
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the impact of CO2 exposure during calcination on the physicochemical and electrochemical properties of LiNi0.8Co0.1Mn0.1O2†
Marc
Nel-lo Pascual
 *a,
Elías Martínez
Moreno
a,
Leif Olaf
Jøsang
b,
Maximiliano
Merlo
a and
Jordi
Jacas Biendicho
*a
*a,
Elías Martínez
Moreno
a,
Leif Olaf
Jøsang
b,
Maximiliano
Merlo
a and
Jordi
Jacas Biendicho
*a
aCatalonia Institute for Energy Research-IREC, Sant Adriá de Besós, 08930 Barcelona, Spain. E-mail: mnello@irec.cat; jjacas@irec.cat
bCerpotech, Kvenildmyra 6, 7093 Heimdal, Norway
First published on 14th November 2024
Abstract
The synthesis atmosphere plays a fundamental role in determining the physicochemical properties and electrochemical performance of NMC811 cathode materials used in lithium-ion batteries. This study investigates the effect of carbonate impurities generated during synthesis by comparing three distinct samples: NMC811 calcined in ambient air, NMC811 calcined in synthetic air to mitigate carbonate formation, and NMC811 initially calcined in ambient air followed by annealing in synthetic air to eliminate carbonate species. Physicochemical characterization through XRD, SEM, FTIR, and TGA techniques revealed noticeable differences in the structural and chemical properties among the samples. Electrochemical assessments conducted via coin-cell testing demonstrate superior performance for materials synthesized in synthetic air, exhibiting an enhanced discharge capacity of 145.4 ± 4.8 mA h g−1 compared to materials synthesized in normal air (109.4 ± 4.3 mA h g−1) at C/10. More importantly, sample annealing in synthetic air after air calcination partially recovers the electrochemical performance of the cathode (142.1 ± 4.6 mA h g−1 at C/10) and this is related to the elimination of carbonate species from the ceramic powder. These findings highlight the importance of controlling synthesis conditions, particularly the atmosphere, to tailor the properties of NMC811 cathode materials for optimal lithium-ion battery performance.
1 Introduction
Li-ion batteries (LIBs) have become the main power source for the next generation of electric vehicles (EVs) because of their high energy density and long cycle life.1,2 Among the state-of-the-art cathode active materials (CAMs) for lithium-ion batteries, the classic LiCoO2 (LCO), and LiFePO4 (LFP) are still at the heart of nearly all portable electronic devices. For instance, while LFP cathodes are known for their safety, cycle life, and thermal stability, they typically offer lower discharge capacities (160–170 mA h g−1),3 which can be a limiting factor for high-demand energy density applications. Similarly, LCO (140–150 mA h g−1),4 provides excellent cycle life but suffers from safety concerns and higher costs due to the extensive use of cobalt. Nevertheless, mixed metal layered oxides, such as LiNiaMnbCocCO2 (a + b + c = 1) with various compositions have acquired popularity in the last years.5For the NCM materials, the general understanding is that Ni provides high capacity but poor thermal stability, that Mn maintains good cycle life and safety, and that Co offers structural stability and high electronic conductivity resulting in a better rate capability.6,7 To boost the energy density and minimize the dependence on Co, current development focuses on Ni-rich compositions.8,9 Nowadays, considerable research effort is devoted to NCM811 (180–200 mA h g−1),10 which is being envisaged as a potential CAM for high-energy battery packs in the next generation of electric cars.11,12
Despite its advantages, NMC811 faces several significant challenges that must be addressed to ensure its widespread adoption and reliable performance. One of the primary issues is that the high nickel concentration can lead to increased susceptibility to microcracking and structural instability during charge and discharge cycles,13 negatively impacting the battery's longevity and performance consistency. Another challenge is the complexity and cost of production; since these materials are also more challenging to synthesize, requiring oxygen-rich calcination conditions, and are more prone to lithium/transition-metal site exchange.14,15 This metal exchange occurs principally between Li and Ni ions due to the similar ionic radii of Ni2+ (0.69 Å) and Li+ (0.76 Å).16
Preparation of Ni-rich NMC cathode material is usually a two-stage process, first involving the formation of the precursor species (co-precipitation, sol–gel, hydrothermal, etc.) or the raw material (spray pyrolysis process) both of them then followed by calcination. Calcination requires temperatures up to 750–900 °C for prolonged dwelling times.17,18 Synthesis conditions play a critical role in the morphology of the final cathode, which strongly affects its rate performance and cyclability. Several competing processes occur at high temperatures, where cation mixing and particle growth depend on calcination temperature and dwelling time.19
The high surface reactivity of Ni-rich positive electrodes can lead to the formation of surface impurity species upon reactions with carbon dioxide and water, which can cause problems during electrode slurry preparation, battery storage, and cycling.20–25 These impurities have been documented to have the potential to compromise the electrochemical performance of the material21,22,25,26 as well as induce CO2 evolution during cycling.27–29 Despite that, the role of carbonates on battery performance is still under debate. On one hand, insulating materials can be produced on the cathode from these surface impurities reacting with the electrolyte leading to increased kinetic barriers in the Li+ insertion/desertion process. On the other hand, Li2CO3 has been explored as an electrolyte additive or as one component of a coating layer to mitigate side reactions at the cathode surface.30,31
In general, three processes can be responsible for the presence of surface carbonates, which include (i) residual impurities stemming from unreacted precursors during synthesis, (ii) a higher equilibrium coverage of surface carbonates required to stabilize the surface of Ni-rich materials after the synthesis process, and/or (iii) impurities formed during ambient storage.20 The vast majority of the reported studies are centered on studying the evolution and effect of carbonate species due to storage on commercial material,25,26,32,33 and there is little literature concerning the presence of this species after the synthesis of the material in atmospheres with different CO2 content.
In this context, this study aims to elucidate the influence of carbonate species on samples produced with different carbonate content. These differences are caused by calcining the raw NMC811 powders in atmospheres with different CO2 contents. Two distinct atmospheres were considered: air with an average of ≈400 ppm of CO2 concentration and commercial synthetic air with CO2 content ≤2 ppm. The former is expected to promote the formation of carbonate species during synthesis, and the latter is aimed at preventing their formation. Furthermore, to gain a better insight into the impact of carbonate species on material performance, a third sample was prepared: the air-calcined sample underwent subsequent annealing in synthetic air at 700 °C for 6 hours to effectively eliminate potential carbonate species. Through extensive physicochemical analyses, including X-ray diffraction (XRD), scanning electron microscopy (SEM), Fourier-transform infrared spectroscopy (FTIR), and thermogravimetric analysis (TGA), the variance in carbonate species content between the samples and their structural impact was evaluated. Electrochemical tests were subsequently conducted to assess the influence of carbonate species on the performance of NMC811.
2 Experimental
2.1 Material synthesis
LiNi0.8Co0.1Mn0.1O2 cathode material was synthesized via spray pyrolysis. The precursor solution was prepared by dissolving designed amounts of transition-metal nitrates in distilled water. The droplets were aerosolized from the precursor solution by a nebulizer inside a pyrolysis furnace (700–1200 °C). The resulting precursor was collected in a stainless-steel container. The mixture was annealed at 850 °C for 6 h in two different atmospheres: air and synthetic air (Linde, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 O2:N2, high purity mixture). The calcination temperature was set at 850 °C since it was selected based on optimization studies conducted through calcination tests. Some concluding results of this study can be observed in Fig. S1 in ESI.† A third sample was prepared to disclose the effect of carbonate species on the material's performance: the air-calcined sample was annealed in a synthetic air atmosphere at 700 °C for 6 hours. This temperature was selected based on thermogravimetric analysis (TGA) of Li2CO3 (Fig. S2†), as it corresponds to the onset of mass loss. The choice of 700 °C, the lowest temperature at which decomposition begins, was made to minimize potential alterations to particle morphology. A diagram of the synthesis process for each of the samples under study is shown in Fig. 1.
:80 O2:N2, high purity mixture). The calcination temperature was set at 850 °C since it was selected based on optimization studies conducted through calcination tests. Some concluding results of this study can be observed in Fig. S1 in ESI.† A third sample was prepared to disclose the effect of carbonate species on the material's performance: the air-calcined sample was annealed in a synthetic air atmosphere at 700 °C for 6 hours. This temperature was selected based on thermogravimetric analysis (TGA) of Li2CO3 (Fig. S2†), as it corresponds to the onset of mass loss. The choice of 700 °C, the lowest temperature at which decomposition begins, was made to minimize potential alterations to particle morphology. A diagram of the synthesis process for each of the samples under study is shown in Fig. 1.
 | ||
| Fig. 1 Graphical representation of the synthesis process for the NMC811 samples. | ||
2.2 Physicochemical characterization
X-ray diffraction (XRD, Cu-Kα radiation: λ = 0.15406 nm) was performed with a Bruker D8 Advance instrument to characterize the crystal structure of the samples. FT-IR analyses were performed with a Bruker Alpha-P instrument. TGA analyses were performed with a PerkinElmer TGA 4000. Finally, microstructure studies were performed with a ZEISS GeminiSEM scanning electron microscope (SEM).Rietveld refinement of NMC crystal structures was performed using GSAS® software. The background was fitted using the Chebyshev-1 function in GSAS®. Structural models involved refining lattice parameters, atomic positions, and occupancies to obtain the best fit. Thermal displacement parameters were fixed at 0.0200Uiso for all the elements and refinements. Structural constraints were added to evaluate Li–Ni site exchange: occupancy on the 3a site and 3b site were fixed to 1, where Li occupancy considering both sites had to be equal to 1.0 and Ni equal to 0.80. Co and Mn occupancies were fixed at 0.1 in all the refinements.
2.3 Electrochemical characterization
To prepare the samples’ cathodes, the NMC811 powders were mixed with carbon black Super-P as a conducting agent and polyvinylidene fluoride (PVDF, MTI, ≥99.5%) as a binder in a ratio of 90:5:5 (wt%). The mixture was then dispersed in N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich, ≥99.0%). All the mixing and dispersing steps were performed using a Mixer Mill MM400 (Retsch). The obtained NMC slurry was coated by applying 150 μm wet thickness on Al foil and dried at 110 °C under vacuum overnight. Finally, the aluminum foil was cut into 12 mm diameter disks to form the cathode. The resulting cathodes had a mass loading of around 3.8 mg cm−2 ± 0.3 mg. Coin cells with dimensions 2032 were fabricated using a Celgard® 2400 membrane as a separator and a lithium disc (∅: 12 mm) as an anode. The electrolyte consisted of 1 mol L−1 LiPF6 solution in EC:DEC (50/50 vol%, Sigma-Aldrich, battery grade). All the operations, except the cutting of the discs and its posterior drying at 110 °C under vacuum, were carried out in an argon-atmosphere glovebox (MBRAUN, <0.1 ppm H2O and O2).
Room temperature (RT) galvanostatic charge–discharge experiments were conducted using a CT-4008T Neware battery testing system. Two testing protocols in the voltage range of 3.0–4.3 V vs. Li/Li+ were used for electrochemical characterization. The cycling protocol included two formation cycles at C/20 (Qmax = 200 mA h g−1), followed by 50 cycles at C/10. Every charge (C/10) was performed in constant current – constant voltage (CC-CV) mode with a C/20 current cutoff at 4.3 V. Electrochemical impedance spectroscopy (EIS) measurements were also conducted on the cycled cells before and after the 50 cycles. Experiments were performed with a BCS-805 potentiostat/galvanostat system from BioLogic in a frequency range from 10 kHz to 100 mHz with an AC amplitude of 10 mV. Data analysis was performed using ZView® software. The second protocol included testing at different C-rates, which consisted of two C/20 formation cycles, followed by 5 cycles at C/10, C/5, C/2, 1C, C/5, and C/2. This second protocol's charge and discharge were carried out in CC mode. At least two replicates were conducted for each of the electrochemical experiments.
3 Results and discussion
NMC811 characterization results have been grouped into two distinct parts. The first part involves a physicochemical investigation of the materials to assess the impact of the gas atmosphere on its morphological, structural, and chemical properties. The second phase of this study focuses on the materials’ electrochemical performance and how it correlates to the previous physicochemical studies.3.1 Physichochemical characterization
NMC811 raw material obtained from spray pyrolysis was calcined at 850 °C for 6 h in two gas atmospheres: air (NMC811 AIR) and synthetic air (NMC811 SA). A third sample was included in these studies to elucidate the effect of carbonates on the NMC811 material. Since more carbonate species will be expected on the NMC811 AIR sample, this was thermally treated a second time at 700 °C for 6 h in synthetic air (NMC811 AIR + SA) to decompose these potential impurities. Fig. 2 presents a series of scanning electron microscopy images, offering a comprehensive view of the ceramic materials’ morphology and particle characteristics under the different gas atmospheres and temperature programs.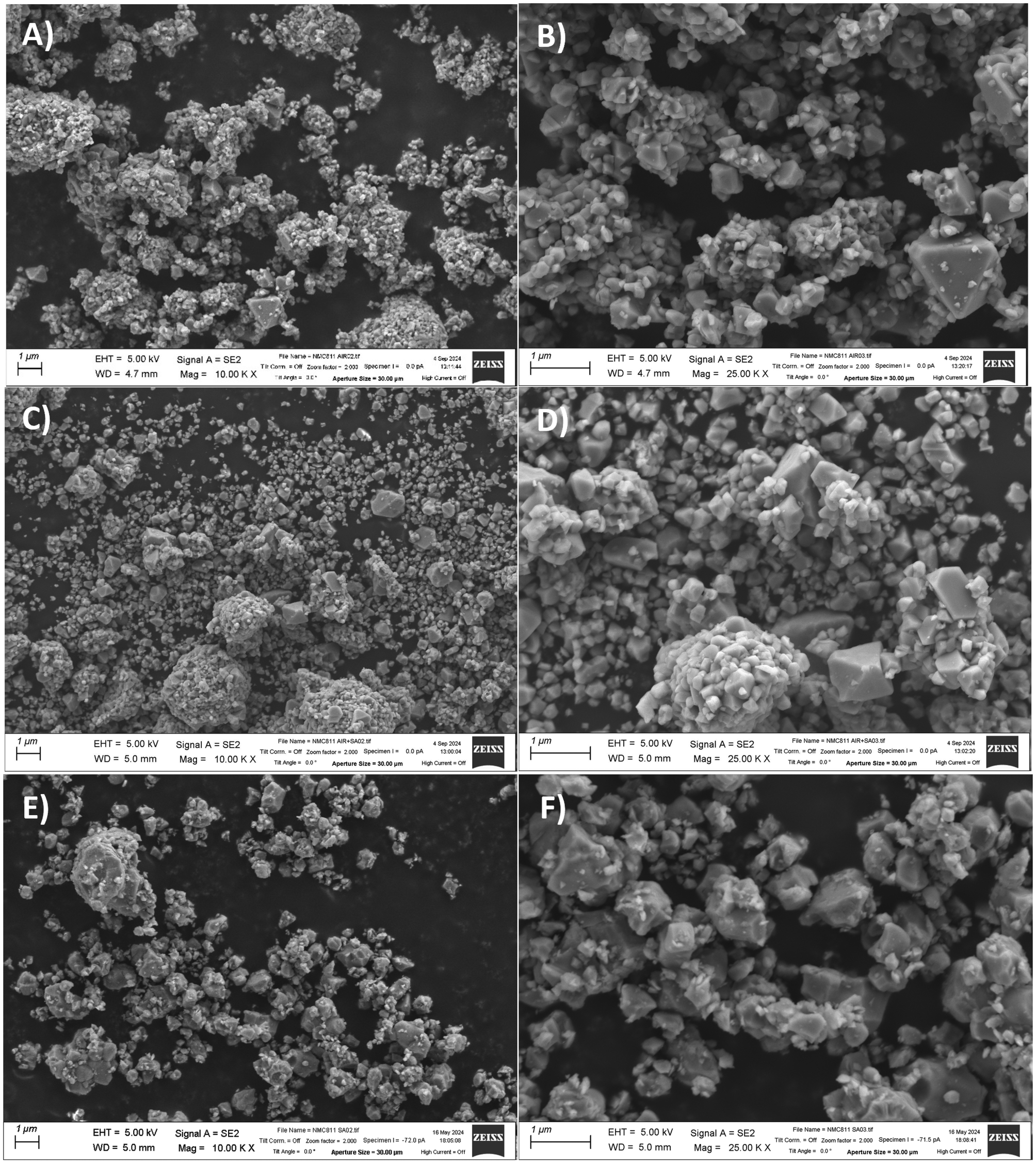 | ||
| Fig. 2 SEM images at ×10000 and ×25000 magnifications of the NMC811 calcined in (A and B) air, (C and D) synthetic air, and (E and F) air and annealed in synthetic air. | ||
All NMC811 samples demonstrated a similar morphology, showing particles between 100 and 500 nm. Some aggregates could be observed with sizes ranging from 1 to 10 μm. It is worth mentioning that the annealing in synthetic air had no significant impact on the morphology of the air-calcined material, as intended. PSD analysis was also performed on the samples to have better insight into the particles’ sizes and distribution. The analysis results are shown in Fig. 3 and Table 1.
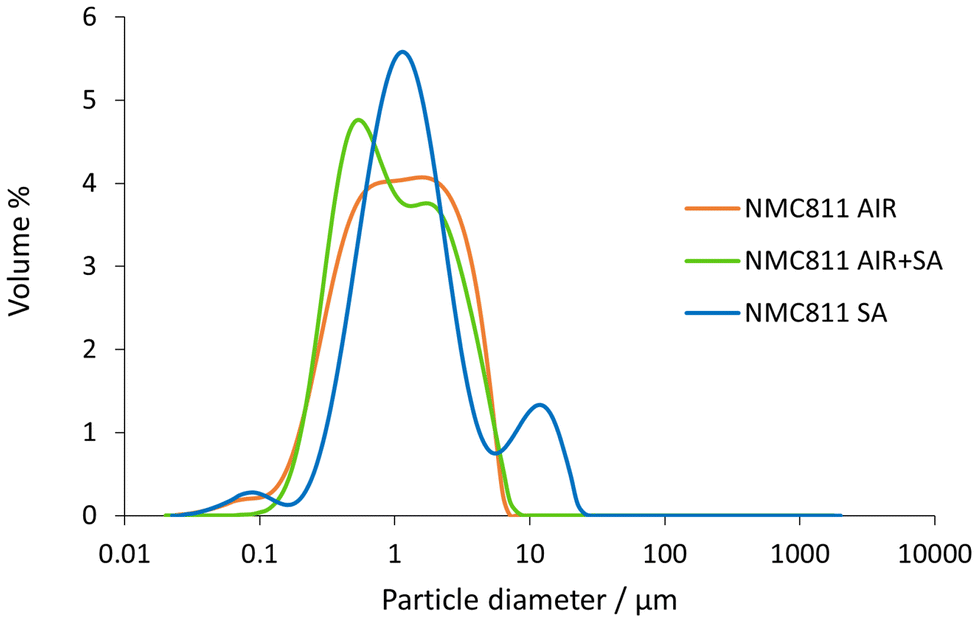 | ||
| Fig. 3 PSD analysis for the NMC811 AIR, AIR + SA, and SA sample powders. | ||
| NMC AIR | NMC AIR + SA | NMC SA | |
|---|---|---|---|
| d (0.1) | 0.315 | 0.336 | 0.418 |
| d (0.5) | 1.132 | 0.973 | 1.170 |
| d (0.9) | 3.675 | 3.500 | 6.942 |
From the analysis, most particles had an average particle size close to 1 μm. The only appreciated difference was that NMC811 SA had a larger aggregation, around 10 μm. Nevertheless, the d(0.9) of the sample indicated that 90% of the particles were below 7 μm, and 50% below 1.2 μm. These values were also valid for NMC811 AIR. The results confirmed SEM's observations, depicting that all materials showed a similar particle size.
XRD measurement was required to determine the impact of each calcination condition on the final crystallographic structure. The three NMC811 samples could show differences in their crystal structure due to Li/Ni cation mixing, carbonate impurities, or even the samples’ crystallinity. The resulting XRD data is shown in Fig. 4.
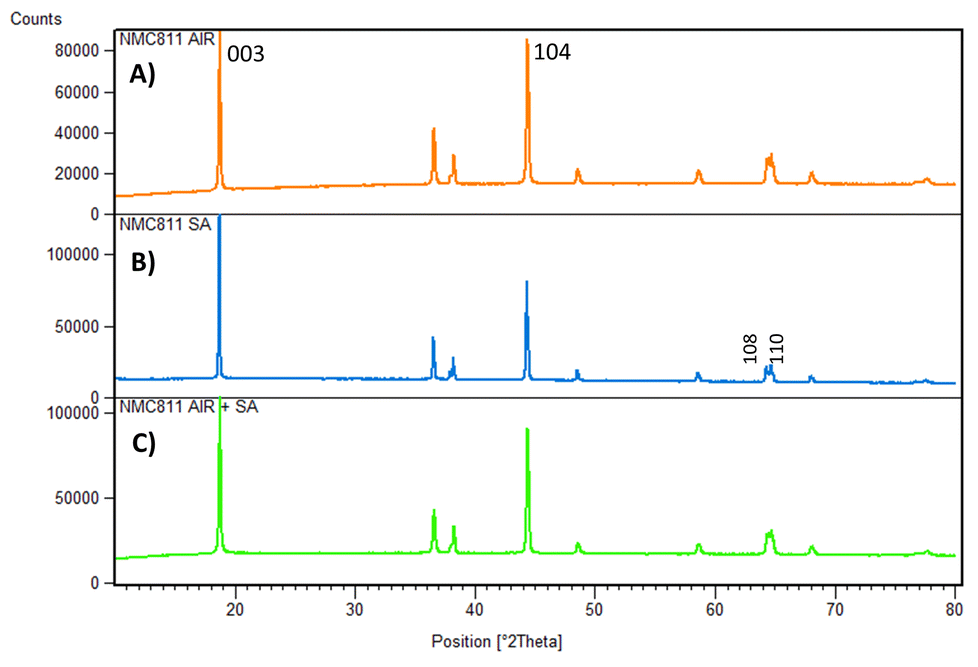 | ||
| Fig. 4 XRD spectra of the NMC811 calcined in (A) air, (B) synthetic air, and (C) air and synthetic air. | ||
All the powders exhibited a well-defined layered structure based on a hexagonal α-NaFeO2 structure with a space group R3![[m with combining macron]](https://www.rsc.org/images/entities/i_char_006d_0304.gif) and no impurity phases. The clear peak splits in the (108)/(110) doublets observed for the calcined sample in NMC811SA indicated a slightly higher crystallinity. The I003/I104 ratio was evaluated since it indirectly indicates cation mixing between Li+ and Ni2+ in the lithium layer. Usually, a lower I003/I104 ratio is related to increased cation mixing. It is important to minimize this crystallographic defect since the Ni2+ ions in the Li layer decrease discharge capacity and impede Li+ ionic diffusivity. Such structural defect is known as the leading cause of poor electrochemical performance for Ni-rich NMC materials.6,31,34
and no impurity phases. The clear peak splits in the (108)/(110) doublets observed for the calcined sample in NMC811SA indicated a slightly higher crystallinity. The I003/I104 ratio was evaluated since it indirectly indicates cation mixing between Li+ and Ni2+ in the lithium layer. Usually, a lower I003/I104 ratio is related to increased cation mixing. It is important to minimize this crystallographic defect since the Ni2+ ions in the Li layer decrease discharge capacity and impede Li+ ionic diffusivity. Such structural defect is known as the leading cause of poor electrochemical performance for Ni-rich NMC materials.6,31,34
From the XRD spectra results, significant differences could be appreciated in these peak intensities’ ratios (Table 2). It has been reported that a quotient of the intensities (I003/I104) below 1.2 describes a significant Li/Ni cation mixing (10% or more), implying a significant impact on the performance of the electrode.35
| Sample | I 003/I104 |
|---|---|
| NMC811 AIR | 0.88 |
| NMC811 SA | 1.17 |
| NMC811 AIR + SA | 1.14 |
Despite that, all materials exhibited values lower than 1.2; a major difference could be observed between the values obtained for the NMC materials calcined or annealed in SA and the just air calcined. The cation mixing improved significantly from the annealing process: from 0.88 (NMC811 AIR) to 1.14 (NMC811 AIR + SA), and the final refined value for the two samples thermally treated in SA was of the same order of magnitude.
As mentioned, a higher I003/I104 ratio indicates lower cation mixing.35 However, the best approach to analyze and quantify this defect is by performing a refinement of the crystal structure using diffraction data. The results of the structural refinement are presented in Table 3.
| NMC AIR | NMC SA | NMC AIR + SA | |
|---|---|---|---|
| a/Å | 2.87845(5) | 2.878614(24) | 2.87682(4) |
| c/Å | 14.19449(16) | 14.21275(8) | 14.19326(14) |
| Volume/Å3 | 101.8515(18) | 101.9942(9) | 101.7277(16) |
| Oxygen, z | 0.25604(12) | 0.25604(8) | 0.25752(9) |
| 3b Li/Ni, occ. | 0.8949(9)/0.1051(9) | 0.9135(6)/0.0865(6) | 0.9132(7)/0.0868(7) |
| 3a Ni/Li, occ. | 0.6949(9)/0.1051(9) | 0.7135(6)/0.0865(6) | 0.7132(7)/0.0868(7) |
| U iso | 0.0200 | 0.0200 | 0.0200 |
| χ 2 | 8.59 | 6.08 | 5.01 |
The refinement results followed the previously evaluated I003/I104 ratios trend. NMC811 AIR material had the highest cation mixing: 10.5%, while the materials thermally treated in synthetic air had cation mixings of 8.7%. It was further demonstrated that the SA treatment appeared to improve the cation mixing phenomenon. Overall, the samples’ cation mixing could be considered high, but this was principally related to the gas atmosphere employed during the calcination.17,18
It is also worth noting that there was a difference between the materials’ lattice parameters. The materials calcined in air showed a more similar c value, which was smaller than that of the synthetic air material. Despite that, there was no significant impact on the cell volume.
Since different atmospheres were used, the samples might differ in their amount of carbonate species on the surface of the material, caused by the reaction between Li2O or LiOH with CO2.26 It has been reported that surface impurities of lithium carbonate are notably involved in parasitic surface reactions36 as well as in CO2 gas evolution during cycling.27,28 To determine the presence of carbonates, an FT-IR analysis was performed. Fig. 5 shows the attained transmission spectra along with the Li2CO3 transmission spectrum for reference.
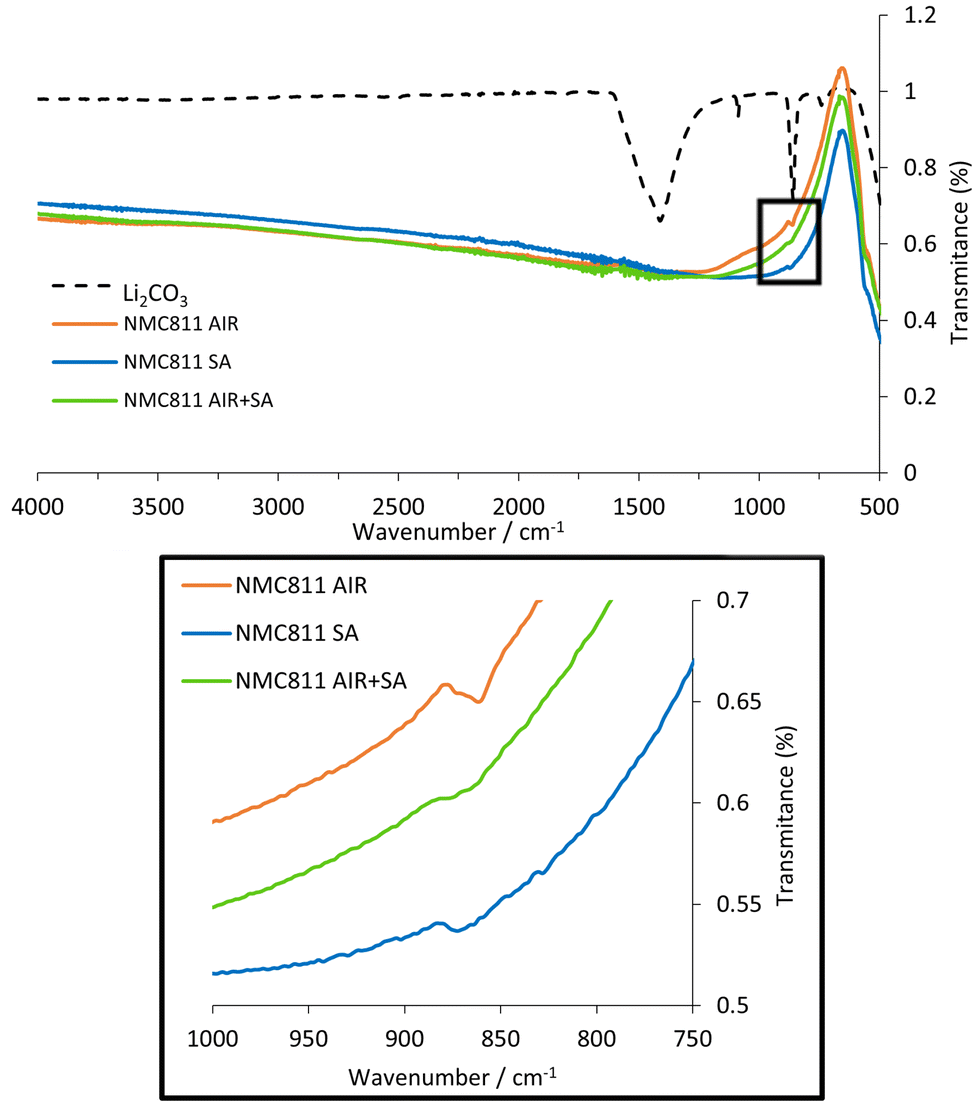 | ||
| Fig. 5 FT-IR spectra of NMC811 AIR, SA, and AIR + SA samples. FT-IR spectrum of commercial Li2CO3 (Alfa Aesar, 99%) is included for comparison. | ||
The most intense absorbance bands for Li2CO3 are observed at 1410–1450 cm−1 corresponding to the CO32− ion asymmetric stretching vibration and the 850–880 cm−1 corresponding to the CO32− ion bending out of plane vibrations.37 All NMC811 sample spectra showed a small absorbance close to 860 cm−1, indicative of the CO32− ion bending out of plane vibrations. Among these spectra, notable differences in signal transmittance could be observed (Fig. 5). The highest intensity was determined in the powder calcined in AIR, most likely due to the greater concentration of CO2 in the calcination gas. Conversely, both NMC811 samples calcined and annealed in SA showed smaller peaks. The presence of carbonates in these two samples was attributed to potential impurities in the gases used or to a reaction with atmospheric CO2, which could have occurred during the brief handling of these materials outside the glovebox.
To obtain a more precise determination of the carbonate content within the materials, TGA analysis was conducted on all three NMC811 materials. The procedure consisted of two identical sequential thermal programs using two different atmospheres: first N2 gas and second synthetic air. The first thermal program with N2 was used to decompose all carbonaceous species. However, with this thermal program, the material could also decompose, releasing O2 gas.15,26 The second thermal program in synthetic air was used to reintroduce the oxygen into the ceramic.15 Taking both programs into consideration, the difference between the initial and final weight corresponded to the liberated CO2 content (Fig. 6).
 | ||
| Fig. 6 TGA results of (A) NMC811 AIR, (B) NMC811 SA, and (C) NMC811 AIR + SA. | ||
The highest content in CO32− species was measured for the sample calcined in the air: 1.2% (w/w). This agreed with the previously shown FT-IR results (Fig. 5). The samples thermally treated in SA showed significantly lower values: 0.1% and 0.3%, respectively. Thus, the annealing in synthetic air at 700 °C for 6 hours substantially reduced the air-calcined sample's carbonate content.
This event was further confirmed upon assessing the thermal decomposition of Li2CO3 using a similar thermal process. In this case, a small sample of this salt was examined using TGA instrumentation, exposing it at 700 °C for 6 h in synthetic air (Fig. 7).
 | ||
| Fig. 7 FT-IR spectra of NMC811 AIR, SA, and AIR + SA samples. FT-IR spectrum of commercial Li2CO3 (Alfa Aesar, 99%) is included for comparison. | ||
After 6 hours of thermal treatment, the sample experienced around 40% weight loss. This value indicated that almost 70% of the carbonate content had been decomposed from the sample. The latter demonstrated that the procedure was sufficient to remove the samples’ carbonate content, which was closer to 1% of the sample weight for NMC811Air.
3.2 Electrochemical characterization
The cathode samples were assembled into half cells and subjected to cycling between 3.0 V and 4.3 V vs. Li+/Li, as depicted in Fig. 8. Two experiments were conducted: C/10 cycling of the materials to assess their stability and capacity retention, and C-rate measurements to correlate the materials’ capacities with different charge and discharge currents. It is important to highlight that after the cycling tests, post-mortem analysis of the samples revealed no significant differences between the fresh and cycled electrodes, as observed through SEM imaging. A direct comparison of the fresh and cycled electrodes is presented in Fig. S3.†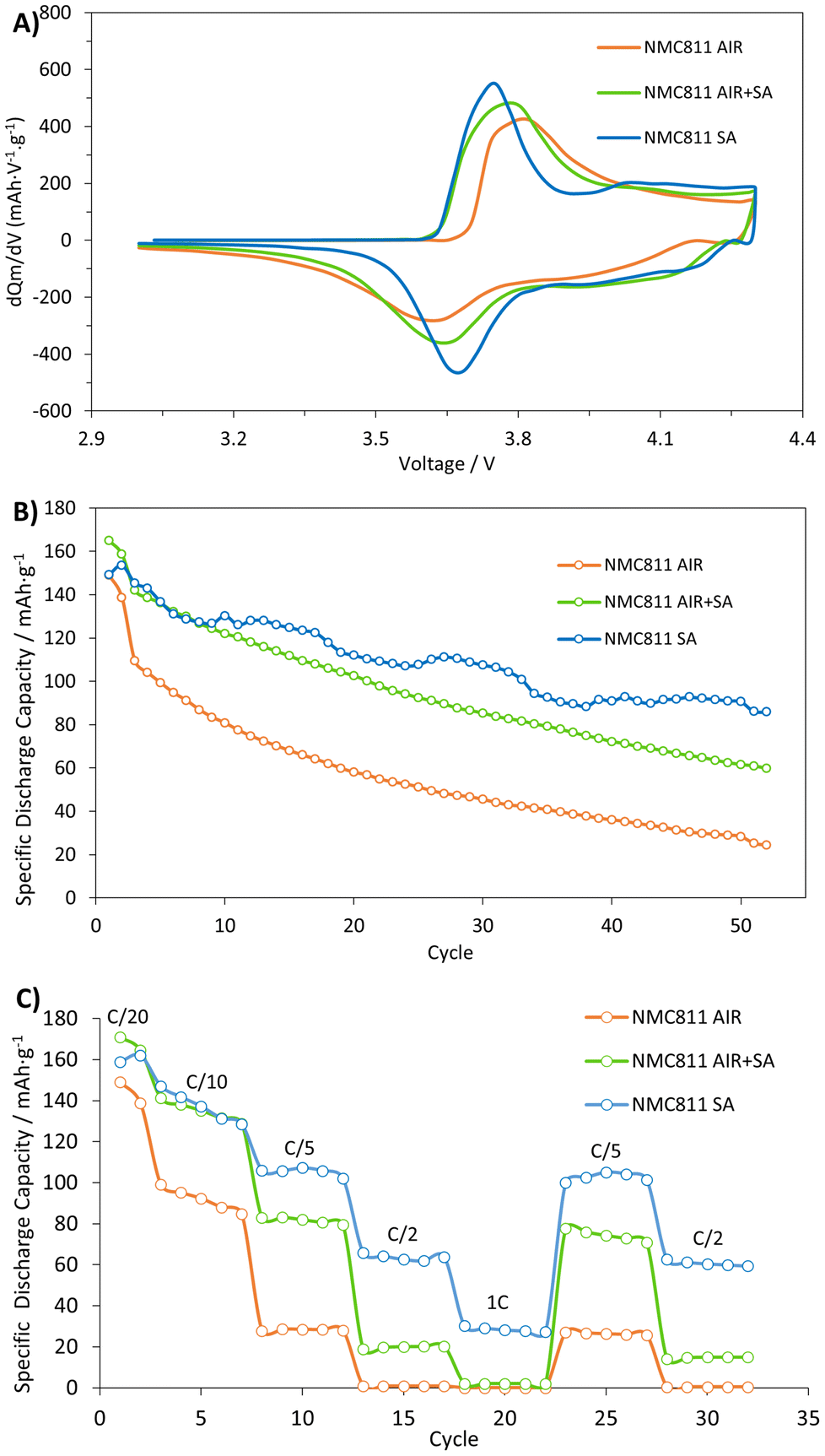 | ||
| Fig. 8 (A) dQm/dV plots of the second formation C/20 cycle, (B) cycle stability at C/10, and (C) C-rate capability from C/20 to 5C of NMC811 calcined in air, in synthetic air (SA), and in air and synthetic air (AIR + SA). | ||
Fig. 8A displays the dQm/dV plots of the second C/20 formation cycle. The primal distinction among the materials is the peak separation observed for the principal contributing peaks on the charge and discharge process. All materials show a similar oxidation peak position around 3.7 V typical of nickel-rich NMC materials, where Ni is the principal element oxidized,13,38–40 Co only starts to oxidize by the end of the peak. At this first potential (∼3.7 V), since Li is being removed, there is a slight volume contraction, causing an anisotropic change in the host lattice, where the a parameter contracts and the c parameter expands. This change in the lattice can be described as a hexagonal (H1) to hexagonal (H2) transition.6,41,42 It is worth noting that the H1 and H2 phases are crystallographically equivalent in terms of space group and atomic sites and are only distinguished by their different unit cell dimensions.
Despite that, the materials heat-treated in SA (NMC811 SA and NMC811 AIR + SA) originate at a lower potential: 3.61 V. On the other hand, the peak for the NMC811 AIR material starts at higher potentials (3.66 V). Conversely, reduction peaks show the opposite trend; the materials calcined and annealed in SA appear at the highest potentials, while the air-calcined material discharges at a lower potential. From these, both materials heat-treated in SA depict larger reversibility. The peak differences for each sample are 70 mV, 156 mV, and 229 mV for NMC811 SA, AIR + SA, and AIR, respectively. Furthermore, these materials show a broader area on their charge and discharge peaks, indicating larger oxidation and reduction capacities.
Upon further charging, a second oxidation peak should be reached at ∼4.2 V. At this point, there is a much more significant volume contraction caused by a drastic decrease in the c parameter. At this point, nickel and cobalt are further oxidized. Nickel experiences the most significant redox, oxidizing up to +3.9 at 4.3 V. Similarly, cobalt also oxidizes but to a lesser extent, from +3 to +3.5. Manganese was found to have an oxidation state of +3.9 for all cells and states of charge.40 The degree of Li depletion and the Ni and Co oxidation trigger different H2–M1–H3 phase transitions. Note that, these transitions are reversible but affect the cycling stability of the layered oxides.13,42
This second peak at ∼4.2 V did not appear for any of the studied materials. Given the possibility that the electrochemical process could be experiencing a significant overpotential, one electrode from each material was tested using the formation protocol (C/20) up to 4.7 V. The resulting graph (see Fig. S4†) shows an oxidation peak for all materials above 4.3 V. This finding supports the overpotential hypothesis, explaining why this peak is not visible in Fig. 8A.
Overall, it could be appreciated that the materials with almost no CO32− presence showed a better charging and discharging performance. The higher charging potential and the lower discharging potential may be attributed to the presence of carbonates, which have been described to have a notorious impact on the SEI14 and on the electrochemical performance.36,43 However, the published information is more related to the content of these species in the electrolyte rather than on the cathode material. In this case, the carbonate species appear to impede an appropriate migration of lithium ions, resulting in a slower kinetic and more inefficient charge transference which causes the observed higher peak splitting outcome. This event will be later evaluated by EIS analysis.
The cycling capabilities of these materials were evaluated (Fig. 8B). After two initial formation cycles at C/20, the cells were cycled at C/10. During the C/20 cycles, the sample with higher carbonate content exhibited the lowest capacity: 148.9 ± 2.9 mA h g−1. In contrast, the samples with lower carbonate content showed higher capacities: 153.5 ± 5.9 mA h g−1 for the SA sample and 165.1 ± 3.8 mA h g−1 for the AIR + SA sample. Such a result agrees with the previous dQm/dV results, where it could be observed that the material with a larger carbonate content delivers lower (anodic and cathodic) peak reversibility. At C/10 the capacities diminished to 109.4 ± 4.3, 145.4 ± 4.8, and 142.1 ± 4.6 mA h g−1 for NMC811 AIR, SA, and AIR + SA, respectively. After 50 cycles at C/10 rates, the NMC811 SA material outperformed NMC811 AIR and NMC811 AIR + SA. Specifically, the AIR, SA, and AIR + SA calcined materials retained 22%, 59%, and 42% of their initial capacity, respectively.
At high C-rates, the evolution pattern of the discharge capacity values follows a similar trend. For instance, at C/5, the material calcined in air shows the lowest capacity: 28.3 ± 2.0 mA h g−1, while AIR + SA and SA materials show significantly superior capacities: 80.8 ± 1.5 mA h g−1 and 105.4 ± 14.6 mA h g−1 respectively. As the C-rate increases, the difference between the SA and AIR + SA increases. At 1C both AIR and AIR + SA have reached their kinetic limit and cannot deliver capacity (≤2mA h g−1). Upon returning to a slower C-rate, specifically C/5, all materials exhibit satisfactory capacity recovery, with values of approximately 91%–97%. The electrochemical performance of the materials, ranked from worst to best, is as follows: NMC811 AIR, NMC811 AIR + SA, and NMC811 SA.
The annealing process with SA results in a notable improvement in the electrochemical properties of the material, though it does not achieve the performance level of the material calcined solely in synthetic air. This improvement is likely due to the elimination of carbonates. Furthermore, a direct correlation has been observed between the elimination of carbonates and the amount of cation mixing. This suggests an interesting hypothesis: the lithium released from the decomposition of carbonates is re-incorporated into the appropriate layer of the NMC811, causing the displacement of nickel atoms to the transition metal layer. Further work could evaluate the evolution of the material's crystalline structure during the annealing process utilizing in situ techniques.
To investigate the electrochemical kinetics of the NCM811 AIR, SA, and AIR + SA electrodes, EIS analysis was conducted before and after 50 charge–discharge cycles at C/10. Fig. 9 presents the Nyquist plots, revealing significant differences between the materials. Notably, the absence of a straight line at a 45° angle in the low-frequency region indicates that no semi-infinite diffusion process occurred, ruling out the use of a Warburg element for accurate data fitting. Consequently, no diffusion coefficients were calculated from the EIS data since these were expected to be significantly biased. Instead, a parallel R-CPE contribution was considered at low frequencies. Therefore, the circuit shown as an insert in Fig. 9A was chosen to fit the impedance data from Nyquist plots. This circuit consists of a resistance (Rs) in series with two RC components (Rhf and Rlf). The high-frequency resistance (Rhf) corresponds to the charge-transfer resistance typically observed in Li-ion coin cells44 and the low-frequency resistance (Rlf) corresponds to the extrapolated value of a non-ideal semicircle intercepting the x-axis at low frequencies.
 | ||
| Fig. 9 Nyquist plots for the NCM811 calcined in air (AIR), in synthetic air (SA), and in air and annealed in synthetic air (AIR + SA), (A) before and (B) after 50 cycles at C/10. The impedance was measured at a fully discharged electrode state. | ||
On the one hand, the series resistance (Rs), which involves the resistance of the electrolyte, connections, and cell components, remained consistent across all samples and experiments, ranging from 7 to 14 Ohms. On the other hand, the Rhf or charge-transfer resistance varied between materials and after cycling. The fitted results are shown in Tables 4 and 5. The highest values were observed in the material calcined in air, which can be attributed to its higher carbonate content. As discussed, carbonates act as a barrier to Li-ion diffusion and negatively affect charge transfer resistance due to their poor electrical conductivity.21,22,25,26
| Sample | R s (Ω) | R hf (Ω) | R lf (Ω) | χ 2 |
|---|---|---|---|---|
| NMC811 AIR | 13.7 | 2840.0 | 3743.0 | 0.086 |
| NMC811 AIR + SA | 12.8 | 1284.0 | 2351.0 | 0.172 |
| NMC811 SA | 10.7 | 120.6 | 4087.0 | 0.024 |
| Sample | R s (Ω) | R hf (Ω) | R lf (Ω) | χ 2 |
|---|---|---|---|---|
| NMC811 AIR | 6.5 | 1695.0 | 8815.0 | 0.110 |
| NMC811 AIR + SA | 10.4 | 1129.0 | 8741.0 | 0.019 |
| NMC811 SA | 11.8 | 215.6 | 3305.0 | 0.036 |
The AIR + SA material exhibited lower charge-transfer resistances compared to the NMC811 AIR material, but higher than the NMC811 material calcined in synthetic air. This suggests that, although most of the surface carbonates were removed, as confirmed by the absence of carbonates in a surface-level analysis (see Fig. 5), residual carbonates still influence this impedance contribution.
Finally, the NMC811 calcined in SA demonstrated the lowest resistances among the three materials, which aligns with its superior cycling performance. Indeed, the magnitude of the charge-transfer resistance or Rhf obtained for the NMC811 calcined in SA before cycling ∼120 Ω indicates a relatively good electrode contact at the coin cell level.45,46 This observation also explains why the other materials exhibited higher overpotentials during oxidation and reduction processes (Fig. 8A). The increased resistance after 50 cycles, from 120.6 to 215.6 Ω observed in Table 5 is a commonly observed phenomenon and indicative of NMC811 degradation after cycling.13
4 Conclusions
Overall, we demonstrate that the calcination gas atmosphere is critical in the physicochemical and electrochemical properties of NMC811 cathode material. Calcination in an air atmosphere produces carbonate species on the material, corresponding to a weight loss of 1.2%, as evidenced by thermogravimetric studies and FTIR analysis. In contrast, materials calcined or annealed in synthetic air showed a mass loss of 0.3% or less. These results conclude that post-treating an NMC811 sample at 700 °C for 6 hours in synthetic air significantly reduces its carbonate content.The removal of carbonate from an NMC811 sample improves its electrochemical performance. The material calcined in air demonstrates a capacity of 109.4 ± 4.3 mA h g−1 (first cycle at C/10) and a retention of 22% after 50 cycles at C/10. In contrast, after annealing in synthetic air, the same material shows a 28% increase in capacity (142.1 ± 4.6 mA h g−1) and a 91% improvement in retention, and a significantly smaller charge-transfer resistance (Table 4). The enhancement in electrical and electrochemical performance can be attributed to, firstly, the elimination of carbonates, which are detrimental to the material's electrochemical performance and, secondly, to the reduction in cation mixing observed in materials calcined in synthetic air by Rietveld refinement of the NMC crystal structure for the samples. The materials with lower carbonate content exhibit similar cation mixing, around 8.7%, which is notably lower than the 10.5% observed in the sample calcined solely in air.
Despite the improvements, powder annealing in synthetic air does not achieve the same electrochemical performance as direct calcination in synthetic air; the material calcined in synthetic air at 850 °C for 6 hours presents the best electrical and electrochemical results, with a capacity of 145.4 ± 4.8 mA h g−1 (first cycle at C/10), a retention of 59% after 50 cycles at C/10, and the lowest charge-transfer resistance ∼120 Ω. The 0.2% weight difference detected by TGA between NMC811 AIR + SA (0.3%) and NMC811 SA (0.1%) may still be too significant to overlook, suggesting that higher annealing temperatures could be explored. However, this approach carries the risk of altering particle morphology, which could complicate data interpretation by making it difficult to isolate the effects of carbonate content from morphological changes.
Author contributions
Conceptualization: M.N. and J.J.; methodology, M.N., M.M., and E.M.; validation, M.N., E.M., and J.J.; formal analysis, M.N and E.M.; investigation, M.N., M.M., and E.M.; resources, M.N, M.M., L.O., E.M., and J.J.; data curation, M.N., and E.M.; writing—original draft preparation, M.N.; writing—review and editing, M.N., E.M., L.O. and J.J.; supervision, M.N., E.M., and J.J.; project administration, M.N., and J.J.; funding acquisition, J.J. All authors have read and agreed to the published version of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† Raw data that support the findings of this study are available from the corresponding author, upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SPINMATE project, under the grant agreement No 101069712 was Funded by the European Union. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or CINEA. Neither the European Union nor the granting authority can be held responsible for them. The authors thank Generalitat de Catalunya for financial support through the CERCA Program and project 2021-SGR-01581. The authors also thank the support of the Department of Research and Universities of the Generalitat de Catalunya through the project M2E. J. J. B. acknowledges the fellowship RYC2021-034994-I, funded by MICIU/AEI/10.13039/501100011033 and the European Union “NextGenerationEU/PRTR”.References
- Y.-K. Sun, Z. Chen, H.-J. Noh, D.-J. Lee, H.-G. Jung, Y. Ren, S. Wang, C. S. Yoon, S.-T. Myung and K. Amine, Nat. Mater., 2012, 11, 942–947 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- C. M. Julien, K. Zaghib, A. Mauger and H. Groult, Adv. Chem. Eng. Sci., 2012, 02, 321–329 CrossRef CAS.
- P. Ramadass, J. Power Sources, 2002, 111, 210–220 CrossRef CAS.
- R. M. Salgado, F. Danzi, J. E. Oliveira, A. El-Azab, P. P. Camanho and M. H. Braga, Molecules, 2021, 26, 3188 CrossRef CAS PubMed.
- H.-J. Noh, S. Youn, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2013, 233, 121–130 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- H. Sun and K. Zhao, J. Phys. Chem. C, 2017, 121, 6002–6010 CrossRef CAS.
- M. D. Radin, S. Hy, M. Sina, C. Fang, H. Liu, J. Vinckeviciute, M. Zhang, M. S. Whittingham, Y. S. Meng and A. V. der Ven, Adv. Energy Mater., 2017, 7, 1602888 CrossRef.
- J. Xu, F. Lin, M. M. Doeff and W. Tong, J. Mater. Chem. A, 2017, 5, 874–901 RSC.
- F. Lin, D. Nordlund, Y. Li, M. K. Quan, L. Cheng, T.-C. Weng, Y. Liu, H. L. Xin and M. M. Doeff, Nat. Energy, 2016, 1, 15004 CrossRef CAS.
- J. Zheng, Y. Ye, T. Liu, Y. Xiao, C. Wang, F. Wang and F. Pan, Acc. Chem. Res., 2019, 52, 2201–2209 CrossRef CAS PubMed.
- T. Li, X.-Z. Yuan, L. Zhang, D. Song, K. Shi and C. Bock, Electrochem. Energy Rev., 2020, 3, 43–80 CrossRef.
- J. Zhao, W. Zhang, A. Huq, S. T. Misture, B. Zhang, S. Guo, L. Wu, Y. Zhu, Z. Chen, K. Amine, F. Pan, J. Bai and F. Wang, Adv. Energy Mater., 2017, 7, 1601266 CrossRef.
- M. S. Idris and A. R. West, J. Electrochem. Soc., 2012, 159, A396–A401 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- P. Swart, J. Dewulf and A. Biernaux, J. Cleaner Prod., 2014, 84, 391–399 CrossRef CAS.
- Y. Li, X. Li, Z. Wang, H. Guo and J. Wang, J. Energy Chem., 2018, 27, 447–450 CrossRef.
- J. M. Tarascon, W. R. McKinnon, F. Coowar, T. N. Bowmer, G. Amatucci and D. Guyomard, J. Electrochem. Soc., 1994, 141, 1421–1431 CrossRef CAS.
- J. Paulsen and J. H. Kim, US2014054495A1, 2014.
- Z. Chen, J. Wang, J. Huang, T. Fu, G. Sun, S. Lai, R. Zhou, K. Li and J. Zhao, J. Power Sources, 2017, 363, 168–176 CrossRef CAS.
- X. J. Zhu, H. H. Chen, H. Zhan, D. L. Yang and Y. H. Zhou, J. Mater. Sci., 2005, 40, 2995–2997 CrossRef CAS.
- K. Matsumoto, R. Kuzuo, K. Takeya and A. Yamanaka, J. Power Sources, 1999, 81–82, 558–561 CrossRef CAS.
- K. Shizuka, C. Kiyohara, K. Shima and Y. Takeda, J. Power Sources, 2007, 166, 233–238 CrossRef CAS.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132–A141 CrossRef CAS.
- J. Sicklinger, M. Metzger, H. Beyer, D. Pritzl and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A2322–A2335 CrossRef CAS.
- S. E. Renfrew and B. D. McCloskey, J. Am. Chem. Soc., 2017, 139, 17853–17860 CrossRef CAS PubMed.
- T. Hatsukade, A. Schiele, P. Hartmann, T. Brezesinski and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 38892–38899 CrossRef CAS PubMed.
- S. E. Renfrew and B. D. McCloskey, ACS Appl. Energy Mater., 2019, 2, 3762–3772 CrossRef CAS.
- R. Wang, X. Li, Z. Wang, H. Guo, T. Hou, G. Yan and B. Huang, J. Alloys Compd., 2015, 618, 349–356 CrossRef CAS.
- W. Liu, P. Oh, X. Liu, M. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef CAS PubMed.
- Z. Fang, M. P. Confer, Y. Wang, Q. Wang, M. R. Kunz, E. J. Dufek, B. Liaw, T. M. Klein, D. A. Dixon and R. Fushimi, J. Am. Chem. Soc., 2021, 143, 10261–10274 CrossRef CAS PubMed.
- A. C. Martinez, S. Grugeon, D. Cailleu, M. Courty, P. Tran-Van, B. Delobel and S. Laruelle, J. Power Sources, 2020, 468, 228204 CrossRef CAS.
- J. Li, G. Liang, W. Zheng, S. Zhang, K. Davey, W. K. Pang and Z. Guo, Nano Mater. Sci., 2022, 25899651 Search PubMed.
- P. Teichert, G. G. Eshetu, H. Jahnke and E. Figgemeier, Batteries, 2020, 6, 8 CrossRef CAS.
- J. P. Pender, G. Jha, D. H. Youn, J. M. Ziegler, I. Andoni, E. J. Choi, A. Heller, B. S. Dunn, P. S. Weiss, R. M. Penner and C. B. Mullins, ACS Nano, 2020, 14, 1243–1295 CrossRef CAS PubMed.
- P. Pasierb, S. Komornicki, M. Rokita and M. Rkas, J. Mol. Struct., 2001, 596, 151–156 CrossRef CAS.
- H. Kobayashi, Y. Arachi, S. Emura, H. Kageyama, K. Tatsumi and T. Kamiyama, J. Power Sources, 2005, 146, 640–644 CrossRef CAS.
- H. Li, M. Cormier, N. Zhang, J. Inglis, J. Li and J. R. Dahn, J. Electrochem. Soc., 2019, 166, A429–A439 CrossRef CAS.
- C. D. Quilty, P. J. West, W. Li, M. R. Dunkin, G. P. Wheeler, S. Ehrlich, L. Ma, C. Jaye, D. A. Fischer, E. S. Takeuchi, K. J. Takeuchi, D. C. Bock and A. C. Marschilok, Phys. Chem. Chem. Phys., 2022, 24, 11471–11485 RSC.
- W. Li, J. Reimers and J. Dahn, Solid State Ionics, 1993, 67, 123–130 CrossRef CAS.
- B. Dong, J. J. Biendicho, S. Hull, R. I. Smith and A. R. West, J. Electrochem. Soc., 2018, 165, A793 CrossRef CAS.
- A. Wuersig, W. Scheifele and P. Novák, J. Electrochem. Soc., 2007, 154, A449 CrossRef CAS.
- M. Gaberscek, J. Moskon, B. Erjavec, R. Dominko and J. Jamnik, Electrochem. Solid-State Lett., 2008, 11, A170 CrossRef CAS.
- Z. C. Huertas, D. Settipani, C. Flox, J. R. Morante, T. Kallio and J. J. Biendicho, Sci. Rep., 2022, 12, 137 CrossRef CAS PubMed.
- K. Rajappa Prakasha, J. Grins, A. Jaworski, T. Thersleff, G. Svensson, L. O. Jøsang, A. D. Dyrli, A. Paulus, D. De Sloovere, J. D'Haen, M. K. Van Bael, A. Hardy, H. Avireddy, J. R. Morante and J. Jacas Biendicho, Chem. Mater., 2022, 34, 3637–3647 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04146a |
| This journal is © The Royal Society of Chemistry 2024 |