
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of cyclobutane-fused chromanones via gold-mediated photocatalysis†‡
Vladislav A.
Voloshkin
 a,
Marco
Villa
b,
Ekaterina A.
Martynova
a,
Marek
Beliš
a,
Kristof
Van Hecke
a,
Paola
Ceroni
*b and
Steven P.
Nolan
*a
a,
Marco
Villa
b,
Ekaterina A.
Martynova
a,
Marek
Beliš
a,
Kristof
Van Hecke
a,
Paola
Ceroni
*b and
Steven P.
Nolan
*a
aDepartment of Chemistry, Centre for Sustainable Chemistry, Ghent University, Krijgslaan 281, S-3, 9000 Ghent, Belgium. E-mail: Steven.Nolan@ugent.be
bDepartment of Chemistry “Giacomo Ciamician”, Center for Chemical Catalysis–C3, University of Bologna, Via Selmi, 2, 40126 Bologna, Italy. E-mail: Paola.Ceroni@unibo.it
First published on 21st February 2024
Abstract
Energy transfer (EnT) photocatalysis has emerged as a valuable tool for constructing complex organic scaffolds via [2 + 2]-cycloaddition reactions. Herein, we present the use of [Au(SIPr)(Cbz)] as a sensitizer for the [2 + 2]-cycloaddition of coumarins and unactivated alkenes. Widely used in EnT catalysis, iridium and organic sensitizers proved less efficient under the examined catalytic conditions. The developed protocol permits the synthesis of cyclobutane-fused chromanones from readily available starting materials. A wide range of alkenes and substituted coumarins, including naturally occurring compounds, were reacted under mild conditions leading to structurally complex products with good functional group tolerance. Mechanistic studies reveal a previously overlooked reaction pathway for energy transfer catalysis involving coumarins.
Introduction
The field of photocatalysis has experienced tremendous growth during the past decade.1–3 It has rapidly become an important tool in organic chemistry allowing the construction of various scaffolds and enabling late-stage functionalization. Despite the dominance of photoredox catalysis employing an electron transfer (ET) mechanism,4–8 in recent years, numerous reports have focused on photocatalytic reactions proceeding via a triplet–triplet energy transfer (TTEnT) mechanism.9–11 Most reports utilizing the latter relate to assembling strained cyclobutane rings. The [2 + 2]-cycloaddition of alkenes is a thermally forbidden process and therefore was initially performed via direct excitation of reaction mixture with powerful UV lamps. However, such methodology is hardly used in modern synthetic chemistry due to several challenges, such as a lack of selectivity, photodegradation and side-reactivity, especially for functionalized substrates. The use of sensitizers overcomes these problems because this permits reactions to be carried out selectively when irradiated with light in wavelength ranges where neither reactant absorbs.Chromanones are privileged scaffolds found in many natural products,12 biologically active molecules, pharmaceutical compounds13,14 and are relevant to the area of flavours and fragrances.15–17 The cyclobutane motif is also often encountered in naturally occurring metabolites18–20 and important in the field of drug discovery.21 However, the assembly of cyclobutane-fused chromanones using a versatile and efficient method, from readily available reagents, represents an unsolved challenge (Fig. 1). There are reports presenting an intermolecular cycloaddition of coumarin and alkenes that describe photolytic excitation using high-pressure mercury lamps (400–500 W).22–28 The investigated scope is narrow and includes mostly alkyl-substituted alkenes and coumarins. Typically, reactions proceed by direct excitation of the reaction mixture with 50-to-100-fold excess of the alkene making these methods synthetically impractical. The Bach group, inspired by work of Lewis and Barancyk,29 have investigated Lewis acid-catalyzed intramolecular [2 + 2]-cycloaddition of one coumarin substrate.30 By conducting the reaction at low temperature and in the presence of 50 mol% of a chiral Lewis acid, the product was formed with an enantiomeric excess of 88%. In 2015, Liu and co-workers reported the intermolecular photocycloaddition of coumarin-3-carboxylates with acrylamides mediated by an iridium sensitizer in acetonitrile.31 An energy transfer reaction mechanism was proposed for this reaction and products were usually obtained in high yields after 24 hours of irradiation. However, the scope was limited to activated coumarin-3-carboxylates and acrylamides or acrylates. During the assembly of this manuscript, Liu and Guo reported the construction of trifluoromethylated cyclobutane scaffolds (3 examples) via a thioxanthone-catalyzed [2 + 2]-cycloaddition with 1-bromo-1-trifluoromethylethene.32 Additionally, two other recent articles focus on EnT photocatalysis involving coumarins. Park and co-workers reported the Ir-catalyzed alkene-alkyne [2 + 2]-cycloaddition.33 Among other compounds, five cyclobutene-fused chromanones, products of cycloaddition of coumarin and diarylacetylenes were synthesized. The Glorius group developed thioxanthone-catalyzed [2π + 2σ]-cycloaddition of coumarins and fused chromanones.34
 | ||
| Fig. 1 Overview of previously reported synthetic strategies leading to cyclobutane-fused chromanones. | ||
We have recently reported on novel NHC-gold carbazolyl sensitizers, possessing high triplet energy (ET) values enabling reactions previously inaccessible with iridium sensitizers.35 Moreover, these gold complexes allowed for a wide range of reactions in commonly-used and environmentally-friendly organic solvents, typically with shorter reaction times than when iridium photocatalysts are used.35,36 Such features showcase gold sensitizers as attractive alternatives to Ir analogues, which are dominant in the area of EnT catalysis. We now report on a simple protocol to access cyclobutane-fused chromanones via gold-catalyzed energy transfer [2 + 2]-cycloaddition between coumarins and various alkenes.
Results and discussion
We initiated our study by reacting coumarin 1a with three equivalents of vinyltrimethylsilane in ethyl acetate in the presence of 2 mol% of [Au(SIPr)(Cbz)] (SIPr = [N,N-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene]; Cbz = carbazolyl) under 365 nm 18 W LED lamp irradiation at room temperature. To our delight, full conversion of coumarin was reached within 1 hour. However, two main products were observed by 1H NMR spectroscopy of the crude reaction mixture. The first was identified as a product generated from a formal [2 + 2]-cycloaddition between coumarin and alkene 2a, the second proved to be the product resulting from the dimerization of coumarin, compound 3 (Table 1). Such dimerization have been extensively studied previously.37 When coumarin is irradiated with 310 nm light, mixtures of isomers of such dimers are formed, while irradiation in the presence of a sensitizer leads to formation of the anti-head-to-head dimer, as the predominant isomer. Consistent with previous reports, we observed anti-head-to-head dimer 3 in the reaction mixture. While performing the same reaction without alkene, full conversion of coumarin into dimer was observed after only 1 hour of irradiation (Scheme S1‡). As the productive [2 + 2] reaction appears to compete with the coumarin dimerization, we proceeded to optimize reaction conditions to maximize the yield of 2a. Increasing the reagent concentration did not affect the product ratio (see Table S1‡). Increasing the number of equivalents of alkene to 10 led to an increase in product yield to 43%. Combining this with the increase of concentration of the reaction mixture to 0.3 M raised the yield to 56% and decreased the amount of dimer to 32%. Previously, in all reports concerning EnT cycloaddition with coumarin longer reaction times had been used. We next performed the same reaction for a longer time, and this led to a significantly higher product yield of 71% after 4 hours of irradiation while the amount of dimer decreased to less than 10% (Table 1, entry 2). Longer reaction times resulted in 88% yield of the product and complete disappearance of the dimer (Table 1, entry 3). This behaviour has not been previously noted, as photolytic reactions have not appeared to be conducted in short reaction times. This observation suggests that, under these reaction conditions, coumarin dimer 3 is initially formed and is slowly converted into the coumarin plus alkene 2a [2 + 2]-cycloaddition product.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Cat. loading (%) | Alkene (eq.) | Time (h) | NMR yield of 2a (%) | NMR yield of 3 (%) |
| 1 | [Au(SIPr)(Cbz)] | 2 | 3 | 1 | 35 | 64 |
| 2 | [Au(SIPr)(Cbz)] | 2 | 3 | 4 | 71 | <10 |
| 3 | [Au(SIPr)(Cbz)] | 2 | 3 | 16 | 88 | — |
| 4 | [Au(IMes)(Cbz)] | 2 | 3 | 16 | 85 | — |
| 5 | [Au(IPr)(Cbz)] | 2 | 3 | 16 | 83 | — |
| 6 | [Au(SIMes)(Cbz)] | 2 | 3 | 16 | 84 | — |
| 7 | [Au(ICy)(Cbz)] | 2 | 3 | 16 | 59 | — |
| 8 | Thioxanthone | 2 | 3 | 16 | 28 | 70 |
| 9 | [Ir(dF(CF3)ppy)2(dtppy)]PF6 | 2 | 3 | 16 | 27 | 71 |
| 10 | Benzophenone | 4 | 3 | 16 | 26 | 74 |
| 11 | [Au(SIPr)(Cbz)] | 2 | 1 | 16 | 64 | 10 |
| 12 | [Au(SIPr)(Cbz)] | 2 | 2 | 16 | 80 | — |
| 13 | [Au(SIPr)(Cbz)] | 1 | 3 | 16 | 87 | — |
| 14 | [Au(SIPr)(Cbz)] | 0.5 | 3 | 16 | 87 | — |

|
||||||

With this intriguing result in hand, we proceeded with further optimization of reaction conditions. Firstly, we screened different catalysts to establish whether this variable affected the reaction outcome. Different NHC-gold carbazolyl complexes performed similarly, with [Au(SIPr)(Cbz)] permitting slightly higher yields (Table 1, entries 4–7). The only exception was [Au(ICy)(Cbz)], which was significantly less efficient, apparently due to lower photo- or chemostability, as signs of decomposition were observed during and after irradiation. A commonly employed iridium sensitizer, having one of the highest ET values, as well as thioxanthone were also examined. Both led to much lower product yields and high yields of dimer under identical conditions (Table 1, entries 8 and 9). These reactions were also performed with their more typical irradiation wavelength of 405 nm leading to the same outcome (see Table S2‡). This suggests that previously reported reactions involving coumarin might proceed through a similar yet unnoticed reaction pathway. Interestingly, the use of benzophenone led to the same outcome as when thioxanthone and the costly iridium sensitizers were used, although requiring higher (4 mol%) catalyst loading (Table 1, entry 10). Next, the effects of the amount of alkene and catalyst loading were evaluated. The use of three equivalents of alkene was found to be the optimal amount (Table 1, entries 11–13). It is possible to decrease the catalyst loading to 0.5 mol% without affecting the product yield (Table 1, entry 14, also see Table S3‡).
With optimized conditions in hand, we began exploring the reaction scope (Scheme 1). The model vinyltrimethylsilane afforded product 2a as a sole regioisomer, in high (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) diastereomeric ratio, in an 81% isolated yield. The same regioisomer was obtained with all examined terminal alkenes. Hex-1-ene and oct-1-ene provided products in 62% (2b) and 75% yields (2c) respectively, with slightly lower d.r. of 4:1. More sterically hindered tetramethylethylene reacted smoothly providing product 2d in an 81% yield. Reaction with an internal alkene, cis-hex-3-ene, led to a mixture of three diastereomers 2e in 90% overall yield. Interestingly, prolonged reaction time and more powerful lamps were required to reach full conversion with hex-3-ene. Next, we explored the reactivity of cyclic alkenes. The reaction with cyclopentene provided product as a mixture of two diastereoisomers with 2f being the major, while [2 + 2]-cycloaddition with cyclohexene unexpectedly led to the mixture of 3 diastereomers, with the minor being an isomer with unfavoured twisted conformation of the hexane ring. Expansion of the cycloalkene ring to cis-cyclooctene led to the formation of all 4 possible diastereomers 2h in a 1:3:4:6 ratio with an 81% overall yield. Subsequently, we investigated functional group compatibility aspects of the reaction. A reaction with vinyl butyl ether was the only reaction, which led to atypical diastereomer 2i as a sole product. Keto and alcohol groups were well tolerated and chromanones 2j and 2l were obtained with 93% and 51% yields, respectively. Tert-butylacrylate, which is considered an activated alkene for [2 + 2]-cycloadditions, also provided product 2k in a good 75% yield. Interestingly, 2-allylphenol reacted with coumarin providing chromanone 2m, while allylbenzene did not react and, in this instance, only dimer 3 was observed. Allyl ester provided product 2n with high diastereoselectivity. Oxirane-containing alkene proved a challenging cycloaddition partner affording 2o with somewhat lower yield as a mixture of two stereoisomers (R and S oxirane) in a 1:1 ratio.
:1) diastereomeric ratio, in an 81% isolated yield. The same regioisomer was obtained with all examined terminal alkenes. Hex-1-ene and oct-1-ene provided products in 62% (2b) and 75% yields (2c) respectively, with slightly lower d.r. of 4:1. More sterically hindered tetramethylethylene reacted smoothly providing product 2d in an 81% yield. Reaction with an internal alkene, cis-hex-3-ene, led to a mixture of three diastereomers 2e in 90% overall yield. Interestingly, prolonged reaction time and more powerful lamps were required to reach full conversion with hex-3-ene. Next, we explored the reactivity of cyclic alkenes. The reaction with cyclopentene provided product as a mixture of two diastereoisomers with 2f being the major, while [2 + 2]-cycloaddition with cyclohexene unexpectedly led to the mixture of 3 diastereomers, with the minor being an isomer with unfavoured twisted conformation of the hexane ring. Expansion of the cycloalkene ring to cis-cyclooctene led to the formation of all 4 possible diastereomers 2h in a 1:3:4:6 ratio with an 81% overall yield. Subsequently, we investigated functional group compatibility aspects of the reaction. A reaction with vinyl butyl ether was the only reaction, which led to atypical diastereomer 2i as a sole product. Keto and alcohol groups were well tolerated and chromanones 2j and 2l were obtained with 93% and 51% yields, respectively. Tert-butylacrylate, which is considered an activated alkene for [2 + 2]-cycloadditions, also provided product 2k in a good 75% yield. Interestingly, 2-allylphenol reacted with coumarin providing chromanone 2m, while allylbenzene did not react and, in this instance, only dimer 3 was observed. Allyl ester provided product 2n with high diastereoselectivity. Oxirane-containing alkene proved a challenging cycloaddition partner affording 2o with somewhat lower yield as a mixture of two stereoisomers (R and S oxirane) in a 1:1 ratio.
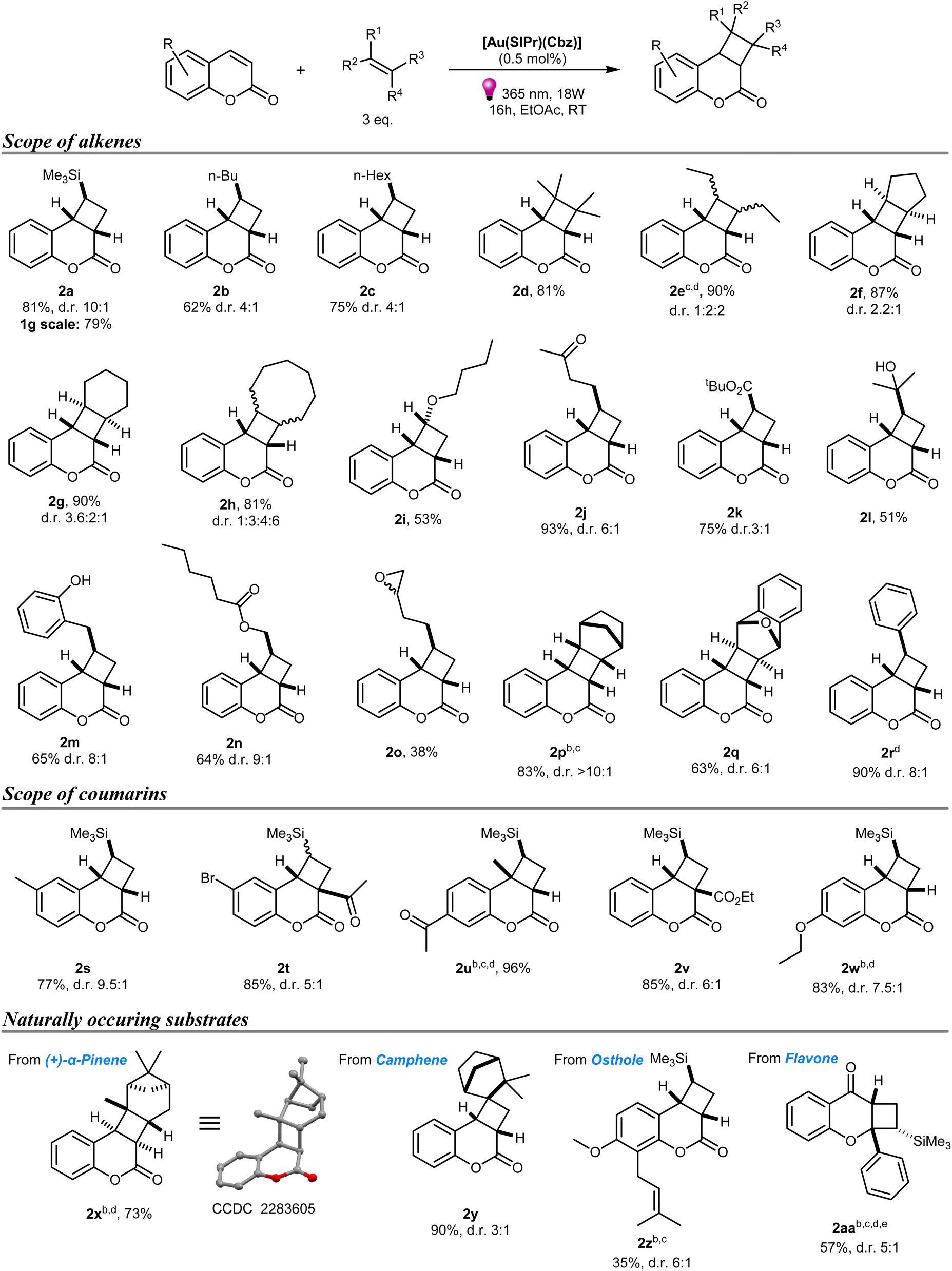 | ||
| Scheme 1 Substrate scope. a Yields of isolated products are reported as combined yields of both diastereomers; d.r. were determined by NMR of the crude; average of two runs. Structure of the major diastereomer is displayed. b 1 mol% [Au(SIPr)(Cbz)]. c 32 hours reaction time. d 30 W + 18 W LED lamps were used. e 6 eq. of alkene. | ||
Simple methodologies for the synthesis of conformationally restricted polycyclic three-dimensional scaffolds are nowadays quite important in the context of bioisosterism.38 We synthesized several of these structures using the present method by reacting coumarin with norbornene and 1,4-dihydro-1,4-epoxynaphthalene. In the case of norbornene, product 2n was isolated in an 82% yield with high diastereoselectivity. A slightly lower yield of 2q was obtained. We also tested whether an aryl-substituted alkene can act as coupling partner. The reaction with styrene provided product 2r in a 90% yield, although styrenes are known to have triplet energy of some 60 kcal mol−1.39
Scaling up of photocatalytic reactions in batch remains one of the challenges in the field, due to significant attenuation of light in the first millimetres of solution surface.40 We performed a 1-gram scale synthesis of 2a using the same experimental setup as in the smaller scale optimization. Full conversion to product was observed after 24 hours and a 79% yield was obtained after isolation.
Next, we explored the scope of various coumarins using vinyltrimethylsilane as the model alkene. Methyl-, bromo-, acyl- and ethoxy-functionalized coumarins were well-tolerated, affording products 2s–w in high yields. Moreover, methyl, acetyl, and ester substituents on the reactive double bond in these coumarins did not significantly interfere with the [2 + 2]-cycloaddition and products were obtained in high yields. Nitro- and hydroxy-substituted coumarins did not afford products even after prolonged irradiation. Decomposition was noticed in the case of the former which is typical for nitroarenes.41 In the case of umbelliferone, the solubility of the substrate was found problematic and could not be circumvented by changing solvent to dichloromethane or methanol (see ESI‡).
Functionalization of naturally occurring molecules or known biologically active molecules is a fascinating strategy used by the pharmaceutical industry. Therefore, we probed naturally abundant terpenes, pinene and camphene, as alkene partners for the [2 + 2]-cycloaddition with coumarin. Despite the rigid and sterically hindered nature of these molecules, cyclization products were obtained. Reaction with (+)-α-pinene afforded product 2x as a sole diastereomer in a 73% yield. Its structure was unambiguously confirmed by diffraction studies on a single crystal.42 Product 2y was obtained in even higher 90% yield with 3:1 diastereomeric ratio when camphene was used as the alkene. Osthole is a naturally occurring coumarin that possesses pharmacological and biological activities, including antitumor activity, anti-inflammatory, neuroprotective and immunomodulatory effects, and suppression of hepatitis.43 Reaction of osthole with vinyltrimethylsilane provided access to cyclobutane-fused chromanone 2z in 35% yield. This lower value can be attributed to the presence of another double bond in the molecule and possible intermolecular cyclization and oligo- or polymerization.
A structural isomer of coumarin – chromone – predictably did not react under our conditions as it has an exceedingly high triplet energy value of 74.8 kcal mol−1. However, the simplest of flavonoids, flavone with an ET value of 62 kcal mol−1 afforded the product 2aa in 57% yield, although an increase of the amount of alkene and irradiation time were needed to achieve this outcome (Scheme 1). Intrigued by this result, we also performed the reactions between flavone and other alkenes under these conditions (Scheme 2). The reaction with hex-1-ene provided product 2ab with d.r. 1.7:1 and 80% yield. Reactions with hex-5-en-2-one and tert-butylacrylate as coupling partners afforded the same regioisomers with 42% and 71% yields, respectively.
 | ||
| Scheme 2 [2 + 2]-cycloadditions with flavone as a substrate. | ||
The synthesized cyclobutane-fused chromanones can be considered late-stage precursors to highly decorated donor–acceptor cyclobutanes by taking advantage of the presence of the lactone ring. As an example, we subjected 2a to alkaline hydrolysis followed by addition of iodomethane to obtain 4a (Scheme 3).
 | ||
| Scheme 3 One-step synthesis of D–A cyclobutane 4a from chromanone 2a. | ||
The slow conversion of coumarin dimer 3 into product observed during the optimization phase of the study was worthy of another look. The photodimerization of coumarin was discovered by Ciamician and Silber in 1902, when sunlight was used to irradiate coumarin dissolved in alcohol for over 2 years.44 The phenomenon of coumarin dimerization under direct irradiation or sensitization has been thoroughly studied.37,45–49
It was shown that under direct irradiation by 300–350 nm light, intermolecular [2 + 2]-cycloaddition of two coumarin molecules occurs. The distribution of head-to-head and head-to-tail syn- or anti-isomers varies as a function of solvent, concentration, and irradiation time. However, if the reaction occurs in the presence of a sensitizer, such as benzophenone, anti-head-to-head dimer forms almost exclusively. It has also been shown that under direct irradiation with <300 nm light, dimers undergo photocleavage to two coumarin molecules.50,51 Intrigued by the fact that in all recent reports devoted to EnT intermolecular [2 + 2]-cycloadditions of coumarins no mention is made of coumarin dimer formation, we conducted a more in-depth study of this reaction to probe the mechanism at play in the present system.
Firstly, we performed control experiments that confirmed the need for a sensitizer and light irradiation for reaction to proceed (Scheme 4). Next, we performed control experiments with isolated coumarin dimer 3, which confirmed that it could be converted into product under reaction conditions. At the same time, the necessity for sensitizer and irradiation was confirmed for reaction between 3 and the alkene to proceed (Scheme S2‡). Absorption spectra of coumarin 1a, coumarin dimer 3, [Au(SIPr)(Cbz)] (Fig. 2) and vinyltrimethylsilane (Fig. S3‡) confirmed that only gold complex absorbs light under the reaction conditions. Previously, we showed that [Au(SIPr)(Cbz)] is irreversibly oxidized in solution and therefore photoredox mechanism is unlikely for this photocatalyst.35
 | ||
| Scheme 4 Control experiments. | ||
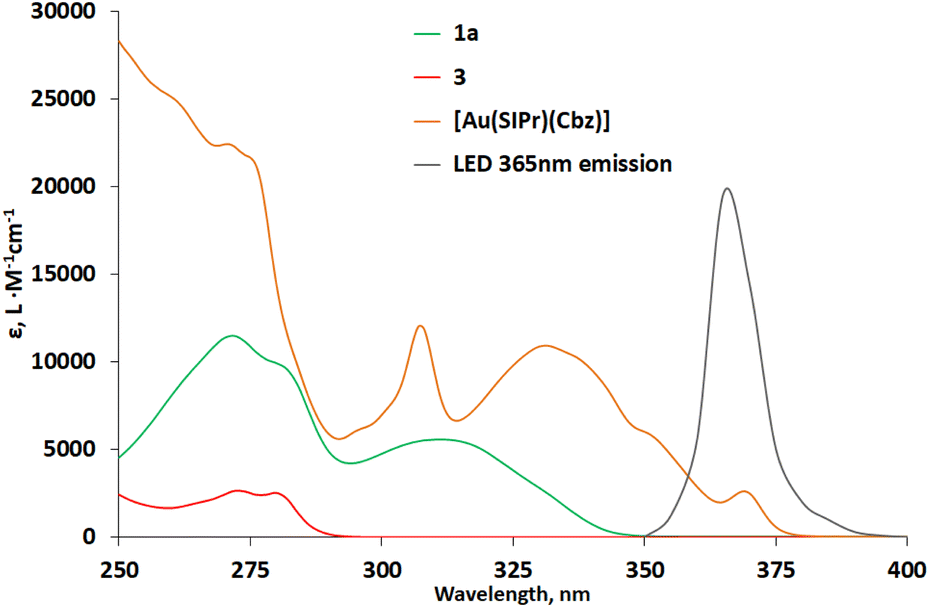 | ||
| Fig. 2 Absorption spectra of 1a, 3, and [Au(SIPr)Cbz]. For clarity emission spectrum of the LED lamp 365 nm is included. | ||
During optimization, we observed the formation of dimers, but the dimer formation was also observed when using an iridium sensitizer and thioxanthone. We also performed reactions in the presence of these sensitizers in acetonitrile, a solvent used in previous reports, to exclude possible solvent effects (Table S2‡). In both cases, the formation of dimer was observed in 59 to 62% yield, although a slightly higher yield of 2a was detected. Therefore, given that typically long irradiation times (>20 hours) had previously been used in photochemical reactions using coumarins, the initial formation of the coumarin dimer 3 and its conversion into final product might have been overlooked.
To gain insights into the photochemical mechanism, we evaluated the quenching of [Au(SIPr)(Cbz)] phosphorescence by Stern–Volmer studies: both coumarin 1a and coumarin dimer 3 quench the [Au(SIPr)(Cbz)] phosphorescence (see Fig. 3 and S6‡), while vinyltrimethylsilane does not (Fig. S5‡). The quenching rate constant in the case of the dimer is more than three orders of magnitude smaller than that of coumarin. Under the reaction conditions in which the coumarin concentration is 0.1 M, the quenching efficiency of the photocatalyst by 1a is more than 99.9%. When the concentration of coumarin is 100 times lower (0.001 M) than that of the dimer 3 (0.1 M) the quenching efficiency by 1a is still high – 95% (Table S4‡). This indicates that the photocatalyst is almost exclusively quenched by the coumarin 1a, even in the presence of 1 mol% of catalyst relative to dimer 3.
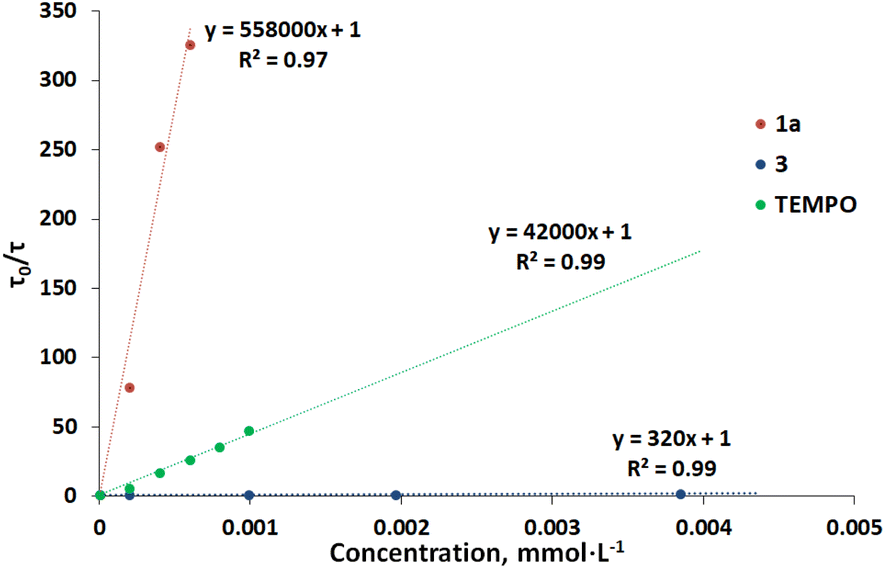 | ||
| Fig. 3 Stern–Volmer plot for catalyst quenching by 1a, 3 and TEMPO. | ||
Since the triplet energy of coumarin (31a*) is close in energy to the phosphorescent excited state of [Au(SIPr)(Cbz)] (see Table S6‡), triplet–triplet energy transfer from the photocatalyst to 1a is likely to occur. The photodimerization mechanism involves the population of the 31a* and the formation of an excimer 3(1a–1a)* that evolves by the [2 + 2]-cycloaddition to the dimer 3, as reported in the literature.37 The quantum yield of the photodimerization of 1a in EtOAc is 0.08, measured upon selective excitation of the [Au(SIPr)(Cbz)] photocatalyst (see Fig. S7 and Table S5‡), a value similar to that reported in the literature for sensitized photodimerization.47 On the other hand, upon selective excitation of the [Au(SIPr)(Cbz)] photocatalyst with dimer concentration of 0.1 M, no photocleavage of the latter is observed (Scheme 5), demonstrating that the photo-stationary state is shifted to the monomer. Therefore, even if the phosphorescent excited state of the photocatalyst is quenched by dimer 3 with the formation of two molecules of monomer, 1a converts back to the dimer via rapid sensitized dimerization. We also confirm that dimer 3 does not convert back to 1a upon heating in solution to 70 °C (Scheme 5).
 | ||
| Scheme 5 Dimer cleavage tests. | ||
Next, we performed reactions involving 1a and 3 under standard conditions in the presence of the well-known radical trap, TEMPO. Coumarin did not react with alkene in the presence of TEMPO, nor was it converted to dimer, with or without alkene in the reaction mixture (Scheme 6, eqn (1) and (2)). Interestingly, when the same reactions were conducted with the coumarin dimer 3, almost full conversion of 3 to coumarin 1a occurred in the presence of TEMPO, regardless of the presence or absence of an alkene (Scheme 6, eqn (3) and (4)). No formation of [2 + 2]-cycloaddition product was observed. To confirm that TEMPO does not cause coumarin dimer cleavage by itself, we performed control experiment without sensitizer in the reaction mixture leading to no conversion of dimer to coumarin (Scheme S3‡). Stern–Volmer quenching of [Au(SIPr)Cbz] phosphorescence by TEMPO revealed that the latter quenches the sensitizer (Table S4‡). However, under the reaction conditions, the sensitizer is mainly quenched by the coumarin, (see ESI‡ for more details) with a quenching efficiency of 94% for 1a and 6% for TEMPO. The dimerization reaction was completely suppressed in the presence of TEMPO because TEMPO does quench not only the sensitizer but also the coumarin triplet excited state 31a*. To estimate the quenching constant of the triplet excited state of coumarin 31a* by TEMPO, we compared the sensitized photodimerization quantum yield of 1a in the absence and presence of TEMPO (see Table S5‡). The quenching constant of 31a* by TEMPO is 7.4 × 108 M−1 s−1, a value higher that the self-quenching constant for coumarin reported in the literature (kq = 3.5 × 108 M−1 s−1 see Table S5‡).47 The quenching efficiency indicates that the triplet excited state of 1a is mainly quenched by TEMPO, in agreement with the experimental observation of dimerization suppression.
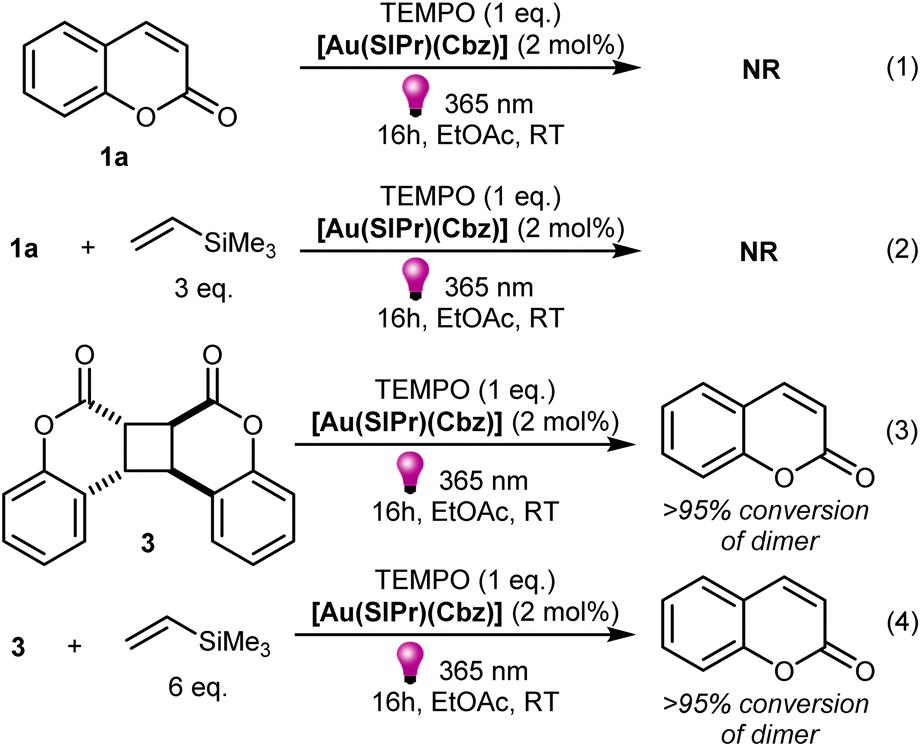 | ||
| Scheme 6 Control experiments in the presence of TEMPO. | ||
Based on these observations, we propose the mechanism presented in Scheme 7 where upon photoexcitation at 365 nm, the phosphorescent 3[Au(SIPr)Cbz]* is populated by intersystem crossing and this state undergoes energy transfer producing a triplet excited state coumarin molecule (31a*). It can then react either with the alkene, affording product 2, or with the coumarin ground state, affording ground state coumarin dimer 3. Coumarin dimer formation proceeds faster than [2 + 2]-cycloaddition with alkene, so that 3 accumulates in the reaction mixture after full conversion of coumarin. Coumarin dimer 3 is the main quencher of the photocatalyst in the absence of coumarin. Reaction between the coumarin dimer excited state and alkene does not occur, otherwise the product of [2 + 2]-cycloaddition would be observed in the presence of TEMPO (Scheme 6, eqn (4)). Although, we did not observe the formation of coumarin 1a from dimer 3 upon excitation, the same reaction in the presence of trapping agent TEMPO (Scheme 6, eqn (3)) provided indirect evidence for the formation of 1a, assumingly in very low quantities. Taking this into account, as well as the possibility of obtaining the product starting directly from the coumarin dimer 3, we suggest the reaction between 31* and alkene can occur due to the slow dimerization reaction because of very low concentration of 31* and a large excess of alkene in the reaction mixture. Thus, coumarin dimer 3 acts as a reservoir of coumarin. We also established that the initial rate of product formation is higher when starting with coumarin as a substrate than when starting with coumarin dimer (see ESI‡), which is in accordance with the proposed mechanism in Scheme 7.
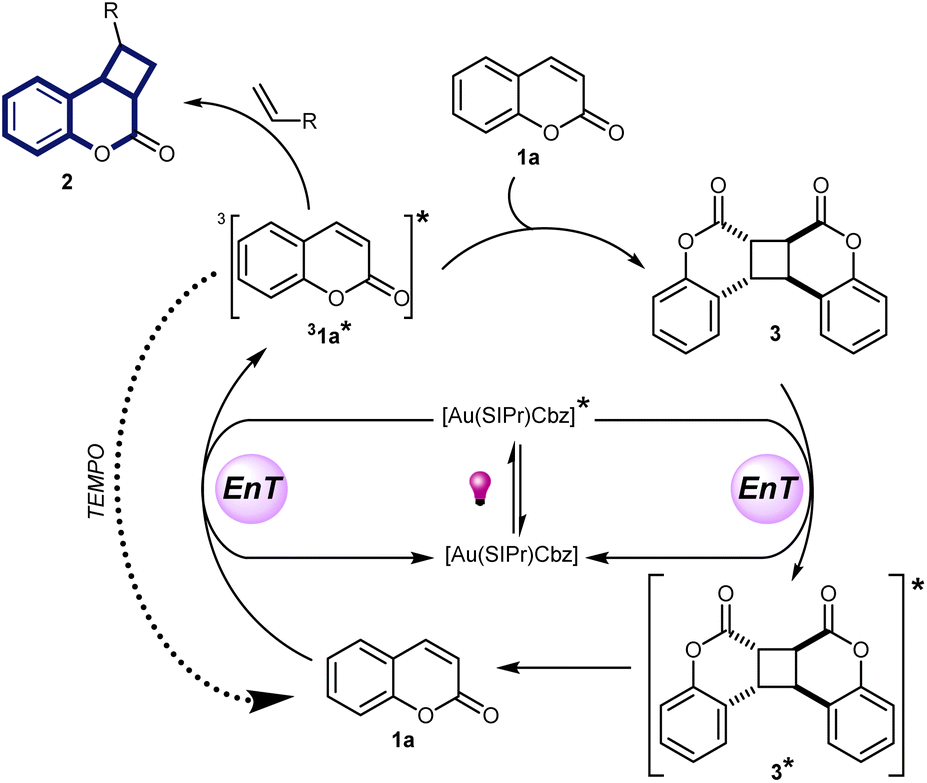 | ||
| Scheme 7 Proposed energy transfer mechanism of the reaction between coumarin and an alkene. | ||
To estimate and compare triplet energies of 1a and 3, emission spectra of these were recorded in rigid matrix at 77 K (Fig. S9‡). The triplet energy of 3 is higher (74.6 kcal mol−1) compared to 1a (63.2 kcal mol−1) and [Au(SIPr)(Cbz)] (67.9 kcal mol−1) (Table S6‡). Therefore, energy transfer between [Au(SIPr)(Cbz)] and coumarin dimer is endergonic. There is one recent example of productive endergonic energy transfer reaction reported by the Glorius group.52 Although we showed that the reaction proceeds with sensitizers possessing even lower ET values (65.5 kcal mol−1 for thioxanthone and 61.8 kcal mol−1 for [Ir(dF(CF3)ppy)2(dtppy)]PF6), the energy transfer mechanism would be even less favourable in these cases.
Therefore, we began to consider the mechanism involving electron transfer between excited photocatalyst and coumarin dimer 3. In one of the reports describing the sensitized photocleavage of 3, an electron transfer (eT) mechanism was proposed. Excited state benzophenone oxidizes the dimer to generate a radical cation 3˙+, which dissociates in 1a and 1a˙+ and then back electron transfer (BeT) between 1a˙+ and the reduced photocatalyst occurs.37 Therefore the dimer is completely cleaved with formation of two molecules of 1a. Although we have previously observed irreversible oxidation in cyclic voltammogram of [Au(SIPr)(Cbz)],35 we reasoned that with fast back electron transfer the EnT–eT–BeT pathway might be possible (Fig. S13 and S14‡). To probe the thermodynamic feasibility of this mechanism we recorded CVs for 1a, 3 and [Au(SIPr)(Cbz)] in THF (see Fig. S10–S12‡).
Based on the obtained redox potentials (Table S7‡) we calculated ΔE of the reactions both in reductive and oxidative quenching cycles with [Au(SIPr)(Cbz)] as a photoredox catalyst. According to the obtained data, excited state [Au(SIPr)(Cbz)] cannot oxidize coumarin dimer 3 making a reductive quenching pathway thermodynamically unfeasible. We did not observe any reduction of coumarin dimer 3 within THF solvent window, which also leads to negative ΔE of oxidative quenching, although it is not possible to determine its exact value. Interestingly, either reductive and oxidative quenching mechanisms are not thermodynamically feasible for the other studied photocatalysts [Ir(dF(CF3)ppy)2(dtppy)]PF6, thioxanthone and benzophenone. Therefore, the dual EnT–eT–BeT pathway appears to be improbable. While the possibility of an endergonic energy transfer pathway involving [Au(SIPr)(Cbz)] exists, additional mechanistic studies are needed to validate or refute this possibility and to elucidate the transformation of coumarin dimer 3 into the product. Furthermore, it is crucial to investigate why the reaction proceeds with sensitizers possessing significantly lower triplet energies.
Conclusions
In summary, we report on the synthesis of cyclobutane-fused chromanones from coumarins via gold-mediated energy transfer catalysis. The reactions proceed in an efficient manner with a wide range of alkenes and coumarins with good functional group tolerance and in a green solvent. Notably, iridium-based sensitizer and thioxanthone were found much less efficient under the same conditions. Products were obtained with naturally occurring alkenes, osthole and flavone. It was also shown that cyclobutane-fused chromanones can be converted into highly functionalized donor–acceptor cyclobutanes in one step. Full conversion of coumarin occurs already after 1 hour of irradiation resulting in a mixture of product and coumarin dimer 3. The latter is slowly converted into product upon further irradiation. Interestingly, although several reports using coumarin as a substrate have appeared, this interesting behaviour of coumarin has not been previously mentioned in the context of sensitized photocatalysis. Control experiments confirmed that this dimer formation also occurs with Ir-based sensitizer and thioxanthone. Further mechanistic investigations including trapping experiments, spectroscopic and electrochemical studies were conducted to provide insights into the reaction mechanism. Redox potentials of the reaction components indicated unlikely electron transfer events. At the same time, energy transfer to coumarin dimer was found to be endergonic. Further investigations into the reaction mechanism are ongoing in our laboratories to shed light on the conversion of coumarin dimer into product and to develop methods taking advantage of this facile [2 + 2]-cycloaddition product formation.Data availability
Data supporting the present study are available in the ESI.‡Author contributions
VAV, EAM and SPN conceived and designed the project. VAV and EAM performed all optimization studies and photocatalytic reactions. Mechanistic studies were conducted by VAV, MV and PC. Photochemical data were analysed by MV and PC. MB and KVH performed the XRD measurements and structure analysis. VAV, EAM and SPN wrote the manuscript with input from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the Special Research Fund (BOF) of Ghent University (project grants to SPN), the iBOF project C3 and The Research Foundation – Flanders (FWO) (G0A6823N and G099319N). PC and MV gratefully acknowledge support from the University of Bologna. We wish to thank Umicore AG for generous gift of materials. We thank Matteo Caruso for help with photochemical measurements and Francis Bru for cyclic voltammetry measurements.Notes and references
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- D. P. Kamat, S. G. Tilve, V. P. Kamat and J. K. Kirtany, Org. Prep. Proced. Int., 2015, 47, 1–79 CrossRef CAS.
- S. Emami and Z. Ghanbarimasir, Eur. J. Med. Chem., 2015, 93, 539–563 CrossRef CAS PubMed.
- S. Kamboj and R. Singh, Arabian J. Sci. Eng., 2022, 47, 75–111 CrossRef CAS PubMed.
- S. C. Rastogi, J. D. Johansen and T. Menne, Contact Dermatitis, 1996, 34, 423–426 CrossRef CAS PubMed.
- T. B. Adams, D. B. Greer, J. Doull, I. C. Munro, P. Newberne, P. S. Portoghese, R. L. Smith, B. M. Wagner, C. S. Weil, L. A. Woods and R. A. Ford, Food Chem. Toxicol., 1998, 36, 249–278 CrossRef CAS PubMed.
- G. A. Burdock, Fenaroli's Handbook of Flavor Ingredients, CRC Press, 6th edn, 2009 Search PubMed.
- V. M. Dembitsky, J. Nat. Med., 2008, 62, 1–33 CAS.
- Y.-Y. Fan, X.-H. Gao and J.-M. Yue, Sci. China: Chem., 2016, 59, 1126–1141 CrossRef CAS.
- J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136–154 RSC.
- M. R. van der Kolk, M. A. C. H. Janssen, F. P. J. T. Rutjes and D. Blanco-Ania, ChemMedChem, 2022, 17, e202200020 CrossRef CAS PubMed.
- J. W. Hanifin and E. Cohen, Tetrahedron Lett., 1966, 7, 5421–5426 CrossRef.
- J. W. Hanifin and E. Cohen, Tetrahedron Lett., 1966, 7, 1419–1424 CrossRef.
- J. W. Hanifin and E. Cohen, J. Am. Chem. Soc., 1969, 91, 4494–4499 CrossRef CAS.
- P. P. Wells and H. Morrison, J. Am. Chem. Soc., 1975, 97, 154–159 CrossRef CAS PubMed.
- N. Yonezawa, S. Nonoyama, K. Saigo and M. Hasegawa, J. Org. Chem., 1985, 50, 3026–3028 CrossRef CAS.
- M. Yasuda, T. Kishi, C. Goto, H. Satoda, K. Nakabayashi, T. Minami and K. Shima, Tetrahedron Lett., 1992, 33, 6465–6468 CrossRef CAS.
- M. Yasuda, T. Kishi, C. Goto, H. Satoda, K. Shima and K. Nakabayashi, J. Photochem. Photobiol., A, 1993, 76, 61–67 CrossRef CAS.
- F. D. Lewis and S. V. Barancyk, J. Am. Chem. Soc., 1989, 111, 8653–8661 CrossRef CAS.
- H. Guo, E. Herdtweck and T. Bach, Angew. Chem., Int. Ed., 2010, 49, 7782–7785 CrossRef CAS PubMed.
- Q. Liu, F.-P. Zhu, X.-L. Jin, X.-J. Wang, H. Chen and L.-Z. Wu, Chem.–Eur. J., 2015, 21, 10326–10329 CrossRef CAS PubMed.
- X. Hu, W. Xu, Y. Liu and H. Guo, J. Org. Chem., 2023, 88, 2521–2534 CrossRef CAS PubMed.
- S. Ha, Y. Lee, Y. Kwak, A. Mishra, E. Yu, B. Ryou and C.-M. Park, Nat. Commun., 2020, 11, 2509 CrossRef CAS PubMed.
- R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 605, 477–482 CrossRef CAS PubMed.
- E. A. Martynova, V. A. Voloshkin, S. G. Guillet, F. Bru, M. Beliš, K. V. Hecke, C. S. J. Cazin and S. P. Nolan, Chem. Sci., 2022, 13, 6852–6857 RSC.
- S. G. Guillet, A. A. Logvinov, V. A. Voloshkin, E. A. Martynova and S. P. Nolan, Org. Lett., 2023, 25, 1403–1408 CrossRef CAS PubMed.
- T. Wolff and H. Görner, Phys. Chem. Chem. Phys., 2004, 6, 368–376 RSC.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022, 610, 81–86 CrossRef CAS PubMed.
- CCDC 2283605 contain the supplementary crystallographic data for this paper.
- M. Sun, M. Sun and J. Zhang, Med. Chem. Res., 2021, 30, 1767–1794 CrossRef CAS PubMed.
- G. Ciamician and P. Silber, Berichte der Deutschen Chemischen Gesellschaft, 1902, 35, 1992–2000 CrossRef.
- G. S. Hammond, C. A. Stout and A. A. Lamola, J. Am. Chem. Soc., 1964, 86, 3103–3106 CrossRef CAS.
- H. Morrison, H. Curtis and T. McDowell, J. Am. Chem. Soc., 1966, 88, 5415–5419 CrossRef CAS.
- R. Hoffman, P. Wells and H. Morrison, J. Org. Chem., 1971, 36, 102–108 CrossRef CAS PubMed.
- X. Yu, D. Scheller, O. Rademacher and T. Wolff, J. Org. Chem., 2003, 68, 7386–7399 CrossRef CAS PubMed.
- K. Muthuramu and V. R. Murthy, J. Org. Chem., 1982, 47, 3976–3979 CrossRef CAS.
- T. Wolff and H. Görner, J. Photochem. Photobiol., A, 2010, 209, 219–223 CrossRef CAS.
- M. Jiang, N. Paul, N. Bieniek, T. Buckup, N. Hampp and M. Motzkus, Phys. Chem. Chem. Phys., 2017, 19, 4597–4606 RSC.
- F. Strieth-Kalthoff, C. Henkel, M. Teders, A. Kahnt, W. Knolle, A. Gómez-Suárez, K. Dirian, W. Alex, K. Bergander, C. G. Daniliuc, B. Abel, D. M. Guldi and F. Glorius, Chem, 2019, 5, 2183–2194 CAS.
Footnotes |
| † Dedicated to Prof. Carl D. Hoff on the occasion o his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2283605. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06675d |
| This journal is © The Royal Society of Chemistry 2024 |