, most current DSC analysis focuses only on the independent thermal behavior of individual components, such as the cathode or anode, while neglecting the critical reactions between the electrode and electrolyte. These reactions, however, are a key contributor to intense exothermic heat release. To understand the exothermic heat from these electrode and electrolyte reactions, the sample chamber must possess high pressure resistance. This allows for reliable thermal measurements under high temperatures conditions. Accelerating rate calorimetry (ARC) complements DSC by evaluating heat evolution in fully assembled cells under adiabatic conditions (Fig. 3b). Unlike the DSC, ARC captures the overall thermal behavior of a fully packed cell, including reactions between the cathode, electrolyte, and anode, thereby providing more realistic, system-level safety insights.59 Typically, ARC results reveal the key thermal runaway thresholds, T1 (self-heating onset temperature), T2 (TR trigger temperature), and T3 (maximum temperature). Therefore, to obtain a holistic understanding of thermal runaway behavior, both DSC and ARC analyses are essential and complementary. In short, while DSC offers high resolution, materials level information on specific decomposition reactions, ARC simulates the thermal behavior of real-world, fully assembled cells. Together, they provide a comprehensive way for evaluating thermal safety in LIBs. Fig. 3c illustrates a schematic representation of the self-extinguish time (SET) measurement process. SET is a straightforward flammability test that quantifies the time required for an ignited liquid electrolyte to self-extinguish, normalized by the electrolyte's mass.60–62 Although widely adopted, it suffers from poor reproducibility due to the lack of standardized definitions for ignition and extinction points. To improve its reliability, Zhang et al.63 proposed the self-extinguishing efficiency (SEE), which normalizes SET against a baseline electrolyte (SET0). Based on these metrics, electrolytes are generally classified as flammable, flame-retardant, or non-flammable. However, both SET and SEE are best used as preliminary screening tools rather than definitive indicators of overall thermal safety. Lastly, Fig. 3d summarizes the advantages and limitations of the aforementioned techniques, providing a guideline for researchers to select the appropriate method for evaluating the thermal characteristics of electrolytes. | ||
| Fig. 3 Schematic diagrams of typical thermal analysis tools (a) DSC, (b) ARC, and (c) SET. (d) A radar chart comparing their characteristics. | ||
3. Electrolytes
A plethora of non-flammable electrolyte designs have been extensively investigated in the realm of electrochemical energy storage systems, particularly focusing on enhancing safety without significantly compromising performance metrics. These designs include a variety of formulations, such as radical scavenging agents, specifically, additives that contain phosphorus or fluoride compounds, which serve to mitigate oxidative processes during electrochemical cycling. In addition, ionic liquids have emerged as a promising alternative due to their unique properties, such as negligible vapor pressure and a wide electrochemical window, further contributing to their appeal as non-flammable electrolytes. High flash point electrolytes also represent a significant advancement in this area, offering improved thermal stability, as illustrated in Fig. 4. Although these innovative strategies enhance thermal stability and safety, they frequently necessitate a careful balancing act between non-flammability and overall electrochemical performance. This trade-off can manifest in several ways, including increased viscosity of the electrolyte solution, which may hinder ionic mobility, and the formation of thick, organic-based SEI layers that can negatively impact charge transfer kinetics. | ||
| Fig. 4 General strategies for flame-retardant or non-flammable electrolytes. | ||
In this section, we will engage in a thorough discussion of various general strategies aimed at developing safer electrolyte systems (Table 1). We will also elaborate on how these strategies contribute to improved thermal stability of the electrolytes while simultaneously examining the conventional methods employed to tackle the resultant challenges associated with performance and safety. Through this comprehensive analysis, we aim to elucidate the intricate relationships between electrolyte composition, thermal characteristics, and electrochemical performances, thus shedding light on future directions for research and development in the field of battery chemistry.
| Electrolyte composition | Thermal stability evaluation | Battery performance improved strategies | Cell performance (configuration, cut-off voltage, discharge capacity, cycle, C-rate, retention) |
|---|---|---|---|
| Fluoride-containing electrolyte | |||
1 M LiFSI/EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC (1:1 v/v%) + FEC 10 wt%36 :DMC (1:1 v/v%) + FEC 10 wt%36 |
250 J g−1 exothermic heat at ∼200 °C in DSC (with charged graphite) | FEC-derived stable SEI layer | |
| 1.0 M LiPF6/EC:methyl (2,2,2-trifluoroethyl) carbonate (FEMC) (3:7 v/v%) + VC (2 wt%)72 |
SET: 0 s g−1|745 J g−1 exothermic heat at 267.8 °C in DSC (with charged NCM622) | FEMC-derived SEI layer to suppress metal dissolution and structural degradation | NCM622‖graphite, 2.5–4.5 V, 186 mAh g−1, 100 cycle, 0.5C, 80% |
| 1.0 M LiPF6/FEC:FEMC:tetrafluoroethylene (TTE) (1:15:4 v/v%)78 |
T 1: 154.3 °C in ARC | TTE-derived SEI layer suppresses oxygen evolution from the layered LCO | LCO‖Li, 3.0–4.6 V, 77.8 mAh g−1, 300 cycle, 0.2C, 70% |
| 1 M LiPF6/methyl 3,3,3-trifluoropropionate (MTFP)80 | No exothermic peak up to 400 °C in DSC (electrolyte only) | Stable fluorinated SEI layer. Good affinity with SBR and CMC binder, high oxidation stability | LCO‖Li, 2.5–4.5 V, 185 mAh g−1, 50 cycle, 25 mA g−1, 92% |
| 1.0 M LiPF6/FEC:EMC:bis(2,2,2-trifluoroethyl) carbonate (TFEC) (3:3:4 v/v%)81 |
SET: 3.34 s g−1|404.2 J g−1 exothermic heat at 219.45 °C in DSC (with charged NCM811)|T3: 446.8 °C in ARC | Uniform CF3-rich SEI layer, high Li reversibility | NCM811‖Li, 2.8–3.3 V, 3 mAh cm−2, 240 cycle, 0.3C/0.5C (charge/discharge), 80% NCM90||Li, 2.7–4.25 V, 1.5 Ah, 98 cycle, 0.2C/0.33C (charge/discharge), 90.02% |
| 1 M LiPF6/FEC:D2 (6:4 v/v%)120 |
1162.0 J g−1 exothermic heat at 298.5 °C in DSC (charged NMC811 and graphite)|T1: 131 °C, T2: 225.6 °C, T3: 770 °C in ARC | Fluorinated SEI layer, high oxidation stability, suppresses other reactions | NMC811‖Gr, 2.9–4.2 V, 1 Ah, 380 cycle, 0.33C, 94.3% |
| Phosphorus-containing electrolyte | |||
| 1.75 M LiDFOB in TEP/DME/HFE (4:1:5 v/v%) + VC (2 wt%)86 |
SET: 6.10 s g−1 | Li solvation sheath tuned with DME, forming Li2O/LiF dual SEI layer to prevent TEP and Li reaction | Coin cell: NCM811‖Li, 2.9–4.4 V, >200 mAh g−1, 200 cycle, 0.3C/0.5C (charge/discharge), 80% pouch cell: NCM811‖Li, 2.9–4.4 V, 0.2 Ah, 140 cycle, 0.2C/0.2C (charge/discharge), 87.9% |
| 1.3 M LiODFB in DEEP + 30% FEC89 | SET: 0 s g−1 | Synergistic effect between conducting salt (LiODFB) and FEC | NCM811‖Li, 2.8–4.5 V, 169.5 mAh g−1, 100 cycle, 1C, 81.8% |
| 1.5 M LiTFSI in PC/TEP (4:1, v/v%) + 2.0 wt% TFANP92 |
No ignition | Li solvation sheath tuned with TFANP, forming LiF-rich SEI layer | NCM622‖graphite, 3.0–4.3 V, 132 mAh g−1, 150 cycle, 0.5C, 99.5% |
| 1.5 M LiNO3 in TEP/FEC (3:1 v/v%)99 |
SET: 1.25 s g−1|delay the flame occurrence in high-temperature test (at 550 °C) | LiNO3-derived Li3N and LiF-rich SEI layer | NCM811‖Li, 3.0–4.3 V, 100 cycle, 0.33C, 96.85% LFP‖Li, 2.5–4.2 V, 100 cycle, 0.33C, 84.99% |
| LiFSI/TMPa/TMS/OTE (1:1.05:1, by molar ratio)114 |
Flash point: 155 °C|no ignition|weak exothermic heat at 220 °C in DSC (with charged NMC811) | Robust SEI layer | NCM811‖Gr, 2.5–4.4 V, 500 cycle, 0.3C, 87.1% |
| Ionic liquids | |||
| 2 M LiTFSI/LiDFOB (8:1, by mole) DFPyrTFSI/DMC (1:1 wt%)102 |
No explosion by nail penetration test | Robust and stable F-rich SEI layer anion-rich solvating structure | NCM622‖Li, 2.8–4.5 V, ∼160 mAh g−1, 300 cycle, 0.33C, 90.3% |
| High flash point | |||
| 1 M LiTFSI/bis(2-methoxyethyl) carbonate (BMEC):EC (7:3 v/v%) + FEC (3 v/v%)118 |
Flash point: 121 °C|SET: 0 s g−1|403.8 J g−1 exothermic heat at 280.4 °C in DSC (with charged NCM811)|delayed T3 (vs. control electrolyte) in ARC | Enhanced ionic conductivity | NMC811‖graphite, 2.7–4.5 V, ∼0.7 Ah, 500 cycle, 0.3C, 91.4% |
3.1 Radical scavenging agent
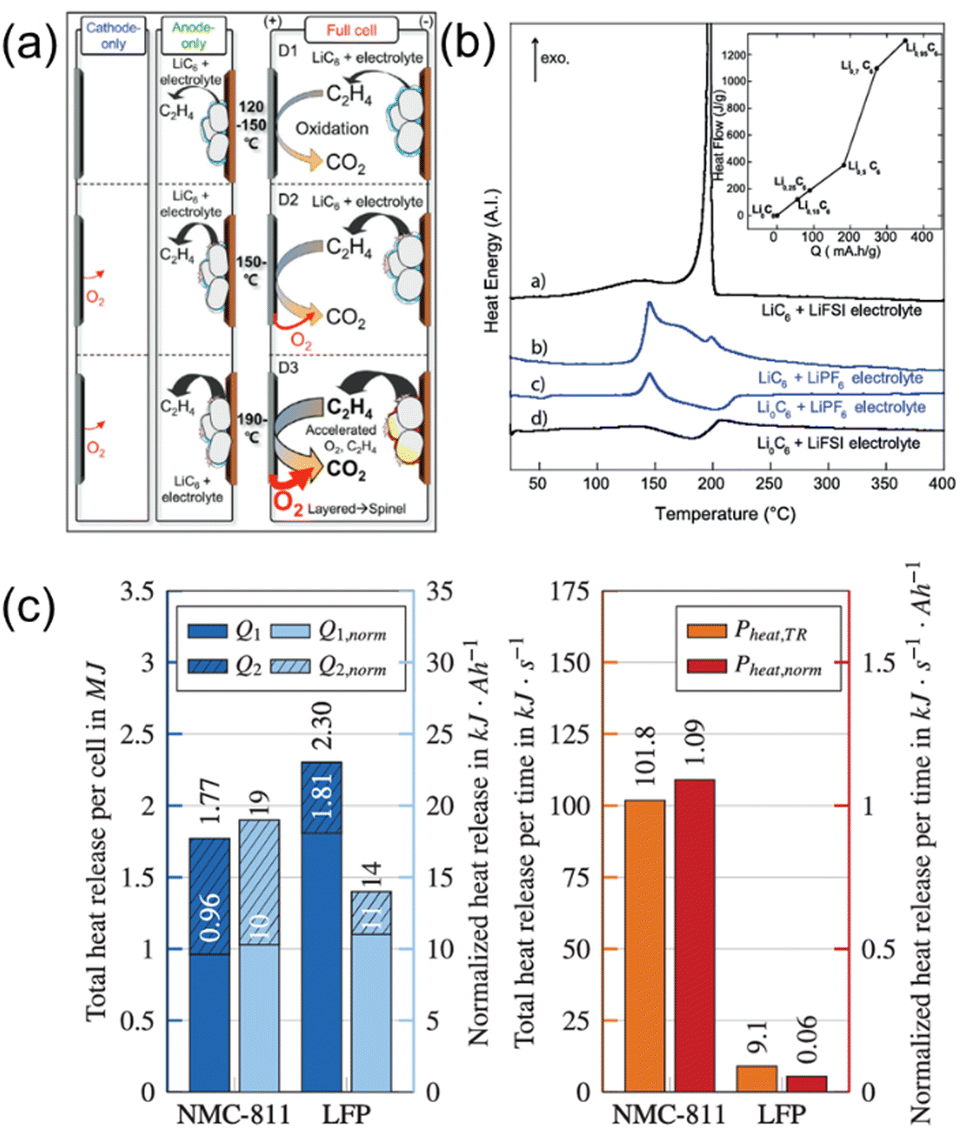
Radical scavengers, including phosphorus- and fluoride-containing additives or co-solvents, are widely used. They preferentially capture radicals, which effectively slows down the combustion reaction of oxygen (Fig. 4). Consequently, the combustion reaction can be delayed until the radical scavengers fully capture the radicals.In both cases, the LiF-rich SEI layer formed from fluorinated solvents exhibits high thermal stability, which leads to a delay in the onset of exothermic reactions. These protective interphases effectively prevent direct contact between the active material and the bulk electrolyte, thereby further suppressing heat generation under elevated temperatures.
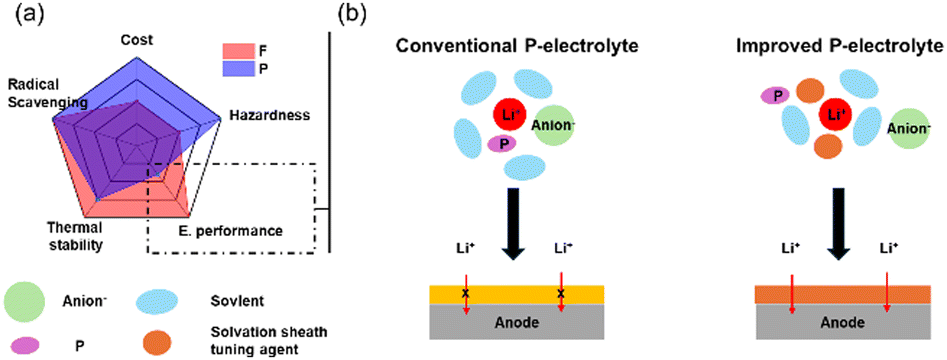
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), which are widely used as flame retardants in liquid electrolytes. The strong radical scavenging capability of these groups helps to inhibit or delay combustion reactions by reacting with reactive oxygen species and suppressing the formation of flammable gases. As a result, the onset temperature of exothermic reactions is elevated, contributing to improved thermal stability. These offer low-cost options, good compatibility with carbonate solvents, high salt concentration, and additive solubility due to their high donor number and ease of synthesis (Fig. 5a).41,82,83 Various phosphate-derived materials have been developed with different alkyl group moieties. In the early stages, trimethyl phosphate (TMP) and triethyl phosphate (TEP) were primarily used.84,85 Later, other alkyl-substituted phosphates such as tributyl phosphate (TBP)86, triphenyl phosphate (TPP)86, dimethyl methyl phosphate (DMMP),87,88 and diethyl ethyl phosphate (DEEP)89, along with cyclic phosphonates90,91 were introduced to enhance flame-retardant properties. However, despite their effectiveness in improving flame retardancy, phosphate-based solvents and additives face critical challenges, particularly their high viscosity, which leads to low ionic conductivity. To address this, they are typically blended with carbonate solvents. TMP, one of the most widely used and effective flame-retardant solvents, exhibits strong coordination with lithium ions and becomes incorporated into the solvation structure, which gives rise to multiple issues. It can induce co-intercalation into the graphite anode, leading to structural degradation and capacity loss, and also promotes the formation of organic-rich SEI layers.92,93 Therefore, for phosphorus-based flame-retardant electrolytes, current strategies should focus on improving cell performance while maintaining their safety benefits (Fig. 5b).
O), which are widely used as flame retardants in liquid electrolytes. The strong radical scavenging capability of these groups helps to inhibit or delay combustion reactions by reacting with reactive oxygen species and suppressing the formation of flammable gases. As a result, the onset temperature of exothermic reactions is elevated, contributing to improved thermal stability. These offer low-cost options, good compatibility with carbonate solvents, high salt concentration, and additive solubility due to their high donor number and ease of synthesis (Fig. 5a).41,82,83 Various phosphate-derived materials have been developed with different alkyl group moieties. In the early stages, trimethyl phosphate (TMP) and triethyl phosphate (TEP) were primarily used.84,85 Later, other alkyl-substituted phosphates such as tributyl phosphate (TBP)86, triphenyl phosphate (TPP)86, dimethyl methyl phosphate (DMMP),87,88 and diethyl ethyl phosphate (DEEP)89, along with cyclic phosphonates90,91 were introduced to enhance flame-retardant properties. However, despite their effectiveness in improving flame retardancy, phosphate-based solvents and additives face critical challenges, particularly their high viscosity, which leads to low ionic conductivity. To address this, they are typically blended with carbonate solvents. TMP, one of the most widely used and effective flame-retardant solvents, exhibits strong coordination with lithium ions and becomes incorporated into the solvation structure, which gives rise to multiple issues. It can induce co-intercalation into the graphite anode, leading to structural degradation and capacity loss, and also promotes the formation of organic-rich SEI layers.92,93 Therefore, for phosphorus-based flame-retardant electrolytes, current strategies should focus on improving cell performance while maintaining their safety benefits (Fig. 5b).
 | ||
| Fig. 5 (a) Radar map to compare the 5 factors between fluoride and phosphorus-based radical scavenging agents. (b) Strategies to improve the poor electrochemical performance of phosphorus agents. | ||
One promising approach to address the limitations of phosphorus in electrochemical performance involves combining fluoride and phosphorus in an electrolyte design.94–96 For example, Jiang et al.94 utilized 4-nitrophenyl trifluoroacetate (TFANP) as an additive (2 wt%) in a phosphate-based electrolyte (1.5 M LiTFSI in PC/TEP; 4:1 v/v%). They found that TFANP modified the solvation structure, reducing the interaction between TEP and lithium ions, which decreased TEP decomposition and led to the formation of a stable SEI and CEI layer on both electrode surfaces. Consequently, NCM622‖Li cells demonstrated higher cycling stability, maintaining 132 mAh g−1 over 150 cycles with a Coulombic efficiency of 99.5%, without compromising fire resistance. Similarly, Wang et al.95 developed a novel fluorinated phosphate solvent, diethyl fluorophosphate (DEFP), featuring a unique P–F bond (2 M LiPF6 in EC/DEC/DEFP). The presence of this P–F bond resulted in a lower LUMO level, aiding the formation of a stable SEI layer, which postponed the SEI decomposition onset temperature from 85.1 °C (TEP electrolyte) to 111.1 °C (DEFP electrolyte) as measured by DSC, indicating improved thermal stability. Furthermore, the NCM811‖Gr pouch cell sustained a discharge capacity of 1.16 Ah over 200 cycles, achieving a capacity retention of 94.2%.
Another strategy involves tailoring the first solvation sheath, including high-concentration electrolytes (HCEs), localized high-concentration electrolytes (LHCEs), and ion-solvent-coordinated (ISC) electrolytes.97–100 The key concept is to minimize the participation of phosphate solvents in SEI formation, allowing only selective solvent molecules or salts to contribute to the development of the SEI layer. For LHCEs,97,98 Zhang's group97 developed a TEP-based LHCE consisting of 1.2 M LiFSI in TEP and bis(2,2,2-trifluoroethyl) ether (BTFE) (1:3 molar ratio). This formulation, tested in NCM622‖Li cells, effectively suppressed dendrite growth by forming a LiF-rich SEI layer, maintained lower viscosity than general phosphate electrolytes, and preserved its non-flammability. Liu's group99 proposed an ISC electrolyte with a similar composition but a general salt concentration rather than HCEs, emphasizing that the molar ratio is more important than the molar concentration of the electrolyte. As a result, LiFSI:TEP electrolytes with a 1:1.5 molar ratio (∼2.2 M) effectively reduce TEP decomposition and mobility, preventing reactions with lithium metal while maintaining high thermal stability. Consequently, no reaction occurs with lithium metal after a week at 60 °C and maintaining a high Coulombic efficiency of 99.3% over 350 cycles in a Li‖Cu cell.
Liao et al.101 recently reported lithium nitrate (LiNO3) as the sole lithium salt, formulating a 1.5 M LiNO3 electrolyte in TEP:FEC (3:1 v/v%). Their findings revealed that NO3− strongly solvates with Li+ rather than TEP, leading to the formation of Li3N and LiNxOy, which are beneficial for suppressing lithium dendrites, enhancing ion conductivity, and preventing gas evolution. This electrolyte demonstrated a SET of 1.25 s g−1, and in a high-temperature test (550 °C), the NCM811‖Li pouch cells showed a delayed flame occurrence of 255 s compared to EC/DEC electrolytes. Furthermore, the electrolyte maintained a 189.1 mAh g−1 discharge capacity with 83.74% retention over 1000 cycles. These results suggest that LiNO3 could be a promising alternative to LiPF6, as its ability to form a Li3N-rich SEI layer enhances safety and longevity. However, its high donor number limits solubility in conventional carbonate electrolytes. Phosphate-based solvents, such as TEP, which have a high donor number, can effectively dissolve LiNO3, making them a viable choice for improving LIBs.
A final approach involves constructing a thermally stable polymeric SEI layer on the anode. For instance, vinylene carbonate (VC) has been widely studied for its ability to form polymeric SEI layers, which improve cycling stability.70,102,103 Moreover, Profatilova et al.70 demonstrated that VC also enhances thermal stability by delaying the onset temperature of exothermic reactions from 176 °C to 232 °C, as confirmed by DSC analysis. Building on this concept, our group reported that adding 5 wt% phosphonium-based polymerizable additives (allylpropylphosphonium TFSI; AP111T) to a DEC/FEC (9:1 v/v%) electrolyte forms a flame-retardant SEI layer while simultaneously improving cycling performance in silicon anodes.37 This polymeric SEI layer significantly reduced exothermic heat by 92%, measuring 208.28 J g−1. Additionally, Si‖Li cells demonstrated stable cycling at 2.12 mA cm−2 over 100 cycles, enhancing both thermal stability and electrochemical performance.
3.2 Ionic liquids
Ionic liquids, a molten salt form with extremely low vapor pressure, non-flammability, and high polarity, exhibit excellent conductivity and wide electrochemical stability, making them a promising candidate to replace conventional carbonate-based solvents.37,104–108 Furthermore, their chemical composition can be easily tailored, as they can be synthesized with various cation (pyrrolidinium,104–107 imidazolium,108 phosphonium,37etc.) and anion (TFSI, FSI, halide, PF6, etc.) combinations.Li et al.104 developed a gem-difluorinated pyrrolidinium ionic liquid electrolyte featuring a dual salt system (LiTFSI/LiDFOB) and DMC as a co-solvent. This formulation enabled stable operation across a wide temperature range (−20 °C to 60 °C) and high cut-off voltages (4.5–4.7 V) in NCM622‖Li cells. The anion-rich solvation structure enhances interphase stability, with IL cations actively interacting with Li+ ions to modify the solvation sheath, promoting the formation of a robust SEI and CEI layer. In addition, DMC is incorporated to mitigate the viscosity increase caused by the IL, ensuring better ion transport properties. The IL-based electrolyte withstands thermal decomposition up to 130 °C, significantly exceeding the thermal limits of conventional carbonate electrolytes. Furthermore, it exhibits exceptional safety under extreme conditions, successfully enduring overcharging at 5.16 V for 2 hours with minimal temperature rise and passing the nail penetration test without explosion, confirming its superior resistance to thermal runaway.
Chatterjee et al.108 proposed a novel imidazolium-based dicationic ionic liquid (DIL), 1,1′-(5,14-dioxo-4,6,13,15-tetraazaoctadecane-1,18-diyl)bis(3-(sec-butyl)-1H-imidazol-3-ium) bis((trifluoromethyl)sulfonyl)imide, as an additive to conventional EC/DMC-based electrolytes for lithium-ion batteries. This DIL exhibited excellent non-flammability, with no ignition observed under direct flame exposure, and provided high thermal stability, as evidenced by the absence of an exothermic peak in DSC analysis. Despite a slight increase in viscosity and a minor decrease in ionic conductivity, the DIL-containing cells showed markedly improved electrochemical performance. Full cells (NMC622‖Graphite) delivered an initial discharge capacity of 142.1 mAh g−1 and ∼90% of capacity over 100 cycles at 0.5C. These enhanced results are attributed to the dicationic moiety's ability to promote the formation of an oxygen-rich SEI layer, which effectively suppresses PF6− decomposition, reduces the generation of flammable gases, and lowers interfacial resistance.
3.3 High flash point electrolyte
A high flash point indicates low ignitability, signifying enhanced safety and reduced flammability. As a result, its SET value tends to be lower, making it a strong candidate for a non-flammable electrolyte.54 In contrast, general carbonate solvents such as EC possess low flash points and undergo thermal decomposition at approximately 150–200 °C. This decomposition leads to the generation of gaseous byproducts within the cell, increasing internal pressure. These gases can subsequently react with flammable components, elevating the fire or explosion risk. Therefore, high flash point electrolytes not only reduce the risk of vapor-phase ignition but also suppress premature gas evolution, contributing to improved cell-level thermal stability under abuse conditions.Gamma-butyrolactone (GBL) has emerged as a promising alternative to conventional electrolytes because it possesses several favorable properties, including a low melting point, a high dielectric constant (39.1), and a high flash point (98 °C).109 Also, due to its structural similarity to EC, it demonstrates good compatibility with the current electrolyte system. However, it displays low ionic conductivity at low temperatures and poor wettability with separators. Furthermore, it creates a thick and inefficient SEI layer on the anode, particularly graphite.109,110 The Hirano group111–114 has extensively studied GBL-based electrolytes as a safe, non-flammable solution with a wide operational temperature range for LIBs. Their research highlights various GBL-based formulations that incorporate flame-retardant additives such as tris(2,2,2-trifluoroethyl) phosphate (TFP), (phenoxy)pentafluorocyclo-triphosphazene (FPPN), and hydrofluoroether (HFE) to improve thermal stability and electrochemical performance. For instance, GBL-TFP electrolytes demonstrated high ionic conductivity (7.40 mS cm−1) and 90.8% capacity retention after 200 cycles at 25 °C, surpassing conventional carbonate-based electrolytes in terms of thermal stability. FPPN-GBL electrolytes exhibited 85.4% retention after 500 cycles and excellent low-temperature performance (90 mAh g−1 at 40 °C). Meanwhile, LiODFB-GBL/HFE electrolytes offered exceptional safety, maintaining 82.2% retention after 500 cycles and sustaining 100 mAh g−1 at −40 °C, making them suitable for extreme environments. XPS and SEM analyses confirmed stable SEI and CEI layer formation, thereby ensuring improved cycle life and interface stability. Designing extended alkyl chains could offer another solution to increase the flash point.115–120 For example, Lee et al.120 developed bis(2-methoxyethyl) carbonate (BMEC), a solvent with a flash point of approximately 121 °C, which is about 90 °C higher than conventional electrolytes such as DMC (18 °C) and DEC (31 °C). As a result, the SET value is 0 s g−1. The B7E3 electrolyte (1 M LiPF6 and 3 vol% of FEC in BMEC:EC; 7:3 v/v%) exhibits superior thermal stability. DSC analysis of fully charged NMC811 cathodes with B7E3 showed that the exothermic decomposition temperature reached 280.4 °C, around 45 °C higher than that of the conventional electrolyte (235 °C) while releasing 2.6 times less heat. Furthermore, mass spectrometry analysis of NMC811/electrolyte mixtures heated to 235 °C demonstrates that B7E3 releases 1.6 times less reactive gas (O2, H2, and CO) compared to the control electrolyte, indicating improved cathode stability. Lastly, the additional ether group in BMEC enhances ion solvation and overall cell performance, as evidenced by an NMC811‖Gr pouch cell (1 Ah) that achieved 91.4% discharge capacity retention over 500 cycles.
4. Challenges and future directions
Despite reports that non-flammable or flame-retardant electrolytes resist ignition, recent studies indicate that these electrolytes alone may not sufficiently suppress battery explosions or prevent thermal runaway.121–123 For instance, Ouyang's group122 found that a fluorinated electrolyte (1 M LiPF6 in FEC/1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl-ether (D2), 6:4 v/v%), known for its flame-retardant properties, exhibited a SET value of 0 s g−1. This electrolyte delayed the TR trigger onset temperature by 20 °C compared to the EC/DMC (1 M LiPF6, 3:7 v/v%) electrolyte. However, the maximum TR temperature remained unchanged at 770 °C, indicating that while the fluorinated electrolyte helps mitigate early TR initiation, it is insufficient to suppress full TR within a cell completely. Moreover, DSC analysis showed that the fluorinated electrolyte slightly delayed the initial exothermic reaction onset temperature by approximately 13 °C in the cathode/electrolyte reaction. Nonetheless, the overall exothermic reaction heat was not significantly reduced. This results from the high oxidative stability of fluorinated electrolytes, which stabilize the cathode but do not prevent O2 release. Consequently, the fluorinated electrolyte cannot fully eliminate cathode/anode crosstalk. Similarly, Zhang et al.81 noted that their FEC/bis(2,2,2-trifluoroethyl) carbonate (TFEC) electrolyte showed a SET value of 3.34 s g−1, confirming its non-flammable nature. When paired with an NCM811‖Li cell, DSC analysis revealed that the onset temperature of the exothermic reaction was delayed by about 20 °C, while the exothermic heat released decreased from 562.3 J g−1 to 401.8 J g−1. Additionally, ARC analysis indicated that its maximum thermal runaway temperature was reduced from 680 °C to 446.8 °C, significantly enhancing thermal stability. However, this was still insufficient to fully suppress TR. Jia et al.121 reported the development of a DFR-E electrolyte, incorporating tris(2,2,2-trifluoroethyl) phosphite (TTFEPi), which achieves non-flammability in both its liquid and vapor phases, featuring a high flash point >160 °C (Fig. 6a). By using TTFEPi as a diluent, EC molecules are effectively separated, forming an LHCE solvation structure that promotes a thin, efficient SEI layer on graphite and improves the cycling stability of LFP‖Gr cells, delivering 90.8 mAh g−1 with 79.9% capacity retention over 300 cycles (Fig. 6b). However, safety tests on 1.2 Ah 18650-type LFP‖Gr cells revealed that, even though the DFR electrolytes are non-flammable, they underperformed compared to the E-baseline electrolyte in terms of cell safety. In a nail penetration test, for example, the E-baseline cell showed a modest temperature increase from 80 to 87 °C and maintained a stable voltage of 3.4 V. In contrast, the cell with the DFR-E electrolyte underwent a rapid temperature spike from 80 to 350 °C within three minutes, with the voltage dropping from 4.0 V to 0 V, indicative of separator melting and a resulting short circuit. Furthermore, DSC measurements on charged LFP and graphite electrodes (obtained from an LFP‖Gr cell) revealed that the reaction between LiFSI and the charged electrodes is the major cause of significant exothermic heat in the 200–240 °C range. In other words, although the non-flammable DFR-E electrolyte did not ignite, it failed to inhibit the LiFSI/electrode reaction (Fig. 6c and d). These results suggest that while non-flammable electrolytes reduce the flammability risk, their contribution to mitigating thermal runaway may be limited during the reactions between lithiated electrode, salt, and electrolyte or cathode–anode cross-talk. These discrepancies between an electrolyte's non-flammability and a cell's thermal behavior highlight the need for further investigation. However, this does not imply electrolyte non-flammability is irrelevant to preventing cell explosions. Rather than focusing solely on an electrolyte-based approach, analyzing thermal behavior in the combined electrolyte and electrodes is crucial, where exothermic reactions are more accurately represented.
 | ||
| Fig. 6 (a) Ignition test for electrolytes and their composition. (b) Cycling plot of LFP‖Gr cells. DSC results for charged (c) graphite and (d) LFP and their electrolytes (adapted from ref. 121, under the terms of the Creative Commons Attribution 4.0 License, CC BY 4.0, https://creativecommons.org/licenses/by/4.0/, copyright 2023, Wiley). | ||
In addition, recent advances in AI-assisted electrolyte design have opened new opportunities for optimizing formulations and selecting materials by predicting and simulating thermodynamic behavior.124–126 Machine learning (ML) approaches have also been employed to predict the risk of thermal runaway and diagnose the thermal status of batteries in real-time.127–129 Furthermore, solid-state electrolytes, including both all-solid-state and gel or polymer-based systems, may serve as an effective strategy to eliminate the flammability concerns associated with conventional liquid electrolytes.130,131 Although challenges such as low ionic conductivity and interfacial issues remain, these systems have promise for improving intrinsic battery safety.
Therefore, we further propose advanced strategies to improve battery safety through liquid electrolyte innovations.
(1) In-depth studies are needed beyond merely evaluating whether a non-flammable electrolyte ignites. Instead, focusing on cell-level interactions between the anode, cathode, and electrolyte to develop more effective designs. Specifically, this includes (i) SEI layers with high thermal stability to suppress gas evolution and (ii) CEI layers that prevent structural collapse in the layered cathode and mitigate oxygen release. (iii) Understanding the salt effect during thermal runaway, such as LiFSI.
(2) Furthermore, prioritizing thermal safety alone may lead to significant performance degradation, necessitating electrolyte designs that enhance safety without sacrificing electrochemical performance. Developing novel solvation sheath structures can help ensure that non-flammable components remain inactive, promoting an anion-derived SEI layer. Incorporating alternative salts (LiNO3) or additives may further enhance both safety and performance.
(3) Standardized guidelines must be established for safety tests that currently lack industry-wide benchmarks, ensuring consistent and reliable evaluations.
5. Conclusion
Ensuring the safety of lithium-ion batteries is not just a challenge; it is an imperative we must address urgently. Mitigating thermal runaway and preventing catastrophic failures, such as fires and explosions—are crucial for the future of energy storage. Significant efforts have been dedicated to developing non-flammable or flame-retardant electrolytes, which feature high flash points and low SET values that are instrumental in reducing ignition risks. Yet, we must recognize that these properties alone are not sufficient to suppress thermal runaway completely, given the intricate nature of its mechanisms and the multitude of reactions that occur simultaneously. Moreover, the challenge of balancing safety improvements with electrochemical performance remains profound. To truly enhance battery reliability, we require a comprehensive strategy. This approach must include the integration of non-flammable solvents, a reduction in electrolyte–electrode reactivity to limit hazardous side reactions, and the implementation of rigorous evaluations under realistic conditions. By embracing these innovative strategies, the lithium-ion battery community can make remarkable strides toward achieving safer energy storage solutions. Let us take bold steps together for a safer, more reliable future in energy technology.Author contributions
Won-Jang Cho: investigation, conceptualization, writing, drawing. Yoo Jeong Huh, Soyeon Choi, Uddhav Kulkarni, Kiran P. Shejale: investigation, editing, reviewing. Gi-Ra Yi: editing, reviewing, supervision.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software, or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
This research was supported by the project titled ‘Development of New Ionic Additives in Liquid Electrolytes for Safety Enhancement of Lithium-Ion Batteries,’ funded by LG Energy Solution, Ltd, the National Research Foundation of Korea (NRF Korea) (2021R1A2C3013800) and the Korea Institute for Advancement of Technology (KIAT) grant funded by the Korea Government (MOTIE) (RS-2024-00419413, HRD Program for Industrial Innovation).Notes and references
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800 CrossRef CAS PubMed.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- T. M. Bandhauer, S. Garimella and T. F. Fuller, J. Electrochem. Soc., 2011, 158, R1–R25 CrossRef CAS.
- X. Feng, S. Zheng, D. Ren, X. He, L. Wang, H. Cui, X. Liu, C. Jin, F. Zhang, C. Xu, H. Hsu, S. Gao, T. Chen, Y. Li, T. Wang, H. Wang, M. Li and M. Ouyang, Appl. Energy, 2019, 246, 53–64 CrossRef CAS.
- J. Wen, Y. Yu and C. Chen, Mater. Express, 2012, 2, 197–212 CrossRef CAS.
- P. V. Chombo and Y. Laoonual, J. Power Sources, 2020, 478, 228649 CrossRef CAS.
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, J. Energy Chem., 2021, 59, 83–89 CrossRef CAS.
- Q. Wang, L. Jiang, Y. Yu and J. Sun, Nano Energy, 2019, 55, 93–114 CrossRef CAS.
- X. Feng, M. Ouyang, X. Liu, L. Lu, Y. Xia and X. He, Energy Storage Mater., 2018, 10, 246–267 CrossRef.
- Q. Wang, P. Ping, X. Zhao, G. Chu, J. Sun and C. Chen, J. Power Sources, 2012, 208, 210–224 CrossRef CAS.
- X. Feng, D. Ren, X. He and M. Ouyang, Joule, 2020, 4, 743–770 CrossRef CAS.
- X. Liu, D. Ren, H. Hsu, X. Feng, G.-L. Xu, M. Zhuang, H. Gao, L. Lu, X. Han, Z. Chu, J. Li, X. He, K. Amine and M. Ouyang, Joule, 2018, 2, 2047–2064 CrossRef CAS.
- K. Deng, Q. Zeng, D. Wang, Z. Liu, G. Wang, Z. Qiu, Y. Zhang, M. Xiao and Y. Meng, Energy Storage Mater., 2020, 32, 425–447 CrossRef.
- R. Gond, W. van Ekeren, R. Mogensen, A. J. Naylo and R. Younsei, Mater. Horiz., 2021, 8, 2913–2928 RSC.
- G. G. Eshetu, S. Grugeon, S. Laruelle, S. Boyanov, A. Lecocq, J.-P. Bertrand and G. Marlair, Phys. Chem. Chem. Phys., 2013, 15, 9145–9155 RSC.
- E. P. Roth and C. J. Orendorff, Electrochem. Soc. Interface, 2012, 21, 45–49 CrossRef CAS.
- A. Yusuf, Z. Li, X. Yuan and D.-Y. Wang, Small Methods, 2022, 6, 2101428 CrossRef CAS.
- Y. Zhang, Y. Lu, J. Jin, M. Wu, H. Yuan, S. Zhang, K. Davey, Z. Guo and Z. Wen, Adv. Mater., 2024, 36, 2308484 CrossRef CAS PubMed.
- K. Zhang, S. Yan, C. Wu, L. Wang, C. Ma, J. Ye and Y. Wu, Small, 2024, 20, 2401857 CrossRef CAS.
- S. W. Kang and Y. Cho, Korean J. Chem. Eng., 2024, 41, 403–409 CrossRef CAS.
- U. Kulkarni, W.-J. Cho, S.-K. Cho, J.-J. Hong, K. P. Shejale and G.-R. Yi, Korean J. Chem. Eng., 2024, 41, 385–402 CrossRef CAS.
- Y. Ou, P. Zhou, W. Hou, X. Ma, X. Song, S. Yan, Y. Lu and K. Liu, J. Energy Chem., 2024, 94, 360–392 CrossRef CAS.
- Z. Wang, J. Yuan, X. Zhu, H. Wang, L. Huang, Y. Wang and S. Xu, J. Energy Chem., 2021, 55, 484–498 CrossRef CAS.
- J. Yamaki, Y. Baba, N. Katayama, H. Takatsuji, M. Egashira and S. Okada, J. Power Sources, 2003, 119–121, 789–793 CrossRef CAS.
- J.-I. Yamaki, H. Takatsuji, T. Kawamura and M. Egashira, Solid State Ionics, 2002, 148, 241–245 CrossRef CAS.
- C. Zu, H. Yu and H. Li, InfoMat, 2021, 3, 648–661 CrossRef CAS.
- L. Kong, Y. Li and W. Feng, Electrochem. Energy Rev., 2021, 4, 633–679 CrossRef CAS.
- S. Jo, S. Seo, S. K. Kang, I. Na, S. Kunze, M. Song, S. Hwang, S. P. Woo, S. Kim, W. B. Kim and J. Lim, Adv. Mater., 2024, 36, 2402024 CrossRef CAS PubMed.
- D. Ren, X. Feng, L. Liu, H. Hsu, L. Lu, L. Wang, X. He and M. Ouyang, Energy Storage Mater., 2021, 34, 563–573 CrossRef.
- D. P. Finegan, E. Darcy, M. Keyser, B. Tjaden, T. M. M. Heenan, R. Jervis, J. J. Bailey, R. Malik, N. T. Vo, O. V. Magdysyuk, R. Atwood, M. Drakopoulos, M. DiMichel, A. Rack, G. Hinds, D. J. L. Brett and P. R. Shearing, Energy Environ. Sci., 2017, 10, 1377–1388 RSC.
- G. G. Eshetu, S. Grugeon, G. Gachot, D. Mathiron, M. Armand and S. Laruelle, Electrochim. Acta, 2013, 102, 133–141 CrossRef CAS.
- W.-J. Cho, S. Lee, U. Kulkarni, J. Jeon, T. Im, Y. K. Jeong, D. S. Hwang, J.-S. Bae, K. Ahn, J. Hong and G.-R. Yi, J. Mater. Chem. A, 2025, 13, 5213–5219 RSC.
- J. Hou, L. Lu, L. Wang, A. Ohma, D. Ren, X. Feng, Y. Li, Y. Li, I. Ootani, X. Han, W. Ren, X. He, Y. Nitta and M. Ouyang, Nat. Commun., 2020, 11, 5100 CrossRef CAS PubMed.
- Y.-S. Park and S.-M. Lee, Bull. Korean Chem. Soc., 2011, 32, 145–148 CrossRef CAS.
- M. Liu, Z. Zeng, Y. Wu, W. Zhong, S. Lei, S. Cheng, J. Wen and J. Xie, Energy Storage Mater., 2024, 65, 103133 CrossRef.
- S. Sharifi-Asl, J. Lu, K. Amine and R. Shahbazian-Yassar, Adv. Energy Mater., 2019, 9, 1900551 CrossRef.
- A. W. Golubkov, D. Fuchs, J. Wagner, H. Wiltsche, C. Stangl, G. Fauler, G. Voitic, A. Thaler and V. Hacker, RSC Adv., 2014, 4, 3633–3642 RSC.
- P. Bai, R. Xu, M. Liu, Z. Jia, Z. Sun, J. Wang, J. Zhang, J. Pei, Q. Zhang, J. Wang, C. Cao, X. Cao, Y. Yang and J. Zhang, ACS Appl. Energy Mater., 2023, 6, 7205–7211 CrossRef CAS.
- Z. Huang, X. Li, Q. Wang, Q. Duan, Y. Li, L. Li and Q. Wang, Int. J. Heat Mass Transfer, 2021, 172, 121077 CrossRef CAS.
- Y. Zhang, S. Cheng, W. Mei, L. Jiang, Z. Jia, Z. Cheng, J. Sun and Q. Wang, Appl. Energy, 2023, 336, 120695 CrossRef CAS.
- H. Sadeghi and F. Restuccia, J. Power Sources, 2024, 603, 234480 CrossRef CAS.
- J. Schöberl, M. Ank, M. Schreiber, N. Wassiliadis and M. Lienkamp, eTransportation, 2024, 19, 100305 CrossRef.
- H. Shen, H. Wang, M. Li, C. Li, Y. Zhang, Y. Li, X. Yang, X. Feng and M. Ouyang, Electronics, 2023, 12, 1603 CrossRef CAS.
- Y. Wang, X. Feng, W. Huang, X. He, L. Wang and M. Ouyang, Adv. Energy Mater., 2023, 13, 22038410 Search PubMed.
- J. Hou, X. Feng, L. Wang, X. Liu, A. Ohma, L. Lu, D. Ren, W. Huang, Y. Li, M. Yi, Y. Wang, J. Ren, Z. Meng, Z. Chu, G.-L. Xu, K. Amine, X. He, H. Wang, Y. Nitta and M. Ouyang, Energy Storage Mater., 2021, 39, 395–402 CrossRef.
- C. Xu, X. Feng, W. Huang, Y. Duan, T. Chen, S. Gao, L. Lu, F. Jiang and M. Ouyang, J. Energy Storage, 2020, 31, 101670 CrossRef.
- Y. Zhang, H. Wang, W. Li, C. Li and M. Ouyang, J. Energy Storage, 2020, 31, 101617 CrossRef.
- K. Xu, S. Zhang, J. L. Allen and T. R. Jow, J. Electrochem. Soc., 2002, 149, A1079–A1082 CrossRef CAS.
- S. Zheng, L. Wang, X. Feng and X. He, J. Power Sources, 2018, 378, 527–536 CrossRef CAS.
- A. Kvasha, C. Gutierrez, U. Osa, I. Meatza, J. A. Blazquez, H. Macicior and I. Urdampilleta, Energy, 2018, 159, 547–557 CrossRef CAS.
- D. Ren, X. Liu, X. Feng, L. Lu, M. Ouyang, J. Li and X. He, Appl. Energy, 2018, 228, 633–644 CrossRef CAS.
- T. Yoon, M. S. Milien, B. S. Parimalam and B. L. Lucht, Chem. Mater., 2017, 29, 3237–3245 CrossRef CAS.
- Y. Wang, D. Ren, X. Feng, L. Wang and M. Ouyang, J. Power Sources, 2021, 514, 230582 CrossRef CAS.
- D. Ouyang, M. Chen, J. Weng, K. Wang, J. Wang and Z. Wang, J. Energy Chem., 2023, 81, 543–573 CrossRef CAS.
- Y. E. Hyung, D. R. Vissers and K. Amine, J. Power Sources, 2003, 119–121, 383–387 CrossRef CAS.
- A. Yang, C. Yang, K. Xie, S. Xin, Z. Xiong, K. Li, Y.-G. Guo and Y. You, ACS Energy Lett., 2023, 8, 836–843 CrossRef CAS.
- S. Hess, M. Wohlfahrt-Mehrens and M. Wachtler, J. Electrochem. Soc., 2015, 162, A3084 CrossRef CAS.
- M. Zhang, J. Xiao, W. Tang, Y. He, P. Tan, M. Haranczyk and D.-Y. Wang, Adv. Energy Mater., 2024, 14, 2401241 CrossRef CAS.
- Y. Wang, Z. Li, Y. Hou, Z. Hao, Q. Zhang, Y. Ni, Y. Lu, Z. Yan, K. Zhang, Q. Zhao, F. Li and J. Chen, Chem. Soc. Rev., 2023, 52, 2713–2763 RSC.
- M. Fang, X. Yue, Y. Dong, Y. Chen and Z. Liang, Joule, 2024, 8, 91–103 CrossRef CAS.
- H. Q. Pham, H.-Y. Lee, E.-H. Hwang, Y.-G. Kwon and S.-W. Song, J. Power Sources, 2018, 404, 13–19 CrossRef CAS.
- R. McMillan, H. Slegr, Z. X. Shu and W. Wang, J. Power Sources, 1999, 81–82, 20–26 CrossRef CAS.
- N.-S. Choi, K. H. Yew, K. Y. Lee, M. Sung, H. Kim and S.-S. Kim, J. Power Sources, 2006, 161, 1254–1259 CrossRef CAS.
- I. A. Shkrob, J. F. Wishart and D. P. Abraham, J. Phys. Chem. C, 2015, 119, 14954–14964 CrossRef CAS.
- I. A. Profatilova, C. Stock, A. Schmitz, S. Passerini and M. Winter, J. Power Sources, 2013, 222, 140–149 CrossRef CAS.
- H. Q. Pham, E.-H. Hwang, Y.-G. Kwon and S.-W. Song, Chem. Commun., 2019, 55, 1256–1258 RSC.
- G. J. Chung, J. Han and S.-W. Song, ACS Appl. Mater. Interfaces, 2020, 12, 42868–42879 CrossRef CAS PubMed.
- Z. Zeng, X. Liu, X. Jiang, Z. Liu, Z. Peng, X. Feng, W. Chen, D. Xia, X. Ai, H. Yang and Y. Cao, InfoMat, 2020, 2, 984–992 CrossRef CAS.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- T. Yang, S. Li, W. Wang, J. Lu, W. Fan, X. Zuo and J. Nan, J. Power Sources, 2021, 505, 230055 CrossRef CAS.
- T. Achiha, T. Nakajima, Y. Ohzawa, M. Koh, A. Yamauchi, M. Kagawa and H. Aoyama, J. Electrochem. Soc., 2009, 156, A483 CrossRef CAS.
- X. Zhou, D. Peng, K. Deng, H. Chen, H. Zhou and J. Wang, J. Power Sources, 2023, 557, 232557 CrossRef CAS.
- S. Yang, T. Meng, Z. Wang and X. Hu, Energy Storage Mater., 2024, 65, 103177 CrossRef.
- D. Ouyang, J. Guan, X. Wan, B. Liu, C. Miao and Z. Wang, ACS Appl. Mater. Interfaces, 2024, 16, 42894–42904 CrossRef CAS PubMed.
- Y. Ugata, K. Yukishita, N. Kazahaya, S. Takahashi and N. Yabuuchi, Chem. Mater., 2023, 35, 3686–3693 CrossRef CAS.
- S. Zhang, S. Li, X. Wang, C. Li, Y. Liu, H. Cheng, S. Mao, Q. Wu, Z. Shen, J. Mao, H. Pan and Y. Lu, Nano Energy, 2023, 114, 108639 CrossRef CAS.
- Z. Zeng, V. Murugesan, K. S. Han, X. Jiang, Y. Cao, L. Xiao, X. Ai, H. Yang, J.-G. Zhang, M. L. Sushko and J. Liu, Nat. Energy, 2018, 3, 674–681 CrossRef CAS.
- U. Kulkarni, W.-J. Cho, S. Lee, D. S. Hwang, T. Im, Y. K. Jeong, K. Ahn and G.-R. Yi, Korean J. Chem. Eng., 2025, 42, 225–231 CrossRef CAS.
- X. L. Yao, S. Xie, C. H. Chen, Q. S. Wang, J. H. Sun, Y. L. Li and S. X. Lu, J. Power Sources, 2005, 144, 170–175 CrossRef CAS.
- X. Wang, E. Yasukawa and S. Kasuya, J. Electrochem. Soc., 2001, 148, A1058–A1065 CrossRef CAS.
- L. Siyuan, Z. Shichao, C. Shiyang, Z. Xiaoxian, C. Cheng, F. Ma, L. Zhang and Y. Lu, Energy Storage Mater., 2021, 42, 628–635 CrossRef.
- Z. Zeng, B. Wu, L. Xiao, X. Jiang, Y. Chen, X. Ai, H. Yang and Y. Cao, J. Power Sources, 2015, 279, 6–12 CrossRef CAS.
- H. F. Xiang, H. Y. Xu, Z. Z. Wang and C. H. Chen, J. Power Sources, 2007, 173, 562–564 CrossRef CAS.
- L. Jiang, Y. Cheng, S. Wang, Y. Cheng, K. Jin, J. Sun, M. Winter, I. Cekic-Laskovic and Q. Wang, J. Power Sources, 2023, 570, 233051 CrossRef CAS.
- Z.-C. Fu, F.-Y. Bu, Z.-P. Li, T. Wang, J.-N. Deng, H.-B. Zhao, S.-C. Huang, Y.-Z. Wang and M.-J. Chen, Chem. Eng. J., 2024, 479, 147935 CrossRef CAS.
- S. Lei, Z. Zeng, Y. Wu, M. Liu, S. Cheng and J. Xie, Chem. Eng. J., 2023, 463, 142181 CrossRef CAS.
- F. Xu, X. Cai, J. Zhang, L. Zhang, J. Wang, N. Zhang and S. Li, ACS Appl. Energy Mater., 2022, 5, 11370–11378 CrossRef CAS.
- J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama and A. Yamada, Nat. Energy, 2018, 3, 22–29 CrossRef CAS.
- G. Jiang, J. Liu, Z. Wang and J. Ma, Adv. Funct. Mater., 2023, 33, 2300629 CrossRef CAS.
- Y. Wang, Y. Zhao, S. Zhang, L. Shang, Y. Ni, Y. Lu, Y. Li, Z. Yan, Z. Miao and J. Chen, Angew. Chem., Int. Ed., 2024, 63, e202412108 CrossRef CAS PubMed.
- A. Yusuf, V. S. Avvaru, J. D. L. Vega, M. Zhang, J. G. Molleja and D.-Y. Wang, Chem. Eng. J., 2023, 455, 140678 CrossRef CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- X. Cao, Y. Xu, L. Zhang, M. H. Engelhard, L. Zhong, X. Ren, H. Jia, B. Liu, C. Niu, B. E. Matthews, H. Wu, B. W. Arey, C. Wang, J.-G. Zhang and W. Xu, ACS Energy Lett., 2019, 4, 2529–2534 CrossRef CAS.
- L. Xiao, Z. Zeng, X. Liu, Y. Fang, X. Jiang, Y. Shao, L. Zhuang, X. Ai, H. Yang, Y. Cao and J. Liu, ACS Energy Lett., 2019, 4, 483–488 CrossRef CAS.
- C. Shi, X. Huang, J. Gu, Z. Huang, F. Liu, M. Wang, Q. Wang, B. Hong, Z. Zhang, J. Li and Y. Lai, J. Energy Chem., 2023, 87, 501–508 CrossRef CAS.
- C. Liao, L. Han, W. Wang, W. Li, X. Mu, Y. Kan, J. Zhu, Z. Gui, X. He, L. Song and Y. Hu, Adv. Funct. Mater., 2023, 33, 2212605 CrossRef CAS.
- M. Nie, J. Demeaux, B. T. Young, D. R. Heskett, Y. Chen, A. Bose, J. C. Woicik and B. L. Lucht, J. Electrochem. Soc., 2015, 162, A7008–A7014 CrossRef CAS.
- H. Ota, Y. Sakata, A. Inoue and S. Yamaguchi, J. Electrochem. Soc., 2004, 151, A1659–A1669 CrossRef CAS.
- Y. Li, F. Ding, Y. Shao, H. Wang, X. Guo, C. Liu, X. Sui, G. Sun, J. Zhou and Z. Wang, Angew. Chem., Int. Ed., 2024, 63, e202317148 CrossRef CAS PubMed.
- H.-T. Kim, J. Kang, J. Mun, S. M. Oh, T. Yim and Y. G. Kim, ACS Sustainable Chem. Eng., 2016, 4, 497–505 CrossRef CAS.
- S. Lee, K. Park, B. Koo, C. Park, M. Jang, H. Lee and H. Lee, Adv. Funct. Mater., 2020, 30, 2003132 CrossRef CAS.
- B. Yang, C. Li, J. Zhou, J. Liu and Q. Zhang, Electrochim. Acta, 2014, 148, 39–45 CrossRef CAS.
- K. Chatterjee, A. D. Pathak, A. Lakma, C. S. Sharma, K. K. Sahu and A. K. Singh, Sci. Rep., 2020, 10, 9606 CrossRef CAS PubMed.
- D. Belov and D.-T. Shieh, J. Solid State Electrochem., 2012, 16, 603–615 CrossRef CAS.
- F. Gebert, M. Longhini, F. Conti and A. J. Naylor, J. Power Sources, 2023, 556, 232412 CrossRef CAS.
- Y. Gu, S. Fang, L. Yang and S.-I. Hirano, Electrochim. Acta, 2021, 394, 139120 CrossRef CAS.
- Y. Gu, S. Fang, L. Yang and S.-I. Hirano, J. Mater. Chem. A, 2021, 9, 15363–15372 RSC.
- Y. Gu, S. Fang, L. Yang and S.-I. Hirano, ACS Appl. Energy Mater., 2021, 4, 4919–4927 CrossRef CAS.
- P. Shi, S. Fang, J. Huang, D. Luo, L. Yang and S.-I. Hirano, J. Mater. Chem. A, 2017, 5, 19982 RSC.
- P. Isken, C. Dippel, R. Schmitz, R. W. Schmitz, M. Kunze, S. Passerini, M. Winter and A. Lex-Balducci, Electrochim. Acta, 2011, 56, 7530–7535 CrossRef CAS.
- H. Jia, B. Broekhuis, Y. Xu, Z. Yang, D. Kautz, L. Zhong, M. H. Engelhard, Q. Zhao, M. E. Bowden, B. E. Matthews, C. Connor, F. Lin, C. Wang and W. Wu, ACS Appl. Mater. Interfaces, 2025, 17, 6260–6270 CrossRef CAS PubMed.
- X. Cui, P. Zhou, Z. Xu, Q. Liu, Y. Nuli, J. Wang, R. C. V. Lehn and J. Yang, Energy Storage Mater., 2024, 66, 103235 CrossRef.
- R. He, K. Deng, D. Mo, X. Guan, Y. Hu, K. Yang, Z. Yan and H. Xie, Angew. Chem., Int. Ed., 2024, 63, e202317176 CrossRef CAS PubMed.
- Z. Chang, C. Ma, R. Wang, B. Wang, M. Yang, B. Li, T. Zhang, Z. Li, P. Zhao, X. Qi and J. Wang, ACS Appl. Mater. Interfaces, 2024, 16, 18980–18990 CrossRef CAS PubMed.
- J. Lee, A.-R. Jeon, H. J. Lee, U. Shin, Y. Yoo, H.-D. Lim, C. Han, H. Lee, Y. J. Kim, J. Baek, D.-H. Seo and M. Lee, Energy Environ. Sci., 2023, 16, 2924 RSC.
- H. Jia, Z. Yang, Y. Xu, P. Gao, L. Zhong, D. J. Kautz, D. Wu, B. Fliegler, M. H. Engelhard, B. E. Matthews, B. Broekhuis, X. Cao, J. Fan, C. Wang, F. Lin and W. Xu, Adv. Energy Mater., 2023, 13, 2203144 CrossRef CAS.
- J. Hou, L. Wang, X. Feng, J. Terada, L. Lu, S. Yamazaki, A. Su, Y. Kuwajima, Y. Chen, T. Hidaka, X. He, H. Wang and M. Ouyang, Energy Environ. Mater., 2023, 6, e12297 CrossRef CAS.
- R. Mishra, M. Anne, S. Das, T. Chavva, M. V. Shelke and V. G. Pol, Adv. Sustainable Syst., 2024, 8, 2400273 CrossRef CAS.
- B. Wang, H. A. Doan, S.-B. Son, D. P. Abraham, S. E. Trask, A. Janswen, K. Xu and C. Liao, Nat. Commun., 2025, 16, 3413 CrossRef CAS PubMed.
- Z. Yang, W. Ye, X. Lei, D. Schweigert, H.-K. Kwon and A. Khajeh, npj Comput. Mater., 2024, 10, 296 CrossRef CAS.
- J. Li, M. Zhou, H.-H. Wu, L. Wang, J. Zhang, N. Wu, K. Pan, G. Liu, Y. Zhang, J. Han, X. Liu, X. Chen, J. Wan and Q. Zhang, Adv. Energy Mater., 2024, 14, 2304480 CrossRef CAS.
- B. R. D. Goswami, Y. Abdisobbouchi, H. Du, F. Mashayek, T. A. Kingston and V. Yurkiv, J. Power Sources, 2024, 614, 235015 CrossRef.
- Y. Wang, X. Feng, D. Guo, H. Hsu, J. Hou, F. Zhang, C. Xu, X. Chen, L. Wang, Q. Zhang and M. Ouyang, Joule, 2024, 8, 2639–2651 CrossRef CAS.
- K. Masalkovaite, P. Gasper and D. P. Finegan, Nat. Commun., 2024, 15, 8399 CrossRef CAS PubMed.
- Z. Lin, Q. Yao, S. Yang, H. Song, Z. Yu, Z. Li, S. Chen, M. Wang, Z. Wang, G. Zhang, L. Zhang, Z. Yu, X. Song, K. Zhou, W. Li, L. Yu, J. Xu and K. Chen, Adv. Funct. Mater., 2025, 2424110 CrossRef CAS.
- X. Liu, H. Jia and H. Li, Energy Storage Mater., 2024, 67, 103263 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |