
Chemical technology principles for selective bioconjugation of proteins and antibodies
Preeti
Chauhan
,
Ragendu
V.
 ,
Mohan
Kumar
,
Rajib
Molla
,
Surya Dev
Mishra
,
Sneha
Basa
and
Vishal
Rai
*
,
Mohan
Kumar
,
Rajib
Molla
,
Surya Dev
Mishra
,
Sneha
Basa
and
Vishal
Rai
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, 462 066, India. E-mail: vrai@iiserb.ac.in
First published on 14th December 2023
Abstract
Proteins are multifunctional large organic compounds that constitute an essential component of a living system. Hence, control over their bioconjugation impacts science at the chemistry–biology–medicine interface. A chemical toolbox for their precision engineering can boost healthcare and open a gateway for directed or precision therapeutics. Such a chemical toolbox remained elusive for a long time due to the complexity presented by the large pool of functional groups. The precise single-site modification of a protein requires a method to address a combination of selectivity attributes. This review focuses on guiding principles that can segregate them to simplify the task for a chemical method. Such a disintegration systematically employs a multi-step chemical transformation to deconvolute the selectivity challenges. It constitutes a disintegrate (DIN) theory that offers additional control parameters for tuning precision in protein bioconjugation. This review outlines the selectivity hurdles faced by chemical methods. It elaborates on the developments in the perspective of DIN theory to demonstrate simultaneous regulation of reactivity, chemoselectivity, site-selectivity, modularity, residue specificity, and protein specificity. It discusses the progress of such methods to construct protein and antibody conjugates for biologics, including antibody–fluorophore and antibody–drug conjugates (AFCs and ADCs). It also briefs how this knowledge can assist in developing small molecule-based covalent inhibitors. In the process, it highlights an opportunity for hypothesis-driven routes to accelerate discoveries of selective methods and establish new targetome in the precision engineering of proteins and antibodies.
 Preeti Chauhan | Dr Preeti Chauhan received her PhD under Prof. H. Chakrapani (IISER Pune) working on targeted and tunable release of H2S in biological systems. She joined Prof. Rai's team in 2022 to accelerate the translational efforts at SERB Precision Antibodies Engineering Center. |
 Ragendu V. | Ms Ragendu V. received her Integrated MSc from IIRBS, Mahatma Gandhi University, Kerala. She is pursuing her PhD under the supervision of Prof. Rai at IISER Bhopal and developing chemical technologies for homogeneous construction of AFCs and ADCs. |
 Mohan Kumar | Mr Mohan Kumar received his BSc (2016) from Lucknow University and MSc (2018) from NIT Manipur, India. In 2019, he joined Prof. Rai's team at IISER Bhopal and has been developing chemical technologies for the modification of proteins in living systems. |
 Rajib Molla | Mr Rajib Molla obtained his BSc (2016) and MSc (2018) from West Bengal State University. He is pursuing his PhD under the supervision of Prof. Rai at IISER Bhopal. He is applying his experience in organic and organometallic chemistry to develop new chemical methodologies for protein bioconjugation. |
 Surya Dev Mishra | Mr Surya Dev Mishra obtained his MSc in Biotechnology (2014) from BBAU Lucknow, India and MPhil (2018) from Central University of Gujarat, India. He joined Prof. Rai's group at IISER Bhopal in 2018. Currently, he is working on interface of chemistry and biology. |
 Sneha Basa | Ms Sneha Basa received her BSc (2017) from Pune University, Pune, and her MSc (2020) from IIT Madras, Chennai. She is pursuing her PhD under the supervision of Prof. Rai at IISER Bhopal. Her research involves developing chemical technologies for homogeneous construction of AFCs and ADCs. |
 Vishal Rai | Prof. Vishal Rai received his PhD under Prof. I. N. N. Namboothiri (IIT Bombay). After his postdoctoral research with Prof. Andrei Yudin (University of Toronto), he started his independent career at IISER Bhopal in 2011. His research group is developing chemical technologies for the precision engineering of native proteins to enable directed and precision therapeutics. He is the recipient of the CDRI award, SERB-TETRA award, DAE award, CRSI Bronze Medal, and Ramanujan and Swarnajayanti fellowships. He is an invited FRSC, Chemical Science Advisory Board member, ACS Chem. Biol. ECAB member, SERB-PACE coordinator, and founder of Plabeltech Private Limited. |
1. Introduction
Organic chemistry with proteins presents fascinating questions of selectivity for basic research. The rules with small molecules often do not extend to proteins. It promises to be a rich source for new principles of engineering chemical bonds. Such discoveries depend on an unambiguous analysis of products. However, it is not straightforward to analyze an unanticipated protein bioconjugate. Notably, translational demand for such technologies is pre-established. While driving selectivity of bioconjugation with isolated proteins is critical for protein-based therapeutics,1 extending it to a biological milieu could set a platform for small molecule-based precision therapeutics.2–6Hence, ground principles to promote hypothesis-driven research in the field could be extremely valuable. If we imagine proteins as a large multifunctional organic compound, it offers a reaction landscape full of nucleophilic residues. What will happen if an electrophile is added to its aqueous solution? Since protein concentration must be kept low to avoid their self-assembly, its impact on kinetics severely limits the bond formation efficiencies. This hurdle is addressed by enhancing the electrophile's stoichiometry, reactivity, or both. The electrophile would meet a pool of diverse nucleophilic proteinogenic residues at this stage. It is essential to realize that this landscape is much more diverse than a collection of amino acids. For example, a Lys residue in a protein presents a range of reactivity due to different microenvironments, unlike its isolated form. It is one of the significant contributors to the promiscuous reactivity of an electrophile when screened with a proteome. Once a chemoselective electrophile can distinguish between one type of residue from others, it is ready to be examined for site-selectivity. The latter requires it to distinguish a residue from its multiple copies. Hence, single-site protein modification demands a bioconjugation reagent to address a combination of reactivity, chemoselectivity, and site-selectivity (Fig. 1).7 While this is valid for significant cases, chemoselectivity is not a prerequisite for site-selectivity.
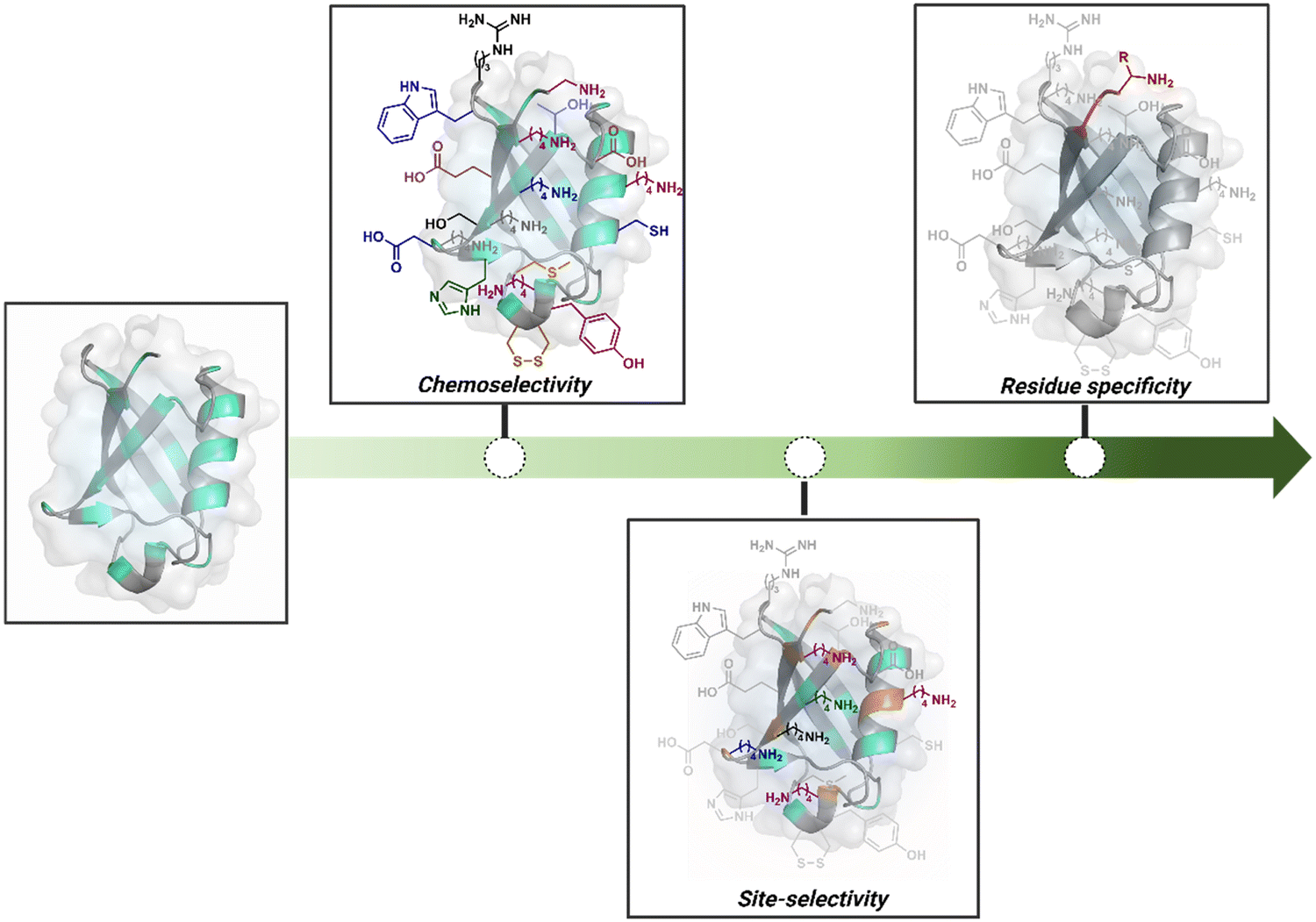 | ||
| Fig. 1 Representative selectivity challenges in the precision engineering of proteins. Chemoselectivity, site-selectivity, residue specificity, and beyond. | ||
The combined effect of a residue's solvent accessibility and microenvironment can make it a reactivity hotspot. Once established, such sites can be targeted via kinetically driven pathways. However, they can change with bioconjugation reagents and their binding preferences. The protein microenvironment is also capable of impacting the reactivity of reagents. Such an inherent character of proteinogenic residues often dictates bioconjugation's overall reactivity and selectivity. The initial efforts towards selective modification of proteins were primarily driven towards targeting such hotspots.
Going beyond such a landscape to establish a modular or residue-specific method can provide access to an entirely new targetome for precision engineering. We can take this a step forward and ask if such control over selectivity can be translated to drive protein specificity within a complex biological milieu.
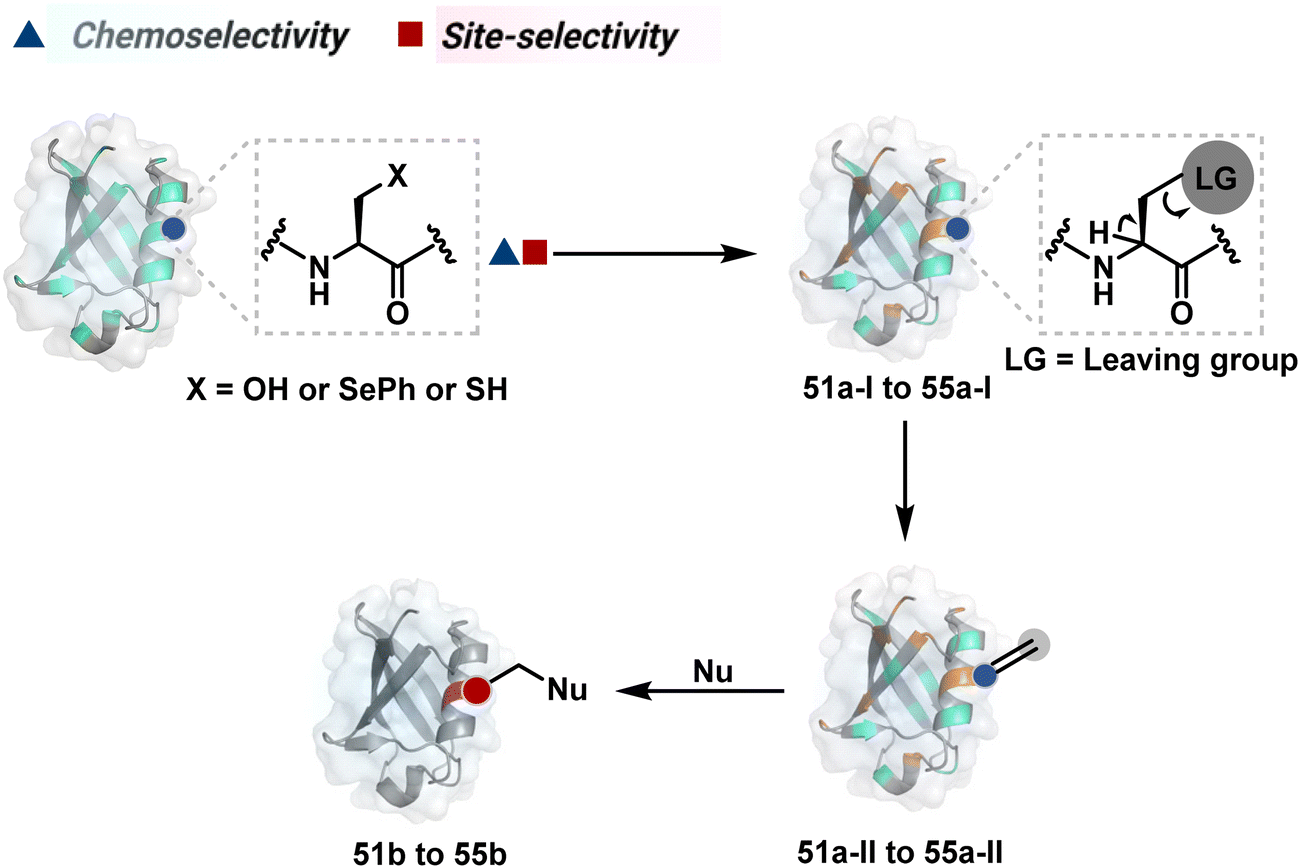
The initial demands relied entirely on heterogeneously labeled protein bioconjugates. Due to a lack of enabling methods, a significant market segment still depends on non-selective routes. Notably, the past few years have witnessed considerable developments in precision technologies. Firstly, reagents and reaction parameters evolved for single-site modification of proteins, where the selectivity attributes are defined in one step. Simultaneously addressing all selectivity parameters allow very few variables to control the precision in this approach. The disintegration of selectivity attributes can provide better control and expand the targetome. We presented the disintegrate (DIN) theory that utilizes a multi-step chemical transformation to disintegrate the selectivity attributes (Fig. 2).8 Such a deconvolution can regulate chemoselectivity, site-selectivity, or protein specificity in distinct steps to offer better control over reaction parameters. Besides, it opens a gateway for a redefined reactivity landscape of proteins (compare routes A, B, C, and D; Fig. 2). In turn, it makes it possible to develop methods where conjugation sites are beyond reactivity hotspots or the inherent reactivity order of proteinogenic residues.
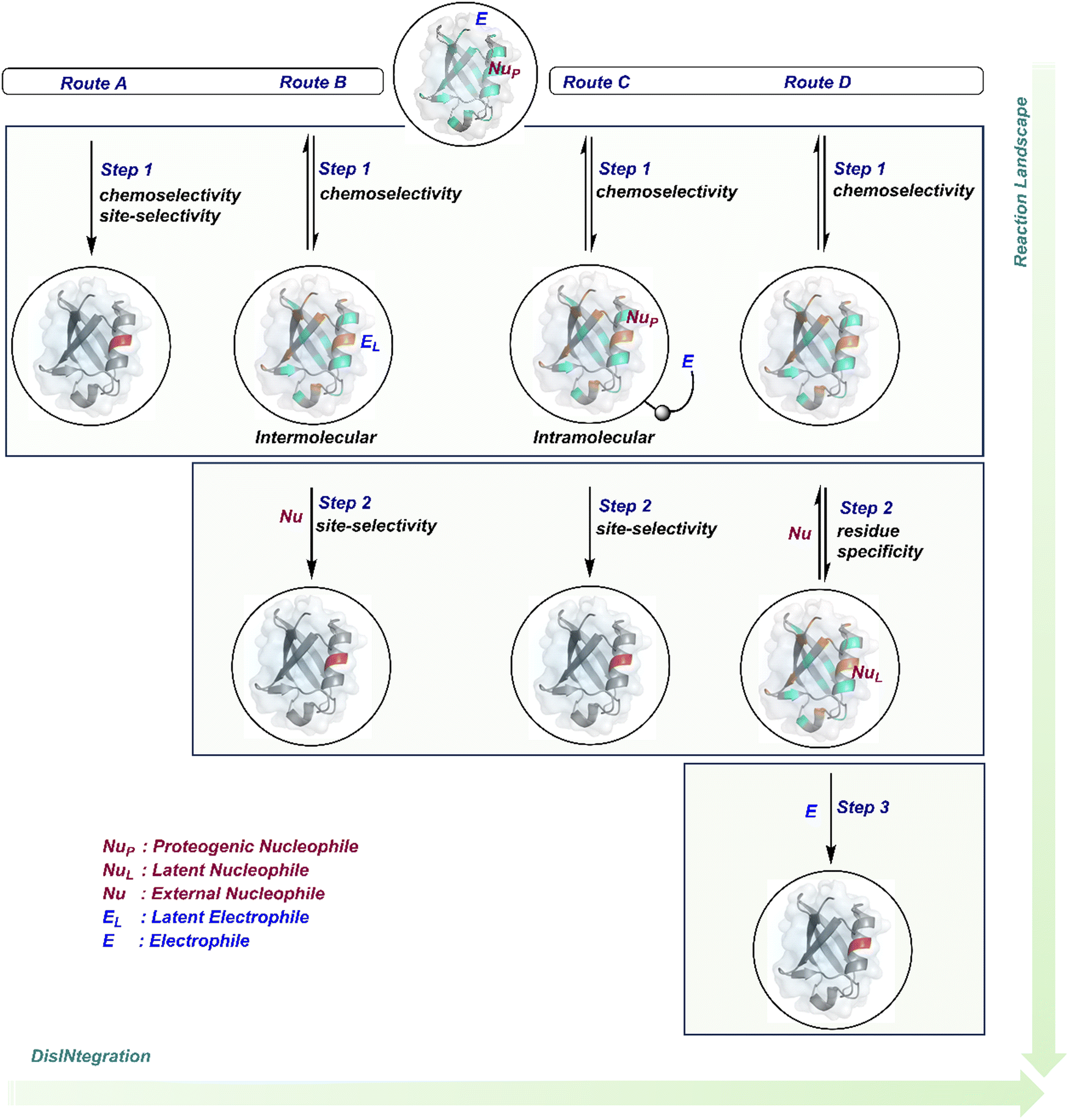 | ||
| Fig. 2 Multi-equilibrium processes disintegrate the selectivity challenges into separate steps. It redefines the reaction landscape to enable single-site protein modification. Route A: single step to target a reactivity hotspot. Route B: two steps to target a reactivity hotspot. Route C: two-step process for simultaneous regulation of chemoselectivity, site-selectivity, modularity, and protein selectivity. Route D: three-step deconvolution of selectivity challenges rendering a residue-specific platform. These representative routes A–D can be extended further to expand the selectivity attributes and targetome within reach of chemical technologies. | ||
This review discusses the selectivity attributes that serve as roadblocks for the precision engineering of proteins.9–11 The discussion extends to chemical methods that can harness reactivity hotspots (route A, Fig. 2). Next, we outline how DIN theory provides a route to redefine reactivity hotspots (route B, Fig. 2).12–14 It expands further to use a combination of steps to deliver modularity and protein selectivity in addition to chemoselectivity and site-selectivity (route C, Fig. 2).15,16 We strengthen the argument with a three-step process disintegrating chemoselectivity and residue-specificity (route D, Fig. 2).17 Such deconvolutions open several dimensions to use a multi-step or multi-equilibrium process to address selective protein bioconjugation. Besides, it offers an opportunity to expand a targetome for precision protein engineering.
Recent developments demonstrate how chemical technologies for the precision engineering of proteins can contribute to the translational demands and knowledge-driven economy.18 We discuss these developments for therapeutic proteins, protein–protein conjugates, and antibody conjugates. Also, the discussion extends to the potential of chemical technologies to realize precision therapeutics with small molecules. Besides, it provides insight into how DIN theory could empower hypothesis-driven research to find solutions to unanswered questions in the field. The routes A and B examples are further subdivided based on residues (Sections 2.1 and 2.2). However, the DIN theory translation for the deconvolution of selectivity attributes is in the early stage. Hence, no such subdivisions exist for routes C and D in Sections 2.3 and 2.4.
2. Disintegration of selectivity challenges
2.1. Route A
Route A comprises a large share of chemical technologies available for protein modification due to the convenience of their design. In this case, site-selectivity is often defined by the residue's availability or frequency and solvent accessibility. While the hydrophobic residues offer poor solvent accessibility, the charged polar residues solvate extensively and offer a large accessible area. Typically, the selectivity is displayed in a narrow range of reaction parameters. Here, the first step decides the chemoselectivity and site-selectivity of bioconjugation, irrespective of the steps that follow. Most examples in this route involve a single irreversible step to yield a product. In others, the subsequent steps regulate the product without having a role in determining selectivity. A few examples proceed with a reversible first step that defines the selectivity, whereas the subsequent reversible or irreversible steps would determine only the product's structure. The generation of high-energy intermediates and their capture in a subsequent step should qualify for route B. However, such intermediates display highly promiscuous reactivity, and net selectivity depends on their generation. We have included such examples in route A. Multiple examples display chemoselectivity but lack site-selectivity. Hence, the precise single-site modification through route A is often non-trivial. Even when achieved, it demonstrates a strong dependency on residue selection and reaction parameters.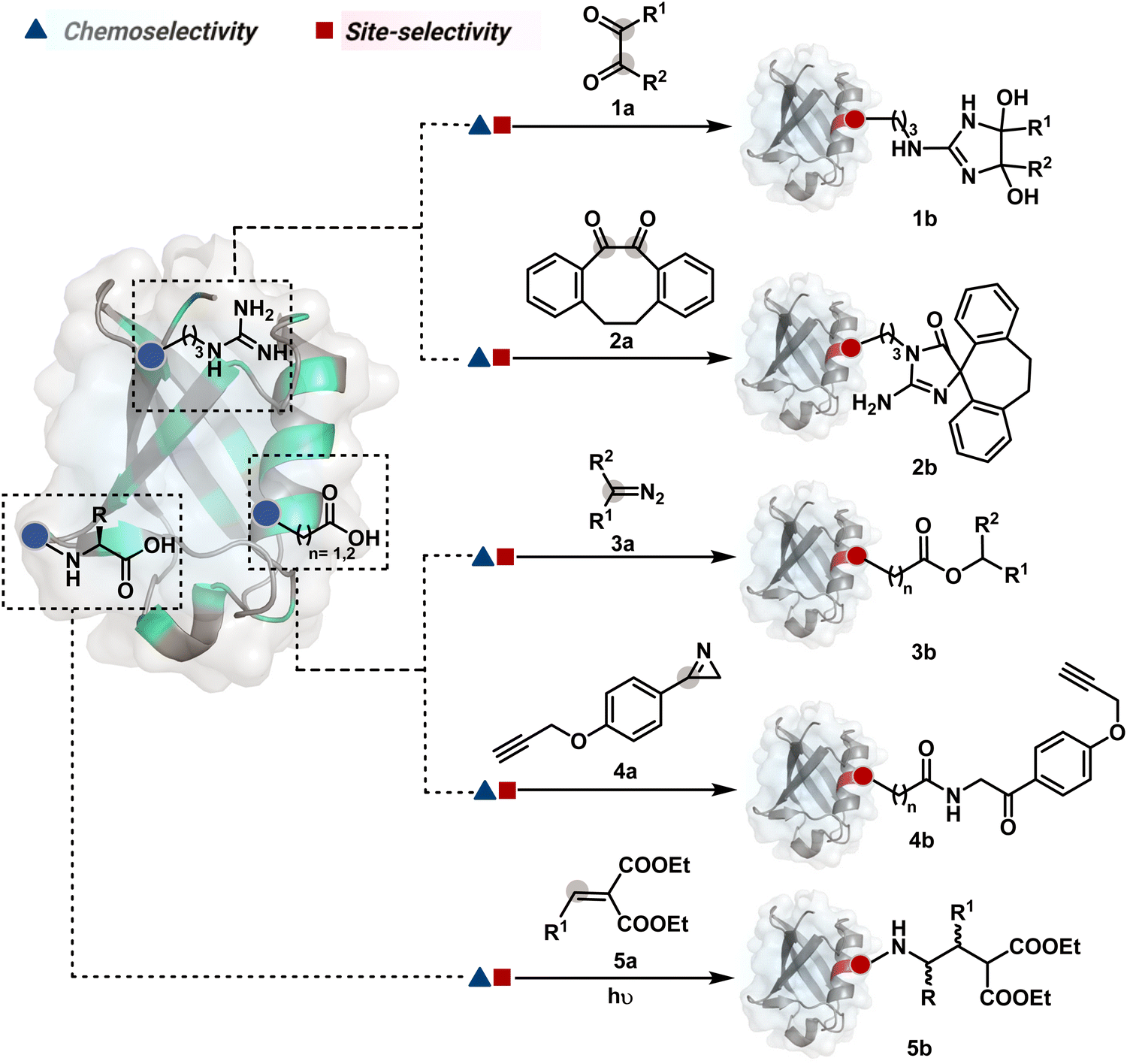 | ||
| Fig. 3 Efforts toward chemoselective and site-selective modification of guanidinium in arginine and carboxylate at C-terminus, aspartic acid, and glutamic acid. | ||
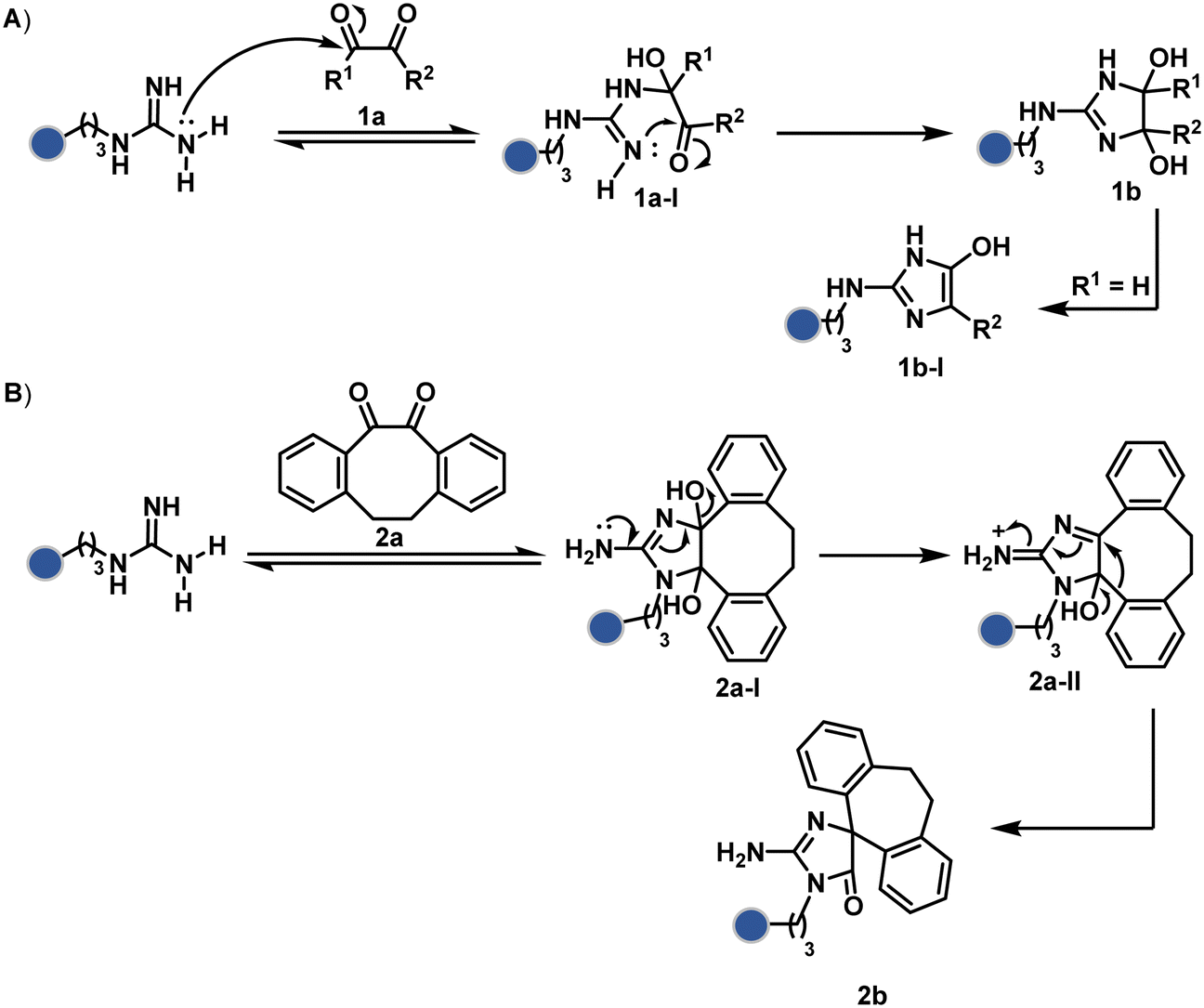 | ||
| Scheme 1 Plausible mechanism for chemoselective and site-selective modification of arginine. The side chain guanidine offers sequential nucleophilic additions where the second step is critical for the irreversible generation of a bioconjugate. | ||
Following the same mechanistic route, 1,2-diketones, especially cyclohexanedione, offer good reactivity with Arg (1a-I, Scheme 1A).24–26 The condensation results in irreversible Arg modification under alkaline conditions. Like oxoaldehydes, the competition from Lys and Cys must also be addressed in these cases. Diketones like dibenzocyclooctendiones (2a) facilitate irreversible Arg modification, although it requires an organic medium.27 It also needs a strong base like Na2CO3 to activate Arg in proteins, e.g., insulin, and enable nucleophilic attack on carbonyl (Scheme 1B). The subsequent condensation and a benzylic acid rearrangement cement the irreversibility. In an interesting translation, a 1,3-diketone-bearing DNA was used to target Arg-rich histone proteins.28 The attempts for Arg modification often face low conversions and a lack of selectivity. It would be interesting to see if disintegration of challenges through alternate mechanistic routes could provide more efficient methods.
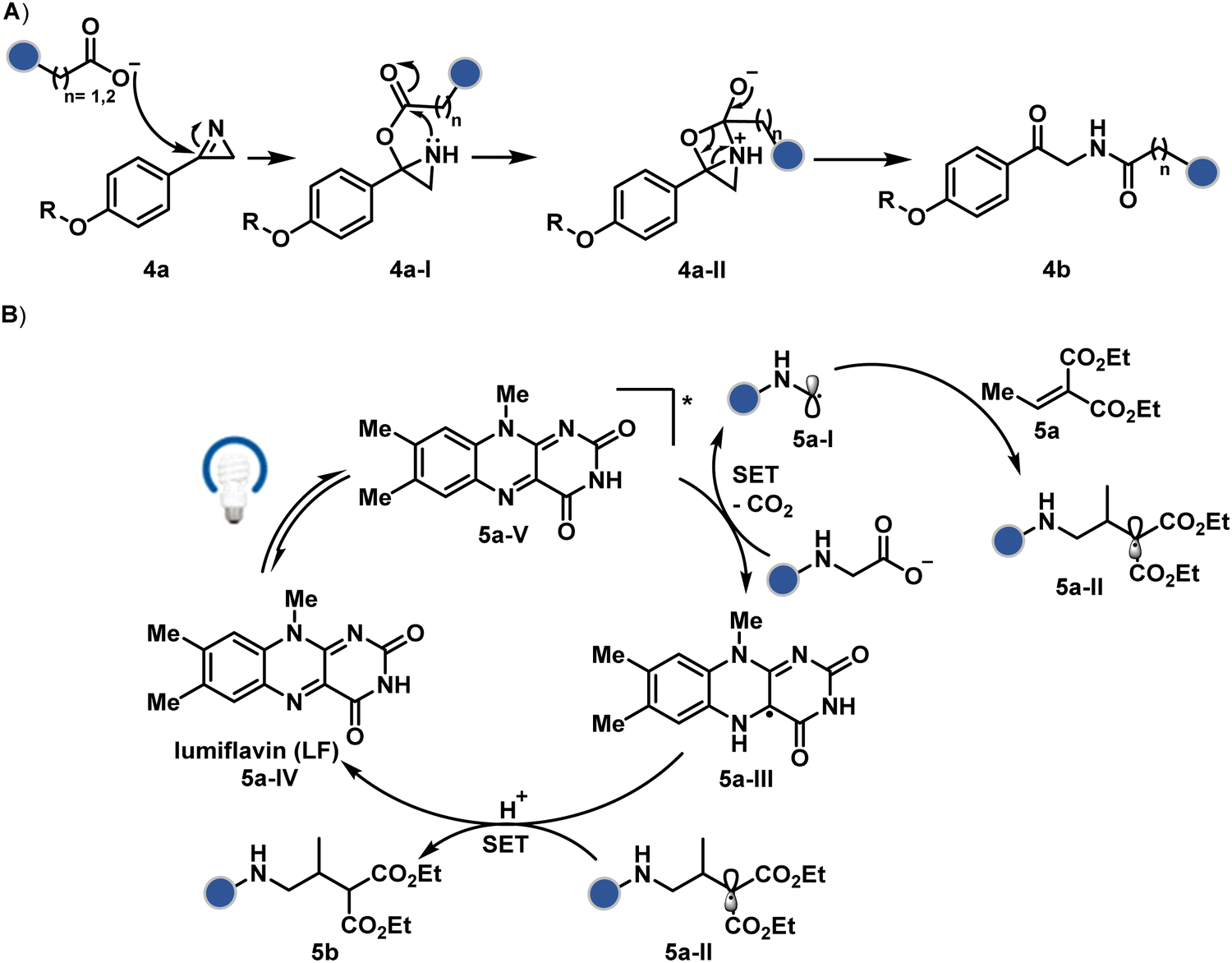 | ||
| Scheme 2 Plausible mechanism for chemoselective and site-selective modification of carboxylate at the C-terminus, aspartic acid, and glutamic acid using (A) alkylation and (B) oxidative decarboxylation. | ||
Another interesting method investigated the oxidation potential difference between the C-terminal and the side-chain carboxylates for their site-selective modification (5b, Fig. 3).32 In particular, a higher barrier of side-chain carboxylates toward oxidation was anticipated to drive C-terminal selectivity. It is primarily due to an unstabilized carbon-centered primary radical in the former, contrary to a heteroatom-stabilized α-amino radical in the latter. The C-terminal intermediate was captured with an α,β-unsaturated carbonyl compound (Scheme 2B). Here, a flavin-based photocatalyst is excited with a 34 W blue light, giving its singlet excited state that undergoes intersystem crossing. The resultant triplet-excited photocatalyst (5a-V) then renders a facile single electron transfer (SET) to the C-terminal carboxylate. Next, its decarboxylation furnishes an α-amino radical (5a-I) stabilized by adjacent nitrogen. This intermediate reacts with a Michael acceptor (5a) to provide an α-acyl radical (5a-II). The subsequent reduction by the photocatalyst generates the corresponding enolate that undergoes protonation to render a bioconjugate (5b, Fig. 3) while regenerating the catalyst. This method yields 44% mono-alkylated product with human insulin, albeit under specific reaction conditions (cesium formate buffer pH 3.5, 8 h). Even though the reaction parameters are incompatible with cells, the approach offers one of the best routes for site-selective carboxylate modification.
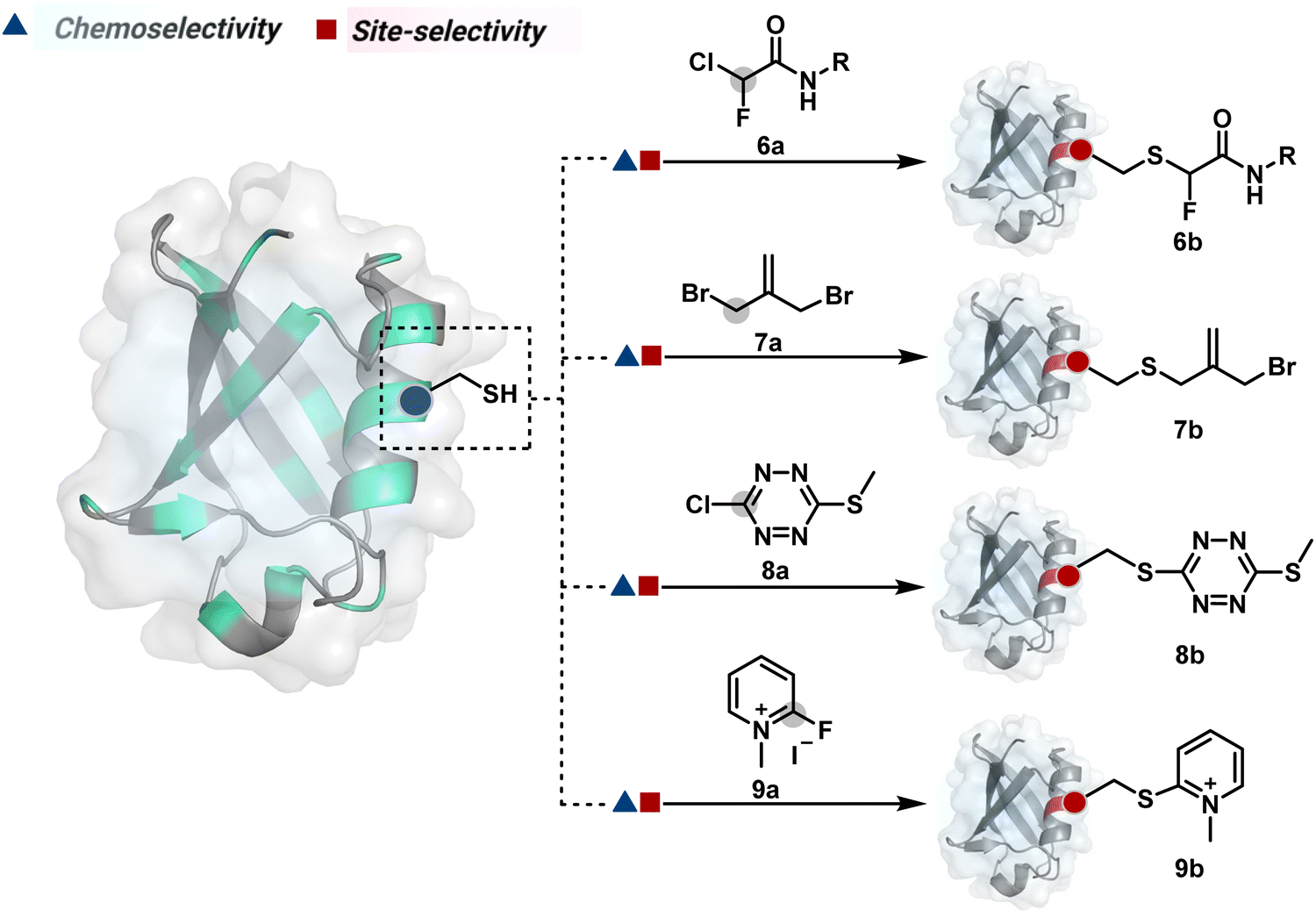 | ||
| Fig. 4 Chemical methods for chemoselective and site-selective modification of cysteine by haloalkanes and haloarenes. | ||
 | ||
| Scheme 3 Mechanistic pathway for cysteine alkylation by haloalkanes and arylation by haloarenes. | ||
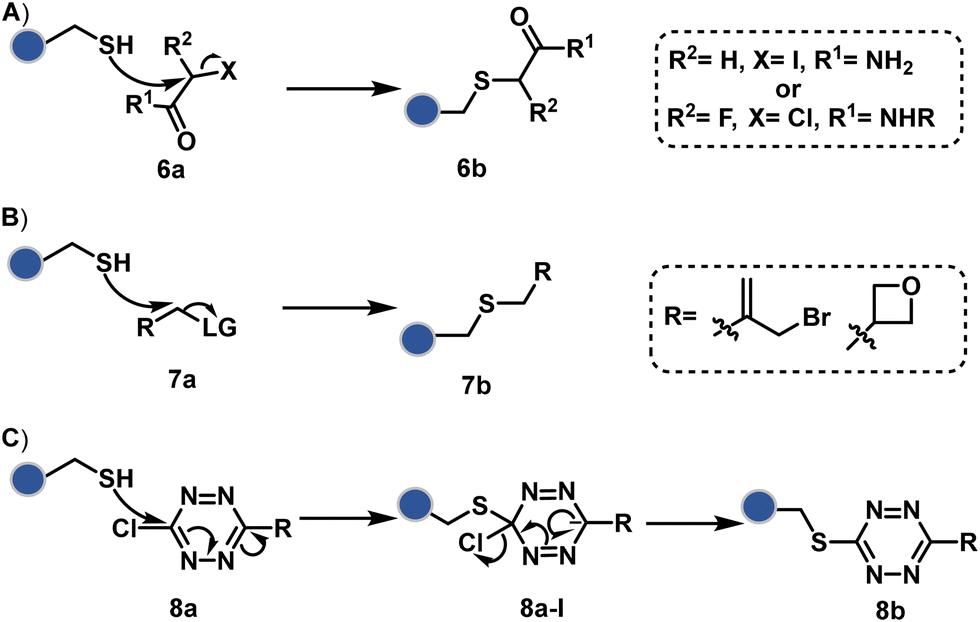
The haloarenes offer a soft electrophilic system that can render thiol selectivity through the SNAr mechanism (Scheme 3C). For example, methylsulfonyl benzothiazoles result in Cys modification.39 However, the competition from Lys must be monitored closely in such cases. Chlorotetrazines follow the same mechanistic pathway, resulting in a tetrazine moiety (8b, Fig. 4) to facilitate dual probe installation by an inverse electron demand Diels–Alder reaction.40 In the addition–elimination mechanism followed by these molecules, when the addition step is rate limiting, the reactions are accelerated if the aromatic ring bears an electron-withdrawing group and a small leaving group. It inspired the use of fluorinated pyridinium salts for rapid Cys arylation under biocompatible conditions (9b, Fig. 4).41
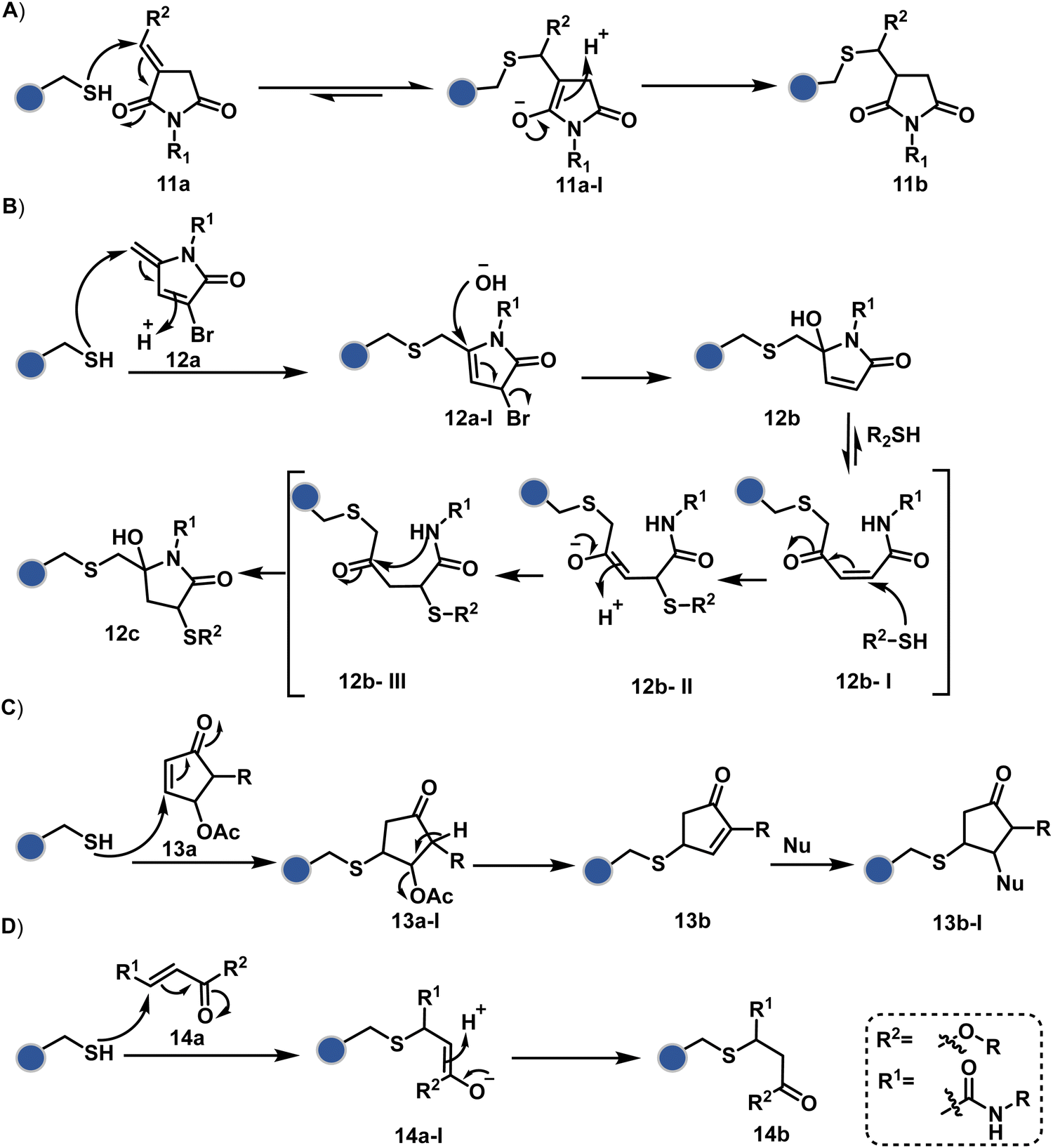
Furthermore, polarized double bonds have led to multiple Cys-selective bioconjugation reagents. The regulation of LUMO energy through electron-withdrawing groups or strain has been examined to tune reactivity and selectivity in such cases. In this perspective, maleimide and its derivatives have created an impactful pool of reagents (Fig. 5).42,43 The thio-Michael addition with a simple maleimide is reversible, hence susceptible to thiol exchanges. Initially, this problem was resolved by engaging subsequent hydrolysis to counter the driving force for C–S bond dissociation.44 Typically a slow process, hydrolysis can also be accelerated through a proximally placed activating group in N-substituted maleimides under mild conditions.45 An alternative way to address the reversibility is redesigning the Michael acceptor to counter the C–S bond dissociation. For example, the exocyclic maleimides offer substantially higher stability than their endocyclic counterparts (11a, Scheme 4A).46 A similar exocyclic double bond in 5-methylene pyrrolones (5MPs) offers thio-Michael addition under neutral pH conditions.47 These electrophilic systems can also be redesigned for dual-conjugate addition by placing a leaving group at an appropriate position.48 For example, the first thio-Michael addition primes the 3-Br substituted 5MP (12a, Scheme 4B) for an elimination reaction, creating an opportunity for second conjugate addition (12b, Fig. 5). The overall process could enable a sequential installation of two probes (12c, Scheme 4B). A similar Cys-selective electrophilic system can also be designed from a cyclopentenone-based reagent to render sequential conjugate additions (13a–13b, Scheme 4C).49,50
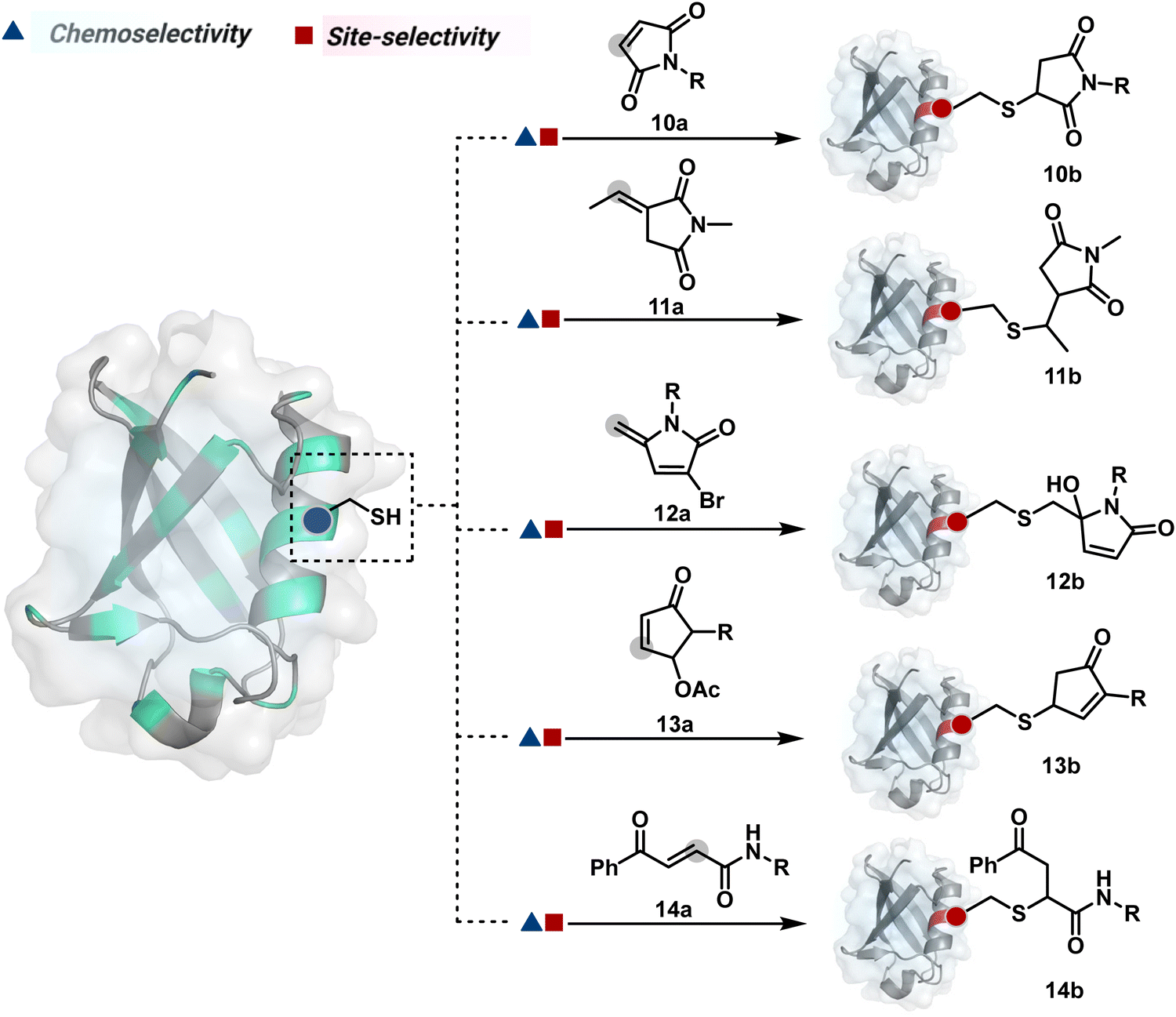 | ||
| Fig. 5 Chemical methods for chemoselective and site-selective modification of cysteine via conjugate addition. | ||
 | ||
| Scheme 4 Plausible mechanism for cysteine modification via conjugate addition. | ||
Also, vinyl sulphone derivatives offer excellent Cys-selectivity. The synthetic flexibility of these systems allows convenient pre-installation of probes without compromising chemoselectivity.51 Besides, their incorporation into strained systems, e.g., azabicyclic vinyl sulfone, could enable late-stage installation of diverse probes through Diels–Alder cycloaddition.52 Similar to other cases, competition from Lys and His becomes more prominent at elevated pH.53 Switching the electron-withdrawing group to amide renders another set of Cys-selective reagents. Like the previous example, the acrylamide handle offers reactivity and selectivity tuning through appropriate functionalities on either side of a polarized double bond. For example, an acrylamide derivative with β-keto functionality (14a, Fig. 5) yields an irreversible Cys-selective bioconjugation (14b, Scheme 4D).54 However, competition from Lys is observed in some instances. Similarly, a polarized double bond with ester is explored for labeling Cys.55 These systems offer opportunities to place functional handles, e.g., azidoacrylate, to install desired probes.
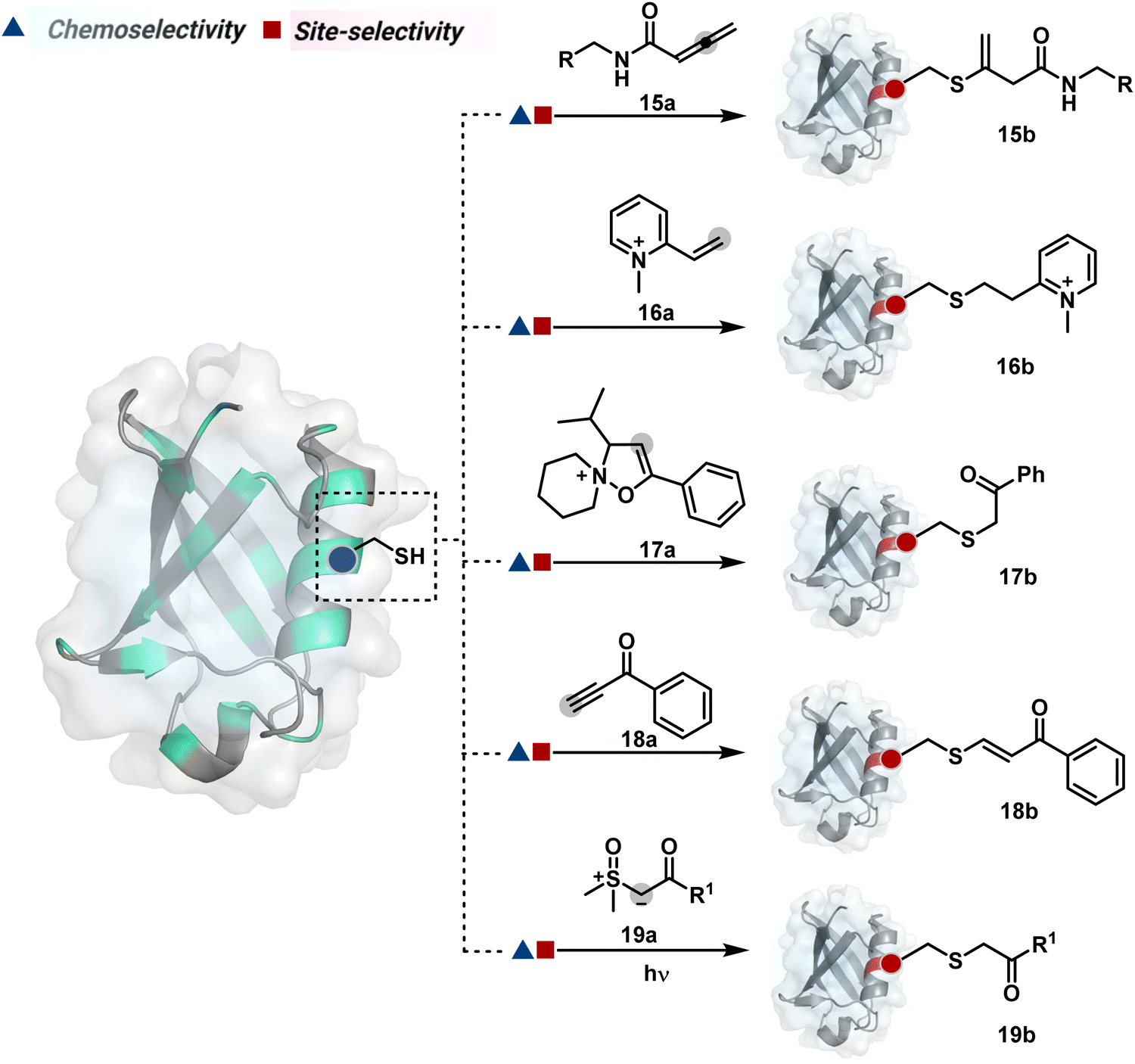
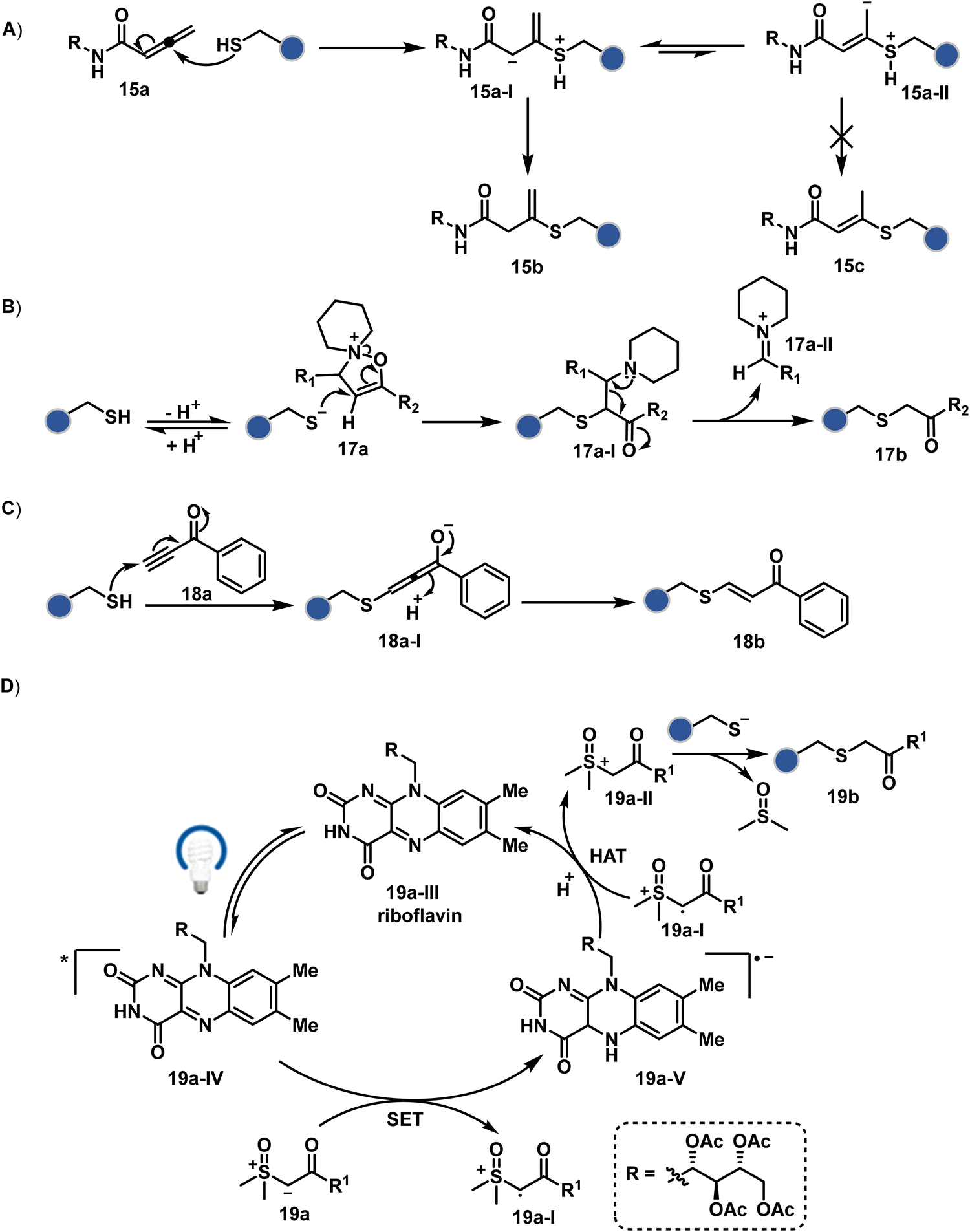
The C-substituted terminal allenamides (15a, Fig. 6) offer a rapid and irreversible chemoselective reaction with thiols.56 It initiates with a conjugate addition, resulting in a kinetically driven product (15bversus15c, Scheme 5A). The double bond can be polarized sufficiently with a quaternized pyridine ring (16a, Fig. 6) to drive thiol addition.57 A suitable vinyl pyridinium reagent can render rapid conversion to bioconjugates with stable C–S bonds. On the other hand, isoxazolinium (17a, Fig. 6) offers a reasonably good reactivity with high Cys selectivity in protein bioconjugation.58 The isoxazole ring opening drives a thiol attack on the double bond. Furthermore, the iminium ion (17a-II, Scheme 5B) release renders a thioalkylated product (17b). Interestingly, it allows photolysis to form thioaldehyde that can return thiol upon subsequent NaBH4 reduction. Along the same lines, the triple bonds polarized with functionalities like amides, esters, and ketones were exploited for Cys modification (18b, Scheme 5C).59 Under basic conditions, the thio-Michael addition forms an intermediate that results in a vinyl sulfide-linked bioconjugate. The electron-withdrawing group and the alkyne position, terminal or internal, offer control over reactivity.
 | ||
| Fig. 6 Additional chemical methods for chemoselective and site-selective modification of cysteine. | ||
 | ||
| Scheme 5 Proposed mechanism for chemoselective and site-selective modification of cysteine through (A)–(C) conjugate addition and (D) alkylation. | ||
The double or triple bonds can be engaged for Cys conjugation through Thiol–Ene or Thiol–Yne Coupling (TEC or TYC).60 The TEC initiates with a light-induced generation of a thiyl radical. Next, its regioselective anti-Markovnikov addition to alkene yields a thioalkyl radical leading to the product. However, this approach comes with a set of side reactions. For example, the Cys glycosylation in BSA with this method was accompanied by disulfide degradation, contributing to heterogeneity.61 Similarly, the anti-Markonikov addition in TYC results in a vinyl sulfide intermediate.62 The latter is prone to attack from an external thiyl radical via a TEC mechanism to form a bioconjugate. Multiple unidentified pathways accompany these reactions and require further investigation. On the other hand, a photo-catalytically generated sulfoxonium ion could render a highly chemoselective click reaction for Cys bioconjugation (19b, Fig. 6).63 The reaction initiates with sulfoxonium ylide (19a, Scheme 5D) and a riboflavin derivative. The latter is photoactivated with 450 nm to promote single electron transfer (SET) and generate a sulfoxonium radical cation (19a-I). Next, it abstracts hydrogen to generate a sulfoxonium intermediate (19a-II) resulting in Cys-alkylation. The method offers remarkably fast reaction kinetics, yet chemoselectivity remains conserved.
Initially, epoxides evolved as one of the most consistent electrophiles for chemoselective His modification.64,65 The nucleophilic addition of the imidazole ring promotes epoxide ring opening, resulting in C–N bond formation. As expected, thiols, amines, and alcohols compete with His, affecting chemoselectivity. The site-selectivity challenge is modest due to a limited number of solvent accessible His residues. The epoxyalkenals were developed anticipating His modification in free Cys-equipped BSA.66 However, such examples require a re-examination of chemoselectivity and site homogeneity with advanced MS-based tools.
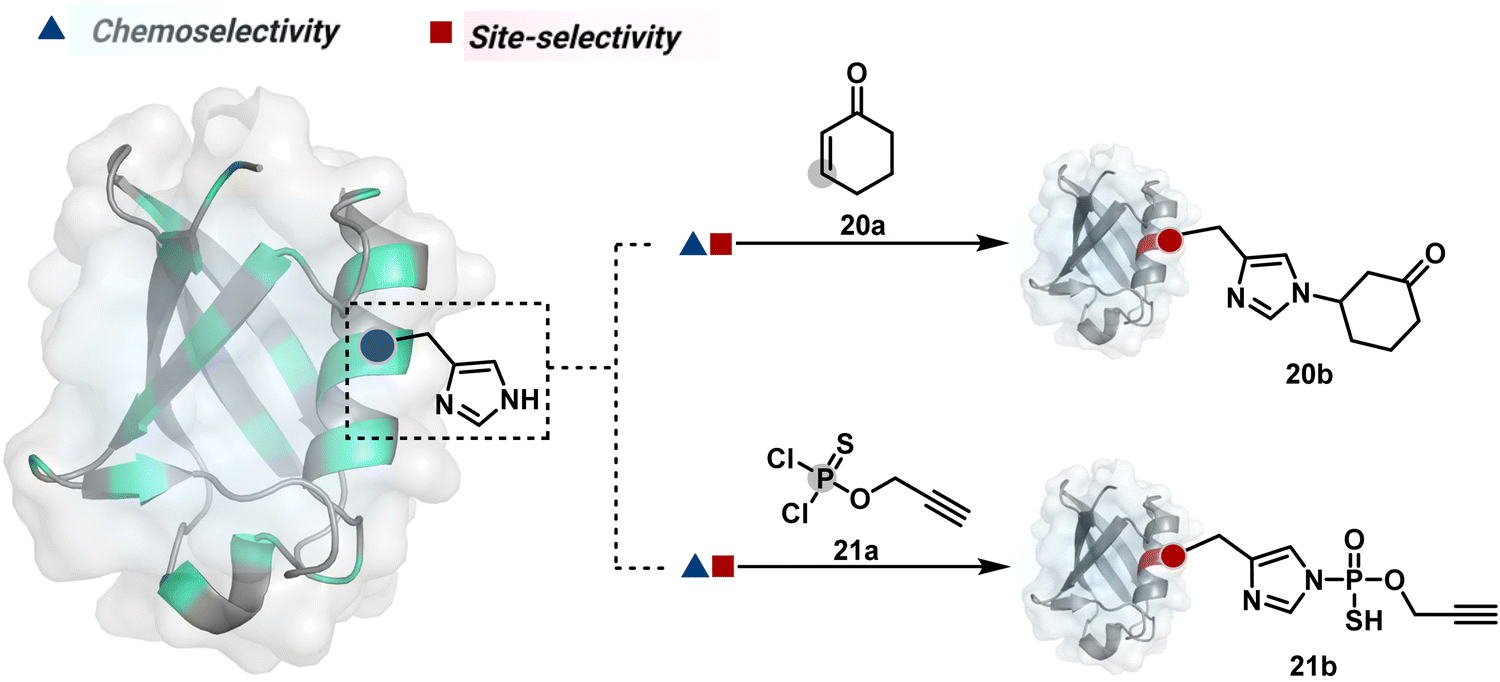
Our group introduced a chemoselective and site-selective strategy for His modification using a ring system like cyclohexenone (20a, Fig. 7).67 The conjugate addition results in a carbonyl used for late-stage installation of probes (Scheme 6A). Single-site modification of structurally diverse proteins was achieved using this strategy. Another method in this perspective involves a thiophosphorodichloridate reagent for P–N bond formation (21a, Fig. 7).68 Here, thiophosphoro alkyne dichloridate (TPAC) rendered high conversion and hydrolysis resistance. At first, an addition–elimination reaction enables the replacement of one chloride by His (Scheme 6B). The hydrolysis of this intermediate (21a-I) renders the final product. The reaction is accelerated under basic conditions. It engages multiple His of RNase A but offers excellent chemoselectivity.
 | ||
| Fig. 7 Chemical methods for chemoselective and site-selective modification of histidine. | ||
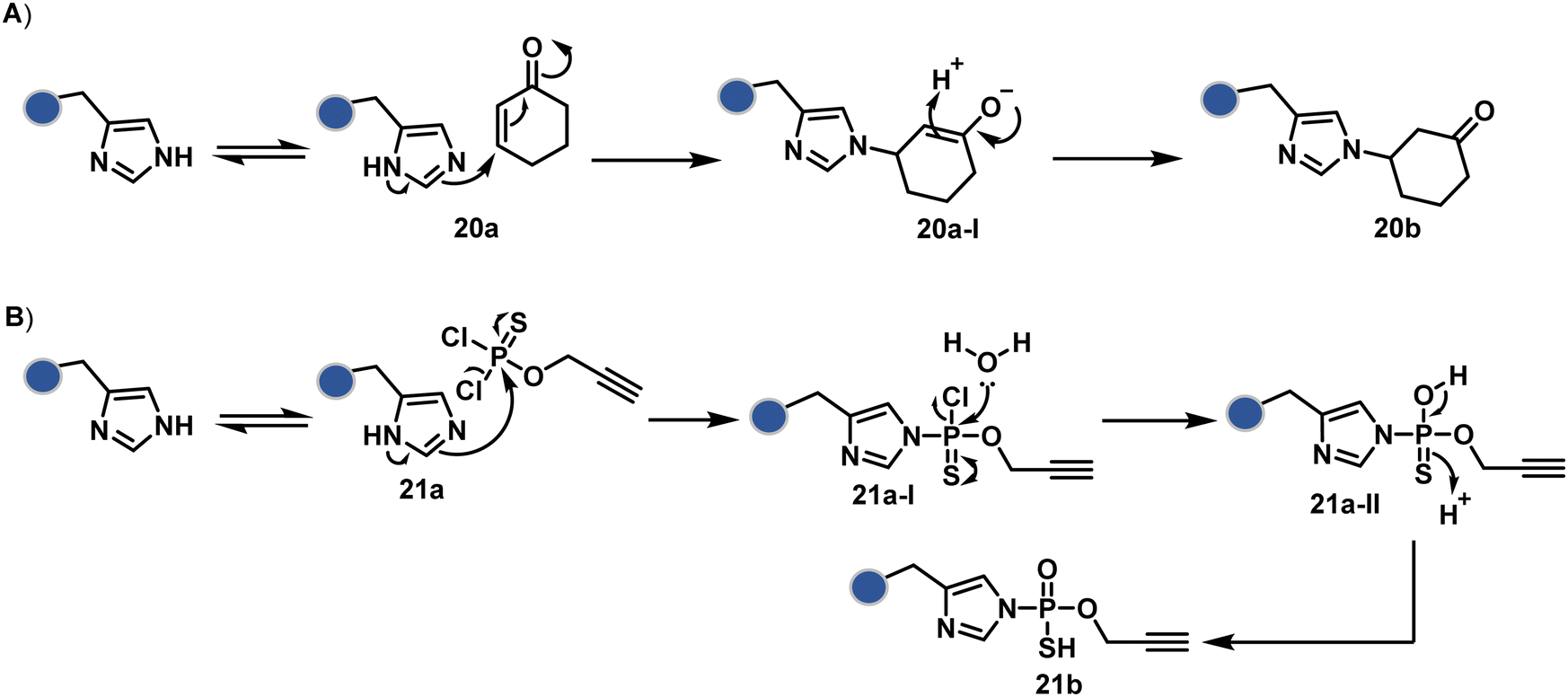 | ||
| Scheme 6 Plausible mechanism for chemoselective and site-selective modification of histidine through (A) N–C and (B) N–P bond formation. | ||
The activated carbonyl compounds offer an excellent building block for amine modification. If Cys presents a competition, it will result in a thioester that can be reverted conveniently after amine-targeted amide formation. Besides, the nature of the leaving group can be regulated to access a range of reactivity. While strong leaving groups, e.g., N-hydroxysuccinimide, give highly reactive electrophiles for higher loading, the reduced reactivity could offer better control over site-selectivity.70 Another conceptually interesting route involves the engagement of amine in the generation of imine. This highly reversible transformation can be locked using intramolecular stabilization through iminoboronates (22a, Fig. 8).70 However, these adducts are reversible in the presence of competing reagents such as fructose, dopamine, or glutathione.
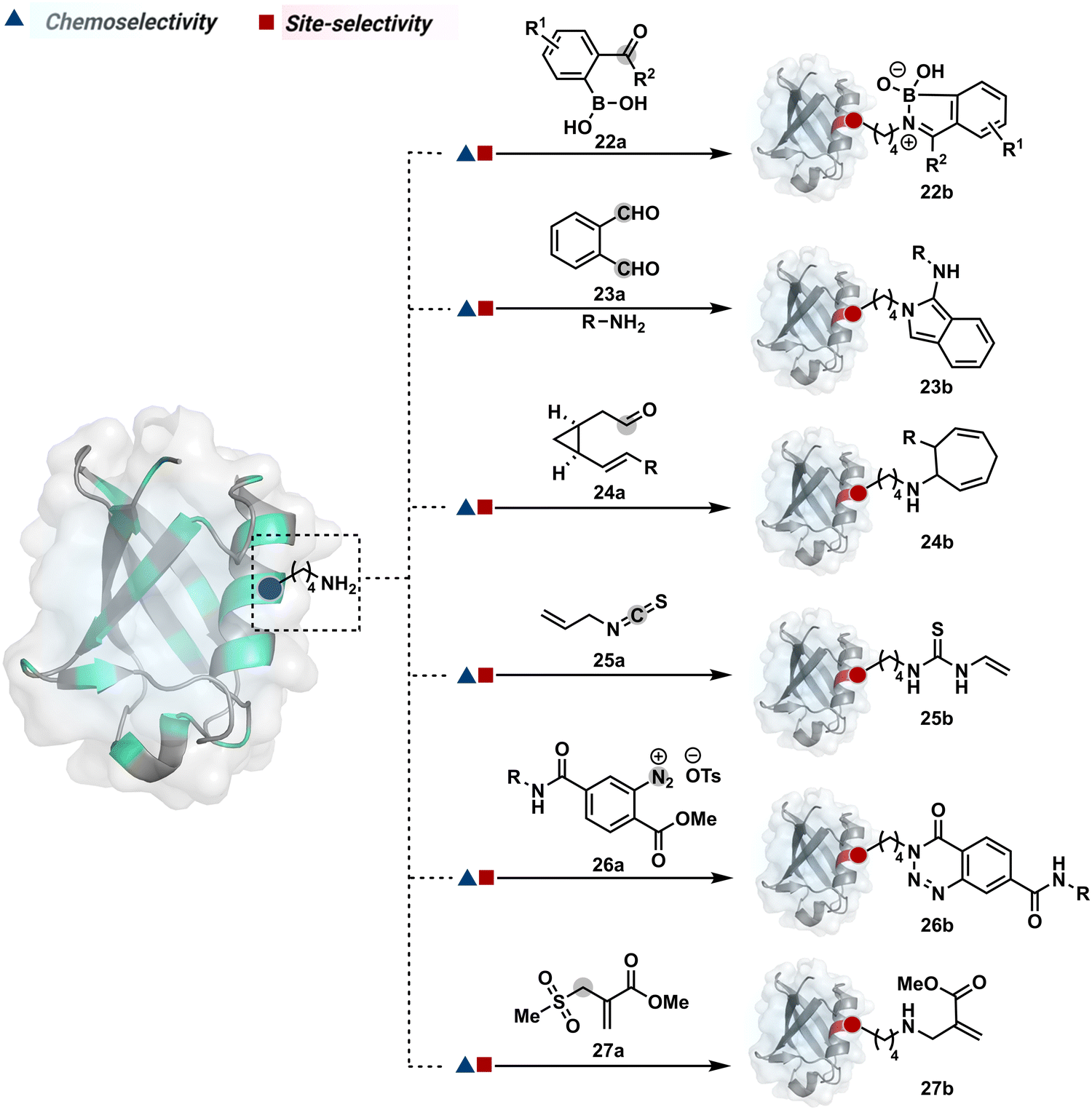 | ||
| Fig. 8 Chemical methods for chemoselective and site-selective modification of lysine. | ||
In another example, hemiaminal (23a-I) generated by amine and aldehyde (23a, Fig. 8) was captured by a proximal aldehyde.71,72 Here, o-phthalaldehyde reacts with a proteinogenic amine to render an acyclic imine (23a-II, Scheme 7) that can react with neighbouring aldehyde resulting in a cyclic iminium ion (23a-III). The subsequent rearrangement forms a stable isoindolin-1-one (23b-II) derivative through a series of steps. As we can see in the mechanism, an external amine can accelerate the process by intercepting the iminium intermediate.73 The involvement of two amines renders 1-aminoisoindole (23b).74 The latter has propensity to tautomerize and generate isoindolin-1-imine (23b-I) that can further hydrolyze to result in isoindolin-1-one. Along the same lines, a Cys placed in proximity of Lys-derived imine can result in an isoindole derivative (23b) in a mechanistically similar process.75 However, the lower relative stability of 1-thiol-isoindole enables facile oxidation under ambient conditions, rendering the isoindoline-1,3-dione (23b-IV) derivative as the final product.
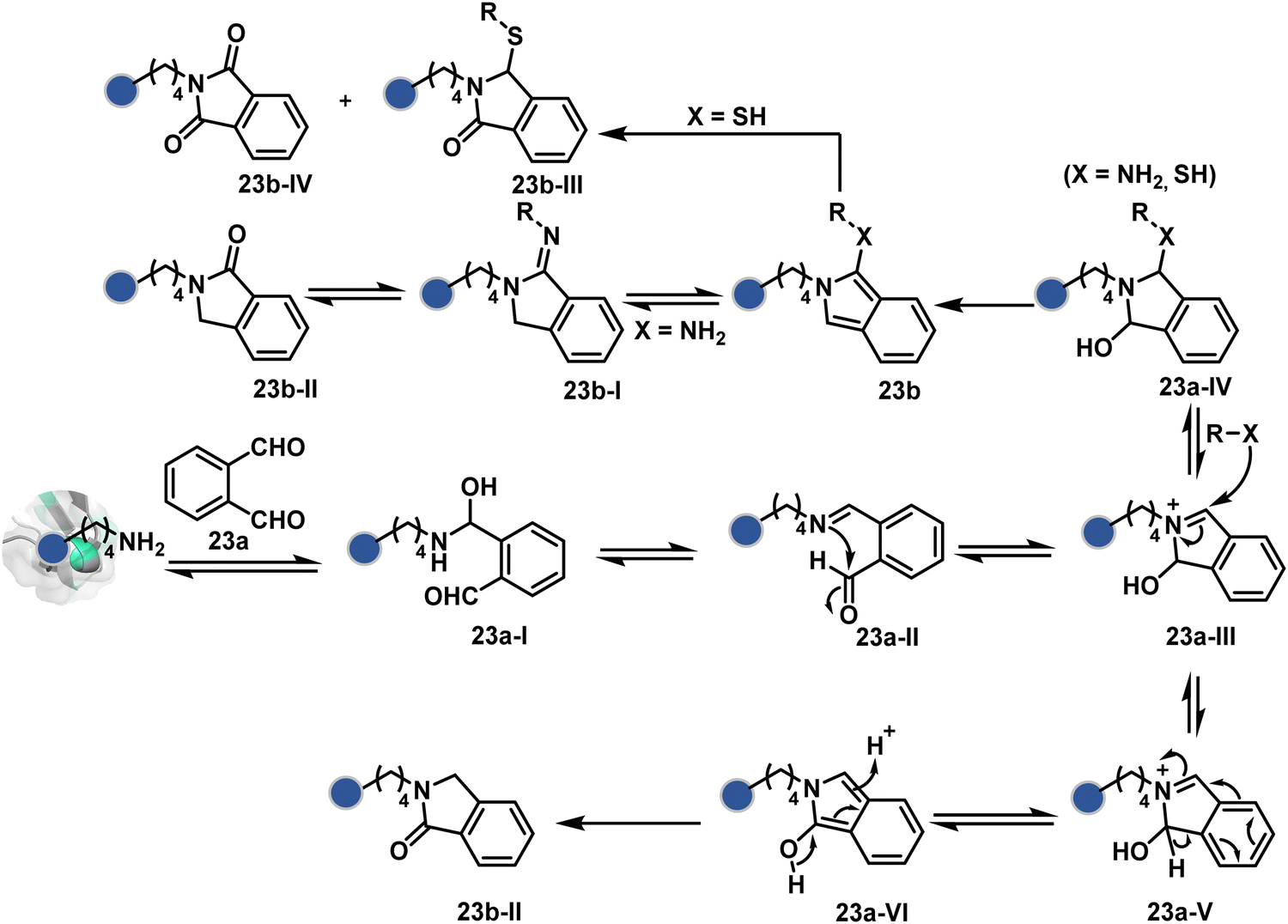 | ||
| Scheme 7 Proposed mechanism for chemoselective and site-selective modification of lysine with o-phthalaldehyde. | ||
In another case, the reversibly generated imine could be engaged in an irreversible pathway through a strain-releasing cycloaddition reaction (24b, Fig. 8, Scheme 8A).76,77 In this case, an irreversible divinylcyclopropane–cycloheptadiene (24a-II) rearrangement with imine could render amine-selective bioconjugation. Importantly, the cycloaddition could be triggered at 40 °C. While chemoselectivity was established in this example, it modifies four amines in the eGFP protein.
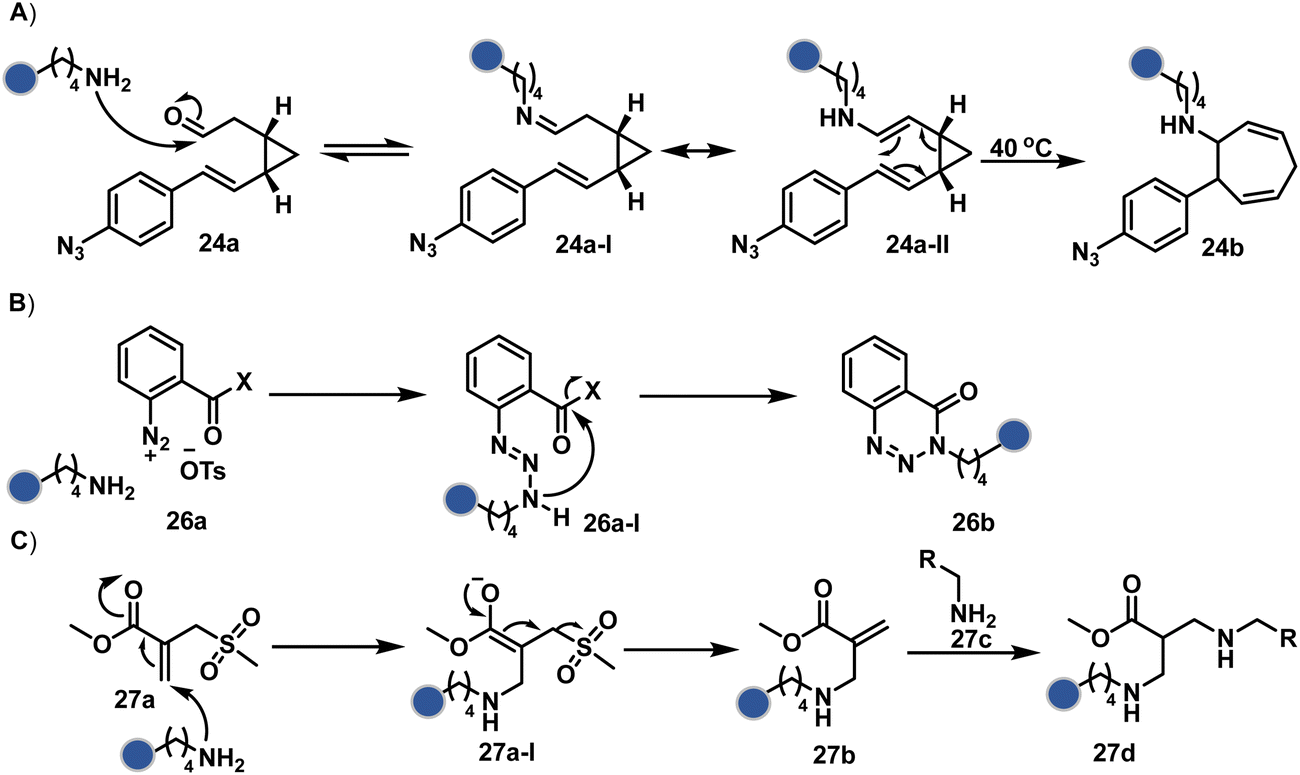 | ||
| Scheme 8 Proposed mechanism for chemoselective and site-selective modification of lysine. The first step defines all the selectivity attributes like other route A examples. | ||
On the other hand, allyl isothiocyanate (AITC, 25a) results in a chemoselective modification of BSA.78 Notably, Cys reacts with isothiocyanate to form a thiocarbamoyl adduct. However, its reversibility allows amine-selective labeling in the bioconjugate. The o-ester substituted arene diazonium salts (26a, Fig. 8) present another interesting case.79 The electrophilic diazonium group reacts with amine, generating an unstable triazine adduct (Scheme 8B). Next, intramolecular capture with the vicinal ester generates a stable benzotriazinone derivative (26b). While chemoselectivity was established, site-selectivity is challenging to control in these transformations. The sulfonyl acrylate reagents (27a, Fig. 8) could result in site-selective Lys modification.80 The residue with the lowest pKa is labeled selectively in the presence of Cys and other copies of Lys. The Michael addition promotes the elimination of methane sulfate to deliver single-site labeling (Scheme 8C). The SNAr route could also promote selective amine modification through reagents such as dichlorotriazine.81
As discussed earlier in this section, fine-tuning pH and reaction parameters could harness Nα–NH2 as a reactivity hotspot for selective modification. At pH 7.2, even a highly electrophilic NHS ester could render N-terminal-specific kinetic labeling if added slowly (28b, Fig. 9, Scheme 9A).82 In this example, RNase A and lysozyme C are tagged with biotin. Since the N-terminal residue is Lys in both cases, the sequencing could not differentiate Nα–NH2 and Nε–NH2. However, other disclosures in this perspective strongly indicate Nα–NH2 modification. In another case, ketene derivatives rendered N-terminal specific modification under mildly acidic conditions (29b, Fig. 9 and Scheme 9B).83
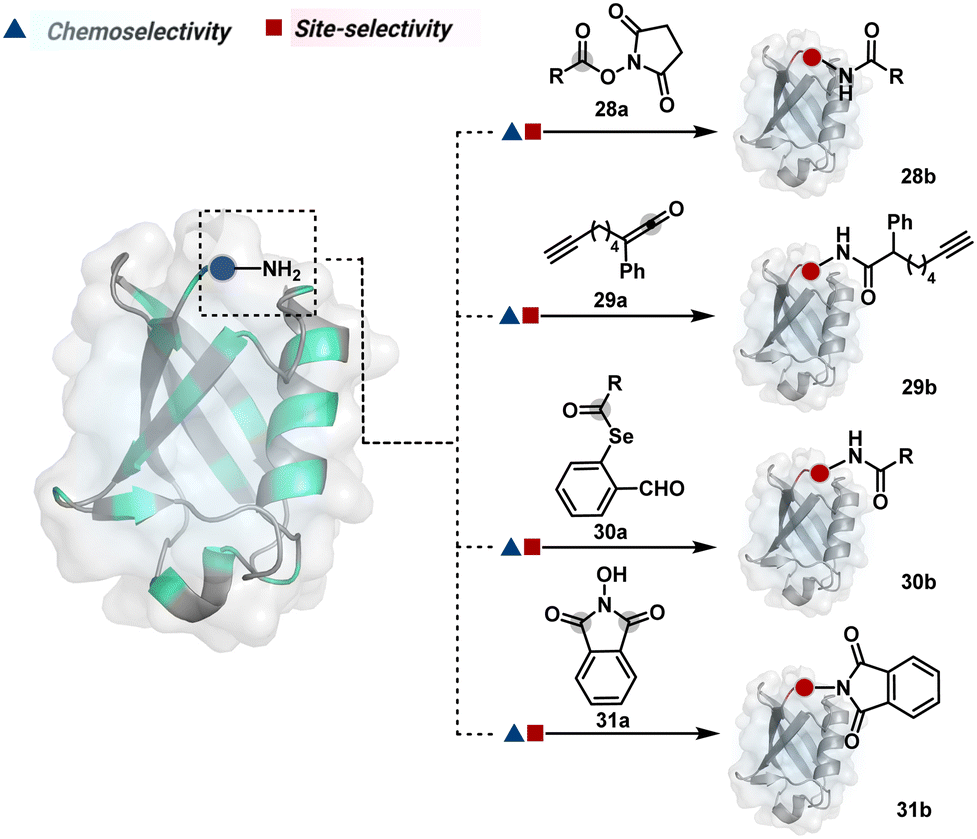 | ||
| Fig. 9 Chemical methods for chemoselective and site-selective modification of N-terminal amine (Nα–NH2). | ||
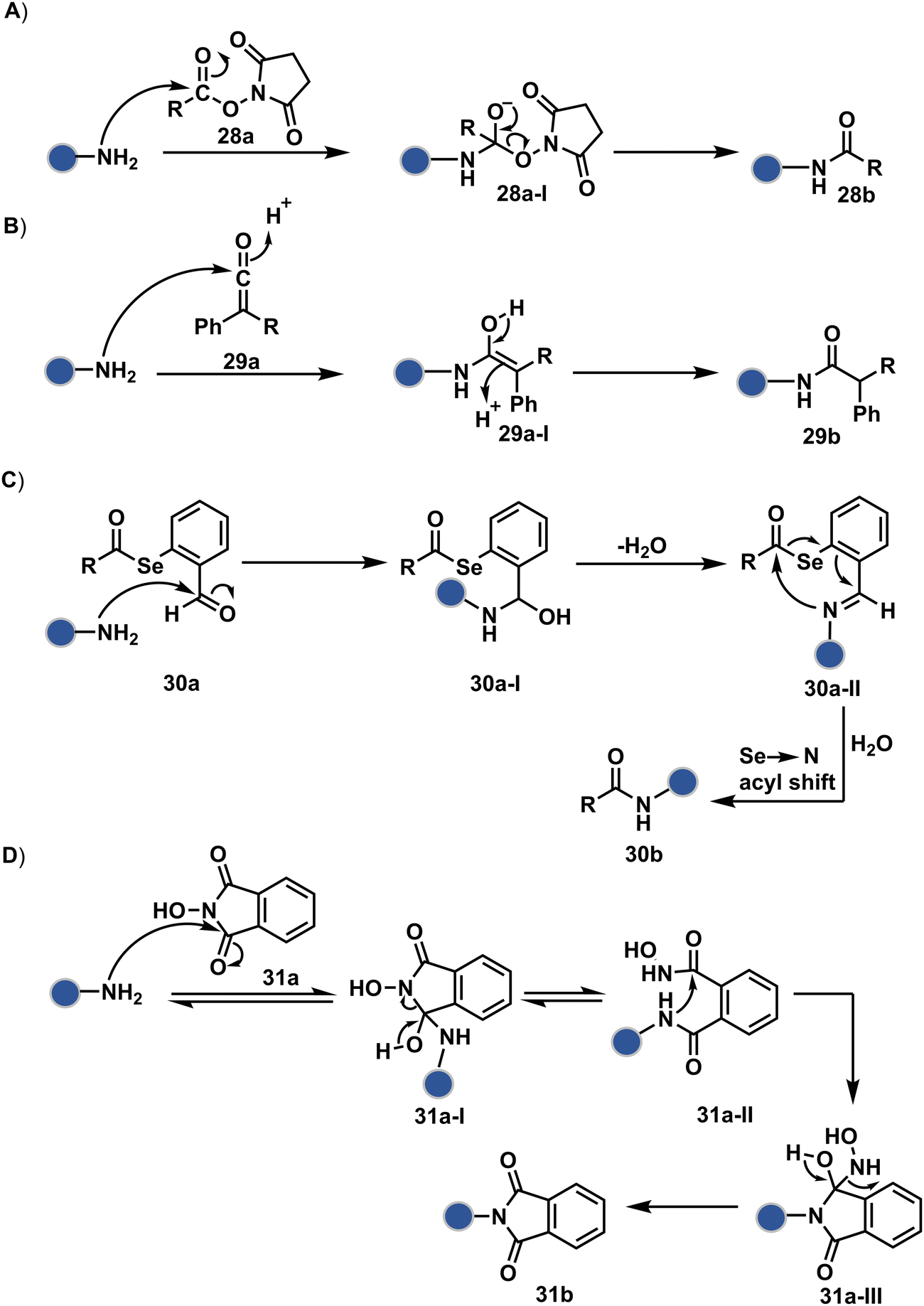 | ||
| Scheme 9 Kinetically preferred chemoselective and site-selective modification of N-terminal amines (Nα–NH2). | ||
The route of intramolecular imine capture also translates well for Nα–NH2 modification. In one of the cases, Nα–NH2-imine with a proximally placed selenium ester moiety results in N-terminal modification (30a, Fig. 9).84 Here, the aldehyde reacts with amine to generate hemiaminal (30a-I) and imine (Scheme 9C). Next, Se to N acyl transfer could deliver a single site labeled ubiquitin bioconjugate with amide linkage (30b). Even though direct acylation cannot be ruled out, control experiments confirm the role of aldehyde in observed kinetics. We established a methodology for single-site N-terminal phthalimidation (31b, Fig. 9).85 The proteinogenic amine reacts with N-hydroxypthalimide and opens the ring (Scheme 9D). The resulting intermediate (31a-II) undergoes an intramolecular nucleophilic addition to generate N-substituted phthalimide while eliminating hydroxylamine.
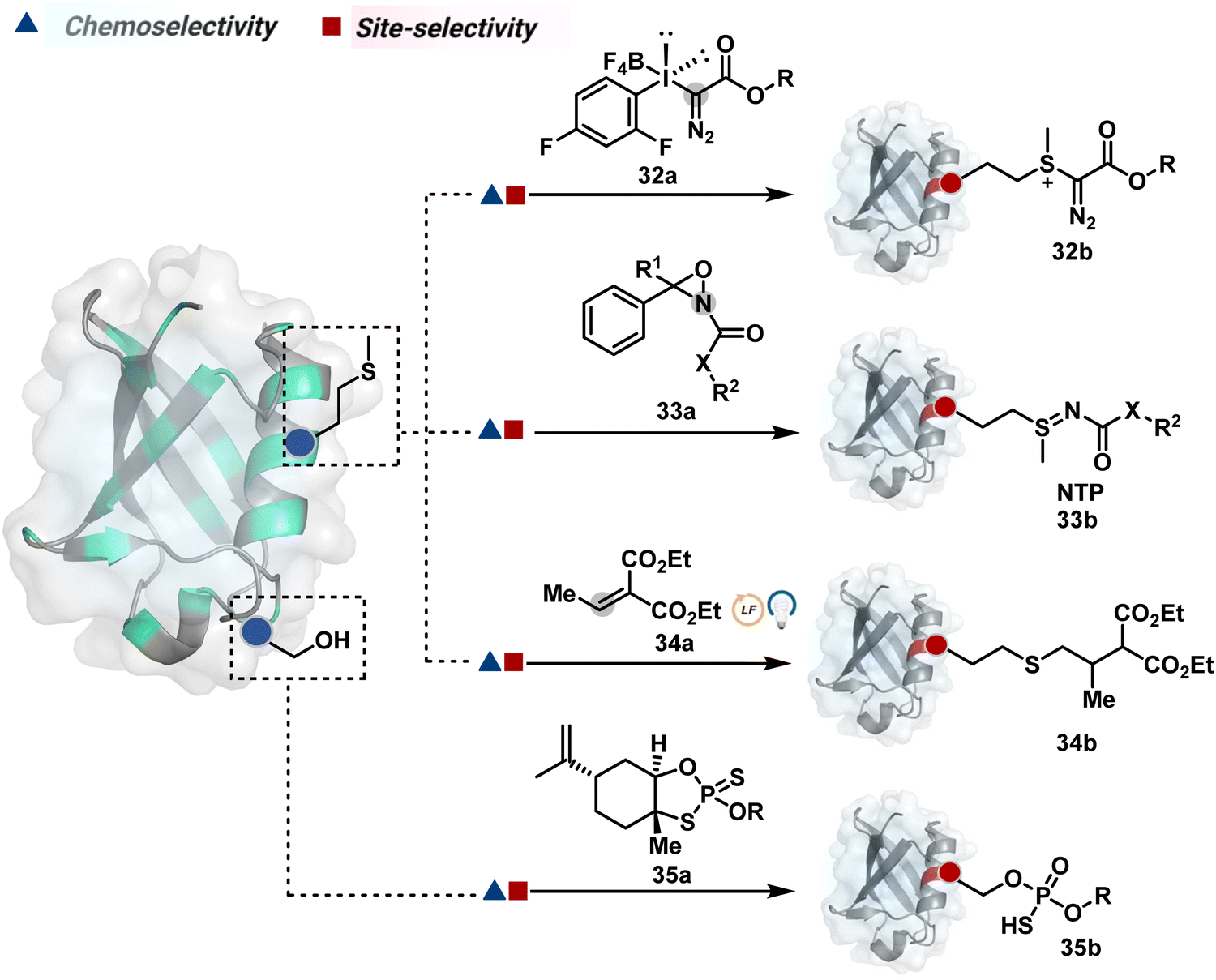 | ||
| Fig. 10 Chemical methods for chemo- and site-selective modification of methionine and serine in proteins. | ||
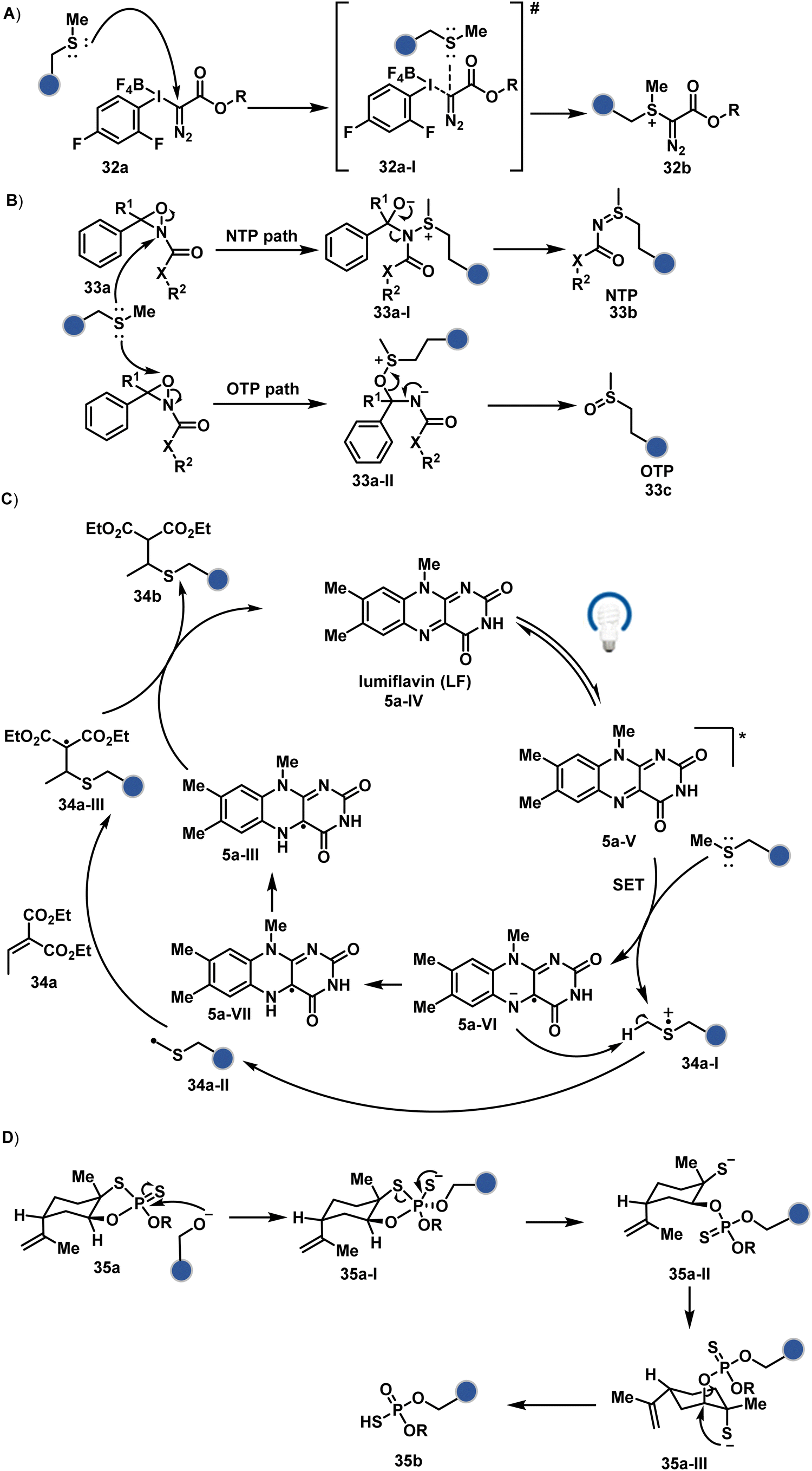 | ||
| Scheme 10 Mechanistically diverse pathways for (A)–(C) methionine and (D) serine-selective bioconjugation. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under acidic (2% TFA) conditions. The protocol restricts the extension of this method to a limited set of structurally robust proteins.
:1) under acidic (2% TFA) conditions. The protocol restricts the extension of this method to a limited set of structurally robust proteins.
 | ||
| Fig. 11 Chemical methods for chemo- and site-selective modification of tryptophan in proteins. | ||
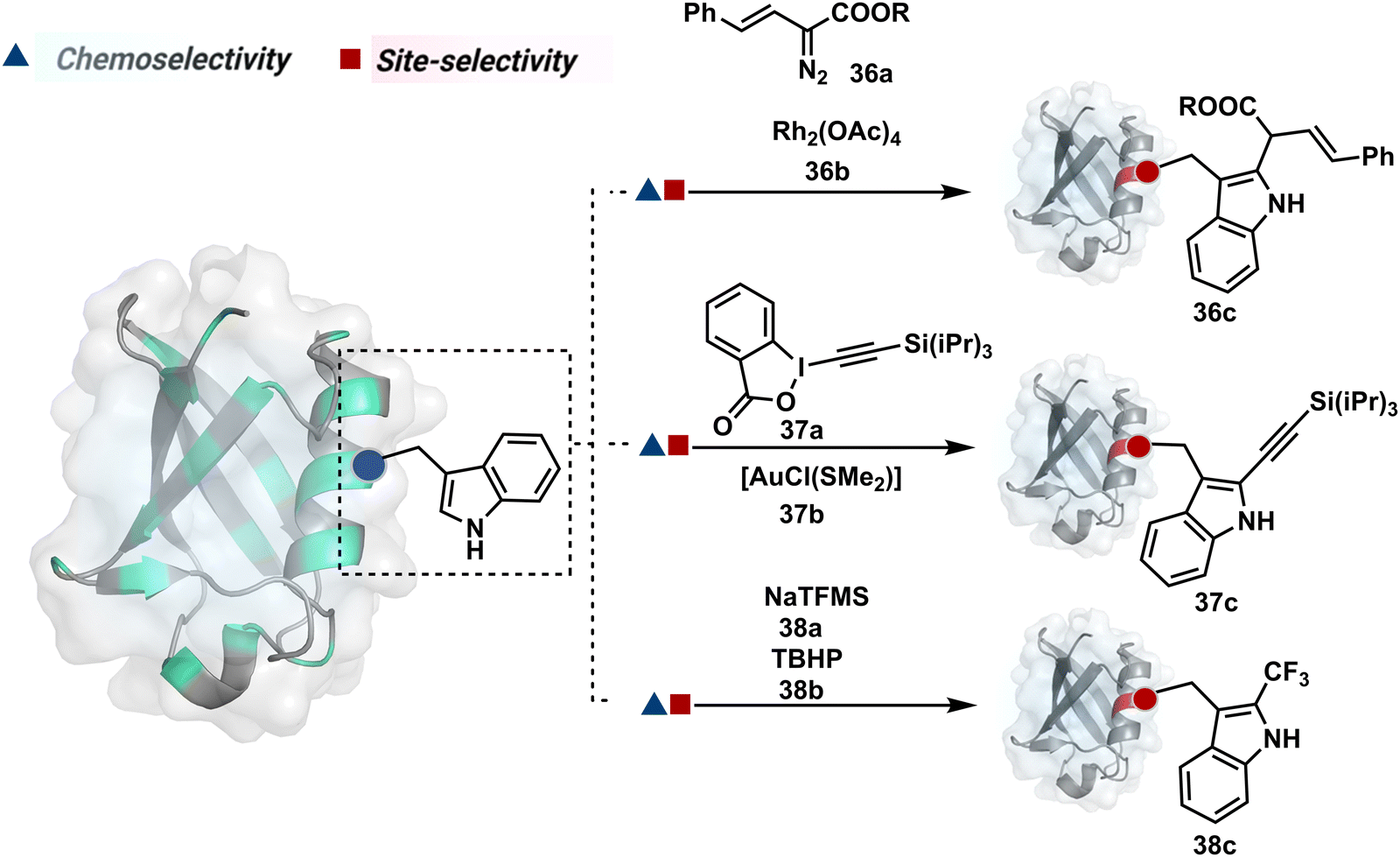
On the other hand, sodium trifluoromethanesulfinate (NaTFMS, 38a, Fig. 11) can promote a chemoselective Trp C-2 trifluoromethylation under physiological conditions through a radical-mediated pathway (Scheme 11A).100 A Pd(0) nanoparticle (PdNP) biohybrid (39b, Fig. 12) and 4-methoxybenzene-diazonium tetrafluoroborate salt (39a) could promote Trp C-2 arylation (39c).101 Alternatively, transition-metal-free Trp-selective bioconjugation has been demonstrated by using an organoradical approach (40c, Fig. 12).102 The in situ generation of an ABNO organoradical (40a) by NaNO2 (40b) and acetic acid led to C–O bond formation at the C-3 position of indole in Trp (Scheme 11B). In another example, the inherent photolability of Trp gave a photochemical approach for the selective modification of proteins by using N-carbamoylpyridinium salts (41a, Fig. 12).103 The photoinduced electron transfer (PET) between Trp and pyridinium salt led to the fragmentation of the pyridinium N−N bond in the N-carbamoylpyridinium reagent to generate an N-centered radical (Scheme 11C). Next, it combines with a Trp radical to give a stable bioconjugate.
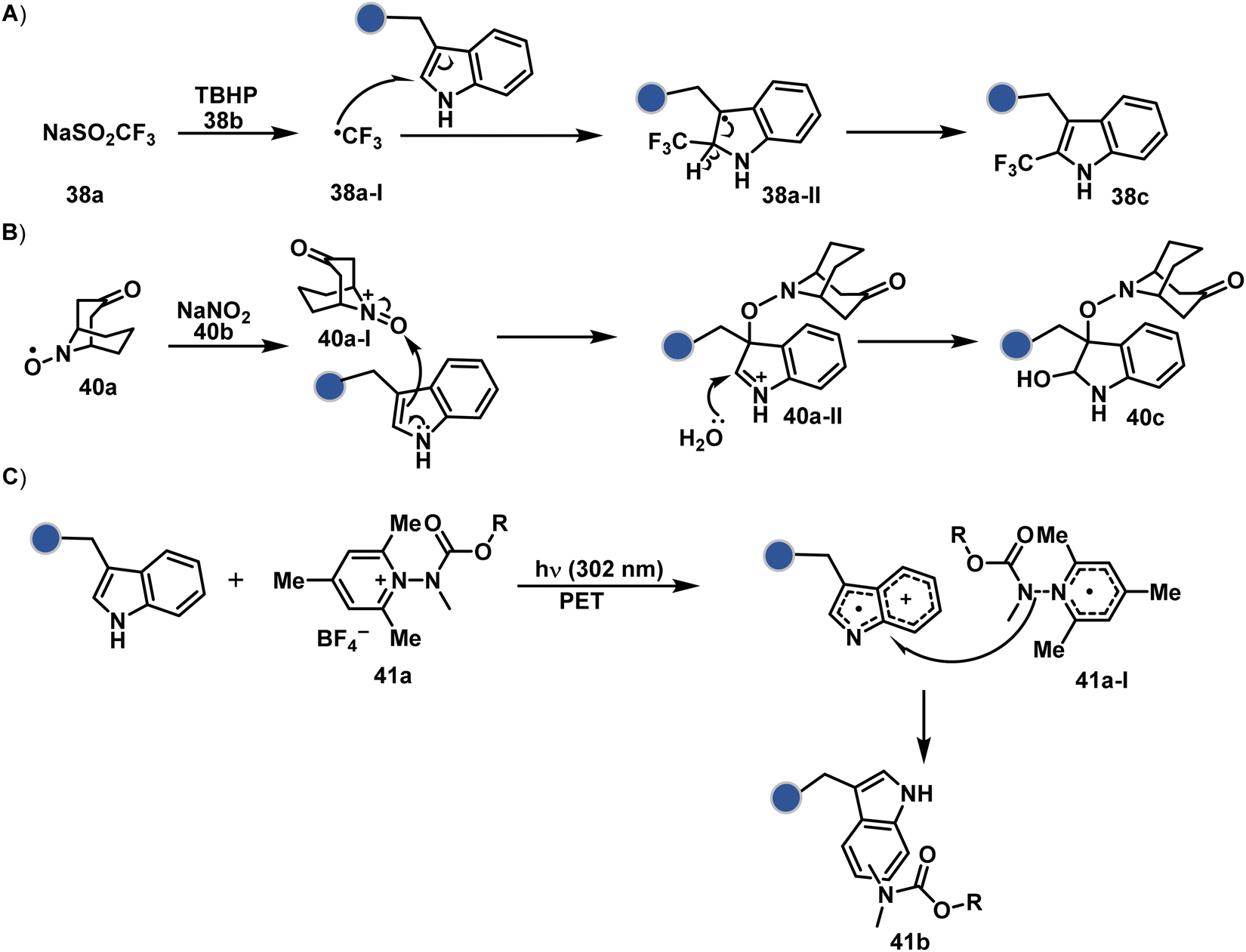 | ||
| Scheme 11 Proposed mechanistic pathways for tryptophan-selective bioconjugation using (A) NaTFMS, (B) ABNO, and (C) N-carbamoylpyridinium salt. | ||
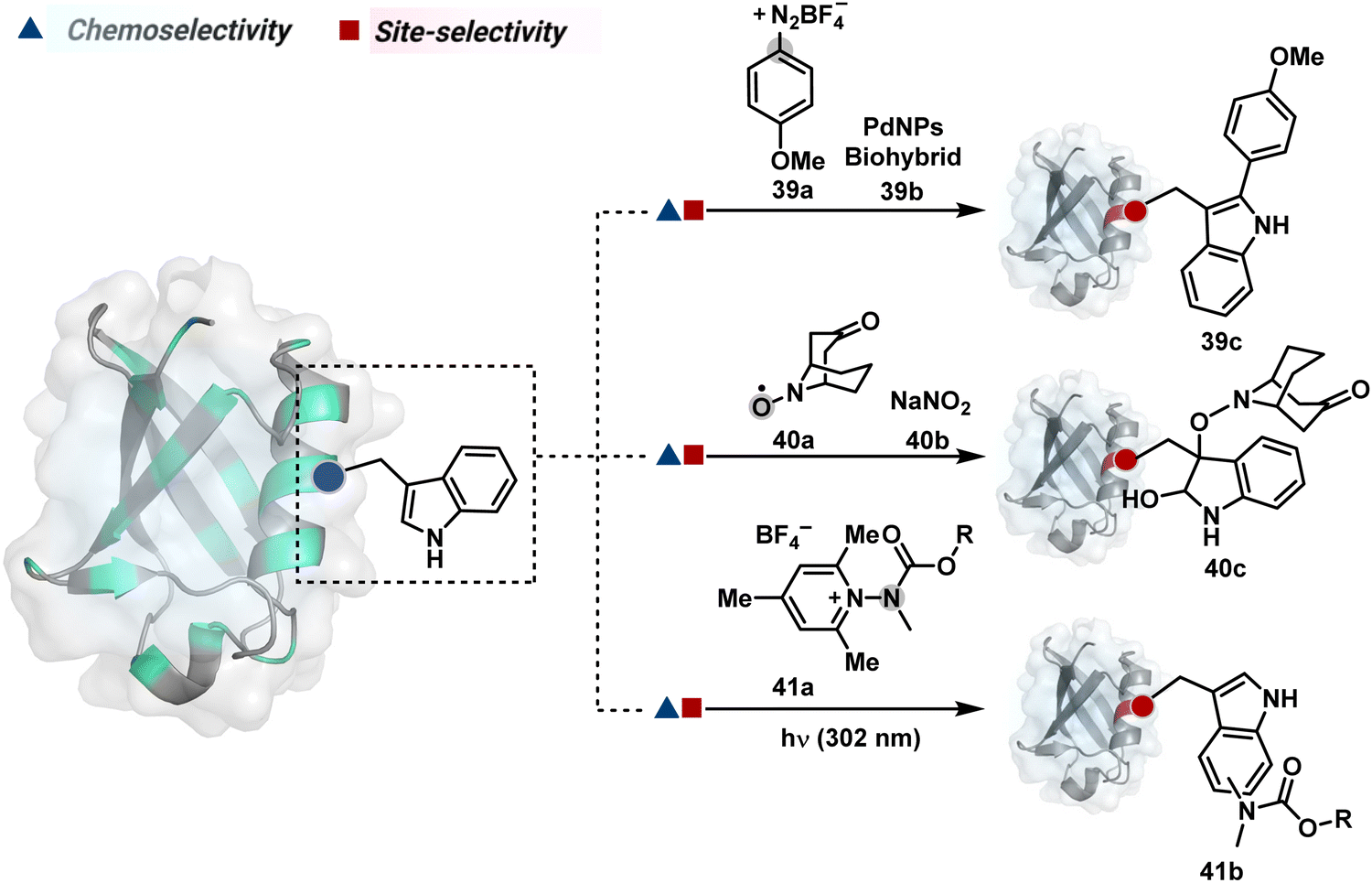 | ||
| Fig. 12 Mechanistically diverse chemical methods for chemo- and site-selective modification of tryptophan in proteins. | ||
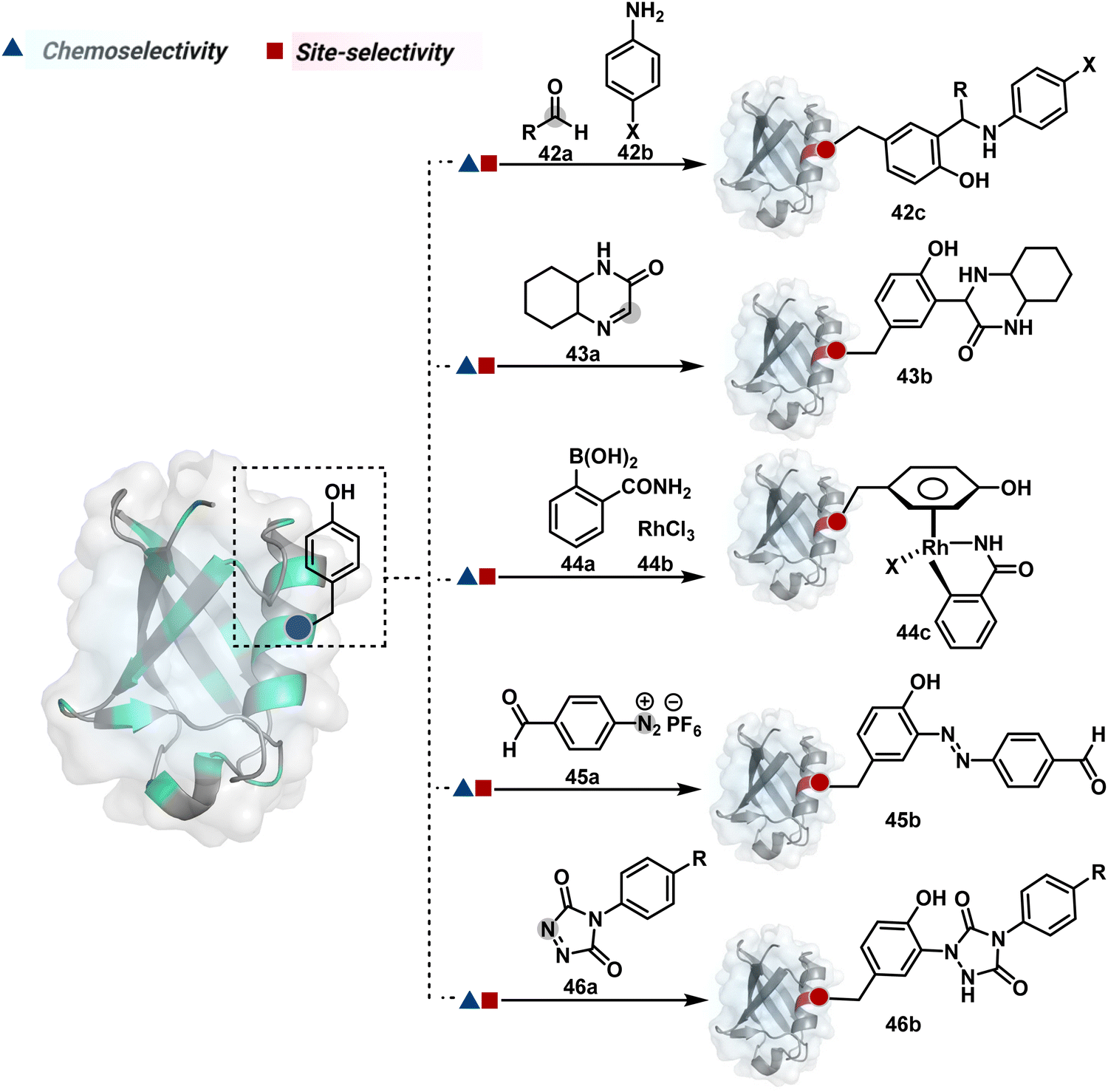 | ||
| Fig. 13 Chemical methods for chemo- and site-selective modification of tyrosine in proteins. | ||
 | ||
| Scheme 12 Proposed mechanistic routes for tyrosine-selective bioconjugation through (A)–(C) ene-type and (D)–(E) addition–elimination reactions. | ||
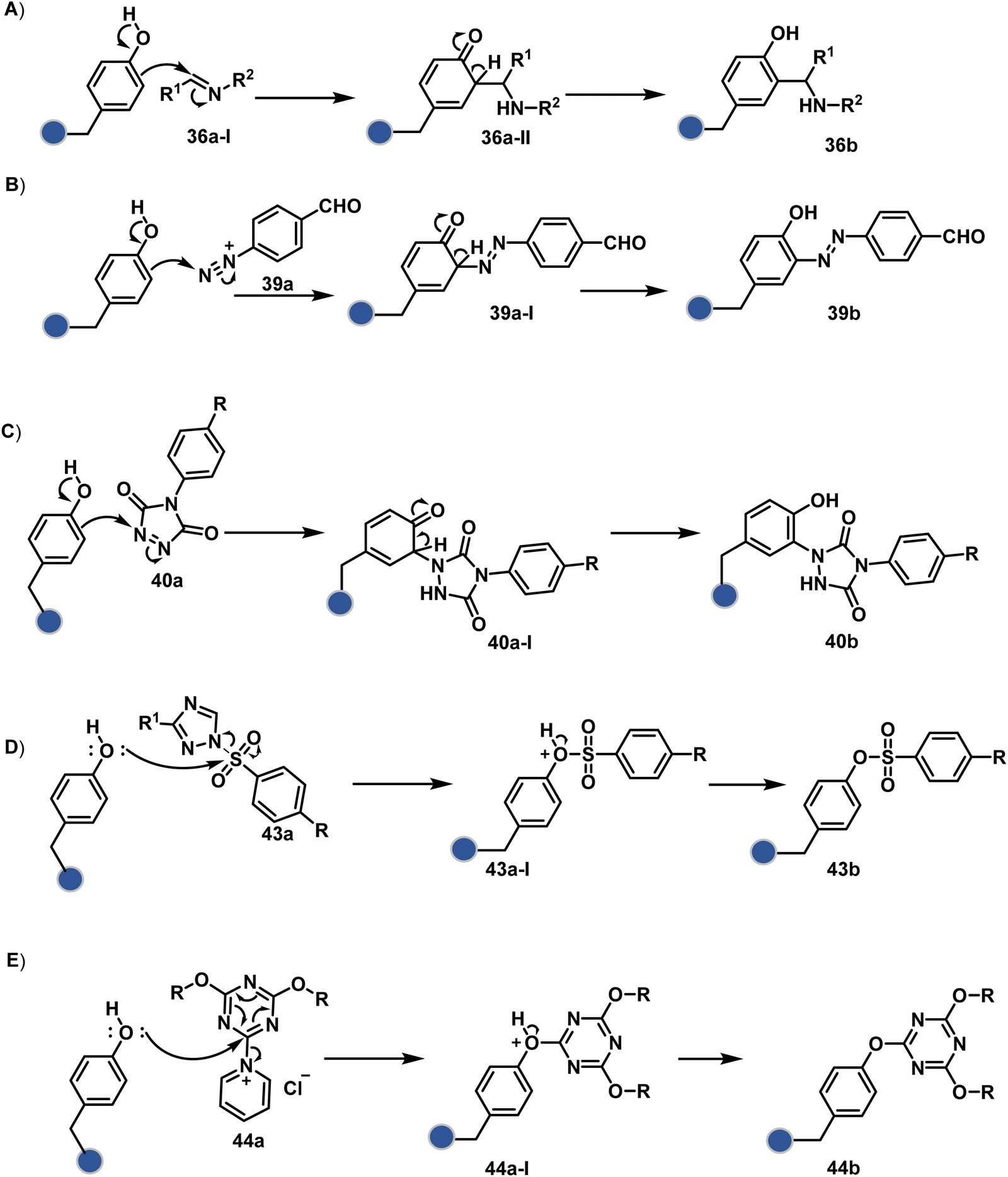
Diazonium reagents are another class of molecules that can enable Tyr modification. It translates well to complex systems of MS2 bacteriophage and tobacco mosaic virus capsids.110,111 The instability of diazo compounds can be addressed by in situ or immediate generation before the reaction. Alternatively, this challenge is addressed using their bench stable version, such as 4-formylbenzene diazonium hexafluorophosphate (FBDP, 45a, Fig. 13).112 The bifunctional FBDP reagent allowed Tyr bioconjugation, installing a biorthogonal aldehyde on the proteins (Scheme 12B). While the chemoselectivity was established with model amino acids, detailed sequencing of protein-based bioconjugates could provide better insights into selectivity attributes. Cyclic diazodicarboxamides (46a, Fig. 13) have also engaged Tyr selectively under physiological conditions.113,114 Again, the ene-type reaction of a phenolic side chain with 4-phenyl-3H-1,2,4-triazoline-3,5(4H)-diones (PTADs) results in a stable C–N bond linked adduct (Scheme 12C). The PTADs tend to decompose in water to form an isocyanate that labels primary amines. This side reaction can be addressed by changing the buffer from PBS to 2-amino-2-hydroxymethylpropane-1,3-diol (Tris), where the latter can act as an isocyanate scavenger.115 Alternatively, the problem can be avoided by an electrochemical in situ generation of PTAD from its 4-phenylurazole precursor.116 The mild nature of applied potential ensures that other amino acids in protein and its bioconjugates remain unaltered. However, this approach also faces chemoselectivity challenge from Trp.117 Initially, the off-target physiologically stable Trp conjugates remained unidentified due to their lability under high-energy MS/MS conditions. In fact, PTADs at lower pH can result in exclusive selectivity for Trp over Tyr. Establishing conjugation sites with MS/MS of such labile conjugates depends on the fragmentation technique. The luminol derivatives (47a, Fig. 14) in the presence of O2/laccase,118 H2O2/hemin,119 H2O2/HRP,120 and H2O2/DNAzyme121 could label Tyr. It is more efficient than PTADs under enzyme catalysed activation and electrochemical activation.122 In a similar mechanistic pathway, electrochemical oxidation of phenothiazine derivatives could prime them for selective modification of Tyr.123
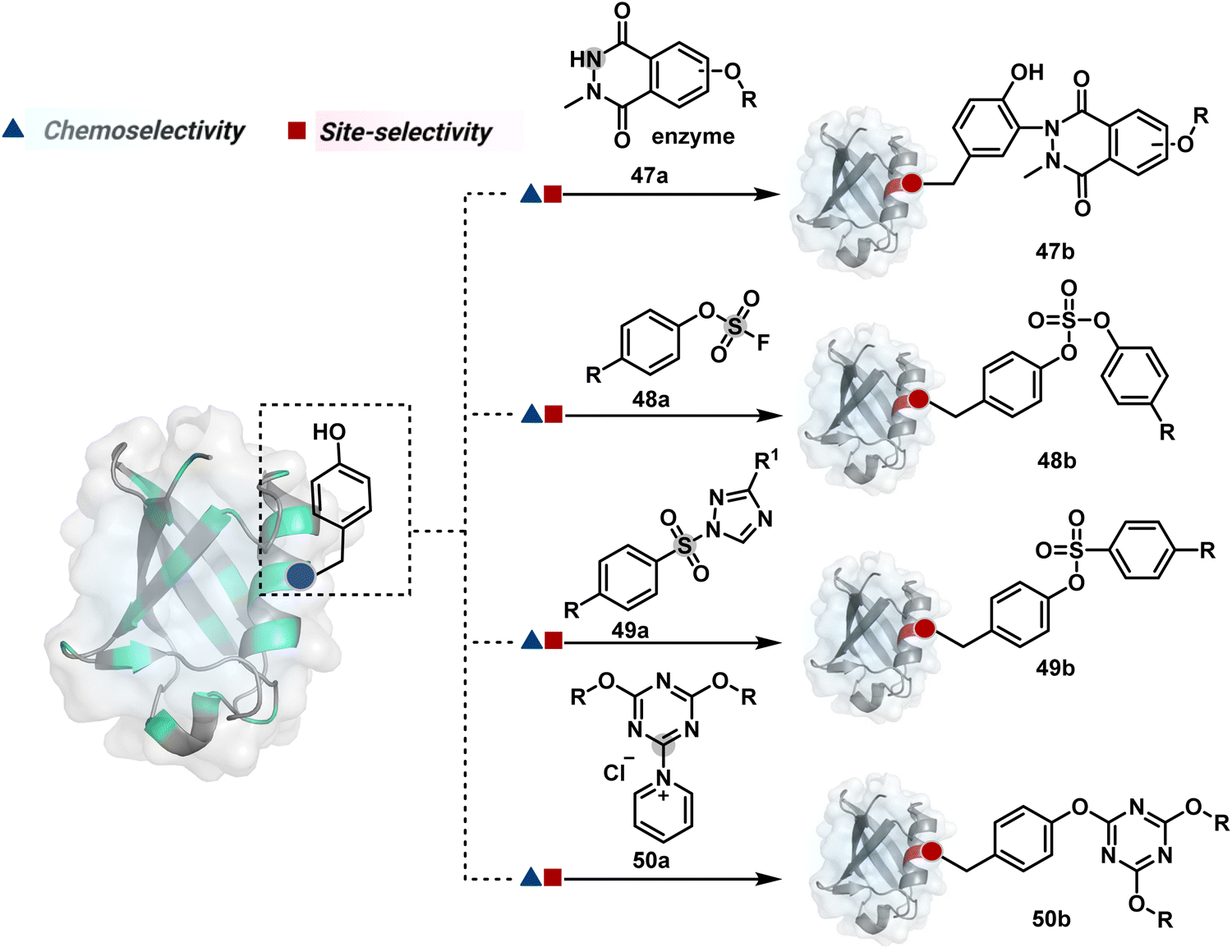 | ||
| Fig. 14 Additional chemical methods for chemo- and site-selective modification of tyrosine in proteins. | ||
In another approach, weak electrophilic systems facilitate Tyr modification using an addition–elimination route (Scheme 12D). For example, sulfonyl fluoride, a promiscuous electrophile, has been utilised for targeting proteins since 1963 through sulfur fluoride exchange (SuFEx) chemistry.124,125 Arylfluorosulfate derivatives (48a, Fig. 14) have been established for probing Tyr in proteome analysis126 and selective Tyr bioconjugation127 using SuFEx chemistry. Similarly, Tyr preference for sulfur-triazole exchange (SuTEx) chemistry was established with human proteome (49b, Fig. 14).128 In this case, the change of fluorine with triazole improves Tyr selectivity with respect to Lys. The sulfur exchange chemistry does not offer exclusive selectivity towards Tyr. Along these lines, 1,3,5-triazine pyridine (50a, Scheme 12E) reagents have been developed for selective Tyr bioconjugation.129 It would be interesting to see if these methods can be tuned further for exclusive Tyr selectivity to deliver a homogeneous bioconjugate with a structurally diverse set of proteins.
 | ||
| Fig. 15 General overview of dehydroalanine enabled selective modification of proteins. | ||
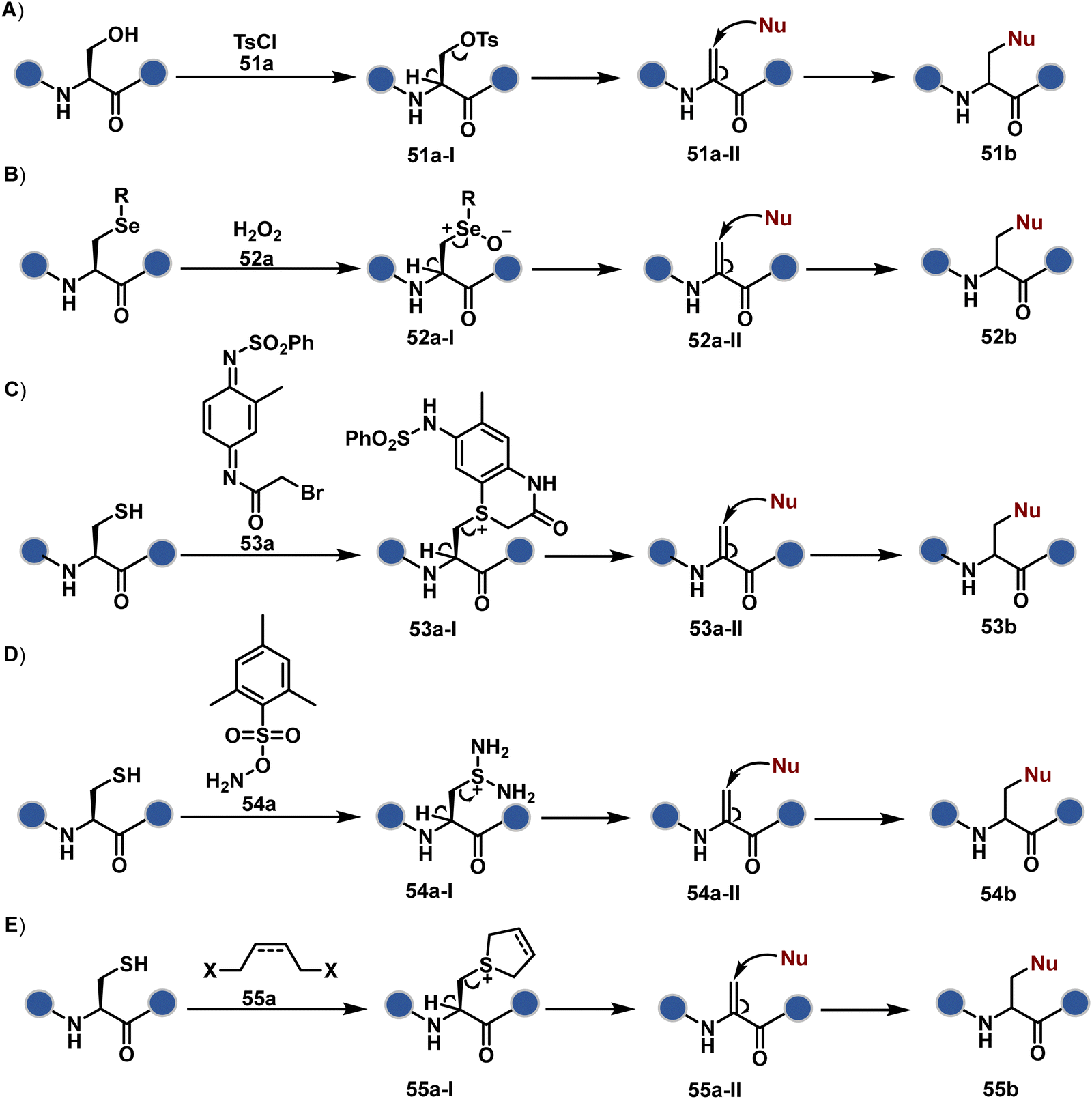 | ||
| Scheme 13 Proposed mechanistic pathways for chemo- and site-selective generation of Dha. | ||
Later, Cys residue was transformed into Dha by using 2-methyl-N1-benzenesulfonyl-N4-bromoacetylquinonediimide (Scheme 13C). Here, thiol dialkylation generates sulfonium salt (53a-I), which is eliminated to yield Dha.135 Also, thiol dialkylation operates conveniently with 1,4-diiodobutane,131 2,5-dibromohexanediamide (DBHDA),136 and methyl-2,5-dibromopentanoate (MDBP)130 for selective generation of Dha. In a mechanistically different approach, selective bis-amination of sulphur by O-mesitylenesulfonyl hydroxylamine (MSH) followed by Cope-type elimination results in Dha (Scheme 13D).137–139 The biorthogonal nature of Dha has been harnessed for various PTM installations on protein via thia-Michael,140,141 aza-Michael,142,143 and radical-mediated addition.143
The methods of route A have addressed chemoselectivity to a reasonable extent. However, only a few offer exclusive site-selectivity or protein specificity. Their extension to large proteins, antibodies, or complex biological mixtures amplifies the challenges. It could benefit from parameters that reduce the number of competitors. The small-molecule non-covalent ligands are valuable in this perspective. The selectivity of their interaction with specific protein domains could restrict the accessibility of electrophilic reagents (Fig. 16). The ligand–protein combinations such as benzenesulfonamide and carbonic anhydrase or biotin and avidin have been utilized to establish a proof of concept. Affinity-based labeling facilitated by reversible covalent or non-covalent binding can significantly minimize the number of competing residues. Such bioconjugation reagents offer a remarkably enhanced probability of executing chemoselective and site-selective labeling. This method is intriguing as rapidly growing drug-targetome data can accelerate it further.
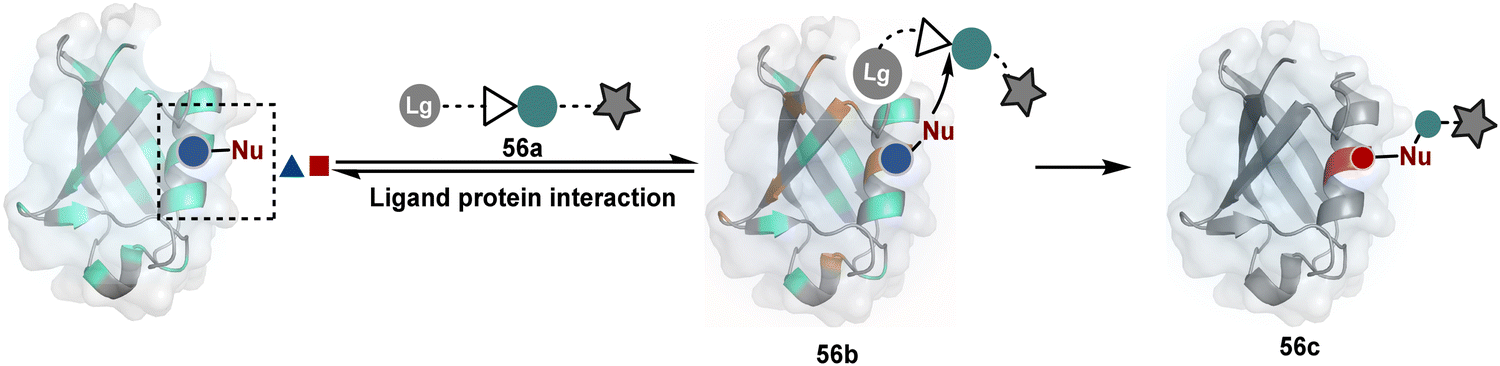 | ||
| Fig. 16 General overview for ligand-directed selective modification of proteins. | ||
One of the earliest examples demonstrates the modification of a His residue in human carbonic anhydrase II (hCA II) through the reaction of an epoxide bound to a benzenesulfonamide ligand.144 The same combination attached through a hydrazone-based linker offered additional flexibility.145,146 It enabled cleaving off the ligand through a hydrazone/oxime exchange reaction post-bioconjugation to regain enzyme activity. While this route offers synthetic flexibility, several probes of interest can be attached through this exchange reaction. The ligand-directed tosyl (LDT) chemistry is an additional entry in this category. Affinity ligands such as benzenesulfonamide for hCA II protein147 or SLF for FKBP12 protein148 were anchored to phenylsulfonate ester (tosylate). It offers control over site-selectivity, resulting in His alkylation in the target proteins. Notably, the localization can overpower the inherent chemoselectivity order. Hence, depending on the reagent design, Tyr and Glu residues were labeled in addition to His. The slow reaction kinetics and low yield in intracellular protein labeling are significant drawbacks of the LDT method. Some of these were addressed by switching the reactive group from tosylate to alkyloxyacyl imidazole (LDAI).149 This approach exhibited a rapid and site-selective Lys modification of membrane-bound folate receptors in live cells. A similar approach has been employed to localize a Ru-complex ([Ru(bpy)3]2+) and enable single electron transfer photocatalysis for selective Tyr modification.150 The strategy also works well for a biotin–avidin pair for selective Lys labeling through O-nitrobenzoxadiazole (O-NBD)151 and diazotransfer with imidazole-1-sulfonyl azide.152 Some of these reagents are susceptible to enzymatic degradation, making them unsuitable for endogenous protein labeling. A sterically hindered dibromophenyl benzoate (LDBB) improves its biological stability.153 The reagent was utilized to label the Lys residue in an endogenous hCA II protein. Ligand-directed N-sulfonyl pyridone (LDSP) reported on similar lines is utilized for FRET-based biosensors to report intracellular ligand binding events.154 Furthermore, ligand-directed N-acyl-N-alkyl sulfonamide (LDNASA) chemistry is an additional entity in this category.155 It demonstrates fast reaction kinetics and good stability under biological conditions. In general, proteins that are unable to specifically bind to small molecule ligands cannot be labeled using this strategy. Also, the presence of a reactive nucleophilic residue in a ligand can render deactivation of the cleavable electrophile through an intramolecular reaction. Affinity-guided 4-dimethylaminopyridine (DMAP) or AGD chemistry was developed to address a few of these shortcomings. A ligand tethered to DMAP first binds to a target protein, then assists the transfer of an acyl group from activated ester to a proximal nucleophilic amino acid residue. It allows for catalyst-based traceless modification of proteins and expands the targetome. Labeling of glycoproteins on live cell surfaces,156 selective modification of HER2 and EGFR,157 or acylation of histones on native chromatin158,159 are some examples of AGD chemistry. Affinity-guided oxime (AGOX) chemistry using pyridinium oxime and N-acyl-N-alkylsulfonamide reagent has also been reported in this category.160 The ligand–protein interaction specificity greatly restricts competitors and promotes route A for selective protein modification in biological settings such as live cells. However, the dependency on protein–ligand combinations for selective labeling significantly limits the range of proteins or sites in selected proteins that can be modified using this approach.
The concept extends to irreversible covalent inhibitors for the precision engineering of proteins. For example, site-selective covalent modification of KRAS mutants such as G12C, G12R, or G12S is now recognized for cancer immunotherapy. The KRAS G12C mutant modified using ARS1620 through the reaction of Cys with the α,β-unsaturated reaction center provides ARS1620 modified peptides in the MHC-I complex (haptens).161 The haptenated MHC-I complex acts as a tumor-specific neoantigen, which is then recognized by a bispecific T-cell engager to elicit an immune response against KRAS G12C cells. An α,β-diketoamide tagged with a switch II ligand for the KRAS protein was used for synthesizing targeted covalent inhibitors by labeling the Arg12 residue.162 A β-lactone-containing inhibitor was used for modifying KRAS G12S mutants.163
2.2. Route B
As we noticed in examples of route A, chemoselectivity and site-selectivity are often addressed in the same step during protein bioconjugation. It severely limits options to rectify any encountered issues. Even when selectivity is achieved in such cases, it is displayed in a narrow range of reaction parameters. In turn, it impacts batch-to-batch reproducibility. An alternate way to address the challenge could be to use a protection–deprotection approach. However, such a route is often inefficient with proteins. The resolution of chemoselectivity and site-selectivity in distinct steps could allow a better handle to regulate the overall precision. It could be achieved by combining reversible reaction(s) with an irreversible transformation rendering the end product. For example, a reversible reaction could lead to chemoselective generation of intermediates. In the next step, an irreversible reaction must address the site-selectivity. Such a combination of reversible–irreversible reactions could render unique reactivity orders based on the choice of reagents or reaction parameters (Fig. 17). It can provide access to distinct sites for bioconjugation.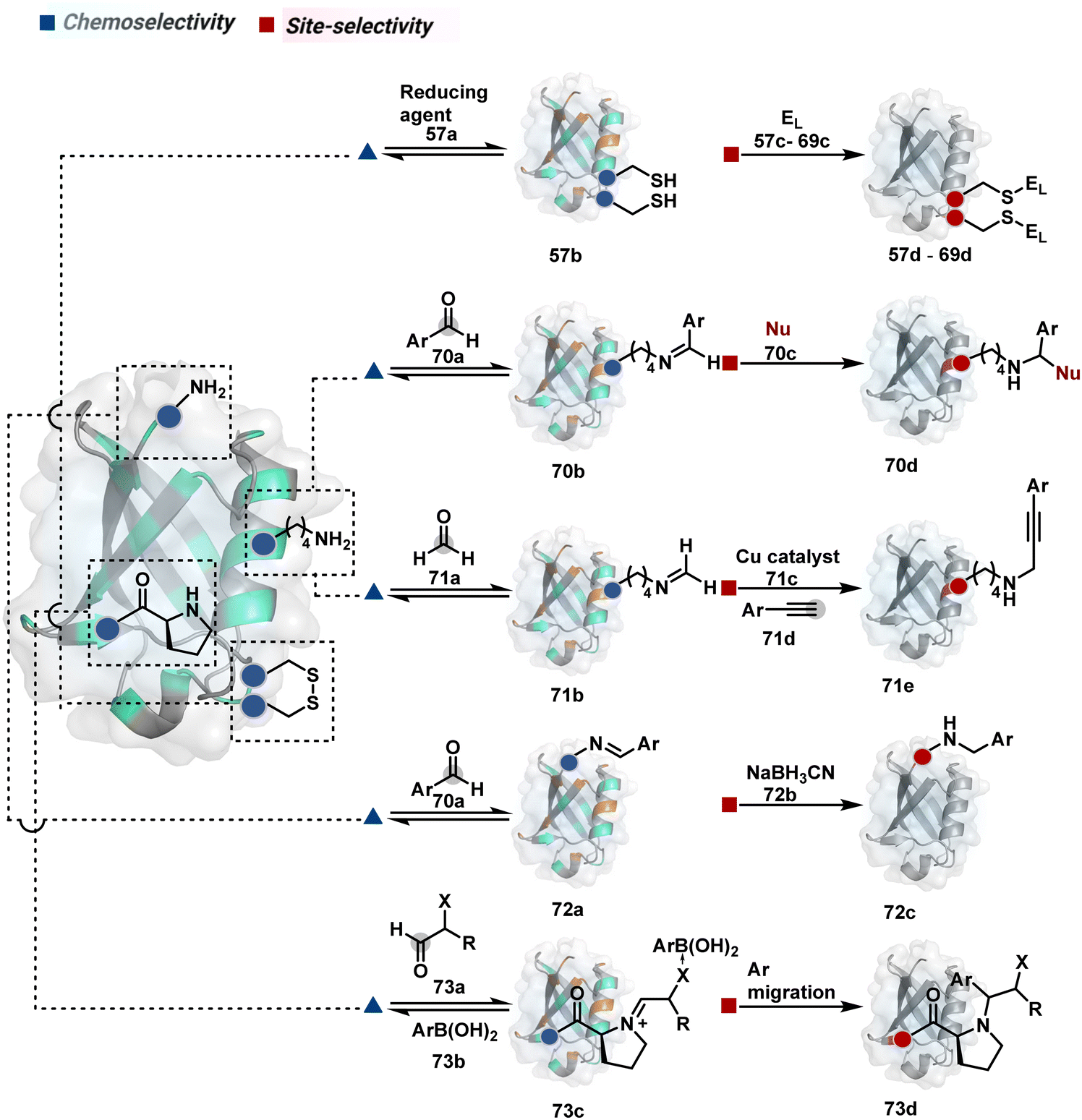 | ||
| Fig. 17 Methods for disintegrating chemoselectivity and site-selectivity to drive single-site protein bioconjugation. While the former involves an intermolecular reversible reaction, the latter engages an intermolecular irreversible pathway. | ||
The high reactivity of thiol has led to the development of multiple Cys-selective reagents. From this pool, a bis-alkylating sulfone reagent (57c, Scheme 14) enables the insertion of a desired probe between two sulphur atoms while rebridging it with a three-carbon chain (57c-I to 57d).166–169 However, analytical challenges made it difficult to validate the conjugation sites. The evolution of mass spectrometric techniques facilitates obtaining a better insight into the selectivity in recent efforts. Still, confirmation of rebridging is only feasible if a mass change is associated with the transformation. The current efforts have allowed this route to be utilized to install PEG, FRET probes, imaging probes, toxins, bio-affinity tags, and dual or multiple probes into proteins, including monoclonal antibodies.170–179 The bis-alkylating sulfone reagent comprises an α,β-unsaturated carbonyl compound equipped with a β′-leaving group. At first, a chemoselective disulfide reduction renders thiol groups (57b). One of them undergoes 1,4-addition. This Michael adduct can generate another α,β-unsaturation by eliminating p-toluenesulfinic acid (57c-II). The latter could then react with the second thiol group to reconnect two sulfur centers through a three-carbon bridge (57d, Scheme 14).
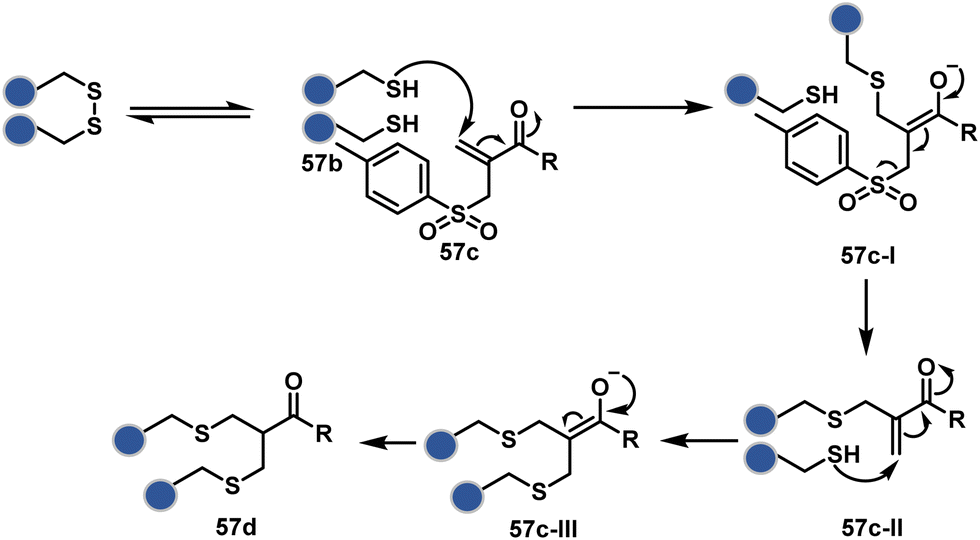 | ||
| Scheme 14 Thiol bis-alkylation with a sulfone reagent for disulfide rebridging. | ||
A similar deconvolution of chemoselectivity and site-selectivity has been explored with other bis-electrophilic systems. The maleimide derivatives, including mono-bromomaleimide (58c, Scheme 15A),180–182 dibromomaleimide (59c, Scheme 15B),183–185 and dithiophenolmaleimide (59c, Scheme 15B),186–190 were examined for harnessing the potential of disulfide rebridging (58d–59d). Another Cys-selective motif, bromopyridazinedione (60c, Scheme 16A),191,192 inspired using dibromopyridazinedione (61c, Scheme 16B)192–199 derivatives for efficient disulfide rebridging (60d–61d). Here, the bioconjugation utilizes two consecutive addition–elimination steps, while two pyridazine nitrogen centers offer sites for pre-installation of two independent probes. The synthetic manoeuvrability of these bioconjugation reagents allows engineering a biorthogonal handle, such as strained alkyne, to install probes. Additionally, this route allowed the synthesis of relatively homogeneous antibody–drug conjugates. Along the same lines, Cys-selective 3-oxetane bromides (62c, Scheme 17A) inspired using 3,3-bis(bromomethyl)oxetane (63c, Scheme 17B) for disulfide rebridging (62d–63d).37,38 Here, a thiol-selective alkylation creates an opportunity for another round of alkylation.
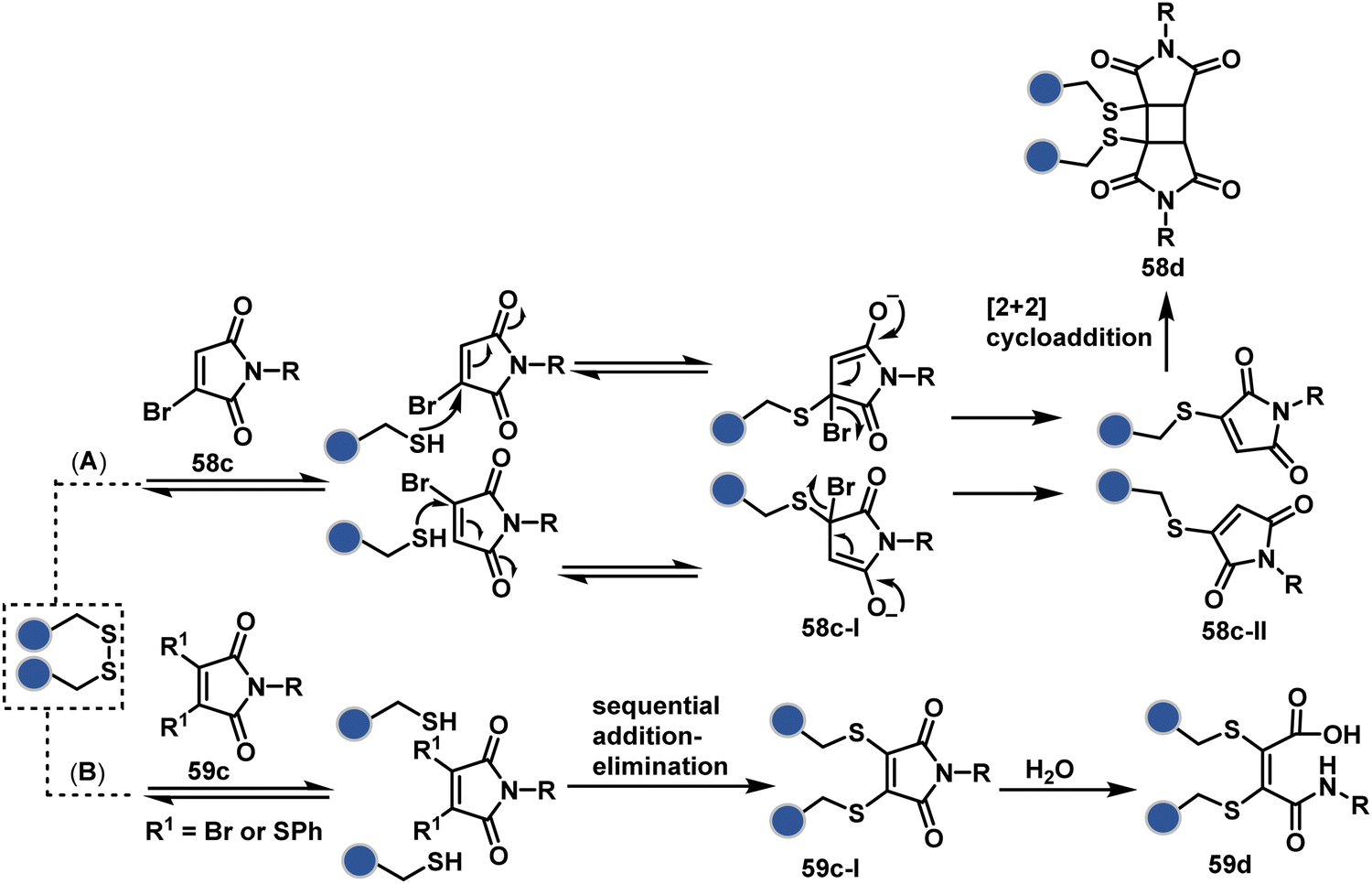 | ||
| Scheme 15 Disulfide rebridging with a maleimide derivative through an addition–elimination process. | ||
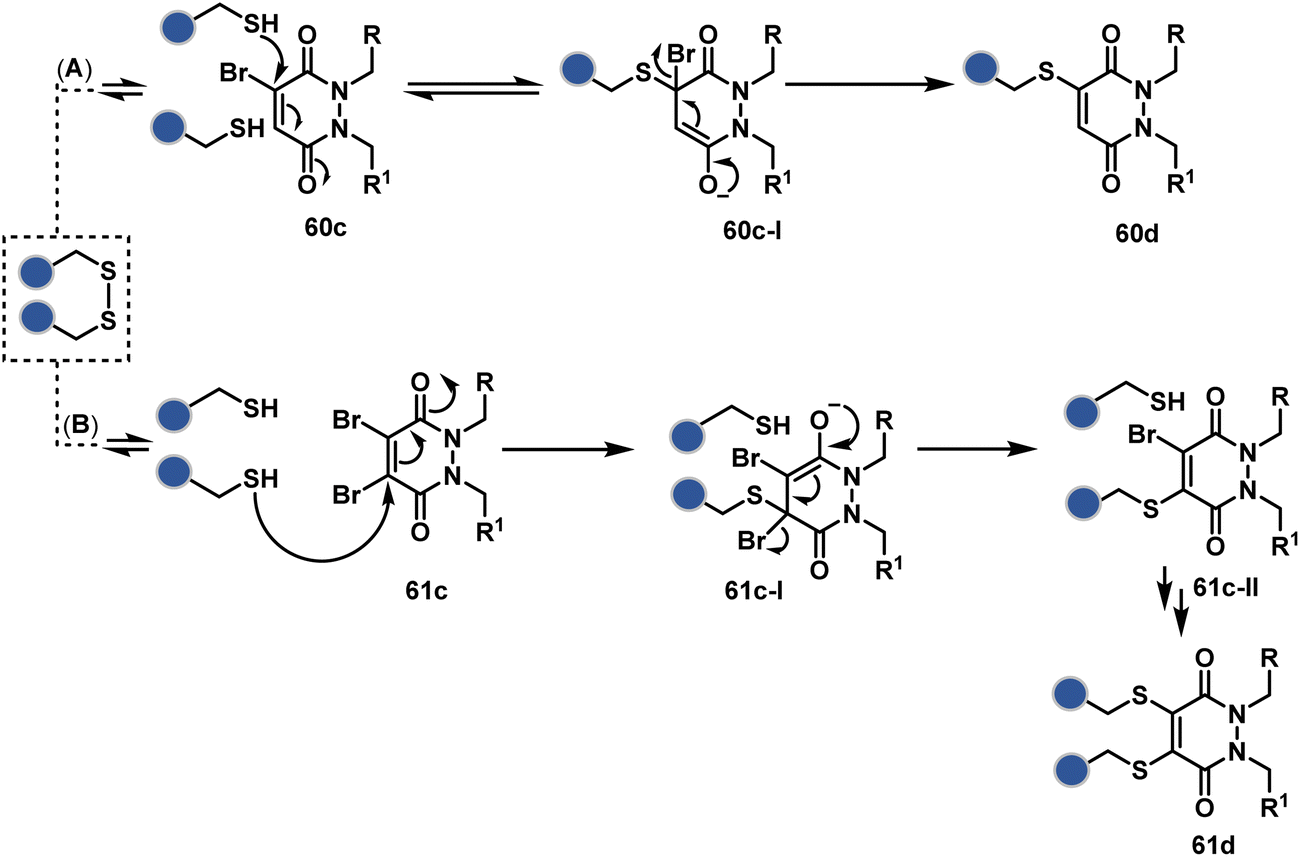 | ||
| Scheme 16 Addition–elimination sequence enabled disulfide rebridging. | ||
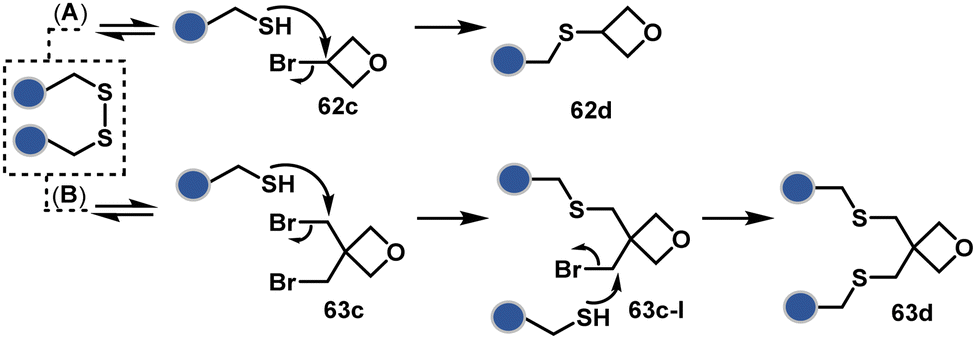 | ||
| Scheme 17 Oxetane-mediated disulfide rebridging in proteins. | ||
On the other hand, unsaturated bonds with subtle polarization could also enable disulfide modification. For example, a photochemical activation of reduced disulfide offers an efficient thiol–yne coupling reaction (64c–64d, Scheme 18A).200 It generates an alkene (64c-II) that enables rebridging through thiol–ene coupling (64d). This UV-initiated thiol–yne conjugation was demonstrated with an antigen-binding fragment (Fab) of a monoclonal antibody (mAb). The sequential process utilizes 365 nm UV light in the presence of lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, catalyst) as a radical initiator (Scheme 22). On the other hand, rebridging can also be achieved through a bis-electrophilic system through two independent thiol–ene reactions. The electron-deficient divinylpyrimidine (65c, Scheme 18B) provides adequately polarized double bonds to react with thiols to deliver homogeneous antibody conjugates (65d).201–203 The thioether linkage in these bioconjugates displays high serum stability over seven days. The synthetic flexibility of bioconjugation reagents allows preinstallation or late-stage installation of diverse probes.
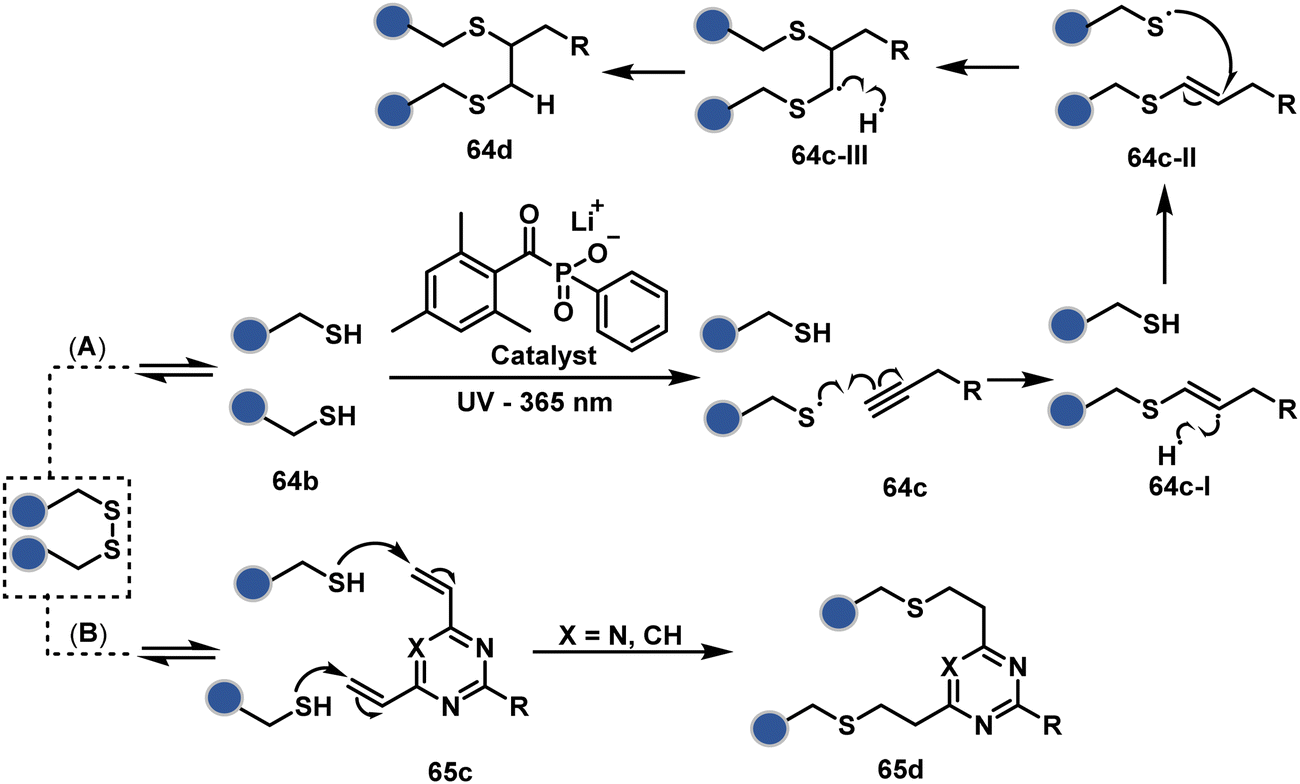 | ||
| Scheme 18 Thiol–yne coupling and divinyl-pyrimidine reaction for disulfide rebridging. | ||
In a mechanistically similar approach, the polarized alkyne in ethynyl P(V) electrophiles (66c, Scheme 19A) could result in chemoselective modification of thiols generated from disulfide reduction.204–206 However, the alkene in the product (66d) did not promote a subsequent thiol–ene reaction. Later, diethynyl-phosphinate derivatives (67c, Scheme 19B) could render parallel thiol–yne reactions, resulting in disulfide rebridging in a monoclonal antibody (67d).207
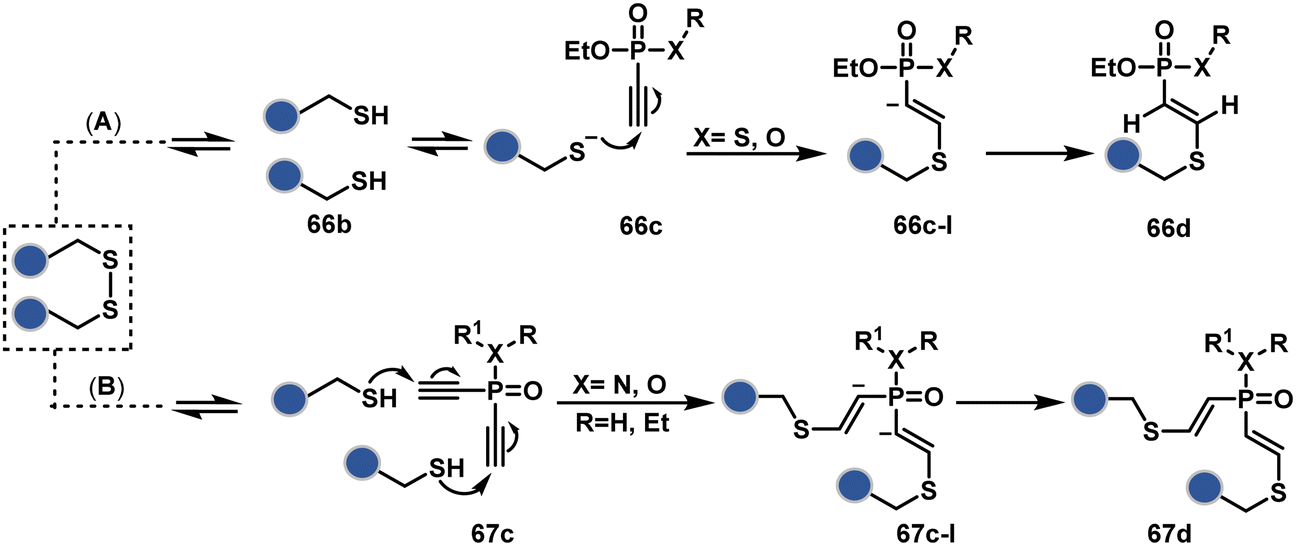 | ||
| Scheme 19 Ethynylphosphonites and diethyl-vinylphosphonite mediated disulfide rebridging. | ||
Another set of polarized alkynes, 3-arylpropiolonitrile (APN, 68c, Scheme 20A), offers excellent Cys-selectivity (68d).208,209 Like the previous case, olefin did not display reactivity with the second molecule of Cys. Hence, an aryl ring was used to construct a bis-electrophilic system, 3,3′-arylene-dipropiolonitrile (ADPN, 69c, Scheme 20B).210 While it delivers disulfide rebridging (69d), the aryl ring offers an additional substituent to provide synthetic flexibility for the installation of probes.
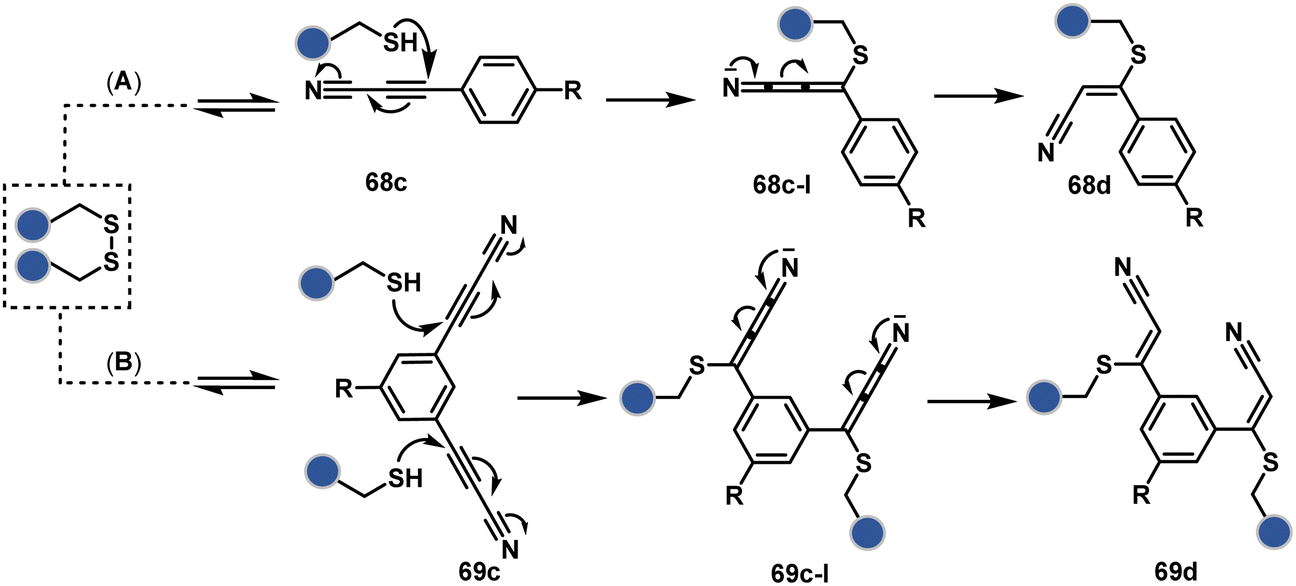 | ||
| Scheme 20 APN mediated disulfide rebridging. | ||
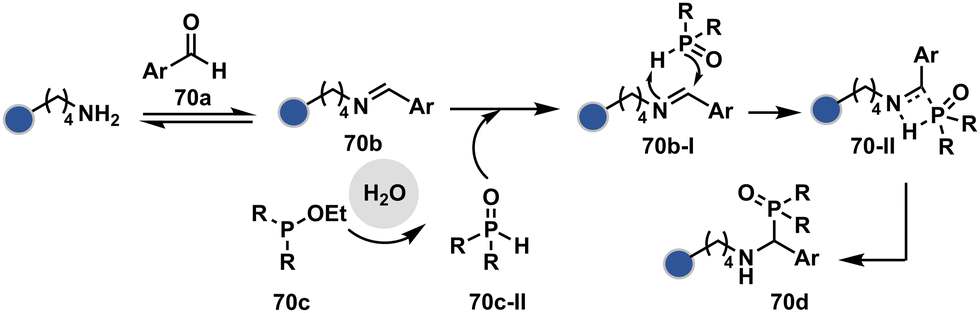 | ||
| Scheme 21 Chemoselective generation of imine creates an opportunity for the site-selective phospha-Mannich reaction and single-site protein modification. | ||
Alternatively, a Cu-acetylide complex (71d-I, Scheme 22) can capture imines (71a-I) in a site-selective irreversible reaction.14 Notably, the N-terminus amine forms an imine that does not engage with an external nucleophile. It is primarily because an intramolecular reaction with the proximal amide bond forms an imidazolidinone adduct. Even though it is a reversible reaction, it is good enough to block N-imine reactivity towards an external nucleophile. While the site-of-conjugation remains the same with some proteins, it changes in others with respect to the imine captured in phospha-Mannich reactions.
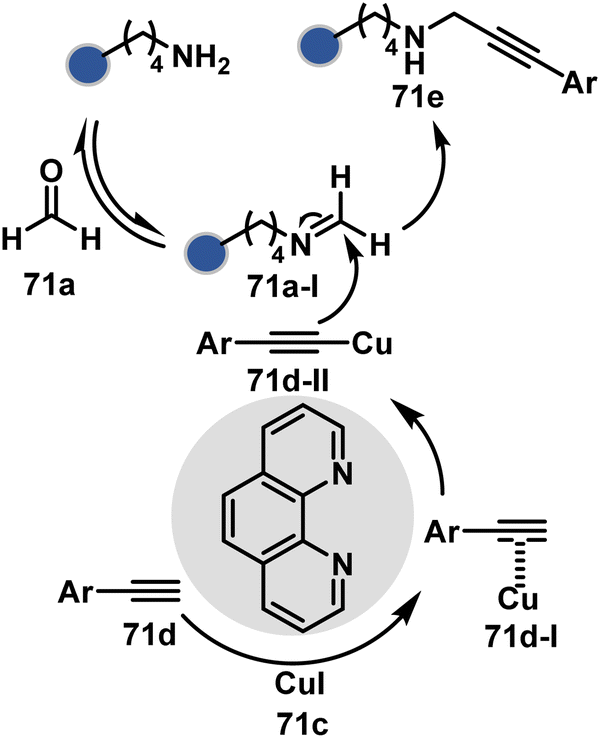 | ||
| Scheme 22 Chemoselective generation of imine creates an opportunity for site-selective A3 coupling in the presence of Cu and ligands. | ||
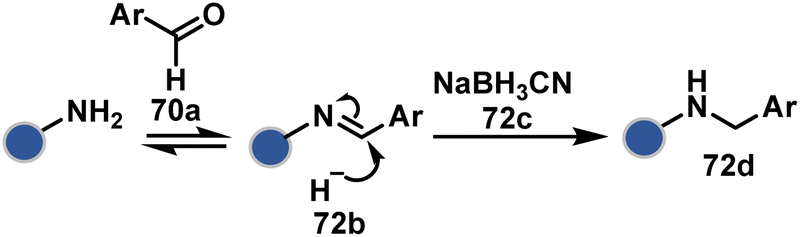 | ||
| Scheme 23 Chemoselective generation of imine creates an opportunity for site-selective addition of hydride. | ||
An organoboron reagent (73b, Scheme 24) efficiently stabilizes the N-Pro-imines (73c).211 Again, the first step involves reversibly generating an iminium ion. The aldehyde also bears a proximal nucleophilic group that can coordinate with boron to release the nucleophile that can react irreversibly with the iminium through a cyclic transition state (73c-I). While selectivity control is challenging with amines, its high frequency ensures that successful methods can find proteome-wide applications.
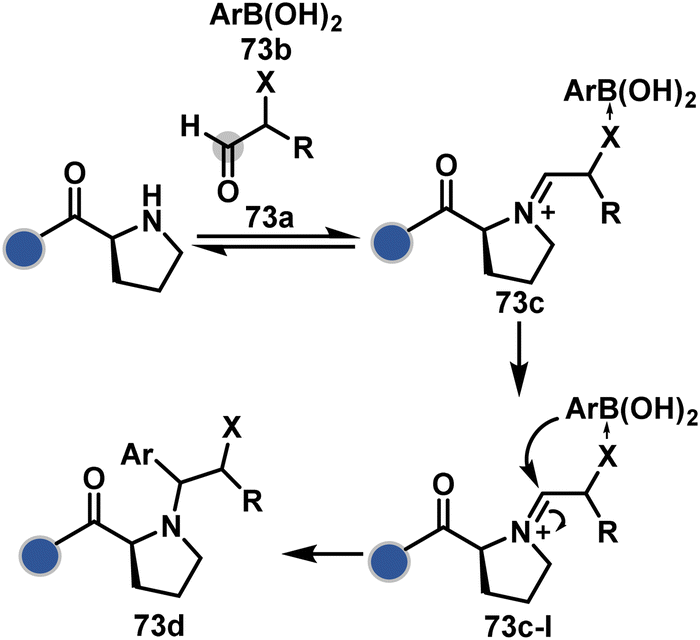 | ||
| Scheme 24 Chemoselective generation of iminium creates an opportunity for the single site Petasis reaction. | ||
In general, the disintegration of selectivity attributes offers precise labeling of unique sites and provides access to a new targetome. Here, the chemoselectively generated intermediate in the first step plays a significant role in defining site-selectivity in the irreversible step. If it is a highly reactive intermediate, control over site-selectivity becomes a significant challenge. For example, a photocatalyzed generation of a His or a Trp-specific radical cation would make single-site bioconjugation difficult in the presence of its multiple copies.103,212 The highly reactive non-selective radical or carbene-based intermediates have been extensively used for interactome analysis. However, the same inherent character makes them inappropriate to drive site-selectivity.213–215
2.3. Route C
The reactive intermediate generated in route B engages one proteinogenic residue in a given transformation. The protein's inherent nature would regulate the relative reactivity of such reaction centers. What if we change the role of such reversible covalent bonds that would not participate directly in an irreversible reaction? In such a case, they can serve as linchpins. Besides, they can be designed to direct another functional group that can engage a proximal residue for irreversible modification. In such cases, two residues of a protein, their relative position, and the match with the placement of functionalities in the reagent would govern the conjugation sites. The change in reagent design should then facilitate the selection of a unique residue pair. Such a modular platform could render control over the regulation of the modification site.Such a method would require a systematic sequence of reactions with a pair of functional groups. At first, a rapid chemoselective reversible reaction should construct linchpins (74b–81b, Fig. 18). It should be able to place the second functional group at a proximal position. If the latter finds an appropriate residue in the microenvironment, it should result in a relatively slow, irreversible reaction. Since the latter would proceed through an intramolecular pathway, it could benefit from kinetic advantage over non-selective background reactions following an intermolecular route. We argued that if such an approach is successful, it can provide guiding principles to mark functional group combinations as signatures for precise modification (74c–81c, Fig. 18). This concept was realized through the Linchpin-Directed Modification (LDM®) platform to target a diverse residue pair in a protein rendering single site modification. The approach can be extended to two functionalities placed on the N-terminus residue. Alternatively, it can be redesigned to mark three residue signatures for bioconjugation.
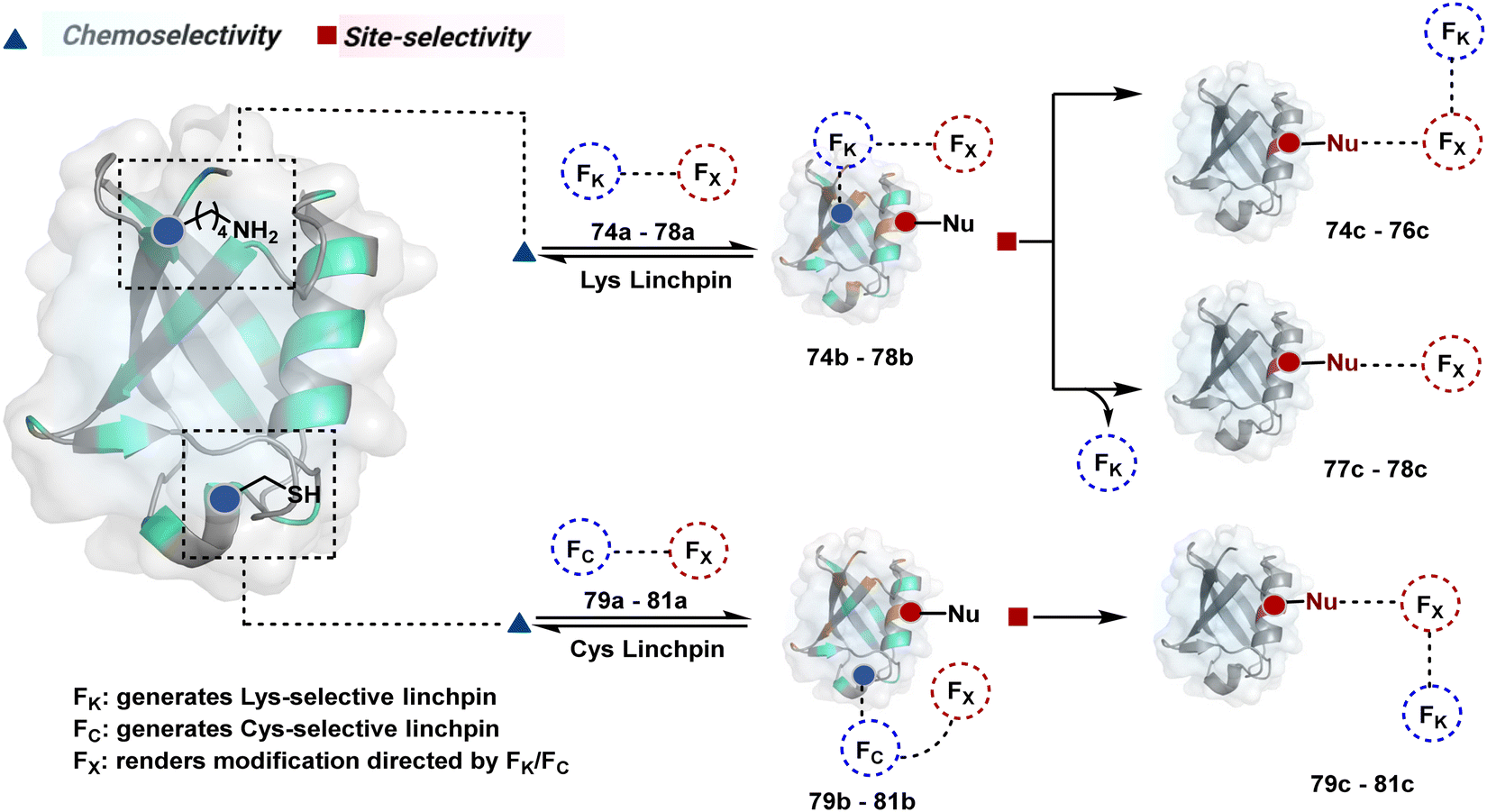 | ||
| Fig. 18 General overview for linchpin-directed modification of proteins. An intermolecular chemoselective linchpin formation at a residue (e.g., Lys or Cys) regulates the proximity of a second electrophile for single-site irreversible modification. | ||
Identifying functional group handles to generate linchpins was initially addressed using o-hydroxybenzaldehyde. It renders a rapid and reversible intermolecular reaction with all solvent-accessible Lys residues to form imines (74b–78b, Scheme 25). The proximal hydroxyl group instils additional stability in these imines. Besides, it dramatically impacts the reactivity of imine towards nucleophiles and ensures that it does not participate directly in an irreversible reaction. This attribute is critical to avoid the route B pathways with imine. Hence, it is limited to acting as a linchpin and directing another functional group to a target proximal residue. The first example involved o-hydroxybenzaldehyde linked with His-selective epoxide by spacers (74a, Scheme 25A).16 It enabled Lys-based linchpin-directed single-site modification of His residues (LDMK–H) in multiple proteins (74c, Scheme 25A). Next, o-hydroxybenzaldehyde is utilized for a late-stage installation of probes through hydrazone or oxime formation. This method established that it is possible to simultaneously regulate the reactivity with chemoselectivity, site-selectivity, and modularity. Replacing His-selective epoxide with Lys-selective activated ester (75a, Scheme 25B) enabled single-site modification of Lys residues (LDMK–K, 75c, Scheme 25B).15 Importantly, all mechanistic attributes required to maintain the overall selectivity were conveniently translated from LDMK–H to LDMK–K. Switching the linchpin fragment towards the leaving group in another effort ensured that it remained excluded from the bioconjugate (76c, Scheme 25C, and 77c, Scheme 25D).216,217
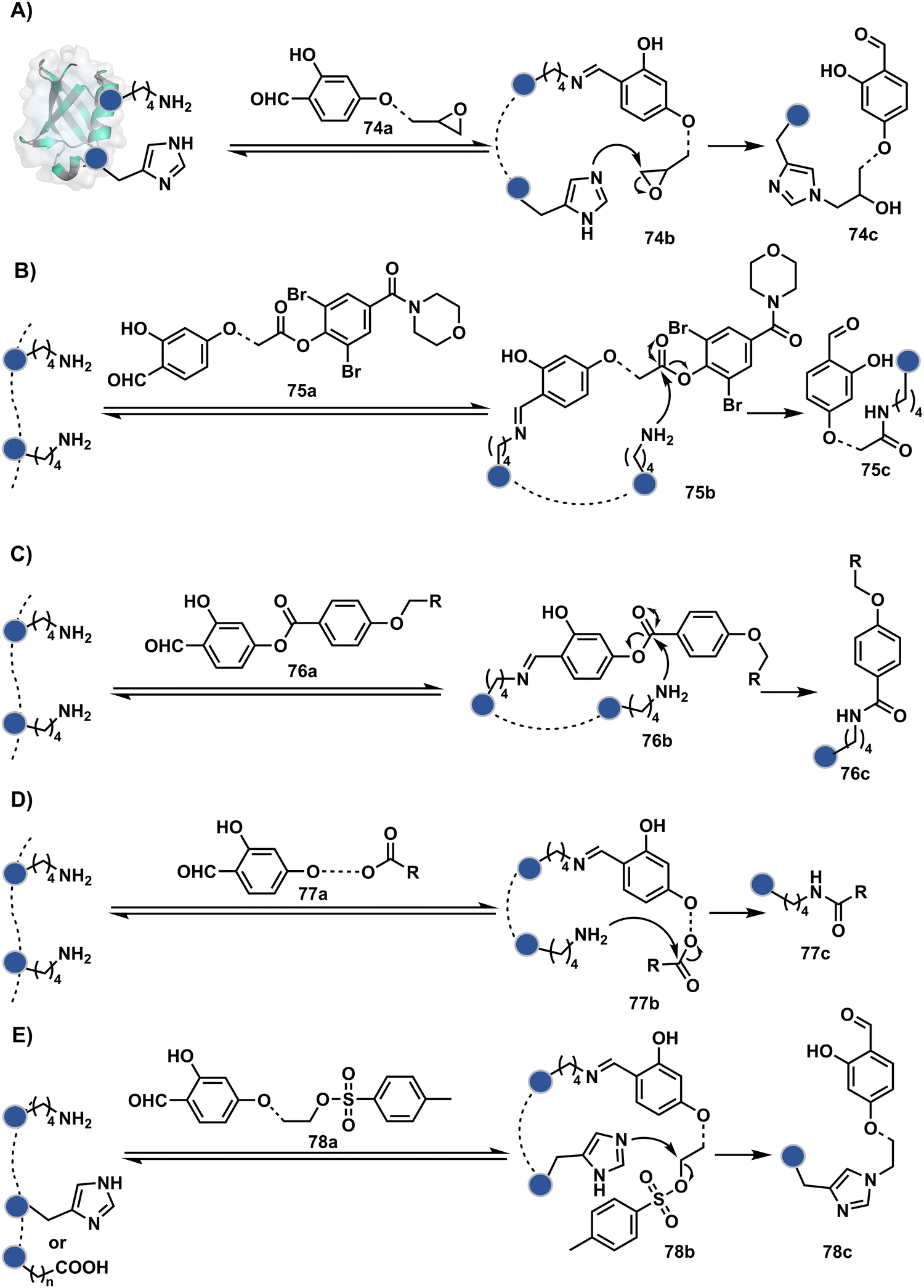 | ||
| Scheme 25 General mechanism for lysine-based linchpin-directed modification. (A) LDMK–H for modification of histidine. (B)–(D) LDMK–K examples for modification of lysine. (E) LDMK–X for modification of histidine or carboxylic acid. | ||
At this point, we asked if the second functional group could be replaced by a non-chemoselective promiscuous electrophile (LDMK–X, 78a, Scheme 25E). If spacers remain flexible throughout the process, a vast conformational space of the second electrophile could engage multiple competitors for irreversible modification. Hence, it should result in heterogeneous protein bioconjugation. In such a Lys-based linchpin-directed modification, a sulfonate ester-based alkylating agent led to the modification of Asp or His (78c, Scheme 25E).218 While the chemoselectivity is promiscuous, the transformation rendered single-site modification. It validates that site-selectivity can be achieved even with non-chemoselective electrophiles. Besides, it indicates that there are factors contributing to spacer conformational restriction in the LDM reagent.
The linchpin-directed modification operated well with multiple proteins. The high-frequency Lys residues offered appropriate options to find a unique pair without compromising selectivity. However, the competitive scale would change drastically in a biological milieu, such as cell lysates or live cells. The substantially elevated number of competitors would make it highly challenging for such methods to target just one pair of residues. Such a technological demand would require restricting the number of linchpins. One way of imposing it is to generate it with a low-frequency residue. For example, Cys can significantly minimize the number of competitors (79b–81b, Fig. 18). Hence, such linchpins can offer an opportunity for protein-specific single-site bioconjugation in a complex biological system. As discussed earlier, electrophiles for rapid Cys-selective were well established. However, the controlled reversibility while rendering a chemically orthogonal handle is non-trivial. Eventually, nitroolefins met all the requirements to serve as a linchpin handle (79a, Scheme 26A).219 The Cys-targeted thio-Michael addition enabled a site-selective acylation of proximal Lys (LDMC–K, 79c, Scheme 26A). A subsequent retro-Michael followed by a retro-Henry reaction drives the linchpin release and renders an aromatic aldehyde under mild conditions for orthogonal late-stage transformations. In this case, molecular dynamics simulations confirmed that the LDMC–K reagents are conformationally flexible. The linchpin formation promotes the interaction of the spacer with the protein surface, forcing it to adopt a rigid conformation. It limits access to the acylating handle and contributes to a highly specific modification. Besides dual-probe installation, overall control allows a protein-specific modification in a complex biological milieu.
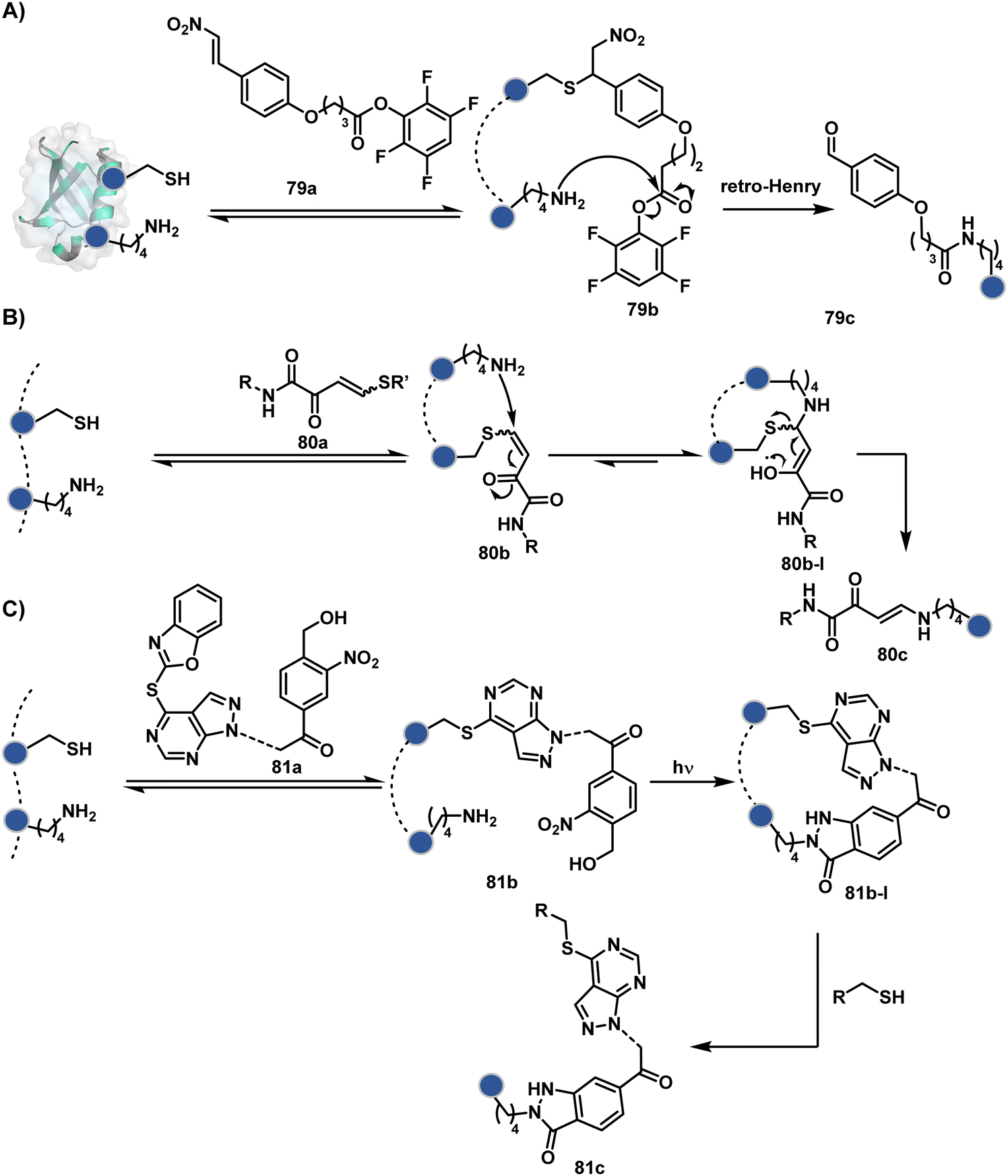 | ||
| Scheme 26 General mechanism for cysteine-based linchpin-directed modification of lysine. | ||
In another example, semioxamide vinylogous thioester-based reagents (80a, Scheme 26B) promote reversible Cys conjugation.220 This linchpin directs an irreversible site-selective modification of Lys (80c). The reversible thiol exchange with Cys promotes the subsequent nucleophilic attack by the proximal Lys. Furthermore, cleavable aryl thioethers coupled with UV-activatable o-nitrobenzyl alcohol (81a, Scheme 26C) could direct the selective labeling of Lys (81b-I).221 Here, the thiol exchange recovers the Cys residue (81c). A strategy has also been reported recently to utilize the LDM concept stepwise. A genetically engineered Histone H3 containing Cys reacts with a cyclooctyne-linked maleimide.222 Its subsequent reaction with an azide equipped with an acetyl donor could facilitate the site-selective acetylation of a proximal Lys residue.
The platform can be conveniently extended to targeting three-residue signatures for single-site modification (Scheme 27).223 It requires two linchpin-directed reagents to generate the reactive functionality for an irreversible modification of a third residue in their proximity. We demonstrated that a linchpin-directed proelectrophile (82b, Scheme 27) coupled with a linchpin-directed nucleophilic catalyst (82a) could generate an acylating reagent. This Linchpin-Directed Catalysis, LDC, offered access to selective modification of Lys distinct from LDMK–K (82d). Another similar method involved reversible boronic acid complexation with glycans to direct a nucleophilic catalyst.224 The linchpin-guided platform inspired the development of 10H-phenoxazine-3,7-dicarboxaldehyde for photoredox catalysed Tyr modification.225 It would be fascinating to find out if this chemo- and site-selective approach can add modularity by incorporating spacers or linkers in these reagents.
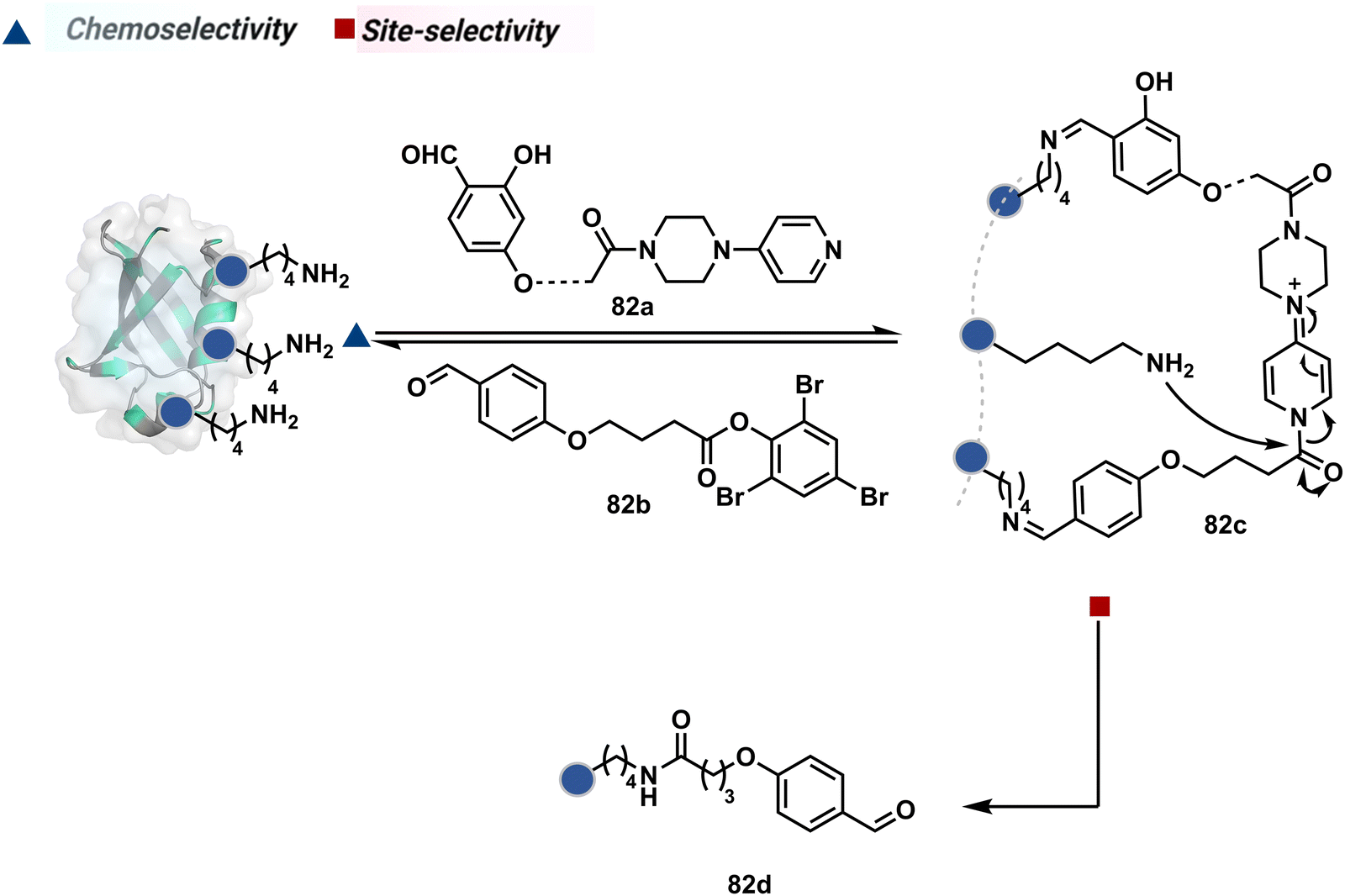 | ||
| Scheme 27 General mechanism for dual linchpin-directed single-site lysine modification by LDC chemistry. | ||
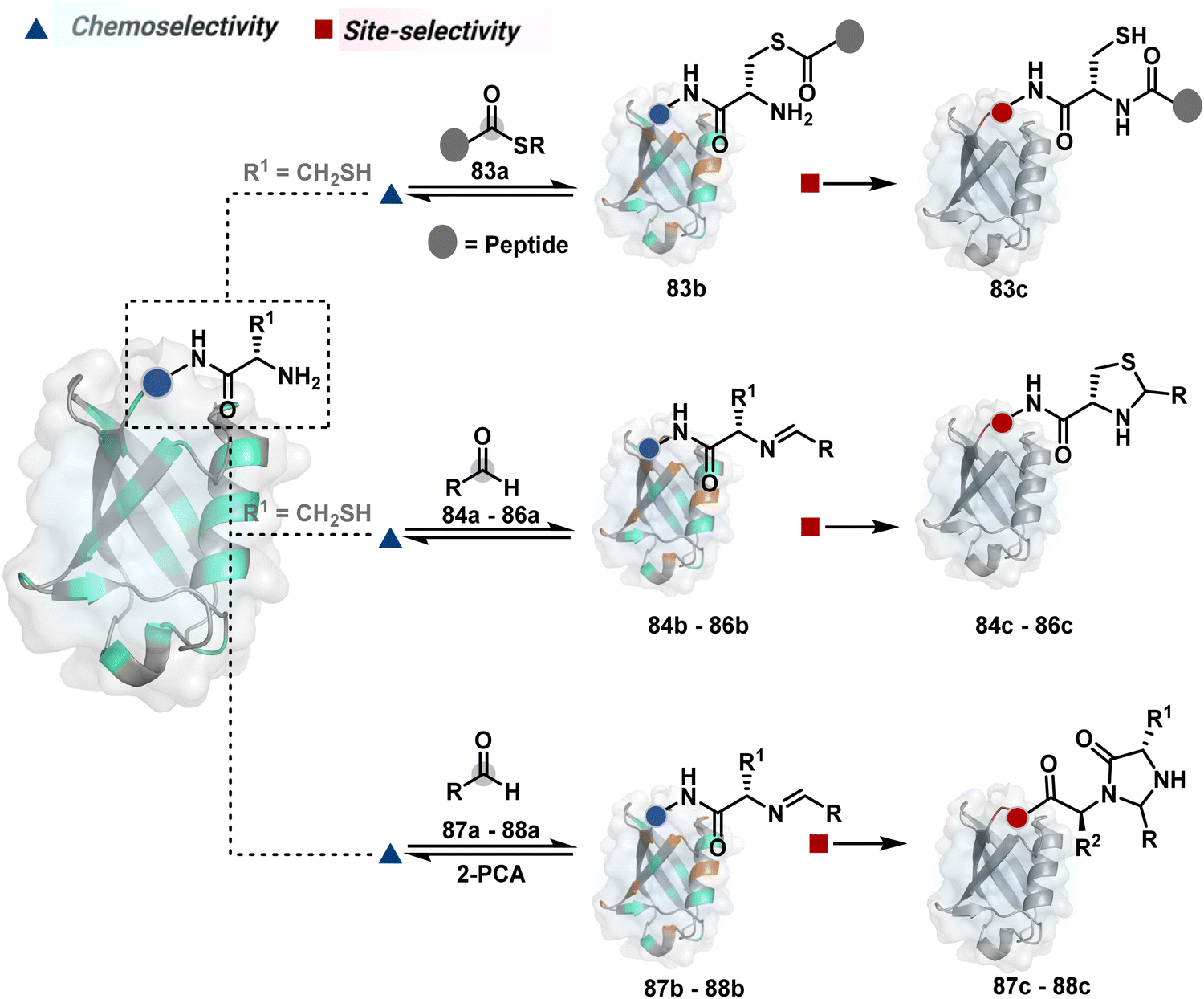
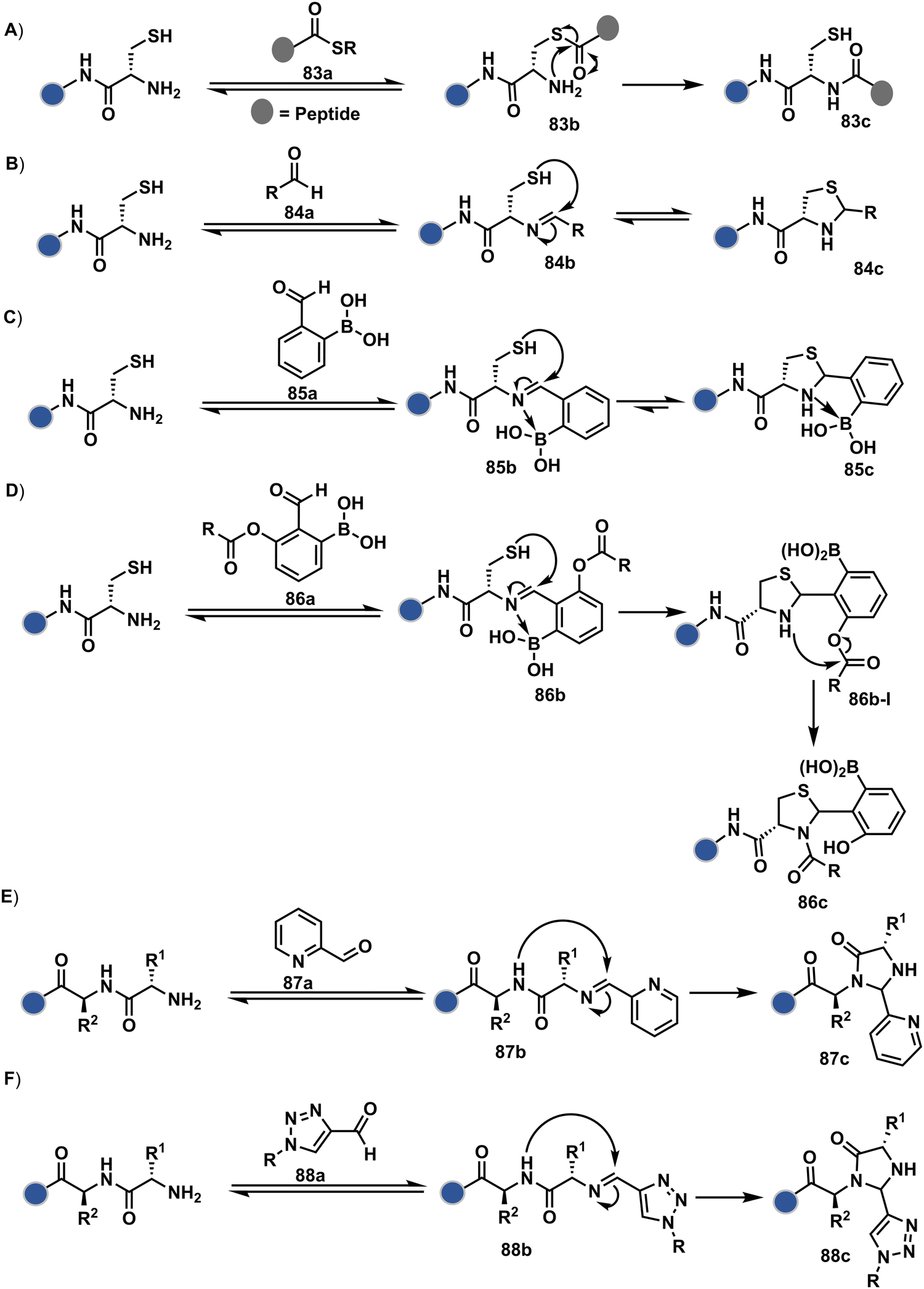
Targeting a pair of functional groups from two residues can be extrapolated for N-terminus-specific or N-residue-specific modification (Fig. 19). Nα–NH2 can be paired with a proximal functionality in such cases. As discussed in routes A and B, unique pKa and high solvent accessibility of Nα–NH2 in multiple native proteins make them an excellent target for preferential kinetic labeling. However, it still requires a method to overcome the challenge of site-selectivity. The N-terminus residue can offer a unique combination of functional groups not offered by any internal position in a protein. Hence, it presents the opportunity for a reversible intermediate generation with one of them, followed by its irreversible capture. For instance, Native Chemical Ligation (NCL) utilizes such a combination for N-Cys modification (83a–83c, Scheme 28A).226,227 At first, the side chain thiol of N-Cys generates a thioester-based intermediate (83b). Next, an intramolecular attack from Nα–NH2 facilitates an irreversible acyl transfer (83c). The thiol group of N-Cys is restored in the bioconjugate.
 | ||
| Fig. 19 General overview of N-terminal or N-terminal residue-specific modification. | ||
 | ||
| Scheme 28 N-terminus residue-specific modification initiates with a reversible generation of imine followed by mechanistically diverse routes. | ||
In another case, a reversible generation of Nα–NH2-imine (84b, Scheme 28B) can engage the side-chain thiol of N-Cys to form thiazolidine (84c).228 However, it is sensitive to pH and often accompanies slow reaction kinetics. Besides, it is not optimal for applications requiring stable bioconjugates. These attributes can be addressed using a proximal boronic acid in the aldehyde (2-formyl phenylboronic acid, 2-FPBA, 85a, Scheme 28C).229,230 The boron activates imine (85b) through a B–N dative bond to promote a rapid formation of a S–C bond (85c, Scheme 28C). Secondly, it stabilizes the thiazolidine via a B–N dative bond. The lone pair on thiazolidine nitrogen can also be engaged through intramolecular acylation (86b-I) to construct stable N-acylthiazolidine bioconjugates (86c, Scheme 28D).231
The N-Cys is rare in native proteins and has to be artificially incorporated. The other N-terminus side chain residues are yet to be explored in this perspective. However, the proximal amide group has effectively been employed to capture imine with specific aldehydes in the absence of N-Cys. For example, Nα–NH2-imine (87b, Scheme 28E) from 2-pyridinecarboxaldehyde (2-PCA, 87a) offers significant reactivity with the proximal backbone amide nitrogen with all amino acids except proline to render imidazolidinone (87c, Scheme 28E).232 The absence of a neighboring amide group for Lys-based Nε–NH2-imine ensures no irreversible adduct is formed at these sites. Furthermore, pyridine likely activates the adjacent amide to promote a cyclization step. Proteins such as aldolase, lysozyme, and thioredoxin were modified using this strategy. 1-H-1,2,3-triazole-4-carbaldehyde (TA4C) has also been reported for N-terminal modification following a similar mechanistic route (88c, Scheme 28F).233
2.4. Route D
The successful selectivity deconvolution in route C encourages one to examine the potential of additional steps in a multi-equilibrium process. It could enable access to unique selectivity attributes. To validate the concept, we examined the Lys-based linchpin formation to direct the proximal generation of a reactive intermediate (Fig. 20).234 The second reversible transformation could site-selectively generate a reactive imine (89c). The subsequent irreversible step could offer flexibility with nucleophiles and render diverse bond constructs in the bioconjugate. The chemical orthogonality of two imines ensures that the linchpin site does not participate in the irreversible pathway.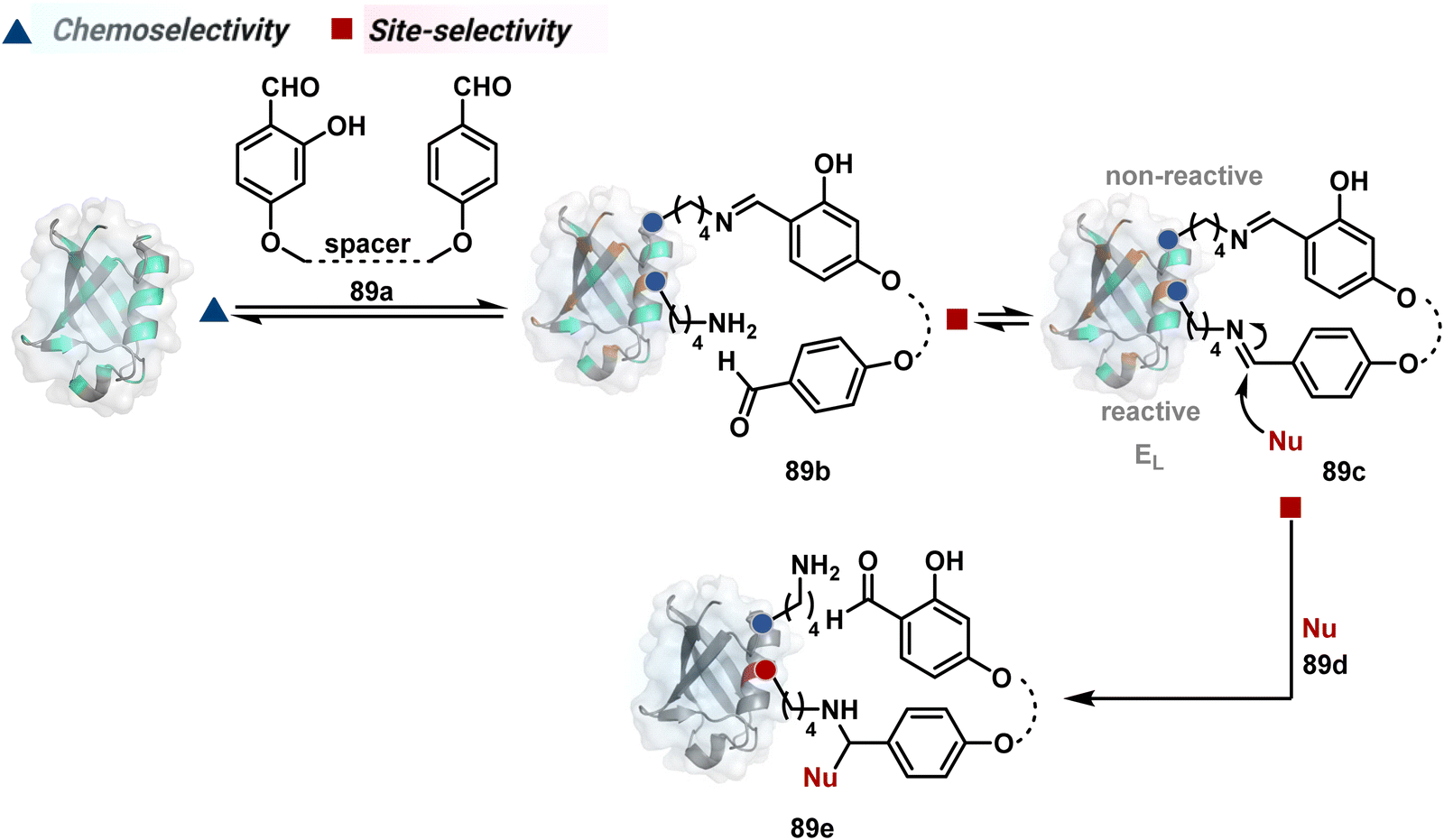 | ||
| Fig. 20 Multistep pathway initiates with chemoselective lysine-based linchpin to direct modification of another lysine. A combination of reversible and irreversible transformations further disintegrates the site-selectivity in two steps. | ||
Additionally, the reversible generation of imines at the N-terminus can promote another reversible reaction involving a proximal Cα–H. In one such case, aldehyde in pyridoxal-5′-phosphate (PLP, 90a, Fig. 21A) forms imine with all accessible primary amines.235 Next, it promotes the N-terminus selective transimination through tautomerization (90c). The second step proceeds site-selectively with Nα–NH2-imine owing to substantially lower pKa of the neighboring α-proton in comparison to Lys-derived Nε–NH2-imine. Finally, the irreversible hydrolysis of glyoxyl imine renders an aldehyde or ketone (90d). The overall transformation offers exclusive N-terminus selectivity without perturbing any internal residue.
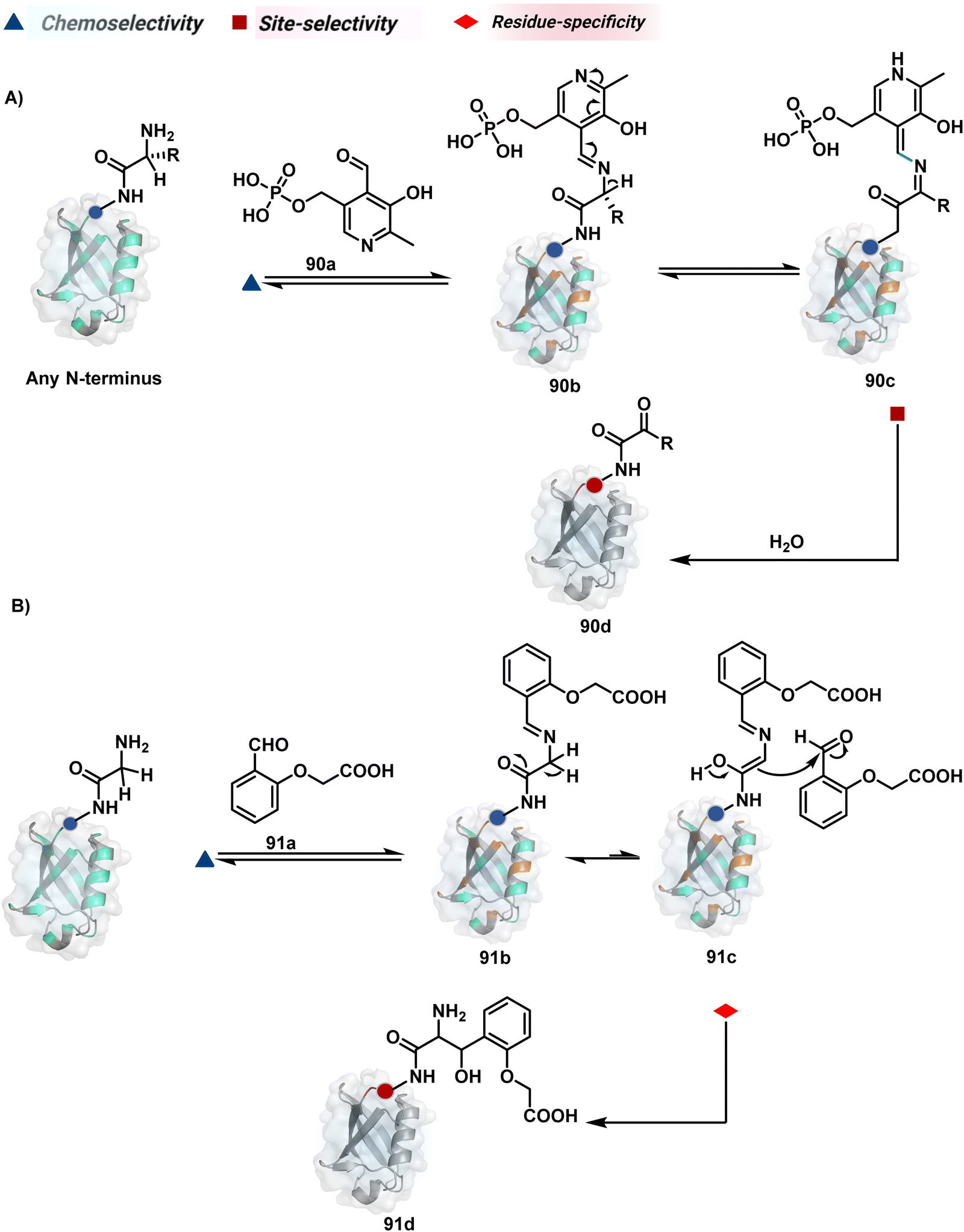 | ||
| Fig. 21 Multistep pathways enable single-site modification of (A) N-terminal residues and (B) N-glycine in proteins. | ||
On the other hand, Nα–NH2-imine from certain aldehydes could promote Cα–H proton shuttling to generate a nucleophilic intermediate (91c, Fig. 21B).17 Notably, the second reversible step proceeds only in the case of N-terminal Gly. The lack of reactivity with its substituted counterparts, i.e., all other proteinogenic amino acids, empowers access to N-Gly residue specificity. Finally, an irreversible reaction with an aldehyde renders an aminoalcohol-based bioconjugate through C–C bond formation (91d). The irreversible pathway remains elusive for all internal residues and Lys-derived imines. The Gly-tag® platform offers flexibility with late-stage installation of probes. The aminoalcohol motif in the bioconjugate can be oxidized under mild conditions to result in glyoxamides.236 Besides, the aminoalcohol C–C bond can be dissociated in a chemically orthogonal transformation in the presence of PLP.237 This residue specificity remains unperturbed even in the presence of diverse functional groups. For example, the azido pyridoxal derivatives could offer N-Gly-specific azidolation of proteins.238
These examples emphasize how multi-equilibrium processes can harness a specific combination of Nα–NH2 and proximal Cα–H as a unique signature to drive the irreversible transformation. Engaging other C–H groups in reversible pathways could create opportunities for new targetome-based residue-specificity in the coming years.
3. Meeting technological demands at the biology–medicine interface
3.1. Protein bioconjugates
The therapeutic protein segment is rapidly expanding and gaining immense attention. However, unfavorable pharmacokinetics and immunogenicity frequently impair the in vivo effectiveness of recombinant protein therapies. Following drug delivery, therapeutic proteins may be promptly broken down by proteases or eliminated by the kidneys, causing a sharp decline in the plasma concentration. A severe immunological response can also result in life-threatening complications like an allergic shock. Anti-drug antibodies, on the other hand, can render the therapeutic protein inactive and result in treatment resistance.The conjugation of protein therapies with long polyethylene glycol (PEG) chains was one of the first systematic uses of protein bioconjugation chemistry to increase their plasma circulation half-life and reduce their immunogenicity in vivo. The lengthy polymer chain improves the hydrodynamic radius, lowering renal filtration while protecting the protein from protease breakdown and identification by the immune system. Many FDA-approved PEGylated protein-based drugs are produced through non-site-selective bioconjugation methods, impacting their attributes. Thus, site-selective methods are in increasing demand for PEGylation of proteins. One example involved N-terminal Ser oxidation to an aldehyde using sodium periodate with subsequent oxime ligation using an aminooxy PEG derivative.239 Proteins like interleukin (IL)-8, granulocyte colony-stimulating factor (G-CSF), and IL-1Ra were PEGylated using this strategy. The proteins retained their activity after modification. PEGylation at the N-terminal of proteins through reductive alkylation of imine formed with the monoaldehyde functionalized PEG chain under slightly acidic conditions has also been used. Using this approach, PEGylation of filgrastim, a human granulocyte colony-stimulating factor (hG-CSF), is widely used to mitigate chemotherapy-related infections.240,241 The pharmacokinetic properties of various interferons (INFs) have been improved upon PEGylation using this method.242,243 PEGylated INFβ-1a is used for treating relapsing multiple sclerosis.244 PEGylated certolizumab, an antibody against tumor necrosis factor (TNF-α), is an example of site-specific modification of C-terminal Cys with PEG-maleimide. It is an anti-inflammatory drug used to treat rheumatoid arthritis, Crohn's disease, and psoriatic arthritis.238 Approaches utilizing PLP-mediated transamination introduce an aldehyde or a ketone at the N-terminus followed by PEGylation using the aminooxy PEG derivative in pH 6.5.235 A single-step site-directed PEGylation can be obtained using a 2-PCA derivative which forms an imidazolidinone ring at the N-terminus (except penultimate N-Pro).232 Selective PEGylation is vital to retain the biological activity of proteins to improve their clinical application.
Along the same lines, labeling biological macromolecules with dyes or radionucleides is immensely important for their diverse application in imaging in modern biology. Conjugating isolated proteins with fluorophores can be used to monitor the localization of labeled proteins or their interactome. Thus, site-selective or site-specific modification of proteins for in vivo applications is vital as it regulates the pharmacokinetics and biodistribution.245,246 Besides, precision engineering becomes immensely important for Förster resonance energy transfer (FRET) applications. The FRET donor and acceptor must not interfere with protein–protein interactions but should be appropriately placed for energy transfer. The commercial kits often rely on chemoselective reagents, including maleimide, sulfone, or haloacetamide derivatives. For applications requiring dual probes, selectivity of the second modification would require a chemically orthogonal handle from an alternative route.247 N-terminal selective protein modification methods can also facilitate such dual labels. For example, the PLP-mediated transamination and oxime formation approach enables the installation of a FRET pair donor at the N-terminus.248 At the same time, the acceptor is tagged using Cys-maleimide chemistry. A similar approach engaging N-residue and Cys was also used for labeling tobacco mosaic virus with chromophores.249
Bioorthogonal chemistry is utilized for selectively labeling proteins in complex mixtures. A copper-catalyzed azide–alkyne cycloaddition (CuAAC) strategy has been applied for tagging anthrax lethal factor with a fluorophore.250 This allowed monitoring its internalization and membrane translocation in a live cell. It provides an interesting approach to monitoring the localization and trafficking of native proteins in living cells.251 Bioorthogonal chemistry can also be extended for nuclear imaging applications. For example, an antibody tagged with a trans-cyclooctene (TCO) ring can selectively react with a radiolabeled variant of a tetrazine ring through an inverse electron demand Diels–Alder (IEDDA) reaction in vivo, enabling the monitoring of the antibody through a PET scanner.252 Another example of PET-based imaging was reported with a reaction of anti-class MHC-II single-domain antibody fragments bearing TCO with 18F-2-deoxyfluoroglucose (FDG).253 This method enabled in vivo imaging of pancreatic tumors with enhanced specificity. The rapid kinetics of the IEDDA reaction makes it suitable for various applications in vivo. However, the trans double bond of the TCO may undergo isomerization, enhancing the errors. Apart from imaging, optical sensors can also be utilized to measure various physiological parameters in living cells. For instance, an acid-sensitive chaperone HdeA inside the periplasm of E. coli was modified with an environment-sensitive fluorophore to form a biosensor for pH detection.254 HdeA forms a plastic-type structure upon lowering pH from 7 to 2, which allows accessibility of the hydrophobic residues to the fluorophore. In turn, it results in an enhanced fluorescence signal. While these examples demonstrate the importance of protein bioconjugation, selective technologies for protein modification in living cells and organisms are needed. In this perspective, ligand-directed protein modification has offered initial solutions for protein-based biosensors,255,256 protein–protein interactions,148 protein activity regulation, and specific targetome enrichment.257
3.2. Antibody bioconjugates
The chemical methods for antibody bioconjugation have contributed immensely to biologics in the last two decades. However, the structural complexity of native antibodies is a level higher than most proteins (Fig. 22). It also amplifies the magnitude of selectivity challenges in their bioconjugation. For these reasons, a heterogeneous antibody conjugate mixture has been considered acceptable in all walks of biology and medicine. However, recent developments in AFCs and ADCs for directed tumor targeting highlight the drawbacks and necessity to change the status quo.18,258–260 For most methods, reaction parameters such as relative stoichiometry and time can regulate the fluorophore or drug-to-antibody ratio (FAR or DAR). However, current benchmarks direct the global efforts to establish new platforms towards homogeneous antibody conjugates. While the chemical bioconjugation with proteins discussed earlier translates to antibodies, there are unmet needs regarding control over selectivity and homogeneity. Besides, structural complexity makes it challenging to establish site-of-conjugation and site-homogeneity. This limitation has led to multiple assumption-based claims in the literature.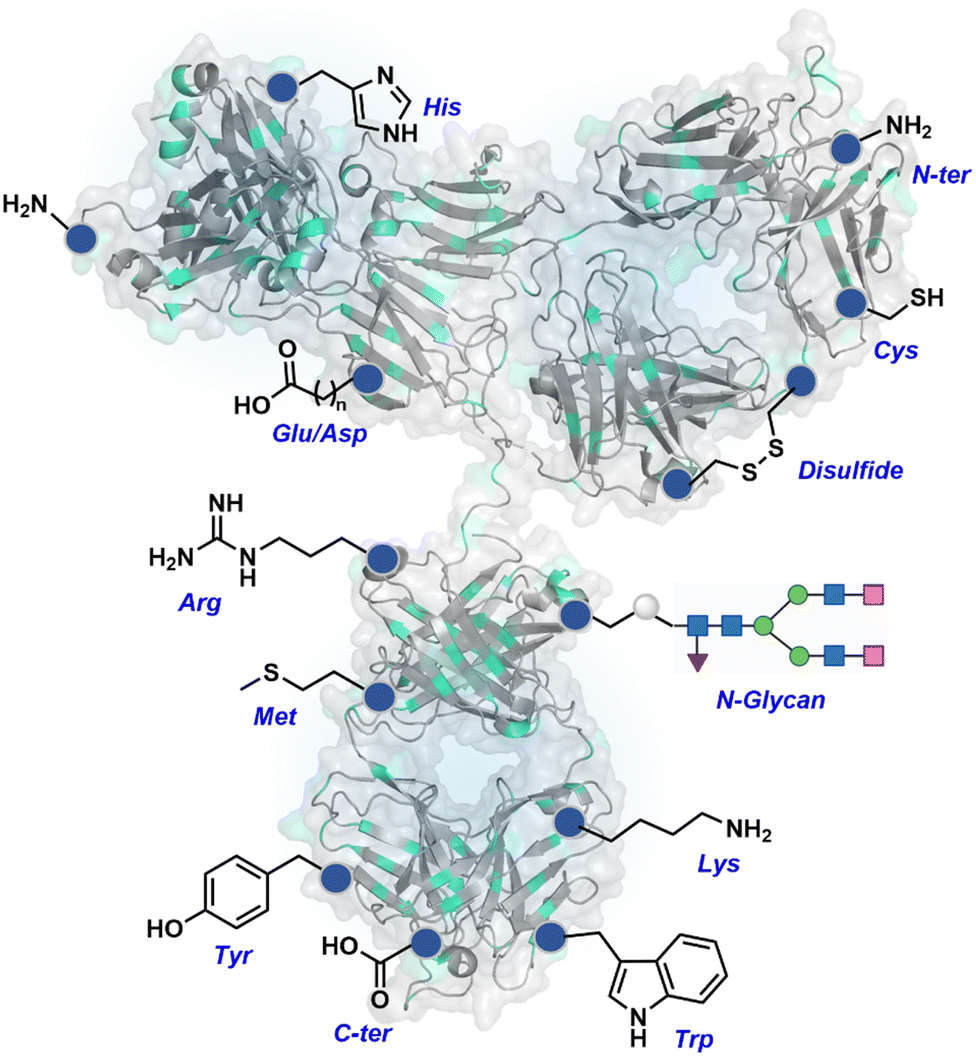 | ||
| Fig. 22 Functional group targetome to construct antibody conjugates include cysteine, lysine, arginine, tryptophan, tyrosine, methionine, glutamic acid, aspartic acid, C-terminus, and asparagine N-glycan. | ||
3.2.1.1. Arginine. The pKa of Arg affects its capability to be used for the synthesis of antibody conjugates under physiological conditions. While the absence of Cys helps, Lys remains a significant competitor. The glyoxal-based reagents have been used to target the guanidine side chain of Arg. The synthetic ease to derivatize them allows the introduction of chemically orthogonal handles. In one such case, an azide introduced in an antibody conjugate facilitates late-stage installation of probes through a strain-promoted alkyne azide-cycloaddition reaction (92c, Fig. 23).261 The genetic insertion of Arg in the dual variable domain of the h38C2 bi-specific antibody allowed its modification with phenylglyoxal conjugated payloads (93b, Fig. 23).262 The covalent bond formation occurred at both Lys and Arg in this case. Arg-specific reagents capable of exclusive chemoselectivity could add substantial value.
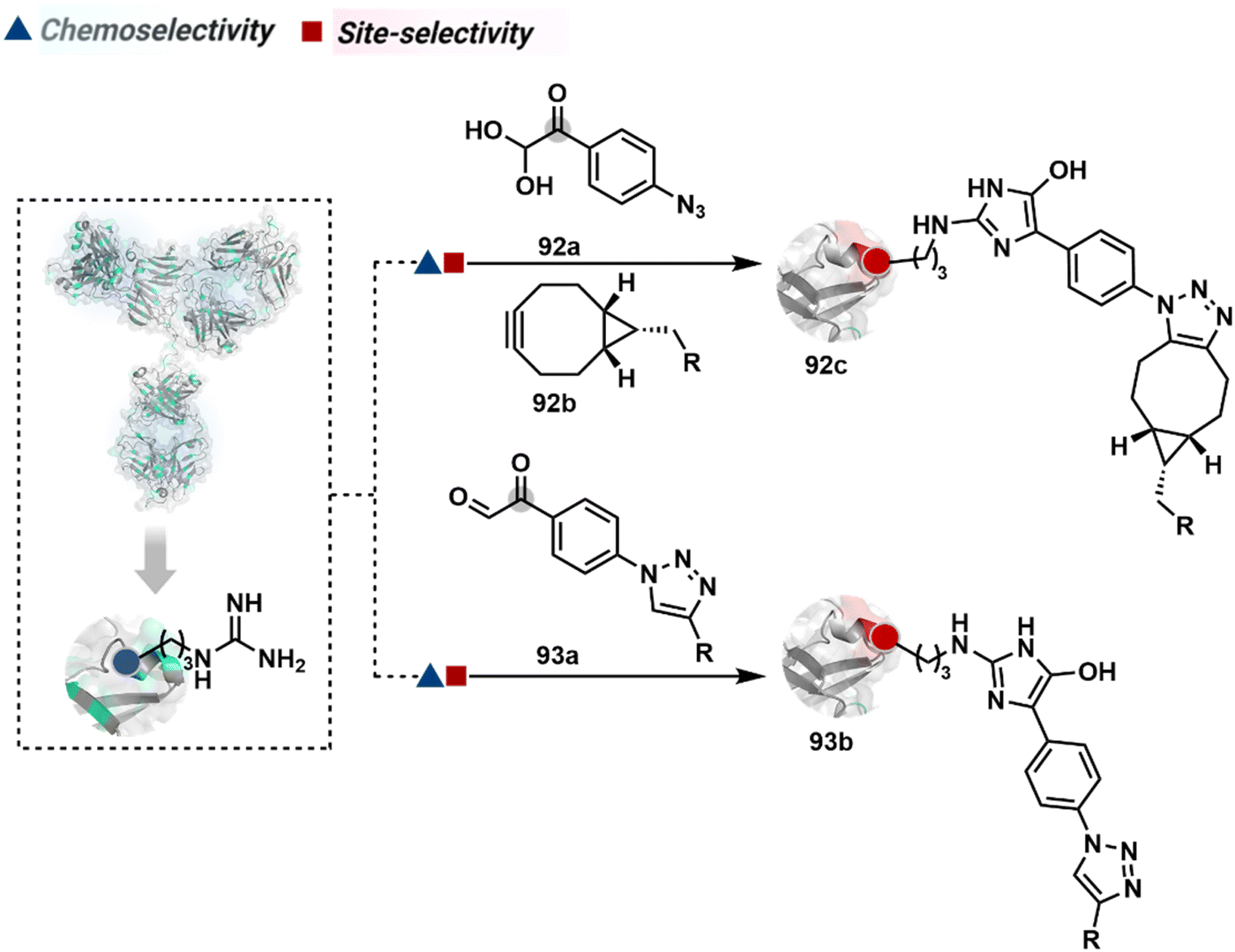 | ||
| Fig. 23 Targeting arginine for synthesis of antibody conjugates. | ||
3.2.1.2. Asparagine (N-glycan). The post-translational glycosylation of antibodies equips them with a highly conserved functionality capable of rendering chemically orthogonal transformations.263,264 Its location, e.g., Asn-297 in the IgG CH2 domain, and stability have attracted attention to its engineering for producing antibody conjugates.265 An initial oxidation can harness its potential for bioconjugation by generating aldehydes. Commonly, sodium periodate or galactose oxidase can facilitate this step.266–268 This single-step irreversible process often produces multiple aldehydes within glycan. In turn, it contributes to heterogeneity after the subsequent installation of probes through hydrazone or oxime formation (94a, Fig. 24).269,270 Besides, extensive oxidation impacts the antigen binding, triggers aggregation, and contributes to batch-to-batch inconsistencies. To address some of these concerns, glycan can be engineered using 1,4-galactosyltransferase and 2,6-sialyltransferase enzymes (95a).271,272 It modifies N-glycan and offers a monosialylated antibody that responds to moderate oxidative conditions. In turn, it improves the homogeneity of antibody conjugates.
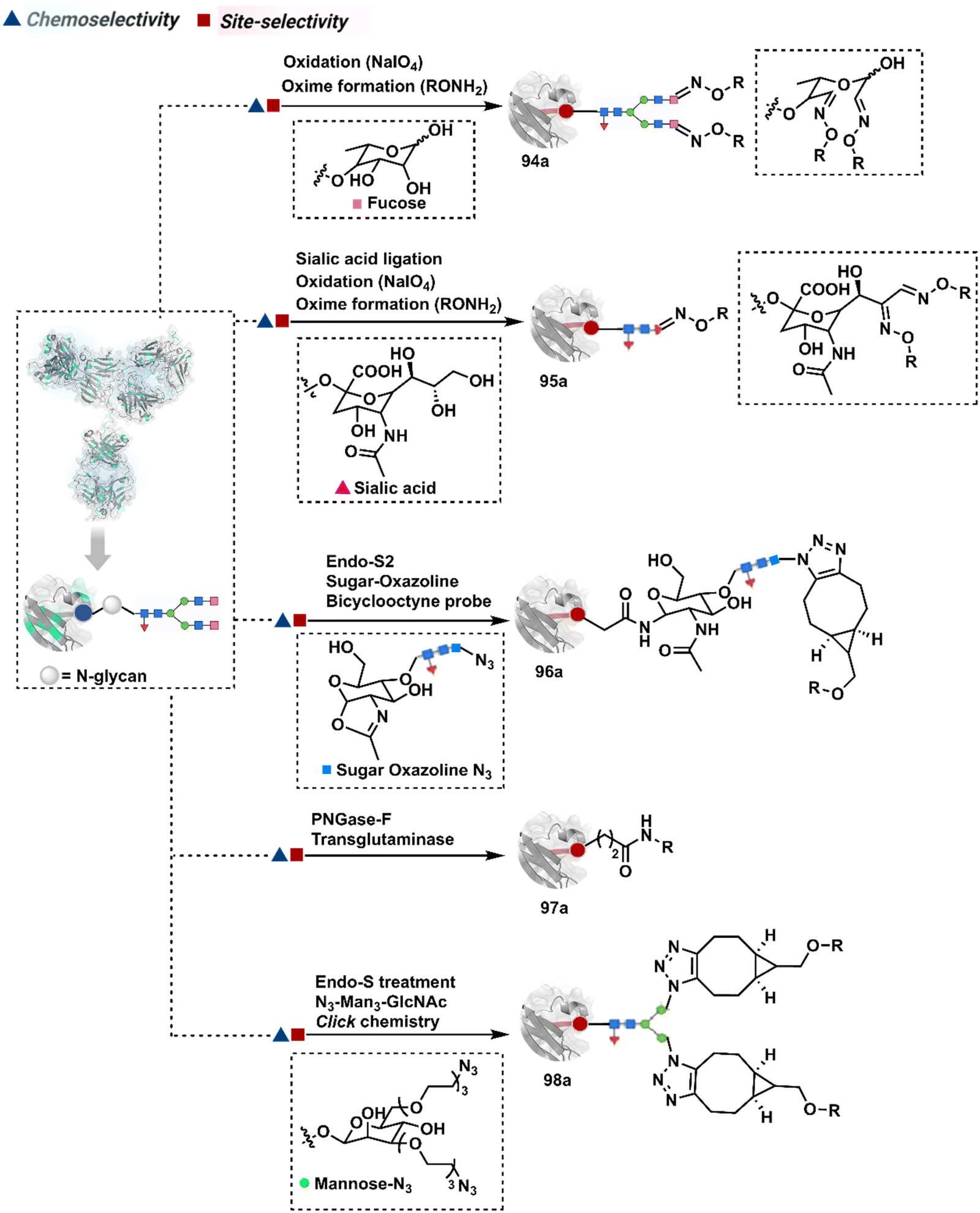 | ||
| Fig. 24 Targeting asparagine N-glycan for synthesis of antibody conjugates. | ||
In another approach, the G0F glycoform of an antibody could produce two aldehydes on N-acetylgalactose (GlcNac) under oxidative conditions.272,273 Alternatively, glycan-specific enzymatic processes can be employed to trim the glycan. For example, endoglycosidase cleaves N-glycan and facilitates transglycosylation to deliver a bioconjugate (96a).265,274–276 Also, enzymatic removal of N-glycan provides free glutamine capable of coupling with an amine-linked probe in the presence of transglutaminase to harness antibody conjugates (97a).277–280 The S. pyogenes endo-S enzyme removes the glycan, generates an oxazolinium intermediate, and enables the polysaccharide hydrolysis. Next, transglycosylation with azide sugar equips it for click chemistry (98a).281–284 The same approach extends to transglycosylation with azide-functionalized sialic acid (99a, Fig. 25).285 The removal of terminal galactose with β-1,4-galactosidase renders homogeneous G0 antibody glycoform. Its enzymatic (1,4-galactosidase-T1-Y289L) glycosylation with N-azidoacetylgalactosamine and C2-keto-galactosamine could offer azide and keto handles for late-stage installation of probes (100a).286–288 The endo-S glycan cutting, GalNAz addition, and subsequent cycloaddition could produce homogeneous antibody conjugates (101a).289 These reagents can also be designed to engage the glycan motif from both the heavy chains in the antibody (102a).290 The N-glycan remodelling can be coupled with other antibody engineering approaches. For example, its implementation to conjugate LacNAc-based drug-linker oxazoline can drive Fc-selective peptide-guided acylation of Lys (103a).291 The deglycosylation-promoted access to antibody conjugates has been employed in radioimmunoassay, PET imaging, and a radioisotope-based study.292–294 The adverse impact of glycan engineering is yet to be fully understood. However, initial investigations highlight the need for methods for their controlled modification.295,296 The oxidative conditions frequently used in these processes often affect the Met residues severely. Furthermore, deglycosylation impacts the stability and half-life of the antibody. It impacts the concentration at which the antibody tends to exhibit self-aggregation.297,298 Besides, glycan composition regulates immunogenicity and function.299
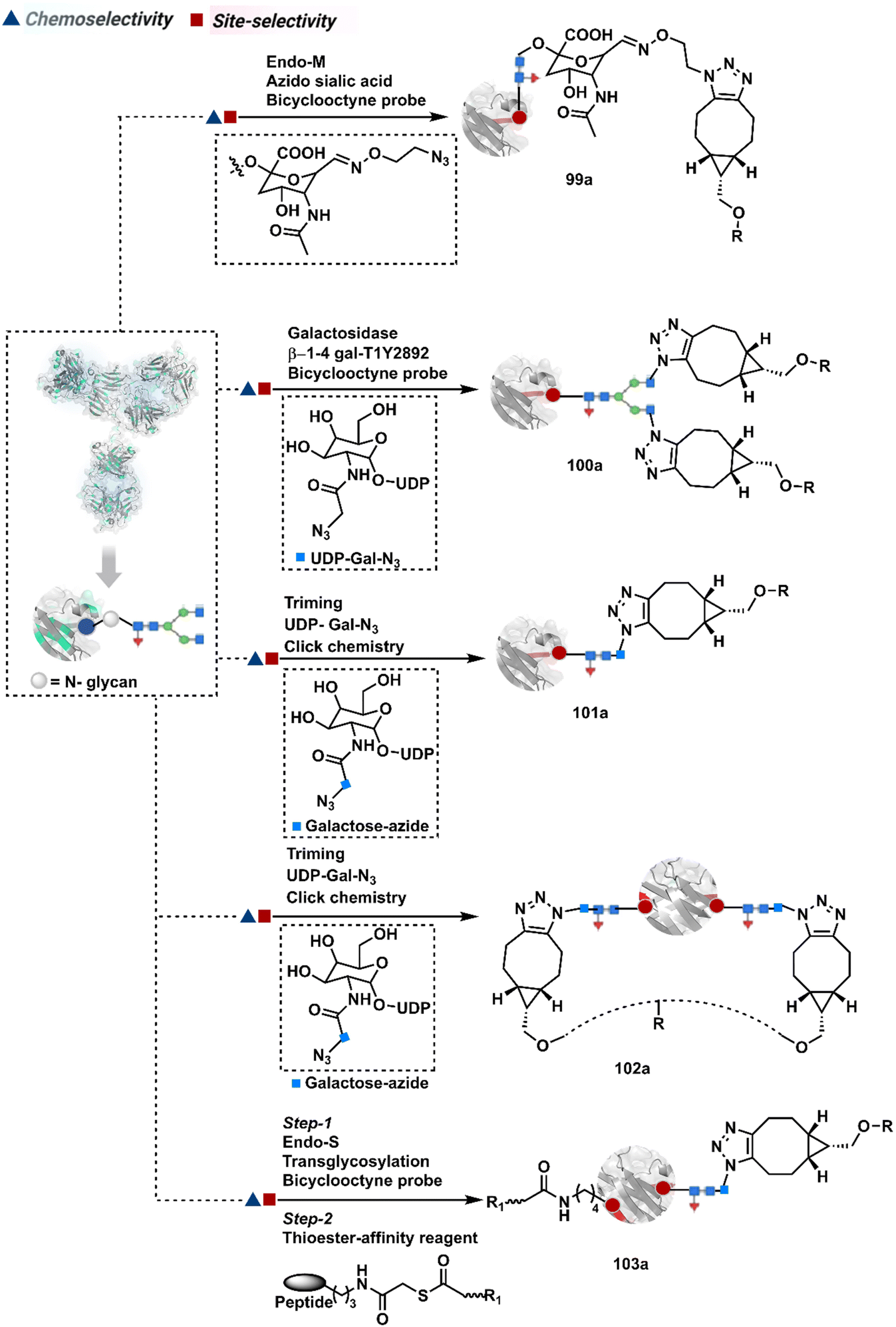 | ||
| Fig. 25 Additional methods targeting asparagine N-glycan for synthesis of antibody conjugates. | ||
3.2.1.3. Cysteine. The low-frequency Cys residue becomes one of the initial choices to bypass site-selectivity challenges. However, native antibodies are devoid of free Cys in general. Its incorporation through site-directed mutagenesis provides one gateway.246,300 Such a route was adapted to render homogeneous THIOMAB-based ADCs with DAR ∼4 while employing maleimide as an electrophilic handle. The thiol-selectivity of maleimide derivatives has been used extensively (104a, Fig. 26).266,301–306 For example, AFCs were constructed using its combination with diverse fluorophores. It was extended to synthesizing multiprobe AFCs to enable FRET-based cellular investigations (105b).307 The polypeptide-maleimide-based reagent also offers a platform for constructing multiprobe antibody conjugates.308
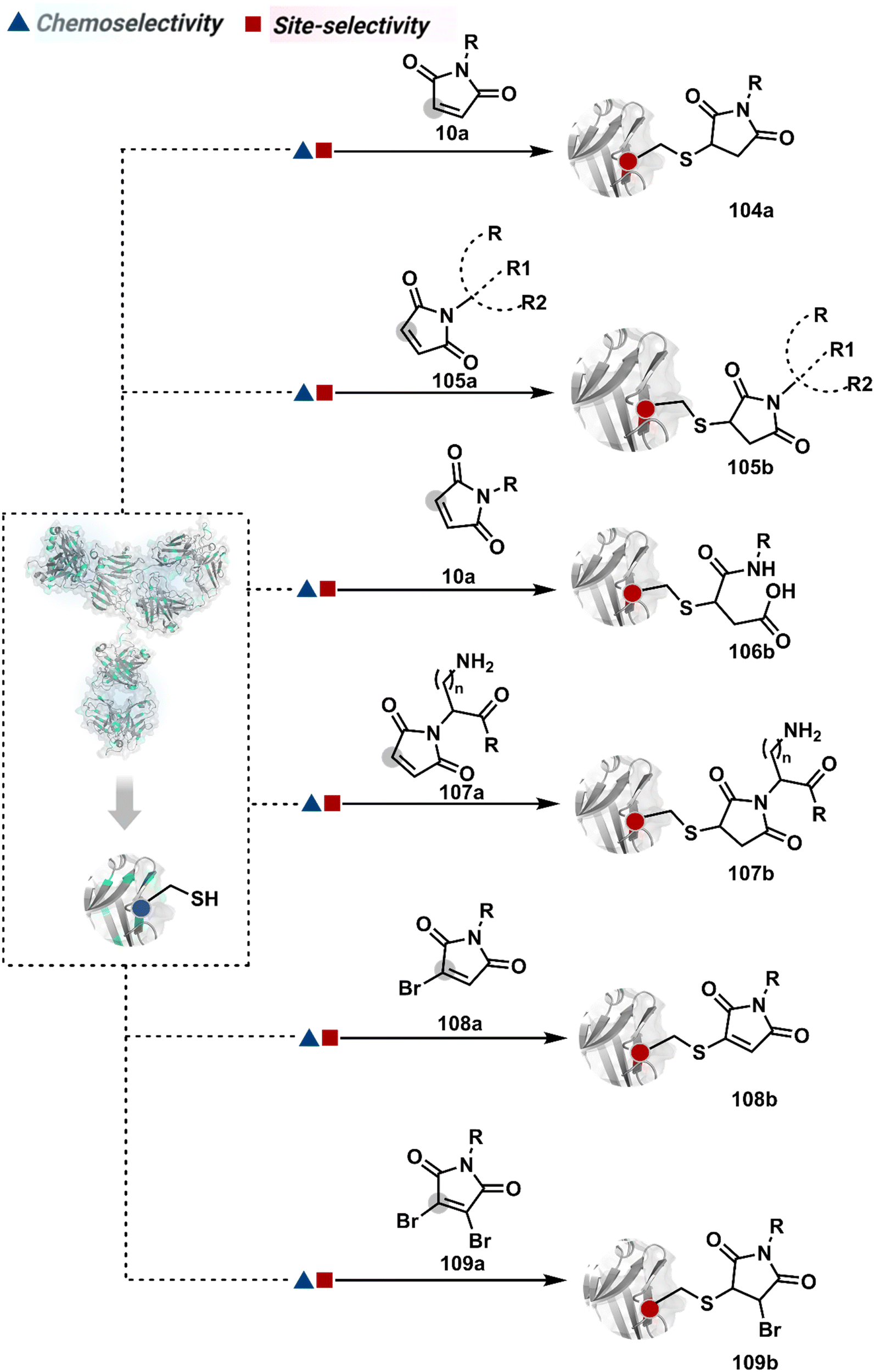 | ||
| Fig. 26 Targeting cysteine for synthesis of antibody conjugates. | ||
The C–S bond of an antibody conjugate is amenable to dissociation, resulting in the release of the probe before reaching the intended target.305 An attempt to resist this retro-Michael addition was made through the hydrolysis of the thiosuccinimide ring (106b).246,309 This late-stage transformation enhances the bioconjugate stability to avoid a non-selective probe release. Furthermore, placing a basic motif in proximity could catalyze the desired hydrolysis and provide better stability, fewer off-target effects, and enhanced in vivo efficacy (107b).46 The C–S bond stability in thio-Michael adducts could also be achieved through an exocyclic olefinic maleimide. Besides mono- and dibromomaleimides, thiobromomaleimide was also able to enhance the stability of thio-Michael adducts (108b–109b).310–312 Among non-maleimide electrophilic systems, phenyloxadiazole sulfone (110a, Fig. 27),313 iodoacetamide derivatives (111a),314N-alkyl vinylpyridine (16a),57 thiol functionalized pyrrolobenzodiazepine (113a),315 and styryl sulfonyl fluoride (114a)316 were able to offer reasonable options.
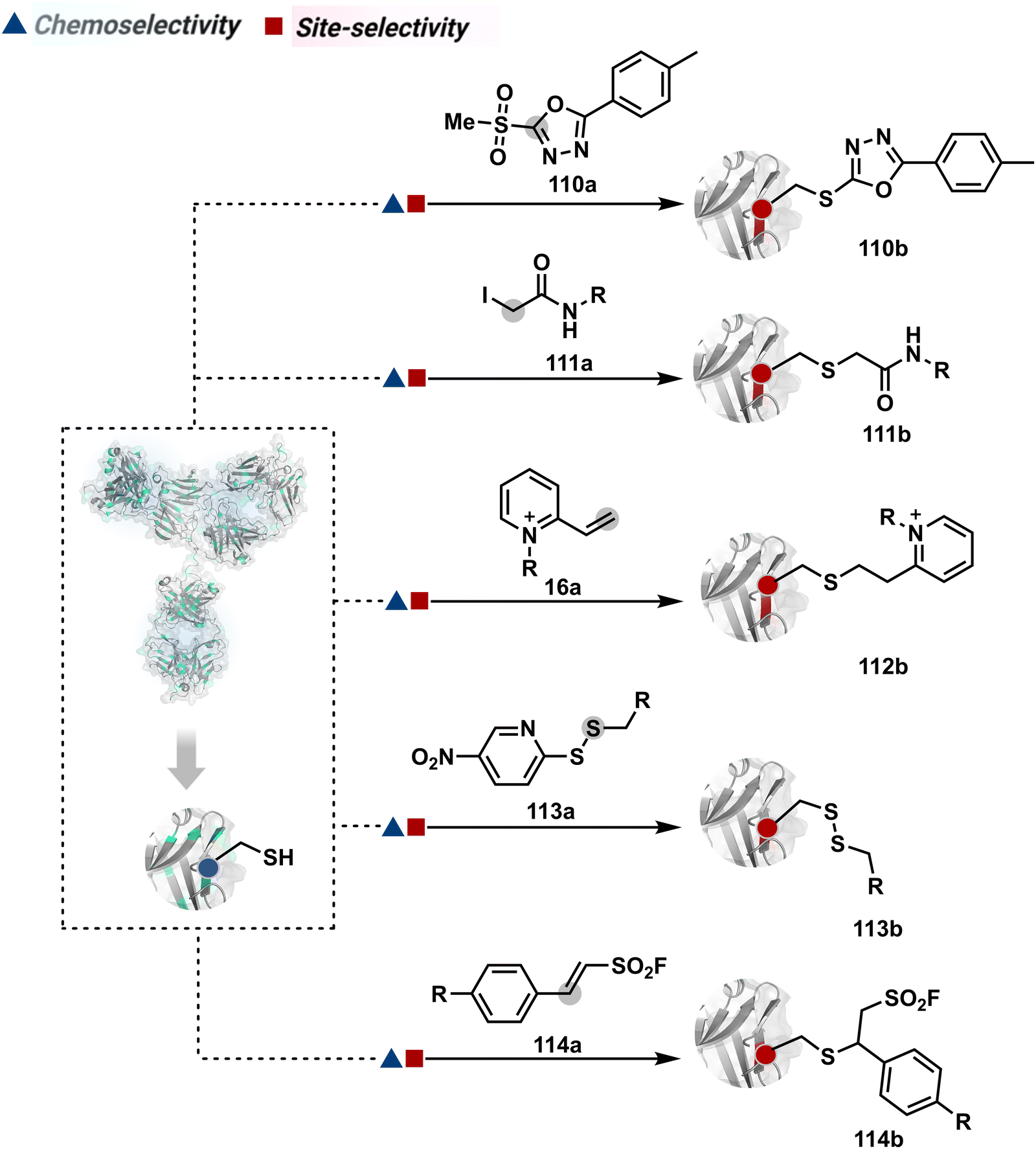 | ||
| Fig. 27 Additional methods targeting cysteine for synthesis of antibody conjugates. | ||
3.2.1.4. Lysine. The primary amines are antibodies' most abundant surface-accessible nucleophilic functional groups. The absence of free Cys provides an uncontested opportunity for their chemoselective targeting through multiple electrophiles, including isothiocyanate (115a, Fig. 28), activated esters (28a), anhydrides (117a), and activated lactams (118a).266,317–319 The NHS ester is the most widely used reagent for lysine conjugation in antibodies.266,320–322 The ease of synthetic manoeuvrability allows the installation of chemically orthogonal handles like alkyne, azide, aldehydes, and hydrazones to place a desired tag in the subsequent step. It has been the core platform for synthesizing clinically approved gemtuzumab ozogamicin (Mylotarg®, DAR ∼3–6)323,324 and mirvetuximab soravtansine (Elahere®).325 A few nucleophilic residues can compete under specific conditions to compromise chemoselectivity.326 However, the high reactivity of NHS ester is the key contributor to the extensive heterogeneity.266 The controlled addition of sub-stoichiometric electrophiles can reduce heterogeneity with conjugation ranging between 0–16. The control over reaction time provides another handle to minimize the extent of heterogeneity.
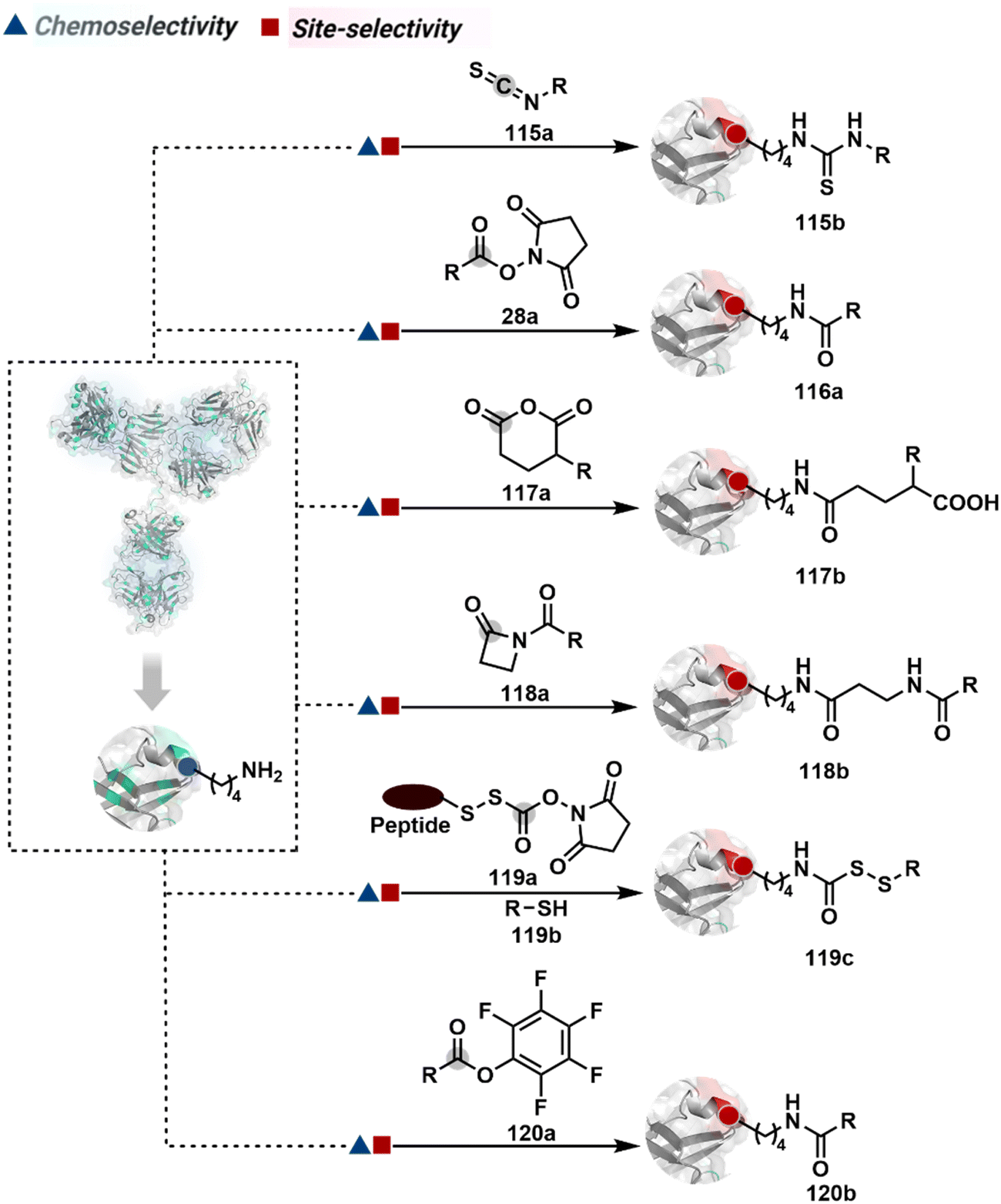 | ||
| Fig. 28 Targeting lysine for synthesis of antibody conjugates. | ||
This chemistry has also been utilized for synthesizing AFCs for various applications. For example, NHS-equipped NIR cyanine derivatives, FNIR-774 and FNIR-Z-759, were conjugated with panitumumab with a FAR ∼2 and ∼5 for in vivo imaging of tumors.327 Furthermore, the chemistry was used to conjugate the ovarian cancer-specific antibody (COC183B2) with IRDye-800CW (COC183B2-800CW) for antibody-guided tumor surgery.328 The bevacizumab and trastuzumab were also conjugated with Alexa Fluor 680 (AF-680) or IRDye-800CW with FAR 0.3 and 1.2, respectively. While reducing the extent of labeling could suppress heterogeneity, it compromises the signal intensity. On the other hand, increased loading contributes to heterogeneity and affects the clearance and other critical quality attributes.329 Installing NHS ester with a directing ligand provides a potential way to improve the homogeneity of an antibody conjugate.330 For example, a peptide-equipped NHS ester was used to synthesize IgG-MMAE conjugates (119c).331–334 Alternatively, the electrophilicity of bioconjugation reagents can be reduced to take advantage of kinetic preferences. In this perspective, pentafluorophenyl ester (120a) was shown to render improved selectivity.335,336 While the overall conversions are relatively lower, such a homogeneous antibody conjugate exhibits better solubility and reduced aggregation. It leads to improved distribution with better pharmacokinetics and tissue availability.
A spectrum of reactivity can also be harnessed through the classical coupling reagents from peptide synthesis. For example, the TSTU or EDC-activated carboxylate derivative has been employed for Lys modification to construct an antibody conjugate for targeted imaging and delivery of toxins (121c, Fig. 29).337,338 On the other hand, a structurally diverse set of Lys-selective electrophiles, isocyanate and isothiocyanate, also offers a broad range of reaction parameters to control the extent of labeling.339 The isothiocyanate labels multiple Lys, rendering a heterogeneous mixture of AFC.340 It would be interesting to see if structural and electronic alterations can retune these electrophiles for better control over selectivity. The electrophilicity spectrum of bioconjugation reagents can also be expanded through acyl fluoride,341 4-azidobenzoyl fluoride (122a), sulphonyl acrylate (27a),80 2-iminothiolane (124a),342 and azaphilones (125a),343 to improve the homogeneity of antibody bioconjugates. The affinity tags or directing ligands could reduce the accessibility of an electrophile to a limited number of Lys residues and offer improved homogeneity of antibody conjugates (126b).344–346
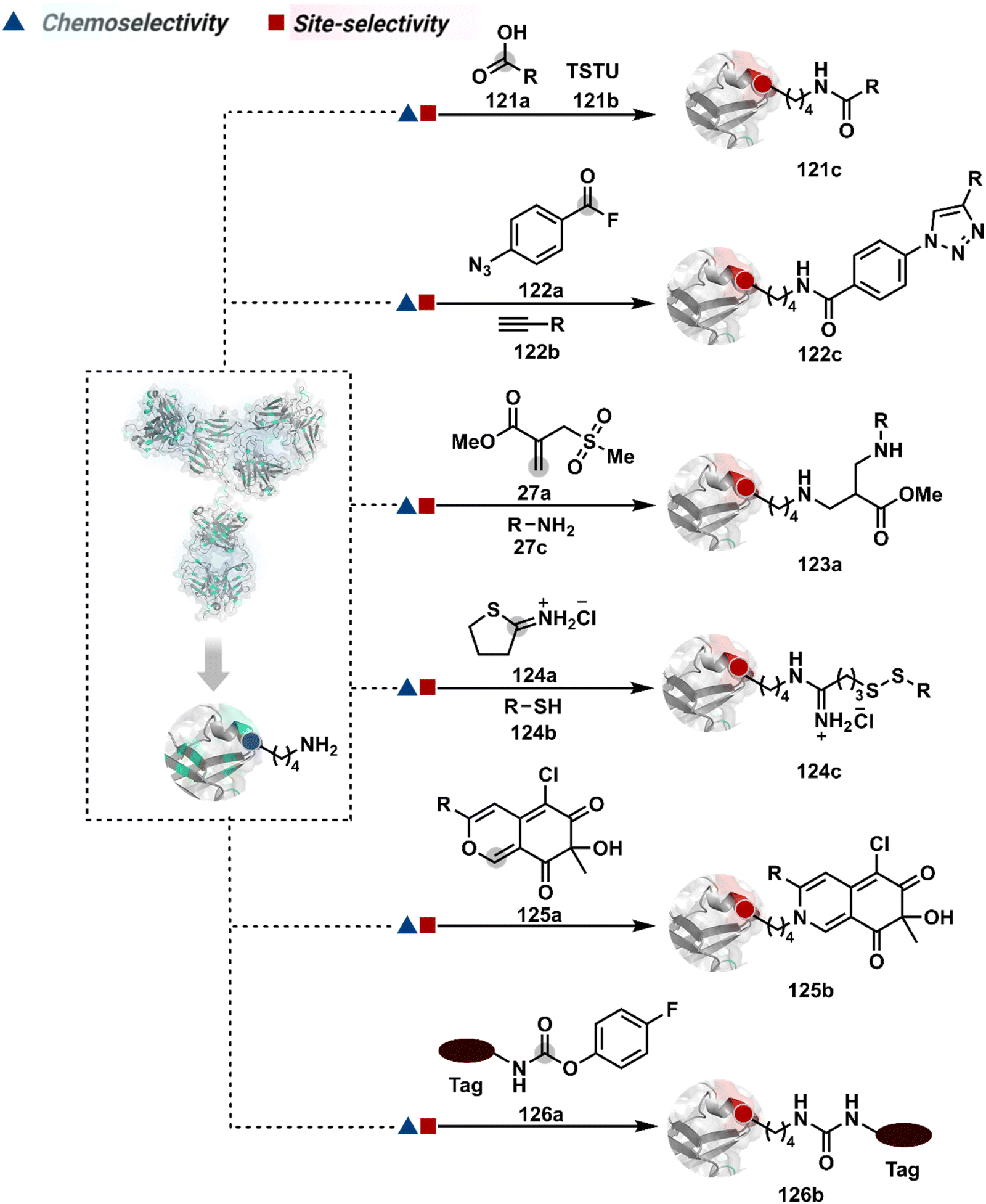 | ||
| Fig. 29 Additional methods targeting lysine for synthesis of antibody conjugates. | ||
3.2.1.5. Methionine. The sensitive nature of Met ensures that it often contributes to side products through undesired pathways. Hence, controlling its selective modification has always been a daunting task. One of the initial successes in this perspective was offered by oxaziridine derivatives to generate a Fab-drug conjugate (127b, Fig. 30).88,347 It would be interesting to see how this method translates toward synthesizing full-length antibody conjugates.
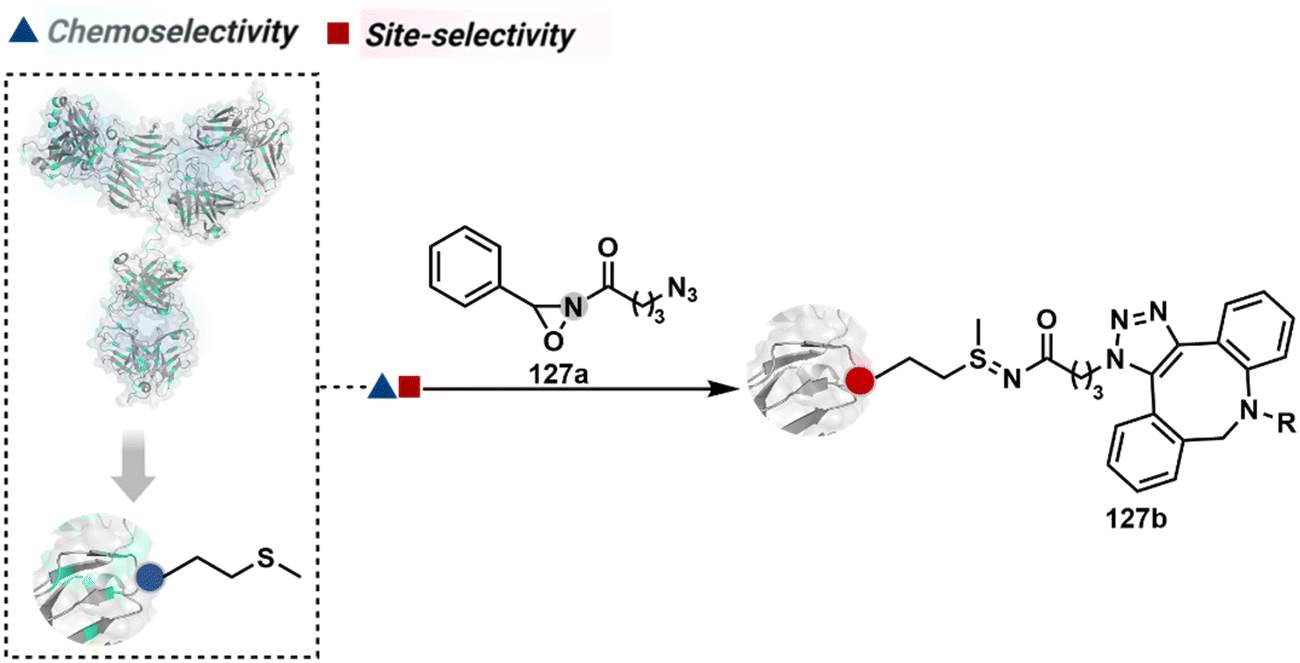 | ||
| Fig. 30 Targeting methionine for synthesis of antibody conjugates. | ||
3.2.1.6. Tryptophan. The low abundance of Trp in antibodies makes it an interesting target for antibody conjugation. However, its inherent reactivity order presents a considerable challenge. A stable organoradical (ABNO) led reaction was established for Trp modification in peptides and proteins (128b, Fig. 31).102 It potentially proceeds through the reaction of a Trp indole residue with an oxoammonium intermediate generated in the presence of NaNO2 (49b) and acetic acid. While exact sites and extent of heterogeneity are not established, the antibody conjugate was functionally validated.348 It would be exciting to see how emerging Trp bioconjugation methods deliver mild and side-reaction-free conditions to address the demand for homogeneous antibody conjugates.
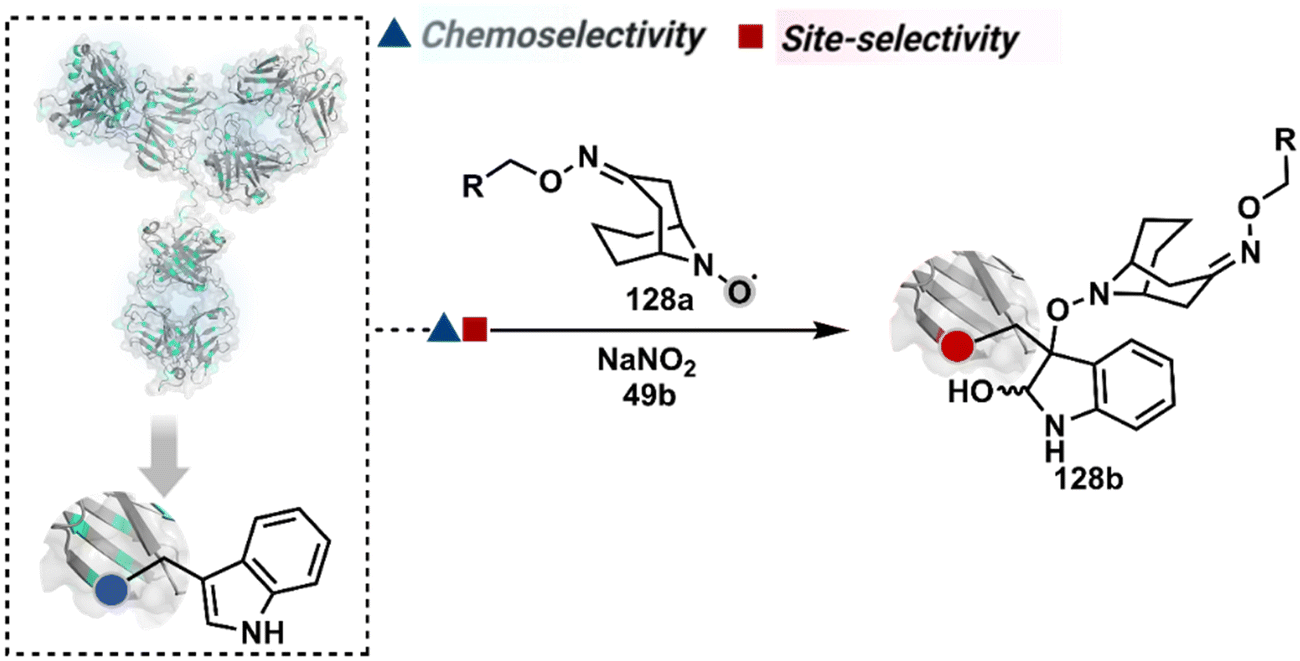 | ||
| Fig. 31 Targeting tryptophan for synthesis of antibody conjugates. | ||
3.2.1.7. Tyrosine. The low solvent accessibility of Tyr makes it a difficult target for bioconjugation. The cyclic diazodicarboxamide, diazo functionalized probe (40a, Fig. 32),113,349 and diazonium hexafluorophosphate derivatives (130a) provide promising Tyr-selective antibody conjugation reagents.112 They can be synthetically maneuvered to engineer chemically orthogonal handles to install probes. Alternatively, the electrochemically activated urazole derivatives could rapidly label Tyr to render an antibody conjugate.116,350,351
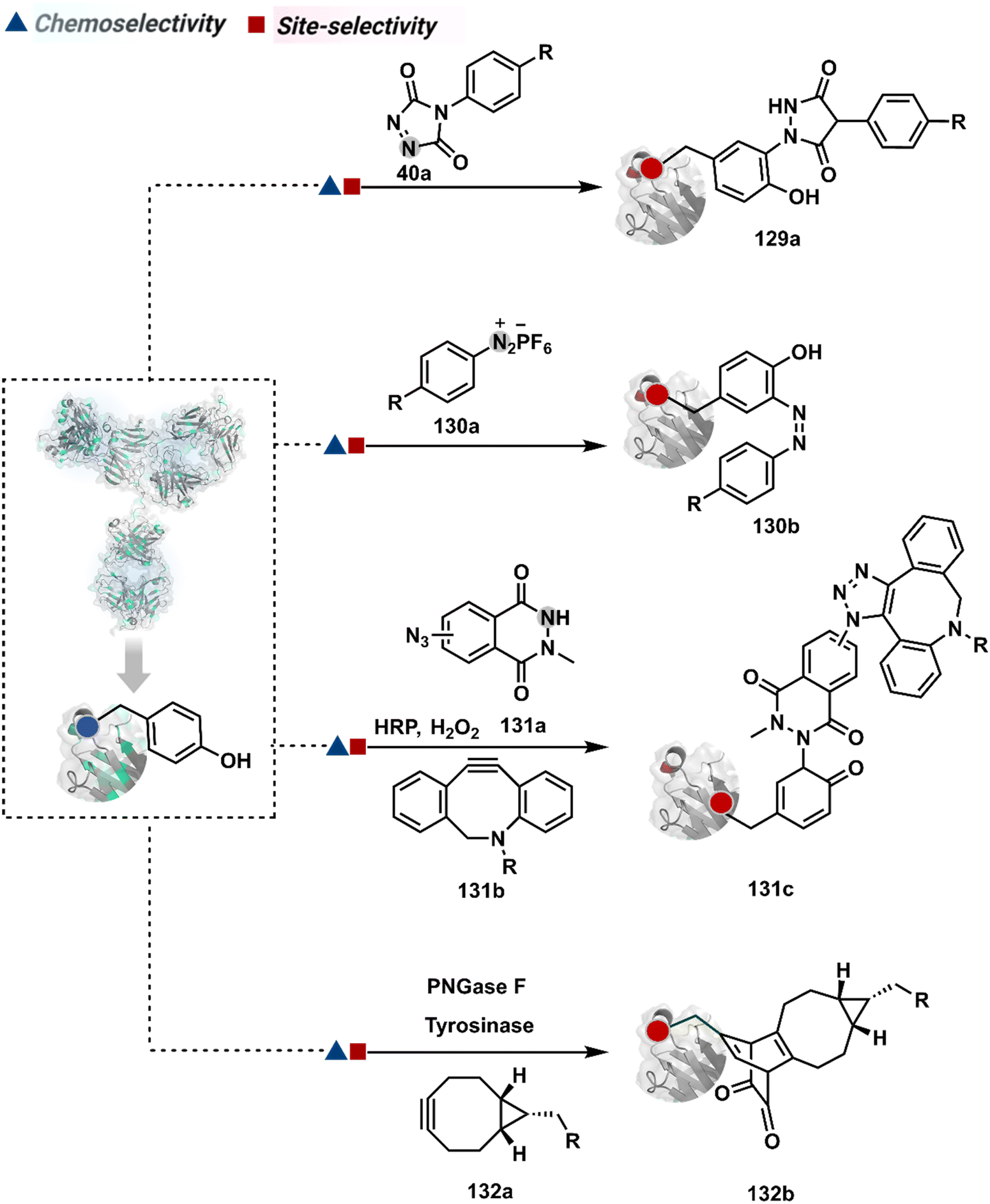 | ||
| Fig. 32 Targeting tyrosine for synthesis of antibody conjugates. | ||
Additionally, HRP and H2O2 can activate it to form an antibody conjugate with N-methylated luminol derivatives (131a).352 The strategy also works to render the conjugates with H2O2/DNAzyme.121 Besides, it can also be activated enzymatically in the presence of tyrosinase to render o-quinone methide.353 Subsequently, these reactive intermediates can be trapped through a cycloaddition to generate an antibody conjugate (132b).353,354 The final step can also be used to install the desirable probe or drug molecule. The residues such as Cys, His, Trp, and Lys present a stiff chemoselectivity challenge for Tyr modification.355
3.2.2.1. Disulfide modification and rebridging. The disulfide provides another valuable target for antibody bioconjugation. Primarily, it is due to the absence of free Cys in native antibodies and a limited number of reduction-prone disulfide groups. Hence, they are reduced chemoselectively to prime it for site-selective modification. Such two-step processes have been harnessed for thiol-selective transformations (Fig. 33).356,357 The concentration of reducing agents, e.g., tris-carboxyethyl phosphine (TCEP), regulates the extent of disulfide reduction.358 In turn, it governs the order of labeling based on the degree of heterogeneity in the combination of thiols. These attributes are reflected in the MS data of antibody conjugates. In recent years, considerable efforts have been made to regulate some of these challenges. In the process, this approach delivered brentuximab vedotin (Adcetris®), a combination of maleimide-equipped MMAE, a protease-sensitive valine-citrulline linker, and anti-CD30 monoclonal antibody (mAb) chimera (133a, Fig. 33).359 Besides, it also led to other clinically approved ADCs, including enfortumab vedotin (Padcev®, DAR 3.8),360 sacituzumab govitecan (Trodelvy®, DAR ∼7.6),361 polatuzumab vedotin (Polivy®, DAR ∼3.5),362 trastuzumab deruxtecan (Enhertu®, DAR ∼8),363 loncastuximab tesirine (Zynlonta®, DAR ∼2.3),362 and tisotumab vedotin (Tivdak®, DAR ∼4).364 The heterogeneity is one of the concerns in maleimide-driven ADCs. The continuous flow reactor has somewhat resolved it and delivered improved batch-to-batch consistencies.365 Besides, the problem of C–S bond reversibility impacting antibody conjugate stability has also drawn considerable attention. Carbonyl acrylate (134a),54 3-arylpropiolonitriles,210 ethynylphosphonamidates (135a),204–206 iodoacetamide derivatives (101a),314 aza-dibenzocyclooctyne or thiol–yne (137a),200,366 vinylphosphonamidates (138a),367 propargyl ether, and alkenyl amide/ester368 have been explored to address this concern. Alternatively, transition metal-based cysteine modification is employed to synthesize antibody conjugates (139b).369 The disulfide exchange was also employed with anti-PD-L1 and trastuzumab after its TCEP reduction, partial oxidation, and disulfide exchange (140b).370,371 The hypervalent iodine-based reagents (141a) offer direct ethynylation of reduced thiol in trastuzumab.372,373
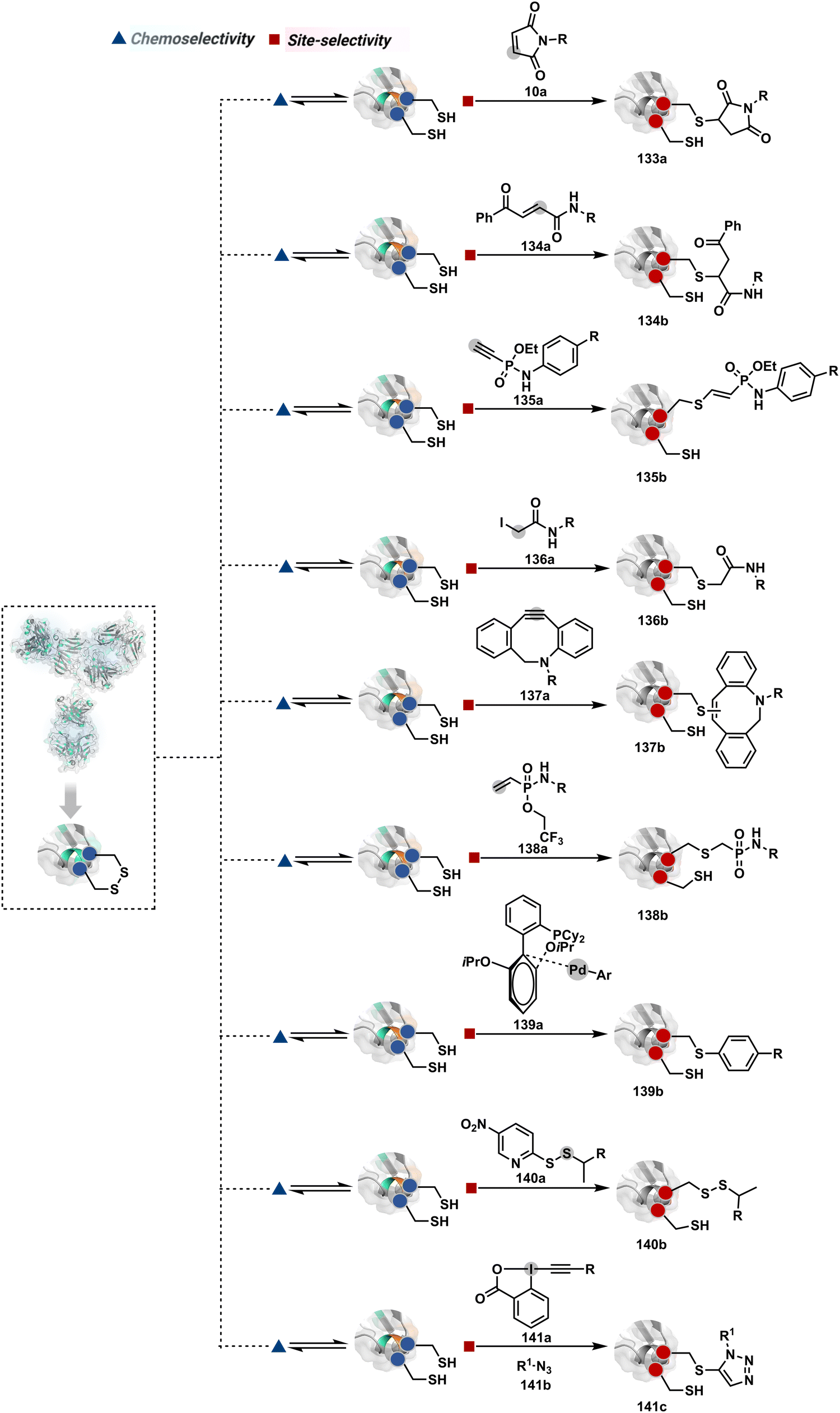 | ||
| Fig. 33 Targeting thiol from chemoselectively reduced disulfide for synthesis of antibody conjugates. | ||
It was also realized that reagents capable of rebridging the disulfide in addition to bioconjugation could avoid structural perturbations in a better way. The 3,4-dihalomaleimide and similar analogs open the opportunity for a sequence of reactions resulting in reconnecting disulfide while installing the payload.190,374 Such dual electrophilic systems can also be created through alternative routes. For example, a bis-sulfone can render rebridging through dual alkylation (142b, Fig. 34).175,375 Alternatively, an appropriately placed sulfone at the α-position of the Michael acceptor creates an opportunity for an addition–elimination–addition sequence to enable disulfide rebridging.174 3-Bromo-5-methylene pyrrolones49 and dibromopyridazinedione derivatives (143a)192,193 follow a similar route. On the other hand, 1,3-dihaloacetone or dichloroacetophenone derivatives (144a),376–378 dibromomethylquinoxaline (145a),375,379 divinylpyrimidines (146a),201,380 divinyltriazines (147a),202 diethynyl phosphinate (148a),207,381 bis-ethynylphosphonamidate (149a),382 dibromomaleimide,184,311,383 3,4-dithiophenoylmaleimide (150a),384 arylene dipropiolonitrile (151a, Fig. 35)210 phenyldivinylsulfonamides (152a),385 2-chloro-2-phenylethanethioester (153a),386 oxSTEF reagents (154a),387 3-bromo-5-methylene pyrrolones (12a),49 and DiPODS (156a, two oxadiazolyl methyl sulfones connected by a phenyl group) were also used for synthesis of disulfide-targeted antibody conjugates (156b).388 In another case, the dithioaryl(TCEP)pyridazinedione facilitated sequential de-bridging and re-bridging (157b).193 Alternatively, a Pt(II) complex with prelinked probes has also been used for rebridging antibodies (158b).389 The data for disulfide reduction-driven pathways indicate that more effort is needed to avoid structural perturbations. It applies to all cases of thiol modification with or without rebridging.
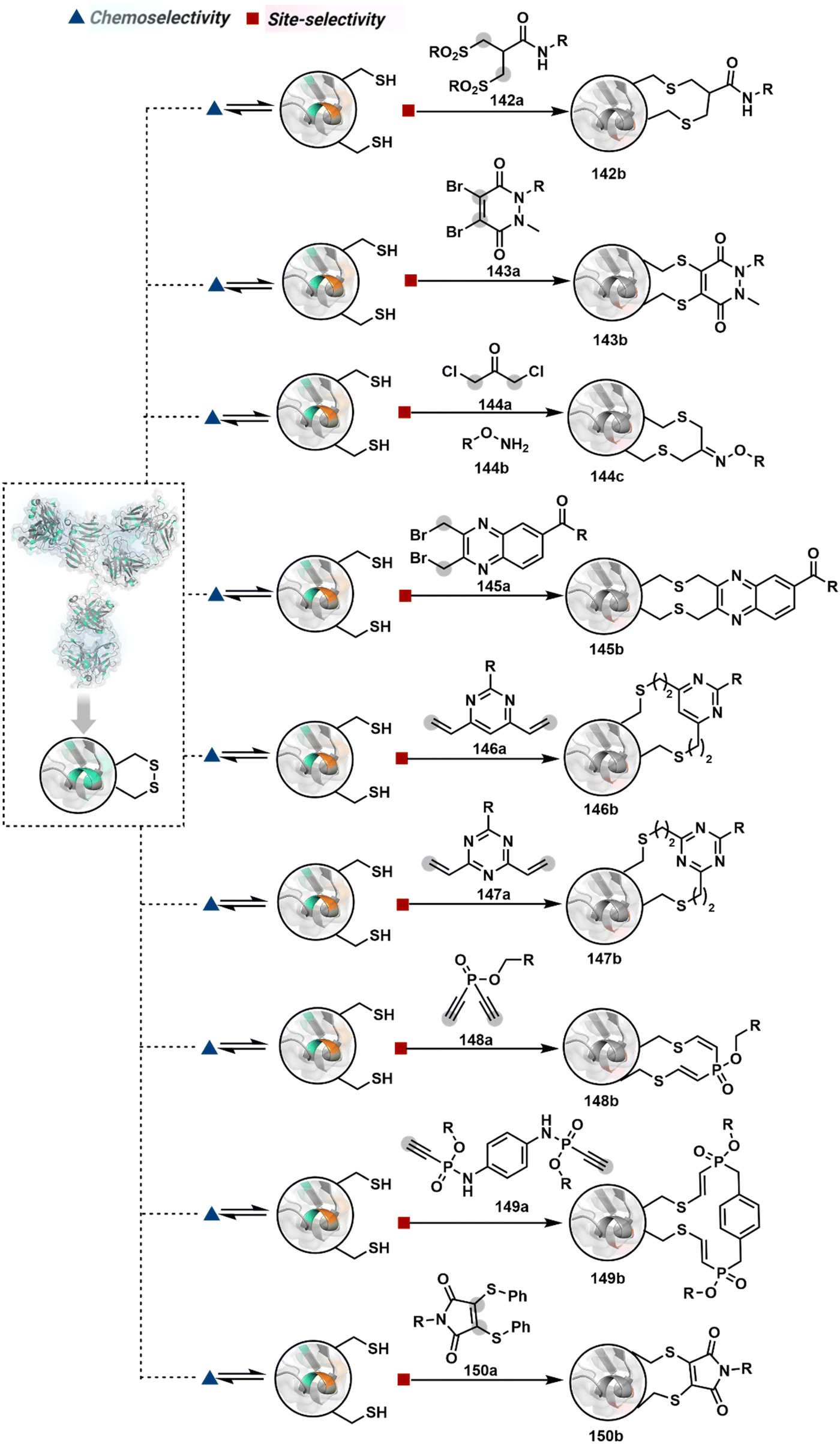 | ||
| Fig. 34 Chemoselective disulfide reduction and rebridging for synthesis of antibody conjugates. | ||
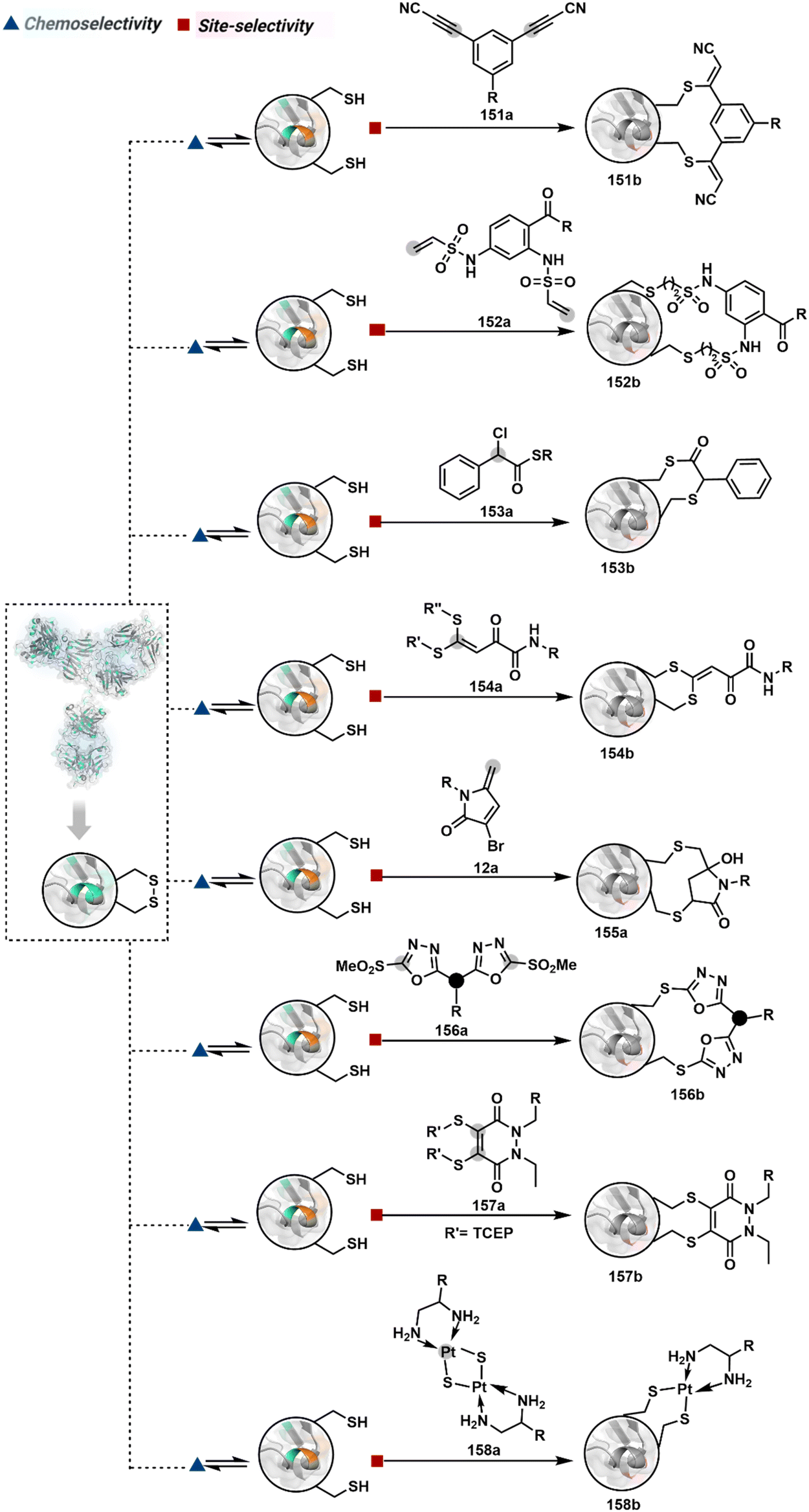 | ||
| Fig. 35 Additional methods involving disulfide reduction and rebridging for synthesis of antibody conjugates. | ||
3.2.2.2. Histidine. The His can be photocatalytically activated in the presence of singlet oxygen to produce an endoperoxide-based intermediate (159b, Fig. 36).390 This electrophilic system in an antibody is primed to react with external nucleophiles such as 1-methyl-4-arylurazole to render a bioconjugate (159d).
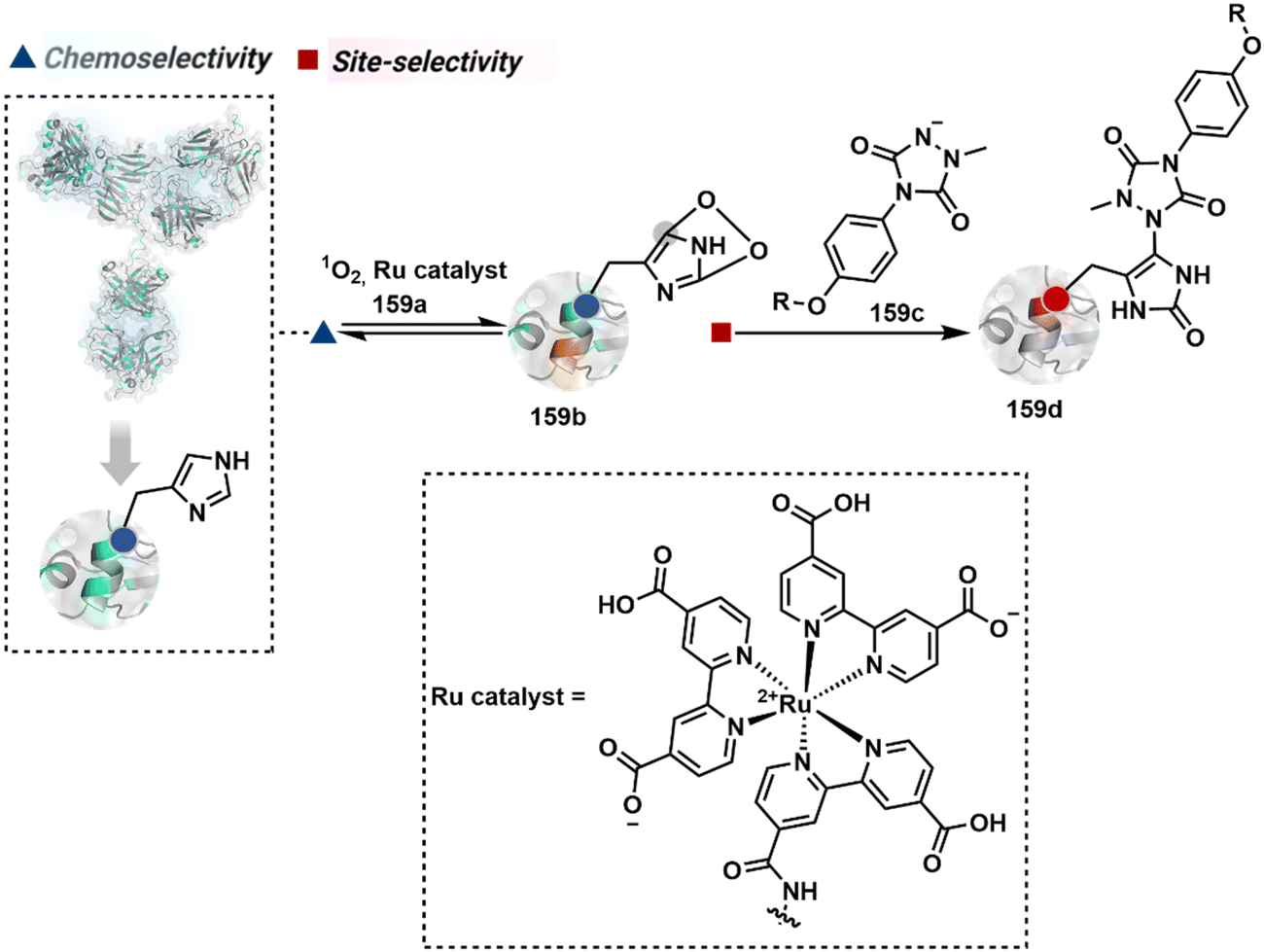 | ||
| Fig. 36 Targeting histidine for synthesis of antibody conjugates. | ||
3.2.2.3. Lysine. The chemoselective reversible generation of imine with Lys residues alters the reactivity landscape of an antibody and creates the opportunity for site-selective irreversible modification. In this perspective, its selective capture by P-centered nucleophiles, such as triethylphosphite, was able to render homogeneous antibody conjugates (160e, Fig. 37).12 While a hydride source prefers to react with N-terminus imine in general,391 the ligand-equipped aldehydes could enable a preferential site-selective irreversible reaction with a Lys-based imine (161b).270,392
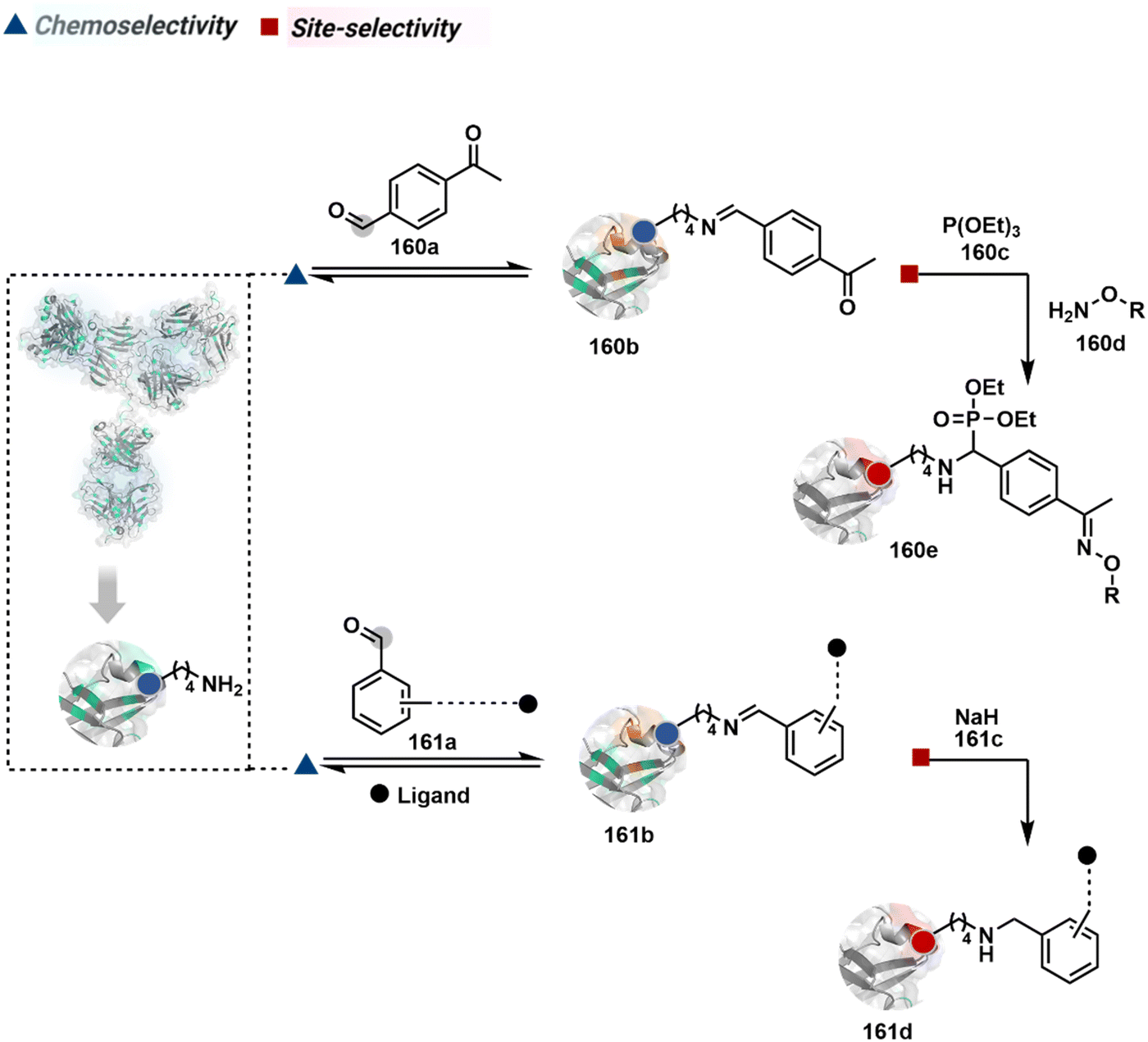 | ||
| Fig. 37 Targeting lysine for synthesis of antibody conjugates (route B). | ||
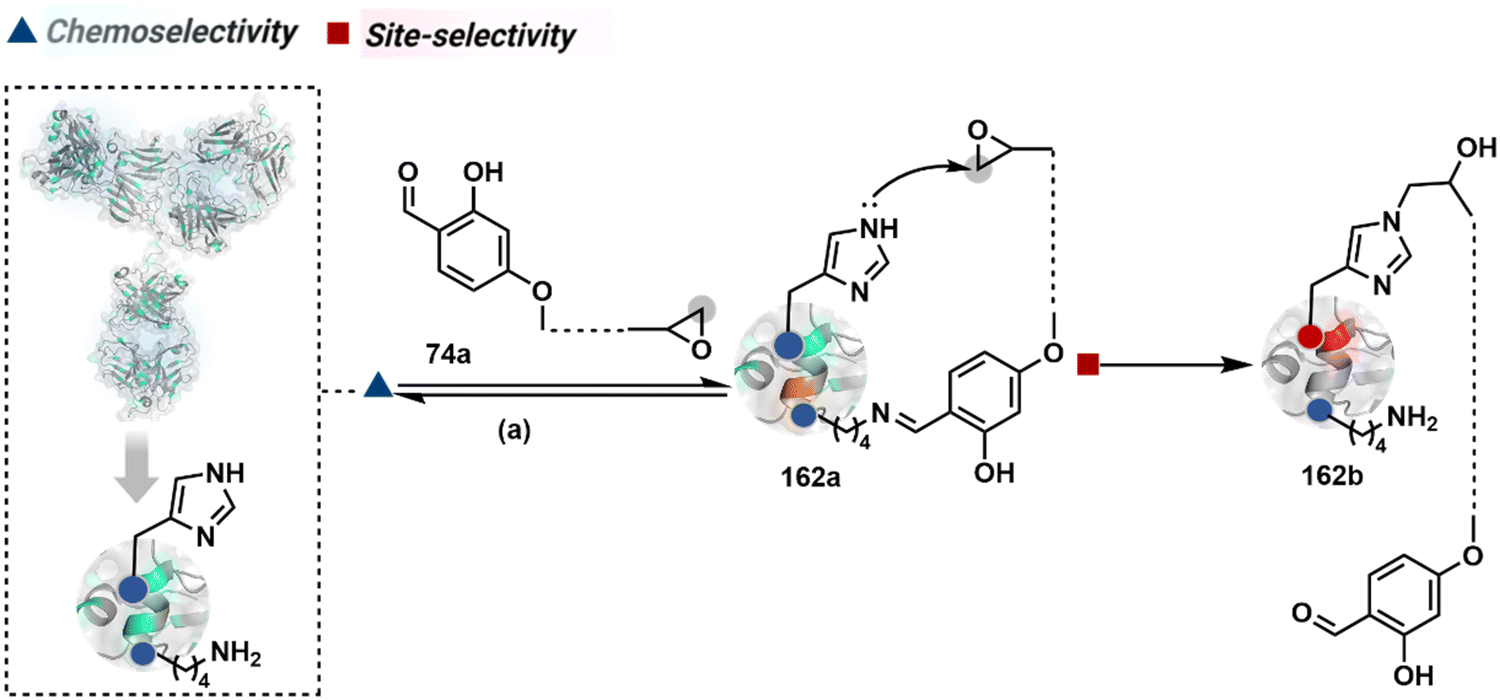 | ||
| Fig. 38 Targeting histidine for synthesis of antibody conjugates (route C). | ||
As discussed earlier, the linchpin-directed modification (LDM®) enables chemoselective and site-selective Lys modification to offer a modular platform for antibody-bioconjugation.15 In one of the representative examples, Lys-directed Lys modification (LDMK–K) followed by oxime formation results in the installation of probes at light chain K169 and heavy chain K395 (163c, Fig. 39). On the other hand, a selected case from Cys-directed Lys modification (LDMC–K) results in light chain K183 and heavy chain K341 modification (164c).219 Swapping the linchpin fragment with the leaving group in LDMK–K reagents also translated well for synthesizing antibody conjugates (165c).216,217,392 In another report, boronic acid-enabled glycans serve as linchpins and direct the alkylation of proximally placed Lys (166c).393 These linchpins could also direct an acylium ion for selective generation of amide with Lys residues.224
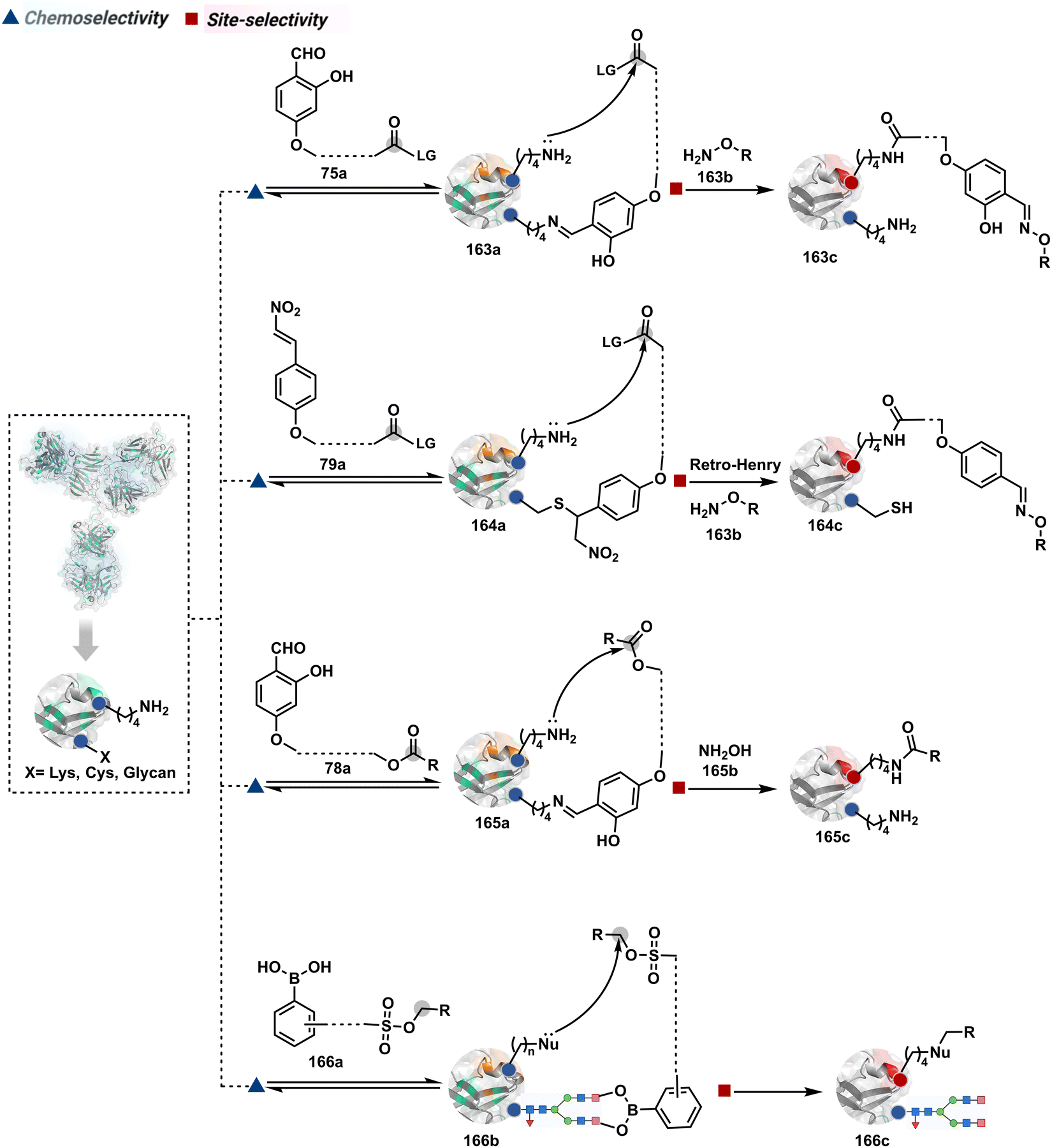 | ||
| Fig. 39 Targeting lysine for synthesis of antibody conjugates (route C). | ||
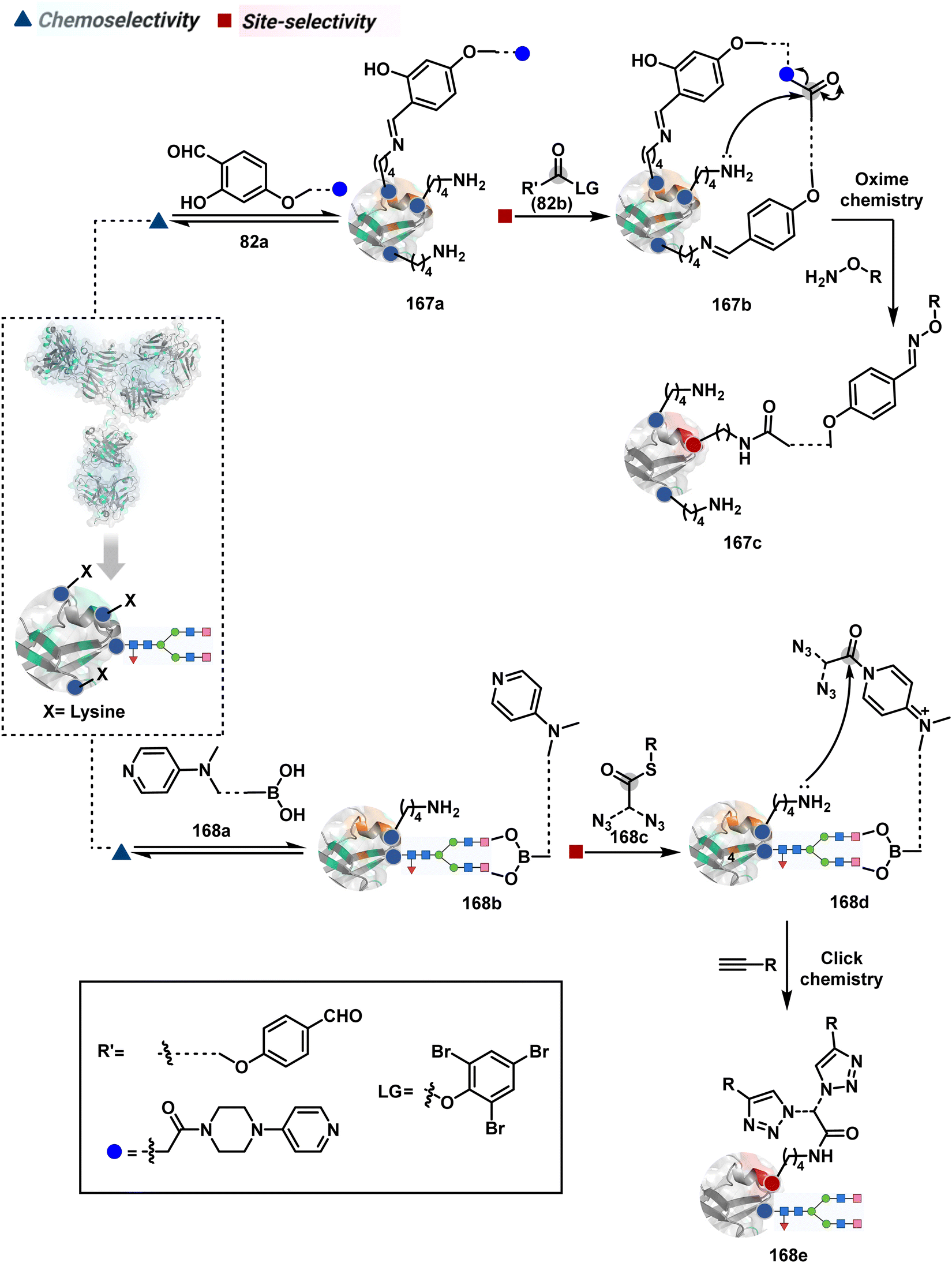 | ||
| Fig. 40 Additional methods targeting lysine for synthesis of antibody conjugates (route C). | ||
Lys-directed Lys modification (LDMK–K) can evolve further to target different residue combinations.223 To harness this capability, the activated acylating agent has to be replaced by its non-activated counterpart. Now, a catalyst can be used to activate it intermolecularly (linchpin-directed catalysis, LDC). However, the same catalyst can also be directed through a linchpin to impose additional restrictions (167b, Fig. 40). While the first approach targets a two Lys signature, the latter offers the potential to target a three Lys signature. In one of the examples, LDC-derived ADC enabled precise installation of probes at light chain K183 and heavy chain K249. Another set of bifunctional reagents is equipped with boronic acid and DMAP moiety at two ends.224 The reaction with a native antibody enables glycan-directed DMAP installation. The latter activates an external acylating reagent to generate an electrophilic center for labeling the antibody. The bioconjugation reagent equipped with an azide facilitates late-stage functionalization with an alkyne-bearing probe to generate homogeneous antibody conjugates (168e).
The N-Cys engineered antibodies chemoselectively generate imine with all accessible amines. However, a proximal thiol is available only for the N-terminal imine. It renders a residue-specific intramolecular capture of N-imine to generate an antibody conjugate (169b, Fig. 41).228 On the other hand, 2-pyridinecarboxyaldehyde driven N-imine of an antibody can react with a penultimate amide bond in the absence of a proximal thiol group to generate imidazolidinone (170b).232,394–396 However, the antibody conjugates in both cases exhibit reversibility upon prolonged incubation in serum.
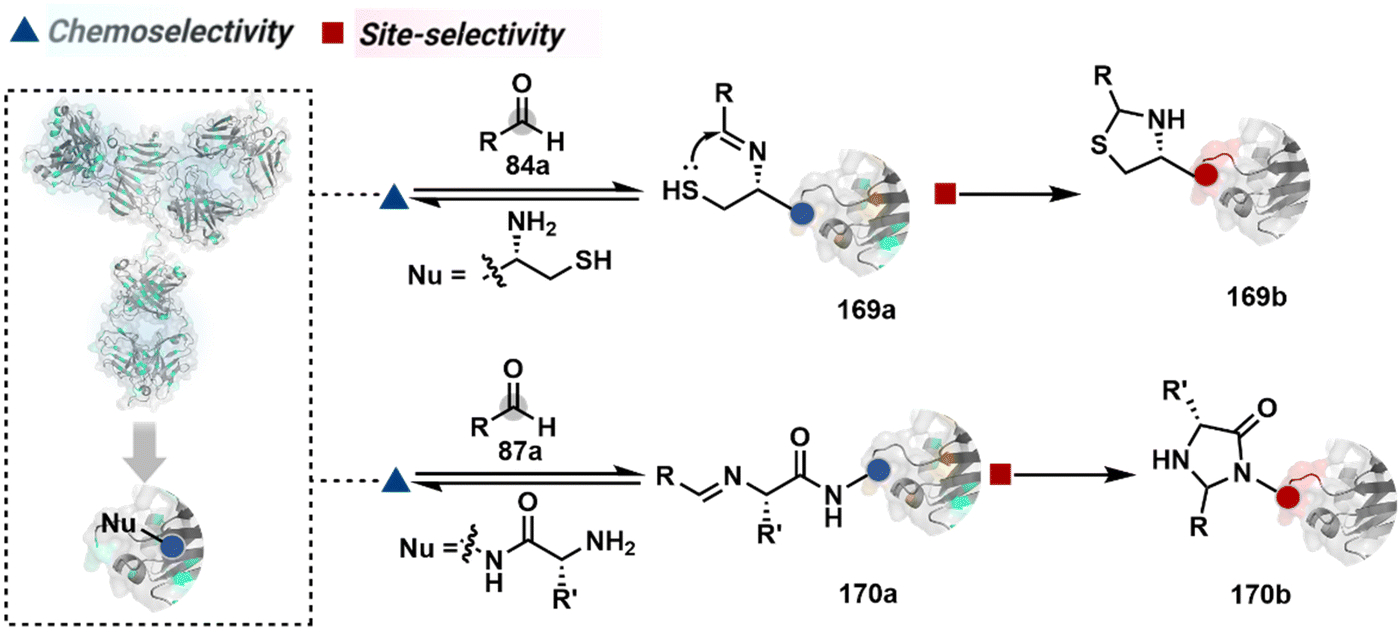 | ||
| Fig. 41 Targeting N-terminal for synthesis of antibody conjugates (route C). | ||
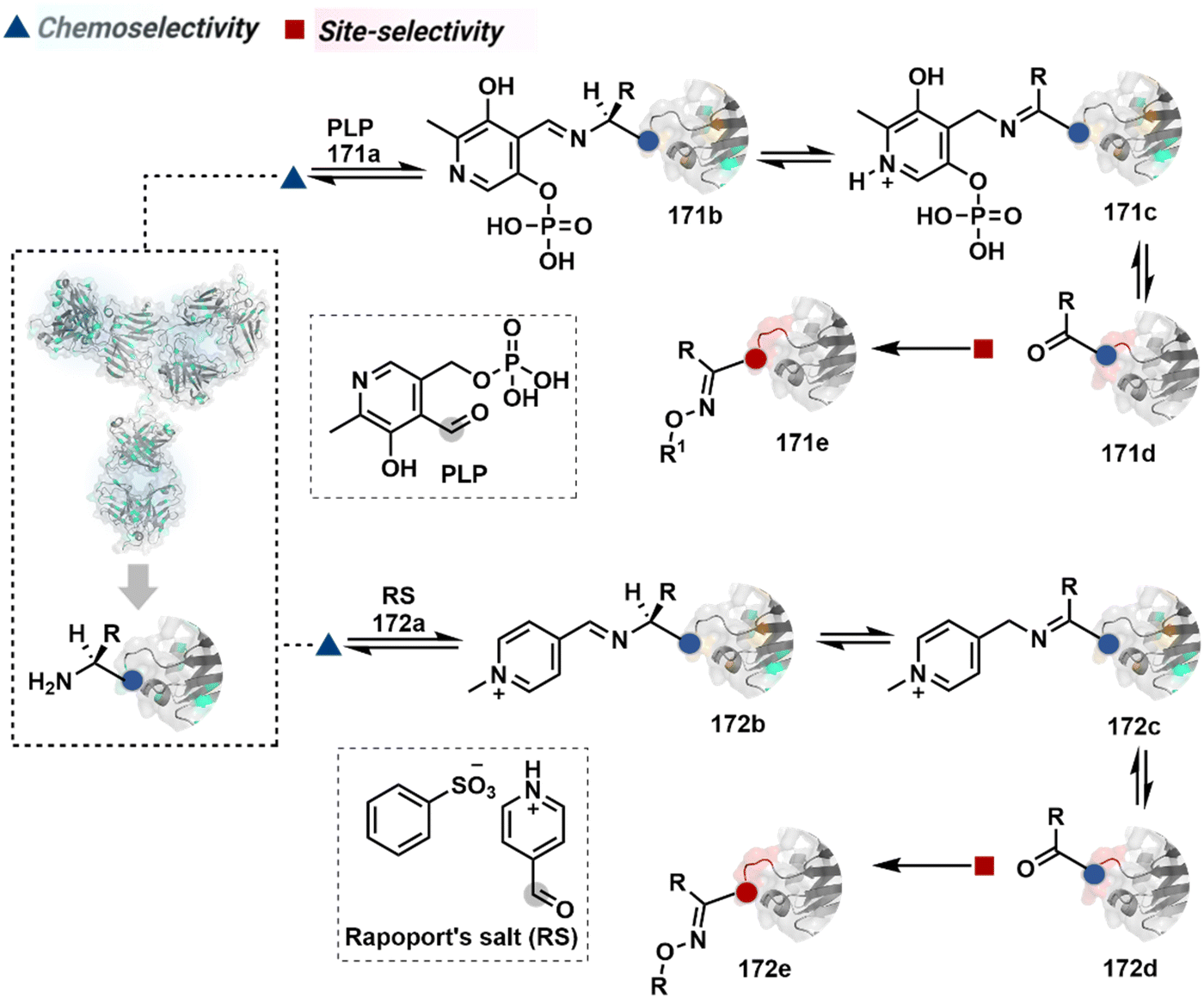 | ||
| Fig. 42 Targeting N-terminal for synthesis of antibody conjugates (route D). | ||
The reversible generation of Lys-based imine could promote isonitrilium ion generation in the presence of an isocyanide. The latter engages a proximal carboxylate in an intramolecular reaction to create an opportunity for the Ugi three-component reaction (173g, Fig. 43).397
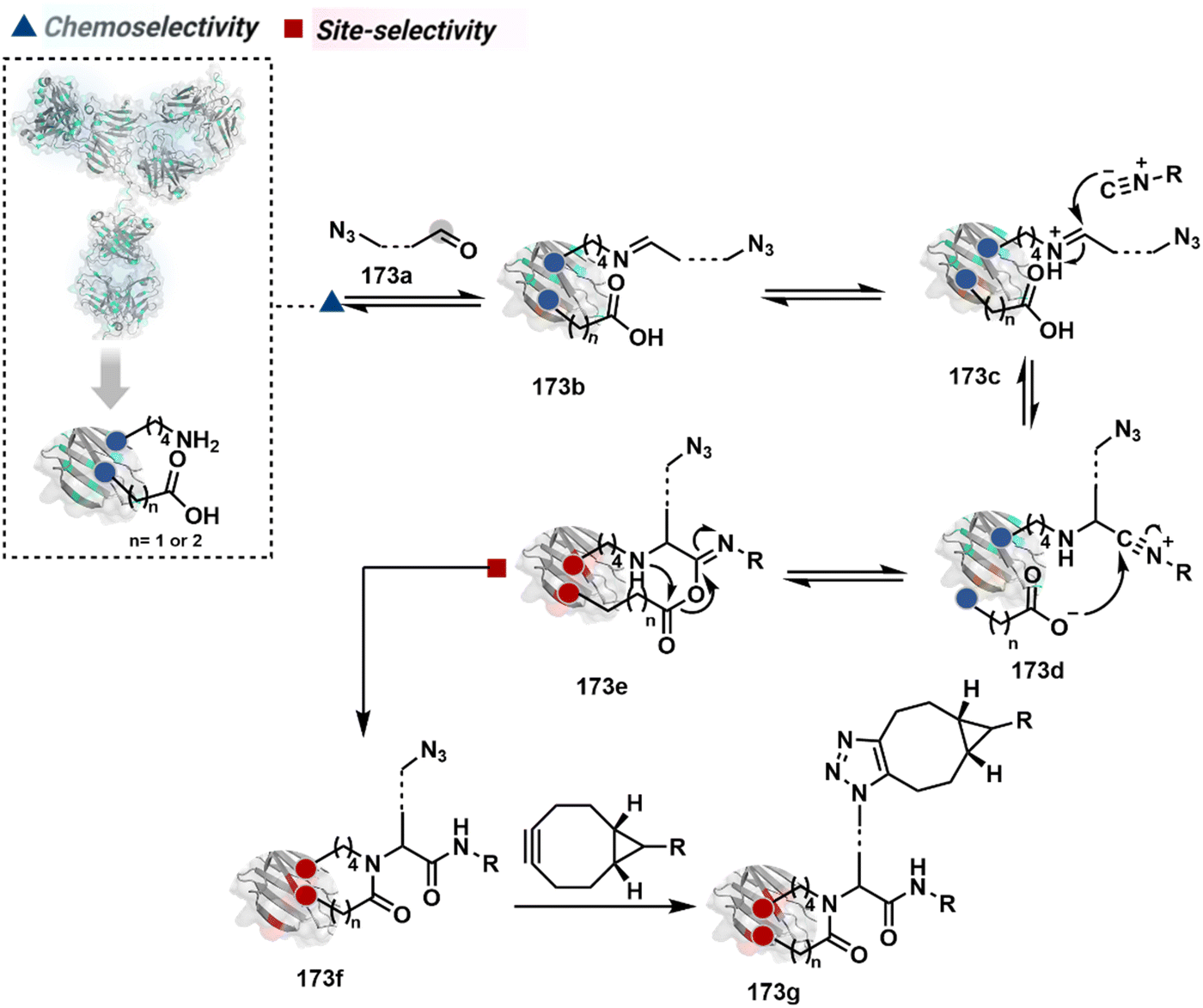 | ||
| Fig. 43 Targeting a combination of amine and carboxylate for synthesis of antibody conjugates (route D). | ||
3.3. Future directions for antibody-conjugates
The production platforms for antibodies, linkers, and toxins have witnessed considerable progress in recent years. The demand for antibodies for diagnostics and therapeutics catalyzed the growth of commercial methods for their production. The development of the other two building blocks benefited immensely from polymer and synthetic organic chemistry. These fields are gradually aligning together to meet antibody-conjugate-specific technological demands. However, linking these three components with control over selectivity presents a substantial challenge. It would be interesting to see how global efforts provide access to a bioconjugation toolbox for the precision engineering of antibody conjugates. An ideal method should deliver complete control over chemoselectivity and site-selectivity. Besides, making it compatible with multiplexing would be valuable for high throughput screening of antibody conjugates. The control over homogeneity and chemical purity will also be critical to address the challenges faced while characterizing these complex molecules. In this modular assembly, the antibody can be altered as per the target antigen, while drug selection defines the specific toxin-mediated inhibitory route. These efforts will be coupled with biologics-centric development of mass spectrometers, proteomics tools, and databases. The growth in pre-clinical and clinical data for ADC-based cancer treatment has also helped acquire knowledge of drug resistance.398–400 The tumors with limited vascular permeability also block the diffusion of ADC, drastically impacting its distribution and efficacy.401 Another complexity emerges from dynamic genetic and epigenetic modifications causing cancer relapse.402 The activity of ADC emerges from both antibody and toxin-mediated pathways.A close look into an ADC's mechanism could provide insight into the technological demands of chemical methods for their engineering. The trastuzumab-emtansine conjugate (T-DM1, Kadcyla) serves as an appropriate example due to the data available for its action and resistance. The antibody (trastuzumab) in T-DM1 operates through multiple pathways after binding with antigen (HER2). The key process initiates with the internalization of the ADC-antigen complex by receptor-mediated endocytosis (Fig. 44). The early endosomes mature with time and deliver the ADC to the lysosome, where degradation releases the toxin component. Consequently, transport of toxins from lysosomes could mediate microtubule depolymerization, leading to mitotic catastrophe and, thus, apoptosis. The antibody-mediated inhibitory pathways could also contribute to overall activity (Fig. 44).403 The binding of ADC to antigen blocks the downstream signalling pathway and extracellular domain (ECD) shedding of antigen. Besides, the FC region induces the antibody-dependent cell-mediated cytotoxicity (ADCC).
 | ||
| Fig. 44 ADC activity and resistance. Receptor mediated endocytosis (pathway 1), inhibition of HER2 signalling pathways and shedding of its extracellular domain (ECD, pathway 2), and antibody-dependent cellular cytotoxicity (ADCC, pathway 3) could be affected by (a) reduced ADC-antigen binding, (b) reduced-antigen expression, (c) signalling through alternate receptors, (d) immune response modulation, (e) recycling ADC-antigen complex to cellular membrane, (f) impaired drug release, (g) altered lysosomal catabolism, (h) drug extrusion through drug-efflux pumps, and (i) signalling dysregulation. | ||
The pre-clinical and clinical data indicate a combination of factors participating in ADC's activity. However, the same feature makes it difficult to pinpoint the exact reason for resistance, which also turns out to be multifactorial. The tumor heterogeneity, reduced ADC-antigen binding, inherent or acquired difference in antigen concentration, and extracellular domain shedding could be potential contributors (Fig. 44a and b).404–406 The adaptation of signalling to alternative receptor tyrosine kinases (RTKs) has also been noted (Fig. 44c).407–409 Furthermore, the immune-mediated cytotoxicity of T-DM1 with a trastuzumab-resistant tumor model indicates that the immune set point of T-DM1 needs further investigation (Fig. 44d).410 The ADC-antigen complex can be recycled to the plasma membrane, diverting it from the desired pathway to the lysosome (Fig. 44e).411 This complex can also meet the same fate if internalized through an alternative caveolin-mediated endocytosis.412 Besides, the lysosomal release of toxin-linker often requires active transport and is affected by receptor concentration (Fig. 44f).413,414 The alterations in the catabolic capability of lysosomes could also adversely impact the release of toxins (Fig. 44g).415 Additionally, a few studies mark elevated drug-efflux transporters as one of the reasons for resistance (Fig. 44h).416 Furthermore, mitotic slippage can cause resistance to mitotic catastrophe in DM1-mediated toxicity.417 More efforts toward understanding the target cell cycle checkpoints promise to add value. Also, genetic mutations can alter the dependence of the signalling pathway on HER2 dimerization to maintain mitotic signals and confer resistance (Fig. 44i).418–421
The complex network of underlying operational principles makes it essential to investigate the mechanistic aspects. Moreover, identifying biomarkers becomes valuable to understanding tumor heterogeneity, potential resistance mechanisms, and the selection of appropriate combination therapy. To accelerate understanding and potential to overcome the resistance, technologies for synthesizing homogeneous antibody-conjugates can play a substantial role. We need platforms that can control site-of-conjugation, residue-of-conjugation, DAR, bond-strength, homogeneity, synthetic manoeuvrability, and flexibility with the number and type of linker-toxin combinations. The knowledge in this field would simultaneously benefit all verticals in the field, including diagnostics, imaging-guided precision surgery, and therapeutics.
3.4. Protein–protein conjugates
Protein–protein conjugates are essential in cellular functions such as signal transduction or native protein activity. For instance, ubiquitination of protein substrates leads them towards degradation via the ubiquitin-proteosome pathway, thereby regulating cellular processes. Various approaches have been investigated in the past to develop well-defined protein–protein conjugates. However, lack of site-selectivity and other constraints hampered the efficiency of these protein–protein conjugation approaches. One way to generate protein–protein conjugates is via “fusion proteins,” wherein pre-designed DNA sequences are employed. The strategy enabled protein purification, imaging, and bifunctional engineered enzyme or antibody production.422–424 Although a powerful technique, it is associated with poor yields, incorrect folding, and poor stability. Besides, the necessity of N-to-C terminal fusion becomes restrictive, where a free N-terminal is critical for biological activity.425 Additionally, some fusion proteins may require separate cell lines for expression.426 It highlights how chemical methods with control over selectivity could contribute to the technological demands of protein–protein conjugates. The preceding examples highlight the efficiency of the bioconjugation toolkit in delivering site-selective therapeutic proteins or antibody–drug conjugates. However, their translation for preparing homogeneous protein–protein conjugates is challenging due to the energy demands of bringing two large molecules close. Nonetheless, a few methods used in this perspective offer a glimpse of how chemical technologies can address challenges in this segment.Primary amines like Lys or Nα–NH2 have also been investigated for conjugating two proteins. Hemoglobin bis-tetramers were produced by acylating Lys residues of deoxyhemoglobin with trimesoyl tris(3,5-dibromosalicylate) followed by aminolysis of the residual ester with 4-azidomethylbenzylamine.438,439 The free azide is then used for a click reaction to form a hemoglobin tetramer with a dialkyne reagent. The altered hemoglobin may act as a circulating oxygen carrier, and has attracted interest for transfusion medicine applications. Thioimidate 2-iminothiolane, a cyclic compound commonly referred to as Traut's reagent, is reported to introduce a free thiol at the Lys residue. Next, it reacts with maleimide installed on a Lys residue of the second protein through bifunctional succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC). This strategy rendered bispecific antibodies for directing stem cells to infarcted myocardium440 or redirecting T-cell cytolytic activity to neuroblastomas.441 This conjugation approach using Traut's reagent and SMCC has also been used for generating bispecific antibodies for cancer therapeutics. However, it requires caution as it is accompanied by retro-Michael addition and thiol exchange issues. A heterobifunctional crosslinker having a photocaged quinone methide tethered to NHS ester has been reported to link proteins and membrane receptors in mammalian cells.442 However, the unselective nature of NHS ester and promiscuity of the generated quinone methide render a highly heterogeneous system. Lys acylation using Pd–protein OAC has also been reported to generate protein–protein heterodimeric conjugates.437 However, metal-based toxicity limits the applicability of this approach for therapeutic purposes. The selectivity control for labeling the first protein is as challenging as earlier examples. However, protein–protein interactions can restrict the number of sites in the second protein that could participate in the reaction.
3.5. Precision therapeutics
Technology for targeting a single site of the protein of interest without affecting other biomolecules in the human body would be an ideal toolbox for small-molecule precision therapeutics. Historically, compounds with electrophilic handles were weeded out, keeping their non-selective irreversible modifications under consideration. That is why early covalent drugs were mostly unintentional, and their mechanism of action was established later. In the last few years, the interest in covalent drugs has reignited.2,448,449 The candidates capable of specificity could present advantages with potency, duration of action, and an opportunity to go for intractable targets. However, non-selectivity will seed toxicity, target assessment, and PK/PD relationship challenges. Even though we are far from finding ideal solutions, recent developments offer covalent candidates with an acceptable combination of reactivity, selectivity, potency, and safety.The advances in chemoproteomic platforms have been one of the facilitators for gaining insights. The electrophilic reagents have been used to map targets in the proteome. The broad-range reactivity, such as in the case of NHS esters, leads to promiscuity. It employs Lys, Ser, Thr, Tyr, and other residues in protein bioconjugation.70 The screening of reagents over the lower range of reactivity benefits chemoselectivity during the target engagement. Such ligandability profiling establishes the correlation between the reactivity and selectivity. At the same time, these experiments are directed toward expanding the targetome. Hence, experimental parameters do not necessarily overlap with the window of selectivity. The probe concentration typically ranges between 200–500 μM in these cases. In contrast, screening candidates for selectivity generally involves their concentration within 1–10 μM.450
Pre-established non-covalent ligands can further empower the control of electrophiles over reactivity and selectivity to reduce the number of targets substantially. Such a ligand-first approach has been the basis of significant developments in the past decade. A complementary route centered around the electrophilic system has also been explored. The emerging knowledge from these approaches highlights the promise of covalent modifiers or bioconjugation reagents to offer precision therapeutics. However, it is still far from ideal, and the off-target engagements highlight the magnitude of the challenge in achieving precision therapeutics. It makes it necessary to understand the reactivity and selectivity features displayed by the covalent modifiers. Each copy of an amino acid experiences a unique microenvironment, impacting its accessibility and reactivity. Hence, a set of chemoselective electrophiles for a residue, such as Cys451 or Lys,452 would have unique targets within a proteome besides the common ones. The current approaches (Fig. 45) have primarily addressed all the selectivity attributes in one-step methods (route A).453–456 While ligand-based localization has benefited in reducing the off-target hits, a substantial distance still needs to be covered towards specificity. It would be interesting to see how the disintegration of selectivity attributes using DIN theory could empower the rational design of covalent drugs and amplify selectivity. The control over sub-cellular distribution, substrate competition, and microenvironment would also benefit the technological demand.
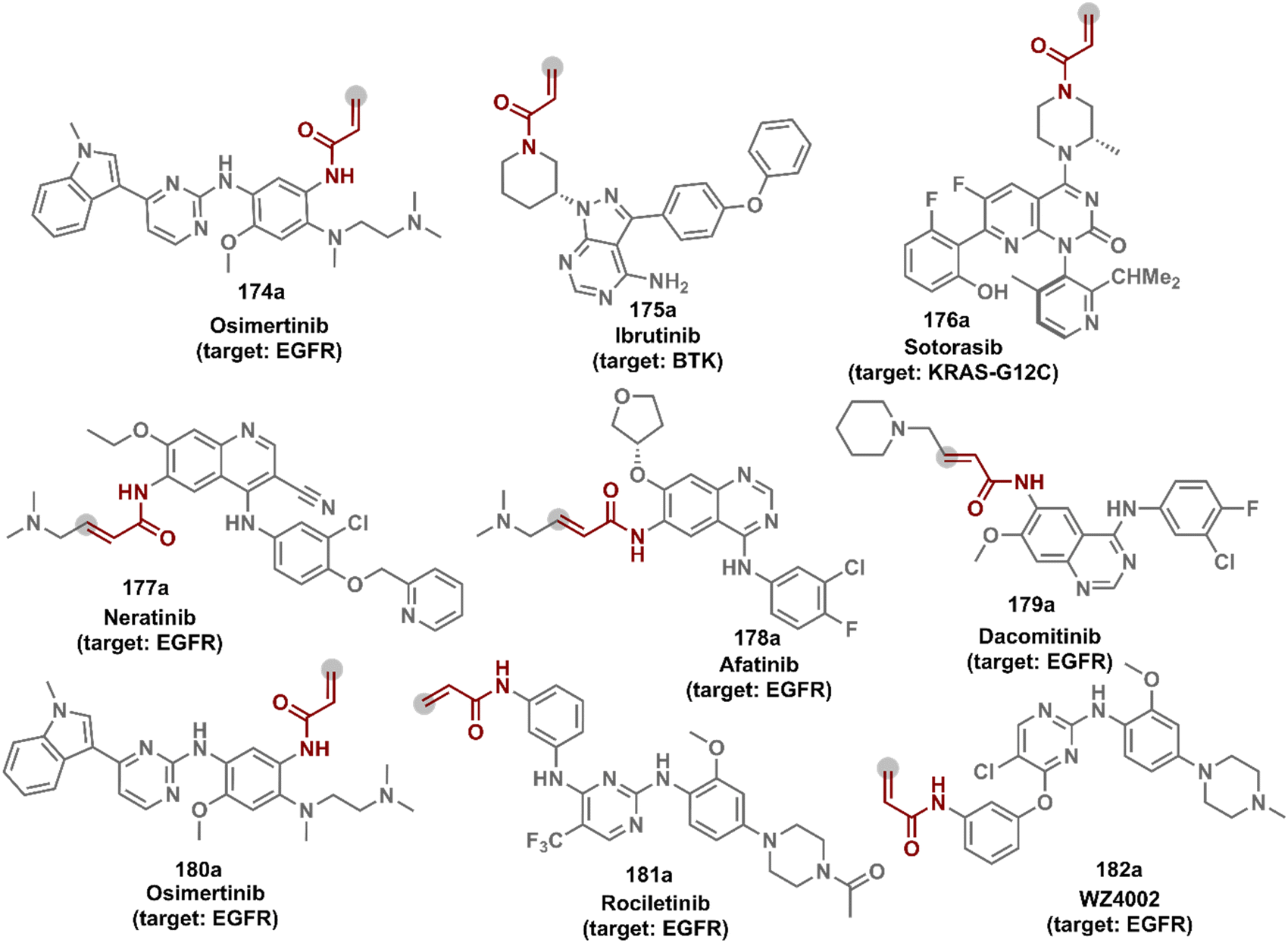 | ||
| Fig. 45 Representative examples of approved irreversible covalent drugs with identified targetomes. | ||
4. Conclusion
Protein bioconjugation has emerged as a significant area of research immensely beneficial at the nexus of chemistry, biology, and medicine. Chemical approaches enabling precision are critical for meeting these technological needs. The diversity of proteins in structure, stability, and other reactivity-defining variables complicates the matter. As a result, hit-and-trial or directed screening remained a popular strategy for protein bioconjugation. The current applications demand well-defined protein conjugates and necessitate a broader array of methods. Understanding the challenges in developing novel strategies for precision protein engineering is the key in this regard.This review discusses the disintegrate (DIN) theory to deconvolute selectivity attributes via a multi-step chemical transformation. It enables the exploration of new reactivity dimensions, predicts potential products, and facilitates hypothesis-driven research. We have classified the examples into four representative categories (routes A–D). Route A involves a classic single-step approach for attaining site-selectivity. Routes B–D demonstrate how deconvolution into multiple steps can empower access to diverse selectivity or specificity attributes. It will be fascinating to see how additional dimensions are added to this repository over time.
The existing developments offer only a glimpse of the organic chemistry landscape with proteins. The DIN theory will drive the investigation of several unique reactive intermediates via reversible transformations in the coming years. For example, we recently demonstrated that disintegrating chemoselectivity into two steps could engage protein–protein interactions to drive site-selective bioconjugation.457 We can anticipate such translations providing access to diverse reactivity orders, resulting in distinct bioconjugation sites, dual or multi-probe installation, and analytically pure bioconjugates.15–17,67,219,223,236,237,458–460 These discoveries can extend the precise bioconjugation methods to low-reactivity residues. It will also be intriguing to examine if DIN theory can enable high-energy intermediates for site-selective modifications contrary to their inherent characteristics. The multi-equilibrim transformations are likely to unravel multiple unchartered dimensions in this perspective. Such expansions will benefit the development of fundamental biology, imaging, and therapeutics. The examples highlight the effectiveness of this approach for selective labeling of therapeutic proteins, homogenous ADCs, AFCs, and protein–protein conjugates. It will be interesting to note if these strategies can strengthen cell–cell conjugation. Such control for protein specificity would also drive the development of covalent inhibitors. The recent advancements provide sufficient proof of principle demonstrating the potential extension of such methods in complex cellular and in vivo systems.
Author contributions
This manuscript was written and approved by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by SERB, DST, DBT, SERB-PACE (IPA/20221/000148), and IISER Bhopal. R. V., M. K., R. M., S. D. M., and S. B. are recipients of a research fellowship from CSIR, PMRF, UGC, and IISER Bhopal. V. R. is a recipient of the Swarnajayanti Fellowship (DST/SJF/CSA-01/2018-19 and SB/SJF/2019-20/01). The figures were made using ChemOffice, PyMol, and Biorender.Notes and references
- S. B. Ebrahimi and D. Samanta, Nat. Commun., 2023, 14, 2411 CrossRef CAS PubMed.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 CrossRef CAS PubMed.
- N. Shindo, H. Fuchida, M. Sato, K. Watari, T. Shibata, K. Kuwata, C. Miura, K. Okamoto, Y. Hatsuyama, K. Tokunaga, S. Sakamoto, S. Morimoto, Y. Abe, M. Shiroishi, J. M. M. Caaveiro, T. Ueda, T. Tamura, N. Matsunaga, T. Nakao, S. Koyanagi, S. Ohdo, Y. Yamaguchi, I. Hamachi, M. Ono and A. Ojida, Nat. Chem. Biol., 2019, 15, 250–258 CrossRef CAS PubMed.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- J. Singh, J. Med. Chem., 2022, 65, 5886–5901 CrossRef CAS PubMed.
- D. Schaefer and X. Cheng, Pharmaceuticals, 2023, 16, 663 CrossRef CAS PubMed.
- N. C. Reddy, M. Kumar, R. Molla and V. Rai, Org. Biomol. Chem., 2020, 18, 4669–4691 RSC.
- P. Chauhan, R. V., M. Kumar, R. Molla, U. V. B. and V. Rai, ACS Cent. Sci., 2023, 9, 137–150 CrossRef CAS PubMed.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef CAS PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2016, 8, 103–113 CrossRef CAS PubMed.
- M. Chilamari, N. Kalra, S. Shukla and V. Rai, Chem. Commun., 2018, 54, 7302–7305 RSC.
- D. Chen, M. M. Disotuar, X. Xiong, Y. Wang and D. H.-C. Chou, Chem. Sci., 2017, 8, 2717–2722 RSC.
- M. Chilamari, L. Purushottam and V. Rai, Chem. – Eur. J., 2017, 23, 3819–3823 CrossRef CAS PubMed.
- S. R. Adusumalli, D. G. Rawale, K. Thakur, L. Purushottam, N. C. Reddy, N. Kalra, S. Shukla and V. Rai, Angew. Chem., Int. Ed., 2020, 59, 10332–10336 CrossRef CAS PubMed.
- S. R. Adusumalli, D. G. Rawale, U. Singh, P. Tripathi, R. Paul, N. Kalra, R. K. Mishra, S. Shukla and V. Rai, J. Am. Chem. Soc., 2018, 140, 15114–15123 CrossRef CAS PubMed.
- L. Purushottam, S. R. Adusumalli, U. Singh, V. B. Unnikrishnan, D. G. Rawale, M. Gujrati, R. K. Mishra and V. Rai, Nat. Commun., 2019, 10, 2539 CrossRef PubMed.
- Z. Fu, S. Li, S. Han, C. Shi and Y. Zhang, Signal Transduction Targeted Ther., 2022, 7, 93 CrossRef CAS PubMed.
- K. Takahashi, J. Biol. Chem., 1968, 243, 6171–6179 CrossRef CAS.
- T. W. Lo, M. E. Westwood, A. C. McLellan, T. Selwood and P. J. Thornalley, J. Biol. Chem., 1994, 269, 32299–32305 CrossRef CAS.
- Y. Nakaya, H. Horinishi and K. Shibata, J. Biochem., 1967, 61, 345–351 CrossRef PubMed.
- M. A. Gauthier and H.-A. Klok, Biomacromolecules, 2011, 12, 482–493 CrossRef CAS PubMed.
- S. M. M. Hensen, W. C. Boelens, K. M. Bonger, R. T. P. Van Cruchten, F. L. Van Delft and G. J. M. Pruijn, Molecules, 2015, 20, 6592–6600 CrossRef CAS PubMed.
- K. Toi, E. Bynum, E. Norris and H. A. Itano, J. Biol. Chem., 1967, 242, 1036–1043 CrossRef CAS PubMed.
- M. S. K. Wanigasekara and S. M. Chowdhury, Anal. Chim. Acta, 2016, 935, 197–206 CrossRef CAS PubMed.
- M. S. K. Wanigasekara, X. Huang, J. K. Chakrabarty, A. Bugarin and S. M. Chowdhury, ACS Omega, 2018, 3, 14229–14235 CrossRef CAS PubMed.
- C.-T. Shih, B.-H. Kuo, C.-Y. Tsai, M.-C. Tseng and J.-J. Shie, Org. Lett., 2022, 24, 4694–4698 CrossRef CAS PubMed.
- D.-L. Leone, M. Hubálek, R. Pohl, V. Sýkorová and M. Hocek, Angew. Chem., Int. Ed., 2021, 60, 17383–17387 CrossRef CAS PubMed.
- N. A. McGrath, K. A. Andersen, A. K. F. Davis, J. E. Lomax and R. T. Raines, Chem. Sci., 2015, 6, 752–755 RSC.
- K. A. Mix and R. T. Raines, Org. Lett., 2015, 17, 2358–2361 CrossRef CAS PubMed.
- N. Ma, J. Hu, Z.-M. Zhang, W. Liu, M. Huang, Y. Fan, X. Yin, J. Wang, K. Ding, W. Ye and Z. Li, J. Am. Chem. Soc., 2020, 142, 6051–6059 CrossRef CAS PubMed.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205–211 CrossRef CAS PubMed.
- R. G. Pearson, Chemical Hardness: Applications from Molecules to Solids, Wiley-VCH, Weinheim, 1997 Search PubMed.
- D. R. Goddard and L. Michaelis, J. Biol. Chem., 1935, 112, 361–371 CrossRef CAS.
- N. J. Davis and S. L. Flitsch, Tetrahedron Lett., 1991, 32, 6793–6796 CrossRef CAS.
- B. Lee, S. Sun, E. Jiménez-Moreno, A. A. Neves and G. J. L. Bernardes, Bioorg. Med. Chem., 2018, 26, 3060–3064 CrossRef CAS PubMed.
- O. Boutureira, N. Martínez-Sáez, K. M. Brindle, A. A. Neves, F. Corzana and G. J. L. Bernardes, Chem. – Eur. J., 2017, 23, 6483–6489 CrossRef CAS PubMed.
- N. Martínez-Sáez, S. Sun, D. Oldrini, P. Sormanni, O. Boutureira, F. Carboni, I. Compañón, M. J. Deery, M. Vendruscolo, F. Corzana, R. Adamo and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2017, 56, 14963–14967 CrossRef PubMed.
- D. Zhang, N. O. Devarie-Baez, Q. Li, J. R. Lancaster, Jr. and M. Xian, Org. Lett., 2012, 14, 3396–3399 CrossRef CAS PubMed.
- C. Canovas, M. Moreau, C. Bernhard, A. Oudot, M. Guillemin, F. Denat and V. Goncalves, Angew. Chem., Int. Ed., 2018, 57, 10646–10650 CrossRef CAS PubMed.
- B. M. Lipka, V. M. Betti, D. S. Honeycutt, D. L. Zelmanovich, M. Adamczyk, R. Wu, H. S. Blume, C. A. Mendina, J. M. Goldberg and F. Wang, Bioconjugate Chem., 2022, 33, 2189–2196 CrossRef CAS PubMed.
- E. Friedmann, D. H. Marrian and I. Simonreuss, Br. J. Pharmacol., 1949, 4, 105–108 CrossRef CAS PubMed.
- E. Friedmann, Biochim. Biophys. Acta, 1952, 9, 65–75 CrossRef CAS PubMed.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS PubMed.
- D. Kalia, S. P. Pawar and J. S. Thopate, Angew. Chem., Int. Ed., 2017, 56, 1885–1889 CrossRef CAS PubMed.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed.
- D. Kalia, P. V. Malekar and M. Parthasarathy, Angew. Chem., Int. Ed., 2016, 55, 1432–1435 CrossRef CAS PubMed.
- Y. Zhang, X. Zhou, Y. Xie, M. M. Greenberg, Z. Xi and C. Zhou, J. Am. Chem. Soc., 2017, 139, 6146–6151 CrossRef CAS PubMed.
- Y. Zhang, C. Zang, G. An, M. Shang, Z. Cui, G. Chen, Z. Xi and C. Zhou, Nat. Commun., 2020, 11, 1015 CrossRef CAS PubMed.
- J. Yu, X. Yang, Y. Sun and Z. Yin, Angew. Chem., Int. Ed., 2018, 57, 11598–11602 CrossRef CAS PubMed.
- J. Morales-Sanfrutos, J. Lopez-Jaramillo, M. Ortega-Muñoz, A. Megia-Fernandez, F. Perez-Balderas, F. Hernandez-Mateo and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2010, 8, 667–675 RSC.
- J. Morales-Sanfrutos, F. J. Lopez-Jaramillo, F. Hernandez-Mateo and F. Santoyo-Gonzalez, J. Org. Chem., 2010, 75, 4039–4047 CrossRef CAS PubMed.
- M. S. Masri and M. Friedman, J. Protein Chem., 1988, 7, 49–54 CrossRef CAS PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- S. Ariyasu, H. Hayashi, B. Xing and S. Chiba, Bioconjugate Chem., 2017, 28, 897–902 CrossRef CAS PubMed.
- A. Abbas, B. Xing and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 7491–7494 CrossRef CAS PubMed.
- M. J. Matos, C. D. Navo, T. Hakala, X. Ferhati, A. Guerreiro, D. Hartmann, B. Bernardim, K. L. Saar, I. Compañón, F. Corzana, T. P. J. Knowles, G. Jiménez-Osés and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2019, 58, 6640–6644 CrossRef CAS PubMed.
- J.-R. Deng, S.-F. Chung, A. S.-L. Leung, W.-M. Yip, B. Yang, M.-C. Choi, J.-F. Cui, K. K.-Y. Kung, Z. Zhang, K.-W. Lo, Y.-C. Leung and M.-K. Wong, Commun. Chem., 2019, 2, 93 CrossRef.
- H.-Y. Shiu, T.-C. Chan, C.-M. Ho, Y. Liu, M.-K. Wong and C.-M. Che, Chem. – Eur. J., 2009, 15, 3839–3850 CrossRef CAS PubMed.
- T. Posner, Ber. Dtsch. Chem. Ges., 1905, 38, 646–657 CrossRef.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem. – Eur. J., 2009, 15, 11444–11449 CrossRef CAS PubMed.
- M. L. Conte, S. Staderini, A. Marra, M. Sanchez-Navarro, B. G. Davis and A. Dondoni, Chem. Commun., 2011, 47, 11086–11088 RSC.
- C. Wan, Z. Hou, D. Yang, Z. Zhou, H. Xu, Y. Wang, C. Dai, M. Liang, J. Meng, J. Chen, F. Yin, R. Wang and Z. Li, Chem. Sci., 2023, 14, 604–612 RSC.
- X. Li, H. Ma, S. Dong, X. Duan and S. Liang, Talanta, 2004, 62, 367–371 CrossRef CAS PubMed.
- X. Li, H. Ma, L. Nie, M. Sun and S. Xiong, Anal. Chim. Acta, 2004, 515, 255–260 CrossRef CAS.
- R. Zamora, M. Alaiz and F. J. Hidalgo, Chem. Res. Toxicol., 1999, 12, 654–660 Search PubMed.
- P. N. Joshi and V. Rai, Chem. Commun., 2019, 55, 1100–1103 RSC.
- S. Jia, D. He and C. J. Chang, J. Am. Chem. Soc., 2019, 141, 7294–7301 CrossRef CAS PubMed.
- S. Kalkhof and A. Sinz, Anal. Bioanal. Chem., 2008, 392, 305–312 CrossRef CAS PubMed.
- C. C. Ward, J. I. Kleinman and D. K. Nomura, ACS Chem. Biol., 2017, 12, 1478–1483 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhang, H. Liu, H. Y. Chow, R. Tian, Y. M. Eva Fung and X. Li, Biochemistry, 2020, 59, 175–178 CrossRef CAS PubMed.
- C. L. Tung, C. T. T. Wong, E. Y. M. Fung and X. Li, Org. Lett., 2016, 18, 2600–2603 CrossRef CAS PubMed.
- B. Li, L. Wang, X. Chen, X. Chu, H. Tang, J. Zhang, G. He, L. Li and G. Chen, Nat. Commun., 2022, 13, 311 CrossRef CAS PubMed.
- X. Chu, B. Li, H.-Y. Liu, X. Sun, X. Yang, G. He, C. Zhou, W. Xuan, S.-L. Liu and G. Chen, Angew. Chem., Int. Ed., 2023, 62, e202212199 CrossRef CAS PubMed.
- Y. Zhang, Q. Zhang, C. T. T. Wong and X. Li, J. Am. Chem. Soc., 2019, 141, 12274–12279 CrossRef CAS PubMed.
- C. Apel, S. S. Hartmann, D. Lentz and M. Christmann, Angew. Chem., Int. Ed., 2019, 58, 5075–5079 CrossRef CAS PubMed.
- C. Apel, M.-A. Kasper, C. E. Stieger, C. P. R. Hackenberger and M. Christmann, Org. Lett., 2019, 21, 10043–10047 CrossRef CAS PubMed.
- T. Nakamura, Y. Kawai, N. Kitamoto, T. Osawa and Y. Kato, Chem. Res. Toxicol., 2009, 22, 536–542 Search PubMed.
- S. Diethelm, M. A. Schafroth and E. M. Carreira, Org. Lett., 2014, 16, 3908–3911 CrossRef CAS PubMed.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- D. Blakeslee and M. G. Baines, J. Immunol. Methods, 1976, 13, 305–320 CrossRef CAS PubMed.
- X. Chen, K. Muthoosamy, A. Pfisterer, B. Neumann and T. Weil, Bioconjugate Chem., 2012, 23, 500–508 CrossRef CAS PubMed.
- A. O.-Y. Chan, C.-M. Ho, H.-C. Chong, Y.-C. Leung, J.-S. Huang, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2012, 134, 2589–2598 CrossRef CAS PubMed.
- M. Raj, H. Wu, S. L. Blosser, M. A. Vittoria and P. S. Arora, J. Am. Chem. Soc., 2015, 137, 6932–6940 CrossRef CAS PubMed.
- R. Singudas, S. R. Adusumalli, P. N. Joshi and V. Rai, Chem. Commun., 2015, 51, 473–476 RSC.
- M. Granold, P. Hajieva, M. I. Toşa, F.-D. Irimie and B. Moosmann, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 41–46 CrossRef CAS PubMed.
- M. T. Taylor, J. E. Nelson, M. G. Suero and M. J. Gaunt, Nature, 2018, 562, 563–568 CrossRef CAS PubMed.
- S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 CrossRef CAS PubMed.
- J. Ohata, L. Krishnamoorthy, M. A. Gonzalez, T. Xiao, D. A. Iovan, F. D. Toste, E. W. Miller and C. J. Chang, ACS Cent. Sci., 2020, 6, 32–40 CrossRef CAS PubMed.
- K. S. Williamson, D. J. Michaelis and T. P. Yoon, Chem. Rev., 2014, 114, 8016–8036 CrossRef CAS PubMed.
- C. Wang, Y.-Y. Jiang and C.-Z. Qi, J. Org. Chem., 2017, 82, 9765–9772 CrossRef CAS PubMed.
- J. Kim, B. X. Li, R. Y. C. Huang, J. X. Qiao, W. R. Ewing and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 21260–21266 CrossRef CAS PubMed.
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CrossRef CAS PubMed.
- J. C. Vantourout, S. R. Adusumalli, K. W. Knouse, D. T. Flood, A. Ramirez, N. M. Padial, A. Istrate, K. Maziarz, J. N. deGruyter, R. R. Merchant, J. X. Qiao, M. A. Schmidt, M. J. Deery, M. D. Eastgate, P. E. Dawson, G. J. L. Bernardes and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 17236–17242 CrossRef CAS PubMed.
- D. Gilis, S. Massar, N. J. Cerf and M. Rooman, Genome Biol., 2001, 2, research0049.1 CrossRef PubMed.
- K. Gevaert, P. V. Damme, L. Martens and J. Vandekerckhove, Anal. Biochem., 2005, 345, 18–29 CrossRef CAS PubMed.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- J. M. Antos, J. M. McFarland, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 6301–6308 CrossRef CAS PubMed.
- M. B. Hansen, F. Hubálek, T. Skrydstrup and T. Hoeg-Jensen, Chem. – Eur. J., 2016, 22, 1572–1576 CrossRef CAS PubMed.
- M. Imiołek, G. Karunanithy, W.-L. Ng, A. J. Baldwin, V. Gouverneur and B. G. Davis, J. Am. Chem. Soc., 2018, 140, 1568–1571 CrossRef PubMed.
- C. Perez-Rizquez, O. Abian and J. M. Palomo, Chem. Commun., 2019, 55, 12928–12931 RSC.
- Y. Seki, T. Ishiyama, D. Sasaki, J. Abe, Y. Sohma, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2016, 138, 10798–10801 CrossRef CAS PubMed.
- S. J. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2020, 142, 9112–9118 CrossRef CAS PubMed.
- N. S. Joshi, L. R. Whitaker and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 15942–15943 CrossRef CAS PubMed.
- D. W. Romanini and M. B. Francis, Bioconjugate Chem., 2008, 19, 153–157 CrossRef CAS PubMed.
- J. M. McFarland, N. S. Joshi and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 7639–7644 CrossRef CAS PubMed.
- J. Guo, J. Wang, J. S. Lee and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 6399–6401 CrossRef CAS PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS PubMed.
- J. Ohata, M. K. Miller, C. M. Mountain, F. Vohidov and Z. T. Ball, Angew. Chem., Int. Ed., 2018, 57, 2827–2830 CrossRef CAS PubMed.
- J. M. Hooker, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 3718–3719 CrossRef CAS PubMed.
- T. L. Schlick, Z. Ding, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 3718–3723 CrossRef CAS PubMed.
- J. Gavrilyuk, H. Ban, M. Nagano, W. Hakamata and C. F. Barbas III, Bioconjugate Chem., 2012, 23, 2321–2328 CrossRef CAS PubMed.
- H. Ban, M. Nagano, J. Gavrilyuk, W. Hakamata, T. Inokuma and C. F. Barbas III, Bioconjugate Chem., 2013, 24, 520–532 CrossRef CAS PubMed.
- H. Ban, J. Gavrilyuk and C. F. Barbas III, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed.
- Q.-Y. Hu, M. Allan, R. Adamo, D. Quinn, H. Zhai, G. Wu, K. Clark, J. Zhou, S. Ortiz, B. Wang, E. Danieli, S. Crotti, M. Tontini, G. Brogioni and F. Berti, Chem. Sci., 2013, 4, 3827–3832 RSC.
- D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mével, D. Deniaud, M. Boujtita and S. G. Gouin, J. Am. Chem. Soc., 2018, 140, 17120–17126 CrossRef CAS PubMed.
- K. W. Decoene, K. Unal, A. Staes, O. Zwaenepoel, J. Gettemans, K. Gevaert, J. M. Winne and A. Madder, Chem. Sci., 2022, 13, 5390–5397 RSC.
- S. Sato, K. Nakane and H. Nakamura, Org. Biomol. Chem., 2020, 18, 3664–3668 RSC.
- S. Sato, K. Nakamura and H. Nakamura, ACS Chem. Biol., 2015, 10, 2633–2640 CrossRef CAS PubMed.
- S. Sato, K. Nakamura and H. Nakamura, ChemBioChem, 2017, 18, 475–478 CrossRef CAS PubMed.
- J. F. Keijzer and B. Albada, Bioconjugate Chem., 2020, 31, 2283–2287 CrossRef CAS PubMed.
- S. Sato, M. Matsumura, T. Kadonosono, S. Abe, T. Ueno, H. Ueda and H. Nakamura, Bioconjugate Chem., 2020, 31, 1417–1424 CrossRef CAS PubMed.
- C. Song, K. Liu, Z. Wang, B. Ding, S. Wang, Y. Weng, C.-W. Chiang and A. Lei, Chem. Sci., 2019, 10, 7982–7987 RSC.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- D. E. Fahrney and A. M. Gold, J. Am. Chem. Soc., 1963, 85, 997–1000 CrossRef CAS.
- W. Chen, J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, J. Am. Chem. Soc., 2016, 138, 7353–7364 CrossRef CAS PubMed.
- E. J. Choi, D. Jung, J.-S. Kim, Y. Lee and B. M. Kim, Chem. – Eur. J., 2018, 24, 10948–10952 CrossRef CAS PubMed.
- H. S. Hahm, E. K. Toroitich, A. L. Borne, J. W. Brulet, A. H. Libby, K. Yuan, T. B. Ware, R. L. McCloud, A. M. Ciancone and K.-L. Hsu, Nat. Chem. Biol., 2020, 16, 150–159 CrossRef CAS PubMed.
- H. Jiang, Q. Zhang, Y. Zhang, H. Feng, H. Jiang, F. Pu, R. Yu, Z. Zhong, C. Wang, Y. M. E. Fung, P. Blasco, Y. Li, T. Jiang and X. Li, Chem. Commun., 2022, 58, 7066–7069 RSC.
- J. Dadová, S. R. G. Galan and B. G. Davis, Curr. Opin. Chem. Biol., 2018, 46, 71–81 CrossRef PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- D. H. Strumeyer, W. N. White and D. E. Koshland, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 931–935 CrossRef CAS PubMed.
- H. Weiner, W. N. White, D. G. Hoare and D. E. Koshland, Jr., J. Am. Chem. Soc., 1966, 88, 3851–3859 CrossRef CAS PubMed.
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849–6851 CrossRef CAS PubMed.
- T. J. Holmes, Jr. and R. G. Lawton, J. Am. Chem. Soc., 1977, 99, 1984–1986 CrossRef PubMed.
- R. I. Nathani, P. Moody, V. Chudasama, M. E. B. Smith, R. J. Fitzmaurice and S. Caddick, Chem. Sci., 2013, 4, 3455–3458 RSC.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS PubMed.
- M. Fernández-González, O. Boutureira, G. J. L. Bernardes, J. M. Chalker, M. A. Young, J. C. Errey and B. G. Davis, Chem. Sci., 2010, 1, 709–715 RSC.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed.
- C. L. Windle, K. J. Simmons, J. R. Ault, C. H. Trinh, A. Nelson, A. R. Pearson and A. Berry, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2610–2615 CrossRef CAS PubMed.
- F. Rowan, M. Richards, M. Widya, R. Bayliss and J. Blagg, PLoS One, 2014, 9, e103935 CrossRef PubMed.
- A. M. Freedy, M. J. Matos, O. Boutureira, F. Corzana, A. Guerreiro, P. Akkapeddi, V. J. Somovilla, T. Rodrigues, K. Nicholls, B. Xie, G. Jiménez-Osés, K. M. Brindle, A. A. Neves and G. J. L. Bernardes, J. Am. Chem. Soc., 2017, 139, 18365–18375 CrossRef CAS PubMed.
- T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W.-L. Ng, R. Raj, S. Faulkner, M. R. J. Vallée, A. Phanumartwiwath, O. D. Coleman, M.-L. Thézénas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, aag1465 CrossRef PubMed.
- G. Chen, A. Heim, D. Riether, D. Yee, Y. Milgrom, M. A. Gawinowicz and D. Sames, J. Am. Chem. Soc., 2003, 125, 8130–8133 CrossRef CAS PubMed.
- Y. Takaoka, H. Tsutsumi, N. Kasagi, E. Nakata and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 3273–3280 CrossRef CAS PubMed.
- H. Wakabayashi, M. Miyagawa, Y. Koshi, Y. Takaoka, S. Tsukiji and I. Hamachi, Chem. – Asian J., 2008, 3, 1134–1139 CrossRef CAS PubMed.
- S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Nat. Chem. Biol., 2009, 5, 341–343 CrossRef CAS PubMed.
- T. Tamura, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 2216–2226 CrossRef CAS PubMed.
- S.-h Fujishima, R. Yasui, T. Miki, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 3961–3964 CrossRef CAS PubMed.
- S. Sato and H. Nakamura, Angew. Chem., Int. Ed., 2013, 52, 8681–8684 CrossRef CAS PubMed.
- T. Yamaguchi, M. Asanuma, S. Nakanishi, Y. Saito, M. Okazaki, K. Dodo and M. Sodeoka, Chem. Sci., 2014, 5, 1021–1029 RSC.
- J. Lohse, L. J. Y. M. Swier, R. C. Oudshoorn, G. Médard, B. Kuster, D.-J. Slotboom and M. D. Witte, Bioconjugate Chem., 2017, 28, 913–917 CrossRef CAS PubMed.
- Y. Takaoka, Y. Nishikawa, Y. Hashimoto, K. Sasaki and I. Hamachi, Chem. Sci., 2015, 6, 3217–3224 RSC.
- K. Matsuo, Y. Nishikawa, M. Masuda and I. Hamachi, Angew. Chem., Int. Ed., 2018, 57, 659–662 CrossRef CAS PubMed.
- T. Tamura, T. Ueda, T. Goto, T. Tsukidate, Y. Shapira, Y. Nishikawa, A. Fujisawa and I. Hamachi, Nat. Commun., 2018, 9, 1870 CrossRef PubMed.
- T. Hayashi, Y. Sun, T. Tamura, K. Kuwata, Z. Song, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2013, 135, 12252–12258 CrossRef CAS PubMed.
- T. Hayashi, Y. Yasueda, T. Tamura, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2015, 137, 5372–5380 CrossRef CAS PubMed.
- Y. Amamoto, Y. Aoi, N. Nagashima, H. Suto, D. Yoshidome, Y. Arimura, A. Osakabe, D. Kato, H. Kurumizaka, S. A. Kawashima, K. Yamatsugu and M. Kanai, J. Am. Chem. Soc., 2017, 139, 7568–7576 CrossRef CAS PubMed.
- T. Ishiguro, Y. Amamoto, K. Tanabe, J. Liu, H. Kajino, A. Fujimura, Y. Aoi, A. Osakabe, N. Horikoshi, H. Kurumizaka, K. Yamatsugu, S. A. Kawashima and M. Kanai, Chem, 2017, 2, 840–859 CAS.
- T. Tamura, Z. Song, K. Amaike, S. Lee, S. Yin, S. Kiyonaka and I. Hamachi, J. Am. Chem. Soc., 2017, 139, 14181–14191 CrossRef CAS PubMed.
- Z. Zhang, P. J. Rohweder, C. Ongpipattanakul, K. Basu, M.-F. Bohn, E. J. Dugan, V. Steri, B. Hann, K. M. Shokat and C. S. Craik, Cancer Cell, 2022, 40, 1060–1069.e1067 CrossRef CAS PubMed.
- Z. Zhang, J. Morstein, A. K. Ecker, K. Z. Guiley and K. M. Shokat, J. Am. Chem. Soc., 2022, 144, 15916–15921 CrossRef CAS PubMed.
- Z. Zhang, K. Z. Guiley and K. M. Shokat, Nat. Chem. Biol., 2022, 18, 1177–1183 CrossRef CAS PubMed.
- S. L. Kuan, T. Wang and T. Weil, Chem. – Eur. J., 2016, 22, 17112–17129 CrossRef CAS PubMed.
- E. B. Getz, M. Xiao, T. Chakrabarty, R. Cooke and P. R. Selvin, Anal. Biochem., 1999, 273, 73–80 CrossRef CAS PubMed.
- S. Mitra and R. G. Lawton, J. Am. Chem. Soc., 1979, 101, 3097–3110 CrossRef CAS.
- S. J. Brocchini, M. Eberle and R. G. Lawton, J. Am. Chem. Soc., 1988, 110, 5211–5212 CrossRef CAS.
- F. A. Liberatore, R. D. Comeau, J. M. McKearin, D. A. Pearson, B. Q. Belonga III, S. J. Brocchini, J. Kath, T. Phillips, K. Oswell and R. G. Lawton, Bioconjugate Chem., 1990, 1, 36–50 CrossRef CAS PubMed.
- R. B. Del Rosario, R. L. Wahl, S. J. Brocchini, R. G. Lawton and R. H. Smith, Bioconjugate Chem., 1990, 1, 51–59 CrossRef CAS PubMed.
- S. Brocchini, S. Balan, A. Godwin, J.-W. Choi, M. Zloh and S. Shaunak, Nat. Protoc., 2006, 1, 2241–2252 CrossRef CAS PubMed.
- S. Shaunak, A. Godwin, J.-W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 312–313 CrossRef CAS PubMed.
- D. S. Wilbur, J. E. Stray, D. K. Hamlin, D. K. Curtis and R. L. Vessella, Bioconjugate Chem., 1994, 5, 220–235 CrossRef CAS PubMed.
- S. Balan, J.-w Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Bioconjugate Chem., 2007, 18, 61–76 CrossRef CAS PubMed.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- P. Bryant, M. Pabst, G. Badescu, M. Bird, W. McDowell, E. Jamieson, J. Swierkosz, K. Jurlewicz, R. Tommasi, K. Henseleit, X. Sheng, N. Camper, A. Manin, K. Kozakowska, K. Peciak, E. Laurine, R. Grygorash, A. Kyle, D. Morris, V. Parekh, A. Abhilash, J.-w Choi, J. Edwards, M. Frigerio, M. P. Baker and A. Godwin, Mol. Pharmaceutics, 2015, 12, 1872–1879 CrossRef CAS PubMed.
- T. Wang, A. Pfisterer, S. L. Kuan, Y. Wu, O. Dumele, M. Lamla, K. Müllen and T. Weil, Chem. Sci., 2013, 4, 1889–1894 RSC.
- T. Wang, D. Y. W. Ng, Y. Wu, J. Thomas, T. TamTran and T. Weil, Chem. Commun., 2014, 50, 1116–1118 RSC.
- T. Wang, N. Zabarska, Y. Wu, M. Lamla, S. Fischer, K. Monczak, D. Y. W. Ng, S. Rau and T. Weil, Chem. Commun., 2015, 51, 12552–12555 RSC.
- L. Xu, M. Raabe, M. M. Zegota, J. C. F. Nogueira, V. Chudasama, S. L. Kuan and T. Weil, Org. Biomol. Chem., 2020, 18, 1140–1147 RSC.
- D. A. Richards, S. A. Fletcher, M. Nobles, H. Kossen, L. Tedaldi, V. Chudasama, A. Tinker and J. R. Baker, Org. Biomol. Chem., 2016, 14, 455–459 RSC.
- R. Malde, M. A. Parkes, M. Staniforth, J. M. Woolley, V. G. Stavros, V. Chudasama, H. H. Fielding and J. R. Baker, Chem. Sci., 2022, 13, 2909–2918 RSC.
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruani, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC.
- F. Bryden, A. Maruani, H. Savoie, V. Chudasama, M. E. B. Smith, S. Caddick and R. W. Boyle, Bioconjugate Chem., 2014, 25, 611–617 CrossRef CAS PubMed.
- C. R. Behrens, E. H. Ha, L. L. Chinn, S. Bowers, G. Probst, M. Fitch-Bruhns, J. Monteon, A. Valdiosera, A. Bermudez, S. Liao-Chan, T. Wong, J. Melnick, J.-W. Theunissen, M. R. Flory, D. Houser, K. Venstrom, Z. Levashova, P. Sauer, T.-S. Migone, E. H. van der Horst, R. L. Halcomb and D. Y. Jackson, Mol. Pharmaceutics, 2015, 12, 3986–3998 CrossRef CAS PubMed.
- M. a Maneiro, N. Forte, M. M. Shchepinova, C. S. Kounde, V. Chudasama, J. R. Baker and E. W. Tate, ACS Chem. Biol., 2020, 15, 1306–1312 CrossRef CAS PubMed.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, Chem. Commun., 2012, 48, 4064–4066 RSC.
- L. Castañeda, A. Maruani, F. F. Schumacher, E. Miranda, V. Chudasama, K. A. Chester, J. R. Baker, M. E. B. Smith and S. Caddick, Chem. Commun., 2013, 49, 8187–8189 RSC.
- F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1525 CrossRef PubMed.
- J. P. M. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC.
- F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC.
- R. J. Spears, C. McMahon, M. Shamsabadi, C. Bahou, I. A. Thanasi, L. N. C. Rochet, N. Forte, F. Thoreau, J. R. Baker and V. Chudasama, Chem. Commun., 2022, 58, 645–648 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- M. T. W. Lee, A. Maruani, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2016, 7, 799–802 RSC.
- M. T. W. Lee, A. Maruani, D. A. Richards, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2017, 8, 2056–2060 RSC.
- A. Maruani, P. A. Szijj, C. Bahou, J. C. F. Nogueira, S. Caddick, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2020, 31, 520–529 CrossRef CAS PubMed.
- C. Bahou, P. A. Szijj, R. J. Spears, A. Wall, F. Javaid, A. Sattikar, E. A. Love, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2021, 32, 672–679 CrossRef CAS PubMed.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
- C. Bahou and V. Chudasama, Org. Biomol. Chem., 2022, 20, 5879–5890 RSC.
- A. Maruani, S. Alom, P. Canavelli, M. T. W. Lee, R. E. Morgan, V. Chudasama and S. Caddick, Chem. Commun., 2015, 51, 5279–5282 RSC.
- N. Griebenow, A. M. Dilmaç, S. Greven and S. Bräse, Bioconjugate Chem., 2016, 27, 911–917 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T. L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- A. J. Counsell, S. J. Walsh, N. S. Robertson, H. F. Sore and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4739–4743 RSC.
- S. J. Walsh, J. Iegre, H. Seki, J. D. Bargh, H. F. Sore, J. S. Parker, J. S. Carroll and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4224–4230 RSC.
- M.-A. Kasper, A. Stengl, P. Ochtrop, M. Gerlach, T. Stoschek, D. Schumacher, J. Helma, M. Penkert, E. Krause, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11631–11636 CrossRef CAS PubMed.
- M.-A. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2019, 58, 11625–11630 CrossRef CAS PubMed.
- P. Ochtrop, J. Jahzerah, P. Machui, I. Mai, D. Schumacher, J. Helma, M.-A. Kasper and C. P. R. Hackenberger, Chem. Sci., 2023, 14, 2259–2266 RSC.
- C. E. Stieger, L. Franz, F. Körlin and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2021, 60, 15359–15364 CrossRef CAS PubMed.
- O. Koniev, S. Kolodych, Z. Baatarkhuu, J. Stojko, J. Eberova, J.-Y. Bonnefoy, S. Cianférani, A. Van Dorsselaer and A. Wagner, Bioconjugate Chem., 2015, 26, 1863–1867 CrossRef CAS PubMed.
- S. Kolodych, O. Koniev, Z. Baatarkhuu, J.-Y. Bonnefoy, F. Debaene, S. Cianférani, A. Van Dorsselaer and A. Wagner, Bioconjugate Chem., 2015, 26, 197–200 CrossRef CAS PubMed.
- O. Koniev, I. Dovgan, B. Renoux, A. Ehkirch, J. Eberova, S. Cianférani, S. Kolodych, S. Papot and A. Wagner, Med. Chem. Commun., 2018, 9, 827–830 RSC.
- Y. E. Sim, O. Nwajiobi, S. Mahesh, R. D. Cohen, M. Y. Reibarkh and M. Raj, Chem. Sci., 2020, 11, 53–61 RSC.
- X. Chen, F. Ye, X. Luo, X. Liu, J. Zhao, S. Wang, Q. Zhou, G. Chen and P. Wang, J. Am. Chem. Soc., 2019, 141, 18230–18237 CrossRef CAS PubMed.
- W. Yu and J. M. Baskin, Curr. Opin. Chem. Biol., 2022, 69, 102173 CrossRef CAS PubMed.
- S.-S. Ge, B. Chen, Y.-Y. Wu, Q.-S. Long, Y.-L. Zhao, P.-Y. Wang and S. Yang, RSC Adv., 2018, 8, 29428–29454 RSC.
- S. Jiang, N. Kotani, T. Ohnishi, A. Miyagawa-Yamguchi, M. Tsuda, R. Yamashita, Y. Ishiura and K. Honke, Proteomics, 2012, 12, 54–62 CrossRef CAS PubMed.
- R. A. Hansen, A. Märcher and K. V. Gothelf, Bioconjugate Chem., 2022, 33, 1811–1817 CrossRef CAS PubMed.
- A. Märcher, J. Palmfeldt, M. Nisavic and K. V. Gothelf, Angew. Chem., Int. Ed., 2021, 60, 6539–6544 CrossRef PubMed.
- D. G. Rawale, K. Thakur, P. Sreekumar, S. T. K., R. A., S. R. Adusumalli, R. K. Mishra and V. Rai, Chem. Sci., 2021, 12, 6732–6736 RSC.
- N. C. Reddy, R. Molla, P. N. Joshi, S. T. K., I. Basu, J. Kawadkar, N. Kalra, R. K. Mishra, S. Chakrabarty, S. Shukla and V. Rai, Nat. Commun., 2022, 13, 6038 CrossRef CAS PubMed.
- B. K. Hansen, C. J. Loveridge, S. Thyssen, G. J. Wørmer, A. D. Nielsen, J. Palmfeldt, M. Johannsen and T. B. Poulsen, Angew. Chem., Int. Ed., 2019, 58, 3533–3537 CrossRef CAS PubMed.
- J. Li, Q.-L. Hu, Z. Song, A. S. C. Chan and X.-F. Xiong, Sci. China: Chem., 2022, 65, 1356–1361 CrossRef CAS.
- C. F. Afonso, M. C. Marques, J. P. M. António, C. Cordeiro, P. M. P. Gois, P. M. S. D. Cal and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2022, 61, e202208543 CrossRef CAS PubMed.
- K. Thakur, S. T. K., S. K. Singh, R. V., D. G. Rawale, S. R. Adusumalli, N. Kalra, S. Shukla, R. K. Mishra and V. Rai, Bioconjugate Chem., 2022, 33, 2370–2380 CrossRef CAS PubMed.
- A. K. Adak, K.-T. Huang, C.-Y. Liao, Y.-J. Lee, W.-H. Kuo, Y.-R. Huo, P.-J. Li, Y.-J. Chen, B.-S. Chen, Y.-J. Chen, K. Chu Hwang, W.-S. Wayne Chang and C.-C. Lin, Chem. – Eur. J., 2022, 28, e202104178 CrossRef CAS PubMed.
- B. X. Li, D. K. Kim, S. Bloom, R. Y. C. Huang, J. X. Qiao, W. R. Ewing, D. G. Oblinsky, G. D. Scholes and D. W. C. MacMillan, Nat. Chem., 2021, 13, 902–908 CrossRef CAS PubMed.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- G. Casi, N. Huguenin-Dezot, K. Zuberbühler, J. Scheuermann and D. Neri, J. Am. Chem. Soc., 2012, 134, 5887–5892 CrossRef CAS PubMed.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Chem. Sci., 2016, 7, 5052–5058 RSC.
- A. Bandyopadhyay, S. Cambray and J. Gao, Chem. Sci., 2016, 7, 4589–4593 RSC.
- K. Li, W. Wang and J. Gao, Angew. Chem., Int. Ed., 2020, 59, 14246–14250 CrossRef CAS PubMed.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS PubMed.
- A. Onoda, N. Inoue, E. Sumiyoshi and T. Hayashi, ChemBioChem, 2020, 21, 1274–1278 CrossRef CAS PubMed.
- T. Sahu, M. Chilamari and V. Rai, Chem. Commun., 2022, 58, 1768–1771 RSC.
- J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, Angew. Chem., Int. Ed., 2006, 45, 5307–5311 CrossRef CAS PubMed.
- T. Sahu, M. Kumar, S. T. K., M. Joshi, R. K. Mishra and V. Rai, Chem. Commun., 2022, 58, 12451–12454 RSC.
- L. Purushottam, U. V. B., D. G. Rawale, M. Gujrati, S. D. Mishra, S. T. K., N. C. Reddy, S. R. Adusumalli, R. K. Mishra and V. Rai, Chem. Sci., 2020, 11, 13137–13142 RSC.
- B. Wang, X. Li, Y. Wang, X. Mao and X. Wang, Chem. Commun., 2022, 58, 7408–7411 RSC.
- H. F. Gaertner and R. E. Offord, Bioconjugate Chem., 1996, 7, 38–44 CrossRef CAS PubMed.
- C. W. Lindsey, ACS Chem. Neurosci., 2014, 5, 250–251 CrossRef PubMed.
- J. K. Dozier and M. D. Distefano, Int. J. Mol. Sci., 2015, 16, 25831–25864 CrossRef CAS PubMed.
- D. P. Baker, E. Y. Lin, K. Lin, M. Pellegrini, R. C. Petter, L. L. Chen, R. M. Arduini, M. Brickelmaier, D. Wen, D. M. Hess, L. Chen, D. Grant, A. Whitty, A. Gill, D. J. Lindner and R. B. Pepinsky, Bioconjugate Chem., 2006, 17, 179–188 CrossRef CAS PubMed.
- B. Podobnik, B. Helk, V. Smilović, Š. Škrajnar, K. Fidler, S. Jevševar, A. Godwin and P. Williams, Bioconjugate Chem., 2015, 26, 452–459 CrossRef CAS PubMed.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, J. Pharm. Sci., 2016, 105, 460–475 CrossRef CAS PubMed.
- A. Perols, H. Honarvar, J. Strand, R. Selvaraju, A. Orlova, A. Eriksson Karlström and V. Tolmachev, Bioconjugate Chem., 2012, 23, 1661–1670 CrossRef CAS PubMed.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- S. Tyagi and E. A. Lemke, Methods Cell Biol., 2013, 113, 169–187 CAS.
- A. P. Crochet, M. M. Kabir, M. B. Francis and C. D. Paavola, Biosens. Bioelectron., 2010, 26, 55–61 CrossRef CAS PubMed.
- M. T. Dedeo, K. E. Duderstadt, J. M. Berger and M. B. Francis, Nano Lett., 2010, 10, 181–186 CrossRef CAS PubMed.
- S. Zheng, G. Zhang, J. Li and P. R. Chen, Angew. Chem., Int. Ed., 2014, 53, 6449–6453 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- M. Z. Brian, K. S. Kuntal, R. Thomas, M. Priya, D. C. Sean, Z. Pat, W. Ralph and S. L. Jason, J. Nucl. Med., 2013, 54, 1389 CrossRef PubMed.
- M. Rashidian, E. J. Keliher, M. Dougan, P. K. Juras, M. Cavallari, G. R. Wojtkiewicz, J. T. Jacobsen, J. G. Edens, J. M. J. Tas, G. Victora, R. Weissleder and H. Ploegh, ACS Cent. Sci., 2015, 1, 142–147 CrossRef CAS PubMed.
- M. Yang, Y. Song, M. Zhang, S. Lin, Z. Hao, Y. Liang, D. Zhang and P. R. Chen, Angew. Chem., Int. Ed., 2012, 124, 7794–7799 CrossRef.
- T. Tamura and I. Hamachi, ACS Chem. Biol., 2014, 9, 2708–2717 CrossRef CAS PubMed.
- T. Ueda, T. Tamura and I. Hamachi, ACS Sens., 2018, 3, 527–539 CrossRef CAS PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- K. Wang, Y. Du, Z. Zhang, K. He, Z. Cheng, L. Yin, D. Dong, C. Li, W. Li, Z. Hu, C. Zhang, H. Hui, C. Chi and J. Tian, Nat. Rev. Bioeng., 2023, 1, 161–179 CrossRef.
- J. S. D. Mieog, F. B. Achterberg, A. Zlitni, M. Hutteman, J. Burggraaf, R.-J. Swijnenburg, S. Gioux and A. L. Vahrmeijer, Nat. Rev. Clin. Oncol., 2022, 19, 9–22 CrossRef CAS PubMed.
- S. Hernot, L. van Manen, P. Debie, J. S. D. Mieog and A. L. Vahrmeijer, Lancet Oncol., 2019, 20, e354–e367 CrossRef CAS PubMed.
- I. Dovgan, S. Erb, S. Hessmann, S. Ursuegui, C. Michel, C. Muller, G. Chaubet, S. Cianférani and A. Wagner, Org. Biomol. Chem., 2018, 16, 1305–1311 RSC.
- D. Hwang, N. Nilchan, A. R. Nanna, X. Li, M. D. Cameron, W. R. Roush, H. Park and C. Rader, Cell Chem. Biol., 2019, 26, 1229–1239 CrossRef CAS PubMed.
- L. Liu, J. Pharm. Sci., 2015, 104, 1866–1884 CrossRef CAS PubMed.
- F. Tang, M. Zhou, K. Qin, W. Shi, A. Yashinov, Y. Yang, L. Yang, D. Guan, L. Zhao, Y. Tang, Y. Chang, L. Zhao, H. Yang, H. Zhou, R. Huang and W. Huang, Nat. Chem. Biol., 2020, 16, 766–775 CrossRef CAS PubMed.
- L.-X. Wang, X. Tong, C. Li, J. P. Giddens and T. Li, Annu. Rev. Biochem., 2019, 88, 433–459 CrossRef CAS PubMed.
- P. Akkapeddi, S.-A. Azizi, A. M. Freedy, P. M. S. D. Cal, P. M. P. Gois and G. J. L. Bernardes, Chem. Sci., 2016, 7, 2954–2963 RSC.
- A. C. Stan, D. L. Radu, S. Casares, C. A. Bona and T. D. Brumeanu, Cancer Res., 1999, 59, 115–121 CAS.
- D. J. O'Shannessy and R. H. Quarles, J. Immunol. Methods, 1987, 99, 153–161 CrossRef PubMed.
- K. A. Sapozhnikova, E. L. Gulyak, V. A. Brylev, V. A. Misyurin, S. D. Oreshkov, A. V. Alexeeva, D. Y. Ryazantsev, M. A. Simonova, E. V. Ryabukhina, G. P. Popova, N. A. Tikhonova, N. A. Lyzhko, A. E. Barmashov, A. V. Misyurin, A. V. Ustinov, V. A. Alferova and V. A. Korshun, Int. J. Mol. Sci., 2023, 24 CAS.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- Q. Zhou, J. E. Stefano, C. Manning, J. Kyazike, B. Chen, D. A. Gianolio, A. Park, M. Busch, J. Bird, X. Zheng, H. Simonds-Mannes, J. Kim, R. C. Gregory, R. J. Miller, W. H. Brondyk, P. K. Dhal and C. Q. Pan, Bioconjugate Chem., 2014, 25, 510–520 CrossRef CAS PubMed.
- K. Zuberbühler, G. Casi, G. J. L. Bernardes and D. Neri, Chem. Commun., 2012, 48, 7100–7102 RSC.
- Faridoon, W. Shi, K. Qin, Y. Tang, M. Li, D. Guan, X. Tian, B. Jiang, J. Dong, F. Tang and W. Huang, Org. Chem. Front., 2019, 6, 3144–3149 RSC.
- W. Shi, W. Li, J. Zhang, T. Li, Y. Song, Y. Zeng, Q. Dong, Z. Lin, L. Gong, S. Fan, F. Tang and W. Huang, Acta Pharm. Sin. B, 2022, 12, 2417–2428 CrossRef CAS PubMed.
- F. Tang, L.-X. Wang and W. Huang, Nat. Protoc., 2017, 12, 1702–1721 CrossRef CAS PubMed.
- Y. Wei, C. Li, W. Huang, B. Li, S. Strome and L.-X. Wang, Biochemistry, 2008, 47, 10294–10304 CrossRef CAS PubMed.
- Y. Anami, C. M. Yamazaki, W. Xiong, X. Gui, N. Zhang, Z. An and K. Tsuchikama, Nat. Commun., 2018, 9, 2512 CrossRef PubMed.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grünberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed., 2010, 49, 9995–9997 CrossRef CAS PubMed.
- P. R. Spycher, C. A. Amann, J. E. Wehrmüller, D. R. Hurwitz, O. Kreis, D. Messmer, A. Ritler, A. Küchler, A. Blanc, M. Béhé, P. Walde and R. Schibli, ChemBioChem, 2017, 18, 1923–1927 CrossRef CAS PubMed.
- A. Ebenig, N. E. Juettner, L. Deweid, O. Avrutina, H.-L. Fuchsbauer and H. Kolmar, ChemBioChem, 2019, 20, 2411–2419 CrossRef CAS PubMed.
- W. Huang, J. Giddens, S.-Q. Fan, C. Toonstra and L.-X. Wang, J. Am. Chem. Soc., 2012, 134, 12308–12318 CrossRef CAS PubMed.
- X. Zhang, C. Ou, H. Liu, S. K. Prabhu, C. Li, Q. Yang and L.-X. Wang, ACS Chem. Biol., 2021, 16, 2502–2514 CrossRef CAS PubMed.
- C. Ou, C. Li, R. Zhang, Q. Yang, G. Zong, Y. Dai, R. L. Francis, S. Bournazos, J. V. Ravetch and L.-X. Wang, Bioconjugate Chem., 2021, 32, 1888–1897 CrossRef CAS PubMed.
- W. Guo, F. Tang, K. Qin, M. Zhou, Z. Le and W. Huang, Carbohydr. Res., 2017, 446–447, 32–39 CrossRef CAS PubMed.
- F. Tang, Y. Yang, Y. Tang, S. Tang, L. Yang, B. Sun, B. Jiang, J. Dong, H. Liu, M. Huang, M.-Y. Geng and W. Huang, Org. Biomol. Chem., 2016, 14, 9501–9518 RSC.
- E. Boeggeman, B. Ramakrishnan, M. Pasek, M. Manzoni, A. Puri, K. H. Loomis, T. J. Waybright and P. K. Qasba, Bioconjugate Chem., 2009, 20, 1228–1236 CrossRef CAS PubMed.
- Z. Zhu, B. Ramakrishnan, J. Li, Y. Wang, Y. Feng, P. Prabakaran, S. Colantonio, M. A. Dyba, P. K. Qasba and D. S. Dimitrov, mAbs, 2014, 6, 1190–1200 CrossRef PubMed.
- W. Shi, J. Zhang, L. Liu, W. Li, Z. Liu, A. Ren, J. Wang, C. Tang, Y. Yang, D. Xu, Q. Huang, Y. Wang, C. Luo, W. Huang and F. Tang, J. Med. Chem., 2023, 66, 1011–1026 CrossRef CAS PubMed.
- R. van Geel, M. A. Wijdeven, R. Heesbeen, J. M. M. Verkade, A. A. Wasiel, S. S. van Berkel and F. L. van Delft, Bioconjugate Chem., 2015, 26, 2233–2242 CrossRef CAS PubMed.
- L. de Bever, S. Popal, J. van Schaik, B. Rubahamya, F. L. van Delft, G. M. Thurber and S. S. van Berkel, Bioconjugate Chem., 2023, 34, 538–548 CrossRef CAS PubMed.
- C. Tang, Y. Zeng, J. Zhang, X. Zheng, F. Tang, X. Yao, Z.-X. Jiang, W. Shi and W. Huang, Bioconjugate Chem., 2023, 34, 748–755 CAS.
- M. L. Jaramillo, T. Sulea, Y. Durocher, M. Acchione, M. J. Schur, A. Robotham, J. F. Kelly, M.-F. Goneau, A. Robert, Y. Cepero-Donates and M. Gilbert, mAbs, 2023, 15, 2149057 CrossRef PubMed.
- B. M. Zeglis, C. B. Davis, R. Aggeler, H. C. Kang, A. Chen, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2013, 24, 1057–1067 CrossRef CAS PubMed.
- P. Adumeau, D. Vivier, S. K. Sharma, J. Wang, T. Zhang, A. Chen, B. J. Agnew and B. M. Zeglis, Mol. Pharmaceutics, 2018, 15, 892–898 CrossRef CAS PubMed.
- K. Zheng, C. Bantog and R. Bayer, mAbs, 2011, 3, 568–576 CrossRef PubMed.
- E. Maverakis, K. Kim, M. Shimoda, M. E. Gershwin, F. Patel, R. Wilken, S. Raychaudhuri, L. R. Ruhaak and C. B. Lebrilla, J. Autoimmun., 2015, 57, 1–13 CrossRef CAS PubMed.
- J. N. Arnold, M. R. Wormald, R. B. Sim, P. M. Rudd and R. A. Dwek, Annu. Rev. Immunol., 2007, 25, 21–50 CrossRef CAS PubMed.
- W. Wang, J. Vlasak, Y. Li, P. Pristatsky, Y. Fang, T. Pittman, J. Roman, Y. Wang, T. Prueksaritanont and R. Ionescu, Mol. Immunol., 2011, 48, 860–866 CrossRef CAS PubMed.
- E. B. Irvine and G. Alter, Glycobiology, 2020, 30, 241–253 CrossRef CAS PubMed.
- T. H. Pillow, J. Tien, K. L. Parsons-Reponte, S. Bhakta, H. Li, L. R. Staben, G. Li, J. Chuh, A. Fourie-O’Donohue, M. Darwish, V. Yip, L. Liu, D. D. Leipold, D. Su, E. Wu, S. D. Spencer, B.-Q. Shen, K. Xu, K. R. Kozak, H. Raab, R. Vandlen, G. D. Lewis Phillips, R. H. Scheller, P. Polakis, M. X. Sliwkowski, J. A. Flygare and J. R. Junutula, J. Med. Chem., 2014, 57, 7890–7899 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- S. Fang, B. M. Brems, E. O. Olawode, J. T. Miller, T. A. Brooks and L. N. Tumey, Mol. Pharmaceutics, 2022, 19, 3228–3241 CrossRef CAS PubMed.
- W. Li, K. H. Veale, Q. Qiu, K. W. Sinkevicius, E. K. Maloney, J. A. Costoplus, J. Lau, H. L. Evans, Y. Setiady, O. Ab, S. M. Abbott, J. Lee, S. Wisitpitthaya, A. Skaletskaya, L. Wang, T. A. Keating, R. V. J. Chari and W. C. Widdison, ACS Med. Chem. Lett., 2019, 10, 1386–1392 CrossRef CAS PubMed.
- K. E. Grier, A. H. Hansen, C. S. Haxvig, X. Li, O. Krigslund, N. Behrendt, L. H. Engelholm, F. Rossi, B. C. Sousa, G. J. Harradence, N. Camper and K. M. Qvortrup, Chem. Commun., 2023, 59, 7240–7242 RSC.
- P. M. S. D. Cal, G. J. L. Bernardes and P. M. P. Gois, Angew. Chem., Int. Ed., 2014, 53, 10585–10587 CrossRef CAS PubMed.
- D. V. Hingorani, M. M. Allevato, M. F. Camargo, J. Lesperance, M. A. Quraishi, J. Aguilera, I. Franiak-Pietryga, D. J. Scanderbeg, Z. Wang, A. A. Molinolo, D. Alvarado, A. B. Sharabi, J. D. Bui, E. E. W. Cohen, S. R. Adams, J. S. Gutkind and S. J. Advani, Nat. Commun., 2022, 13, 3869 CrossRef CAS PubMed.
- B.-C. Lee, C. Chalouni, S. Doll, S. C. Nalle, M. Darwish, S. P. Tsai, K. R. Kozak, G. Del-Rosario, S.-F. Yu, H. Erickson and R. Vandlen, Bioconjugate Chem., 2018, 29, 2468–2477 CrossRef CAS PubMed.
- N. Zacharias, V. N. Podust, K. K. Kajihara, D. Leipold, G. Del Rosario, D. Thayer, E. Dong, M. Paluch, D. Fischer, K. Zheng, C. Lei, J. He, C. Ng, D. Su, L. Liu, S. Masih, W. Sawyer, J. Tinianow, J. Marik, V. Yip, G. Li, J. Chuh, J. H. Morisaki, S. Park, B. Zheng, H. Hernandez-Barry, K. M. Loyet, M. Xu, K. R. Kozak, G. L. Phillips, B.-Q. Shen, C. Wu, K. Xu, S.-F. Yu, A. Kamath, R. K. Rowntree, D. Reilly, T. Pillow, A. Polson, V. Schellenberger, W. L. W. Hazenbos and J. Sadowsky, Chem. Sci., 2022, 13, 3147–3160 RSC.
- D. Shinmi, E. Taguchi, J. Iwano, T. Yamaguchi, K. Masuda, J. Enokizono and Y. Shiraishi, Bioconjugate Chem., 2016, 27, 1324–1331 CrossRef CAS PubMed.
- D. Hwang, N. Nilchan, H. Park, R. N. Roy, W. R. Roush and C. Rader, Bioconjugate Chem., 2022, 33, 1192–1200 CrossRef CAS PubMed.
- O. Feuillâtre, C. Gély, S. Huvelle, C. B. Baltus, L. Juen, N. Joubert, A. Desgranges, M.-C. Viaud-Massuard and C. Martin, ACS Omega, 2020, 5, 1557–1565 CrossRef PubMed.
- J. P. M. Nunes, V. Vassileva, E. Robinson, M. Morais, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 24828–24832 RSC.
- J. T. Patterson, S. Asano, X. Li, C. Rader and C. F. Barbas III, Bioconjugate Chem., 2014, 25, 1402–1407 CrossRef CAS PubMed.
- G. C. Kemp, A. C. Tiberghien, N. V. Patel, F. D'Hooge, S. M. Nilapwar, L. R. Adams, S. Corbett, D. G. Williams, J. A. Hartley and P. W. Howard, Bioorg. Med. Chem. Lett., 2017, 27, 1154–1158 CrossRef CAS PubMed.
- D. Zhang, T. H. Pillow, Y. Ma, J. D. Cruz-Chuh, K. R. Kozak, J. D. Sadowsky, G. D. Lewis Phillips, J. Guo, M. Darwish, P. Fan, J. Chen, C. He, T. Wang, H. Yao, Z. Xu, J. Chen, J. Wai, Z. Pei, C. E. C. A. Hop, S. C. Khojasteh and P. S. Dragovich, ACS Med. Chem. Lett, 2016, 7, 988–993 CrossRef CAS PubMed.
- Q. Wu, Q. Xue, J. Li, Q. Zheng, X. Zhao, W. Li, S. Sun, W. Sha, Y. Yang, Y. Yang and P. Li Jie, CCS Chem., 2023, 0, 1–13 Search PubMed.
- M. B. Skovsgaard, T. E. Jeppesen, M. R. Mortensen, C. H. Nielsen, J. Madsen, A. Kjaer and K. V. Gothelf, Bioconjugate Chem., 2019, 30, 881–887 CrossRef CAS PubMed.
- O. P. Aigbogun, C. P. Phenix, E. S. Krol and E. W. Price, Mol. Pharmaceutics, 2023, 20, 853–874 CrossRef CAS PubMed.
- A. R. Nanna, X. Li, E. Walseng, L. Pedzisa, R. S. Goydel, D. Hymel, T. R. Burke Jr, W. R. Roush and C. Rader, Nat. Commun., 2017, 8, 1112 CrossRef PubMed.
- C. W. Gundlach, Iv, A. Caivano, M. da Graca Cabreira-Hansen, A. Gahremanpour, W. S. Brown, Y. Zheng, B. W. McIntyre, J. T. Willerson, R. A. F. Dixon, E. C. Perin and D. G. Woodside, Bioconjugate Chem., 2011, 22, 1706–1714 CrossRef CAS PubMed.
- A. C. Flor, J. H. Williams, K. M. Blaine, R. C. Duggan, A. I. Sperling, D. A. Schwartz and S. J. Kron, ChemBioChem, 2014, 15, 267–275 CrossRef CAS PubMed.
- Y. Matsuda, K. Yamada, T. Okuzumi and B. A. Mendelsohn, Org. Process Res. Dev., 2019, 23, 2647–2654 CrossRef CAS.
- P. R. Hamann, L. M. Hinman, I. Hollander, C. F. Beyer, D. Lindh, R. Holcomb, W. Hallett, H.-R. Tsou, J. Upeslacis, D. Shochat, A. Mountain, D. A. Flowers and I. Bernstein, Bioconjugate Chem., 2002, 13, 47–58 CrossRef CAS PubMed.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- J. F. Ponte, O. Ab, L. Lanieri, J. Lee, J. Coccia, L. M. Bartle, M. Themeles, Y. Zhou, J. Pinkas and R. Ruiz-Soto, Neoplasia, 2016, 18, 775–784 CrossRef CAS PubMed.
- W. M. McGee, M. Mentinova and S. A. McLuckey, J. Am. Chem. Soc., 2012, 134, 11412–11414 CrossRef CAS PubMed.
- K. Sato, A. P. Gorka, T. Nagaya, M. S. Michie, R. R. Nani, Y. Nakamura, V. L. Coble, O. V. Vasalatiy, R. E. Swenson, P. L. Choyke, M. J. Schnermann and H. Kobayashi, Bioconjugate Chem., 2016, 27, 404–413 CrossRef CAS PubMed.
- J. Chen, C. Zhang, Y. Guo, X. Chang, R. Ma, X. Ye, H. Cheng, Y. Li and H. Cui, World J. Surg. Oncol., 2020, 18, 66 CrossRef PubMed.
- C. Cilliers, I. Nessler, N. Christodolu and G. M. Thurber, Mol. Pharmaceutics, 2017, 14, 1623–1633 CrossRef CAS PubMed.
- S. Kishimoto, Y. Nakashimada, R. Yokota, T. Hatanaka, M. Adachi and Y. Ito, Bioconjugate Chem., 2019, 30, 698–702 CrossRef CAS PubMed.
- Y. Matsuda, C. Clancy, Z. Tawfiq, V. Robles and B. A. Mendelsohn, ACS Omega, 2019, 4, 20564–20570 CrossRef CAS PubMed.
- K. Yamada, N. Shikida, K. Shimbo, Y. Ito, Z. Khedri, Y. Matsuda and B. A. Mendelsohn, Angew. Chem., Int. Ed., 2019, 58, 5592–5597 CrossRef CAS PubMed.
- T. Fujii, Y. Matsuda, T. Seki, N. Shikida, Y. Iwai, Y. Ooba, K. Takahashi, M. Isokawa, S. Kawaguchi, N. Hatada, T. Watanabe, R. Takasugi, A. Nakayama, K. Shimbo, B. A. Mendelsohn, T. Okuzumi and K. Yamada, Bioconjugate Chem., 2023, 34, 728–738 CAS.
- Y. Matsuda, T. Seki, K. Yamada, Y. Ooba, K. Takahashi, T. Fujii, S. Kawaguchi, T. Narita, A. Nakayama, Y. Kitahara, B. A. Mendelsohn and T. Okuzumi, Mol. Pharmaceutics, 2021, 18, 4058–4066 CrossRef CAS PubMed.
- M. P. Luciano, I. Dingle, S. Nourian and M. J. Schnermann, Tetrahedron Lett., 2021, 75, 153211 CrossRef CAS PubMed.
- G. H. Pham, W. Ou, B. Bursulaya, M. DiDonato, A. Herath, Y. Jin, X. Hao, J. Loren, G. Spraggon, A. Brock, T. Uno, B. H. Geierstanger and S. E. Cellitti, ChemBioChem, 2018, 19, 799–804 CrossRef CAS PubMed.
- D. Poplinger, M. Bokan, A. Hesin, E. Thankarajan, H. Tuchinsky, G. Gellerman and L. Patsenker, Bioconjugate Chem., 2021, 32, 1641–1651 CrossRef CAS PubMed.
- D. A. East, D. P. Mulvihill, M. Todd and I. J. Bruce, Langmuir, 2011, 27, 13888–13896 CrossRef CAS PubMed.
- D. S. Wilbur, M.-K. Chyan, H. Nakamae, Y. Chen, D. K. Hamlin, E. B. Santos, B. T. Kornblit and B. M. Sandmaier, Bioconjugate Chem., 2012, 23, 409–420 CrossRef CAS PubMed.
- L. Petri, P. A. Szijj, Á. Kelemen, T. Imre, Á. Gömöry, M. T. W. Lee, K. Hegedűs, P. Ábrányi-Balogh, V. Chudasama and G. M. Keserű, RSC Adv., 2020, 10, 14928–14936 RSC.
- I. Dovgan, S. Ursuegui, S. Erb, C. Michel, S. Kolodych, S. Cianférani and A. Wagner, Bioconjugate Chem., 2017, 28, 1452–1457 CrossRef CAS PubMed.
- B. Li, W. Zhao, X. Zhang, J. Wang, X. Luo, S. D. Baker, C. T. Jordan and Y. Dong, Biorg. Med. Chem., 2016, 24, 5855–5860 CrossRef CAS PubMed.
- S. Yi, S. Wei, Q. Wu, H. Wang and Z.-J. Yao, Angew. Chem., Int. Ed., 2022, 61, e202111783 CrossRef CAS PubMed.
- C. Yu, J. Tang, A. Loredo, Y. Chen, S. Y. Jung, A. Jain, A. Gordon and H. Xiao, Bioconjugate Chem., 2018, 29, 3522–3526 CrossRef CAS PubMed.
- E. von Witting, S. Hober and S. Kanje, Bioconjugate Chem., 2021, 32, 1515–1524 CrossRef CAS PubMed.
- Y. Zeng, W. Shi, Q. Dong, W. Li, J. Zhang, X. Ren, C. Tang, B. Liu, Y. Song, Y. Wu, X. Diao, H. Zhou, H. Huang, F. Tang and W. Huang, Angew. Chem., Int. Ed., 2022, 61, e202204132 CrossRef CAS PubMed.
- S. K. Elledge, H. L. Tran, A. H. Christian, V. Steri, B. Hann, F. D. Toste, C. J. Chang and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5733–5740 CrossRef CAS PubMed.
- H. Tagawa, K. Maruyama, K. Sasaki, N. Konoue, A. Kishimura, M. Kanai, T. Mori, K. Oisaki and Y. Katayama, RSC Adv., 2020, 10, 16727–16731 RSC.
- K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef CAS PubMed.
- M. S. Kang, T. W. S. Kong, J. Y. X. Khoo and T.-P. Loh, Chem. Sci., 2021, 12, 13613–13647 RSC.
- A. S. Mackay, R. J. Payne and L. R. Malins, J. Am. Chem. Soc., 2022, 144, 23–41 CrossRef CAS PubMed.
- S. Sato, M. Matsumura, H. Ueda and H. Nakamura, Chem. Commun., 2021, 57, 9760–9763 RSC.
- J. J. Bruins, J. A. M. Damen, M. A. Wijdeven, L. P. W. M. Lelieveldt, F. L. van Delft and B. Albada, Bioconjugate Chem., 2021, 32, 2167–2172 CrossRef CAS PubMed.
- B. Albada, J. F. Keijzer, H. Zuilhof and F. van Delft, Chem. Rev., 2021, 121, 7032–7058 CrossRef CAS PubMed.
- P. Dennler, E. Fischer and R. Schibli, Antibodies, 2015, 4, 197–224 CrossRef.
- T. Fujii, C. Reiling, C. Quinn, M. Kliman, B. A. Mendelsohn and Y. Matsuda, Explor. Targeted Anti-Tumor Ther., 2021, 2, 576–585 CAS.
- N. K. Damle, Nat. Biotechnol., 2008, 26, 884–885 CrossRef CAS PubMed.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS PubMed.
- R. J. Sanderson, M. A. Hering, S. F. James, M. M. C. Sun, S. O. Doronina, A. W. Siadak, P. D. Senter and A. F. Wahl, Clin. Cancer Res., 2005, 11, 843–852 CrossRef CAS PubMed.
- D. Sussman, L. M. Smith, M. E. Anderson, S. Duniho, J. H. Hunter, H. Kostner, J. B. Miyamoto, A. Nesterova, L. Westendorf, H. A. Van Epps, N. Whiting and D. R. Benjamin, Mol. Cancer Ther., 2014, 13, 2991–3000 CrossRef CAS PubMed.
- D. M. Goldenberg, T. M. Cardillo, S. V. Govindan, E. A. Rossi and R. M. Sharkey, Oncotarget, 2015, 6, 22496–22512 CrossRef PubMed.
- D. Dornan, F. Bennett, Y. Chen, M. Dennis, D. Eaton, K. Elkins, D. French, M. A. T. Go, A. Jack, J. R. Junutula, H. Koeppen, J. Lau, J. McBride, A. Rawstron, X. Shi, N. Yu, S.-F. Yu, P. Yue, B. Zheng, A. Ebens and A. G. Polson, Blood, 2009, 114, 2721–2729 CAS.
- S. Modi, C. Saura, T. Yamashita, Y. H. Park, S.-B. Kim, K. Tamura, F. Andre, H. Iwata, Y. Ito, J. Tsurutani, J. Sohn, N. Denduluri, C. Perrin, K. Aogi, E. Tokunaga, S.-A. Im, K. S. Lee, S. A. Hurvitz, J. Cortes, C. Lee, S. Chen, L. Zhang, J. Shahidi, A. Yver and I. Krop, N. Engl. J. Med., 2019, 382, 610–621 CrossRef PubMed.
- J. T. W. Tong, P. W. R. Harris, M. A. Brimble and I. Kavianinia, Molecules, 2021, 26, 5847 CrossRef CAS PubMed.
- Y. Nakahara, B. A. Mendelsohn and Y. Matsuda, Org. Process Res. Dev., 2022, 26, 2766–2770 CrossRef CAS.
- C. Zhang, P. Dai, A. A. Vinogradov, Z. P. Gates and B. L. Pentelute, Angew. Chem., Int. Ed., 2018, 57, 6459–6463 CrossRef CAS PubMed.
- M.-A. Kasper, M. Glanz, A. Oder, P. Schmieder, J. P. von Kries and C. P. R. Hackenberger, Chem. Sci., 2019, 10, 6322–6329 RSC.
- G. Liu, G. Shi, H. Sheng, Y. Jiang, H. Liang and S. Liu, Angew. Chem., Int. Ed., 2017, 56, 8686–8691 CrossRef CAS PubMed.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- L. He, L. Wang, Z. Wang, T. Li, H. Chen, Y. Zhang, Z. Hu, D. S. Dimitrov, J. Du and X. Liao, J. Med. Chem., 2021, 64, 15716–15726 CrossRef CAS PubMed.
- M. Steiner, I. Hartmann, E. Perrino, G. Casi, S. Brighton, I. Jelesarov, G. J. L. Bernardes and D. Neri, Chem. Sci., 2013, 4, 297–302 RSC.
- R. Tessier, R. K. Nandi, B. G. Dwyer, D. Abegg, C. Sornay, J. Ceballos, S. Erb, S. Cianférani, A. Wagner, G. Chaubet, A. Adibekian and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 10961–10970 CrossRef CAS PubMed.
- E. M. D. Allouche, E. Grinhagena and J. Waser, Angew. Chem., Int. Ed., 2022, 61, e202112287 CrossRef CAS PubMed.
- J. T. Patterson, E. Gros, H. Zhou, G. Atassi, L. Kerwin, L. Carmody, T. Zhu, B. Jones, Y. Fu and G. F. Kaufmann, Bioorg. Med. Chem. Lett., 2017, 27, 3647–3652 CrossRef CAS PubMed.
- N. Forte, V. Chudasama and J. R. Baker, Drug Discovery Today: Technol., 2018, 30, 11–20 CrossRef PubMed.
- L.-H. Wu, S. Zhou, Q.-F. Luo, J.-S. Tian and T.-P. Loh, Org. Lett., 2020, 22, 8193–8197 CrossRef CAS PubMed.
- Q.-Y. Hu and H. Imase, Methods for making conjugates from disulfide-containing proteins, WO2014/083505, 2014 Search PubMed.
- Q.-Y. Hu and M. Allan, Method for making conjugates from disulfide-containing proteins, US2019/0381126, 2019 Search PubMed.
- B. Shi, M. Wu, Z. Li, Z. Xie, X. Wei, J. Fan, Y. Xu, D. Ding, S. H. Akash, S. Chen and S. Cao, Cancer Med., 2019, 8, 1793–1805 CrossRef CAS PubMed.
- A. R. Hanby, S. J. Walsh, A. J. Counsell, N. Ashman, K. T. Mortensen, J. S. Carroll and D. R. Spring, Chem. Commun., 2022, 58, 9401–9404 RSC.
- C. E. Stieger, Y. Park, M. A. R. de Geus, D. Kim, C. Huhn, J. S. Slenczka, P. Ochtrop, J. M. Müchler, R. D. Süssmuth, J. Broichhagen, M.-H. Baik and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2022, 61, e202205348 CrossRef CAS PubMed.
- M.-A. Kasper, L. Lassak, A. M. Vogl, I. Mai, J. Helma, D. Schumacher and C. P. R. Hackenberger, Eur. J. Org. Chem., 2022, e202101389 CrossRef CAS.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- C. Martin, G. Brachet, C. Colas, E. Allard-Vannier, C. Kizlik-Masson, C. Esnault, R. Respaud, C. Denevault-Sabourin, I. Chourpa, V. Gouilleux-Gruart, M.-C. Viaud-Massuard and N. Joubert, Pharmaceuticals, 2019, 12, 176 CrossRef CAS PubMed.
- D. Wei, Y. Jiang, Y. Mao, Z. Xu, J. Chen, X. Gao, J. Li, B. Jiang and H. Chen, Bioorg. Chem., 2023, 134, 106463 CrossRef CAS PubMed.
- A. Chrzastek, I. A. Thanasi, J. A. Irving, V. Chudasama and J. R. Baker, Chem. Sci., 2022, 13, 11533–11539 RSC.
- M. Nisavic, G. J. Wørmer, C. S. Nielsen, S. M. Jeppesen, J. Palmfeldt and T. B. Poulsen, Bioconjugate Chem., 2023, 34, 994–1003 CrossRef CAS PubMed.
- E. Khozeimeh Sarbisheh, G. Dewaele-Le Roi, W. E. Shannon, S. Tan, Y. Xu, B. M. Zeglis and E. W. Price, Bioconjugate Chem., 2020, 31, 2789–2806 CrossRef CAS PubMed.
- N. Gupta, J. Kancharla, S. Kaushik, A. Ansari, S. Hossain, R. Goyal, M. Pandey, J. Sivaccumar, S. Hussain, A. Sarkar, A. Sengupta, S. K. Mandal, M. Roy and S. Sengupta, Chem. Sci., 2017, 8, 2387–2395 RSC.
- K. Nakane, S. Sato, T. Niwa, M. Tsushima, S. Tomoshige, H. Taguchi, M. Ishikawa and H. Nakamura, J. Am. Chem. Soc., 2021, 143, 7726–7731 CrossRef CAS PubMed.
- K. Fukunaga, T. Watanabe, D. Novitasari, H. Ohashi, R. Abe and T. Hohsaka, Chem. Commun., 2018, 54, 12734–12737 RSC.
- M. R. Mortensen, M. B. Skovsgaard, A. H. Okholm, C. Scavenius, D. M. Dupont, C. B. Rosen, J. J. Enghild, J. Kjems and K. V. Gothelf, Bioconjugate Chem., 2018, 29, 3016–3025 CrossRef CAS PubMed.
- C.-H. Chou and P.-C. Lin, Biomacromolecules, 2018, 19, 3086–3095 CrossRef CAS PubMed.
- R. A. Scheck and M. B. Francis, ACS Chem. Biol., 2007, 2, 247–251 CrossRef CAS PubMed.
- K.-L. Wu, C. Yu, C. Lee, C. Zuo, Z. T. Ball and H. Xiao, Bioconjugate Chem., 2021, 32, 1947–1959 CrossRef CAS PubMed.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C.-H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS PubMed.
- C. Sornay, S. Hessmann, S. Erb, I. Dovgan, A. Ehkirch, T. Botzanowski, S. Cianférani, A. Wagner and G. Chaubet, Chem. – Eur. J., 2020, 26, 13797–13805 CrossRef CAS PubMed.
- F. W. Hunter, H. R. Barker, B. Lipert, F. Rothé, G. Gebhart, M. J. Piccart-Gebhart, C. Sotiriou and S. M. F. Jamieson, Br. J. Cancer, 2020, 122, 603–612 CrossRef CAS PubMed.
- F. Loganzo, M. Sung and H.-P. Gerber, Mol. Cancer Ther., 2016, 15, 2825–2834 CrossRef CAS PubMed.
- P. R. Pohlmann, I. A. Mayer and R. Mernaugh, Clin. Cancer Res., 2009, 15, 7479–7491 CrossRef CAS PubMed.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- T. Fojo, Drug Resist. Updat., 2007, 10, 59–67 CrossRef CAS PubMed.
- O. Martínez-Sáez and A. Prat, JCO Oncol. Pract., 2021, 17, 594–604 CrossRef PubMed.
- E. A. Mittendorf, Y. Wu, M. Scaltriti, F. Meric-Bernstam, K. K. Hunt, S. Dawood, F. J. Esteva, A. U. Buzdar, H. Chen, S. Eksambi, G. N. Hortobagyi, J. Baselga and A. M. Gonzalez-Angulo, Clin. Cancer Res., 2009, 15, 7381–7388 CrossRef CAS PubMed.
- F. Loganzo, X. Tan, M. Sung, G. Jin, J. S. Myers, E. Melamud, F. Wang, V. Diesl, M. T. Follettie, S. Musto, M.-H. Lam, W. Hu, M. B. Charati, K. Khandke, K. S. K. Kim, M. Cinque, J. Lucas, E. Graziani, A. Maderna, C. J. O'Donnell, K. T. Arndt and H.-P. Gerber, Mol. Cancer Ther., 2015, 14, 952–963 CrossRef CAS PubMed.
- M. A. Molina, J. Codony-Servat, J. Albanell, F. Rojo, J. N. Arribas and J. Baselga, Cancer Res., 2001, 61, 4744–4749 CAS.
- R. Ghosh, A. Narasanna, S. E. Wang, S. Liu, A. Chakrabarty, J. M. Balko, A. M. González-Angulo, G. B. Mills, E. Penuel, J. Winslow, J. Sperinde, R. Dua, S. Pidaparthi, A. Mukherjee, K. Leitzel, W. J. Kostler, A. Lipton, M. Bates and C. L. Arteaga, Cancer Res., 2011, 71, 1871–1882 CrossRef CAS PubMed.
- M. Luque-Cabal, P. García-Teijido, Y. Fernández-Pérez, L. Sánchez-Lorenzo and I. Palacio-Vázquez, Clin. Med. Insights: Oncol., 2016, 10, 21–30 CAS.
- G. Li, J. Guo, B.-Q. Shen, D. B. Yadav, M. X. Sliwkowski, L. M. Crocker, J. A. Lacap and G. D. L. Phillips, Mol. Cancer Ther., 2018, 17, 1441–1453 CrossRef CAS PubMed.
- P. Müller, M. Kreuzaler, T. Khan, D. S. Thommen, K. Martin, K. Glatz, S. Savic, N. Harbeck, U. Nitz, O. Gluz, M. von Bergwelt-Baildon, H. Kreipe, S. Reddy, M. Christgen and A. Zippelius, Sci. Transl. Med., 2015, 7, 315ra188 Search PubMed.
- C. D. Austin, A. M. De Mazière, P. I. Pisacane, S. M. van Dijk, C. Eigenbrot, M. X. Sliwkowski, J. Klumperman and R. H. Scheller, Mol. Biol. Cell, 2004, 15, 5268–5282 CrossRef CAS PubMed.
- Y.-C. Chung, J.-F. Kuo, W.-C. Wei, K.-J. Chang and W.-T. Chao, PLoS One, 2015, 10, e0133072 CrossRef PubMed.
- H. K. Erickson, P. U. Park, W. C. Widdison, Y. V. Kovtun, L. M. Garrett, K. Hoffman, R. J. Lutz, V. S. Goldmacher and W. A. Blättler, Cancer Res., 2006, 66, 4426–4433 CrossRef CAS PubMed.
- K. Kinneer, J. Meekin, A. C. Tiberghien, Y.-T. Tai, S. Phipps, C. M. Kiefer, M. C. Rebelatto, N. Dimasi, A. Moriarty, K. P. Papadopoulos, S. Sridhar, S. J. Gregson, M. J. Wick, L. Masterson, K. C. Anderson, R. Herbst, P. W. Howard and D. A. Tice, Clin. Cancer Res., 2018, 24, 6570–6582 CrossRef CAS PubMed.
- H. Wang, W. Wang, Y. Xu, Y. Yang, X. Chen, H. Quan and L. Lou, Cancer Sci., 2017, 108, 1458–1468 CrossRef CAS PubMed.
- Y. V. Kovtun, C. A. Audette, M. F. Mayo, G. E. Jones, H. Doherty, E. K. Maloney, H. K. Erickson, X. Sun, S. Wilhelm, O. Ab, K. C. Lai, W. C. Widdison, B. Kellogg, H. Johnson, J. Pinkas, R. J. Lutz, R. Singh, V. S. Goldmacher and R. V. J. Chari, Cancer Res., 2010, 70, 2528–2537 CrossRef CAS PubMed.
- R. Visconti, R. Della Monica and D. Grieco, J. Exp. Clin. Cancer Res., 2016, 35, 153 CrossRef PubMed.
- S. Loibl, I. Majewski, V. Guarneri, V. Nekljudova, E. Holmes, E. Bria, C. Denkert, C. Schem, C. Sotiriou, S. Loi, M. Untch, P. Conte, R. Bernards, M. Piccart, G. von Minckwitz and J. Baselga, Ann. Oncol., 2016, 27, 1519–1525 CrossRef CAS PubMed.
- E. A. Perez, S. L. de Haas, W. Eiermann, C. H. Barrios, M. Toi, Y.-H. Im, P. F. Conte, M. Martin, T. Pienkowski, X. B. Pivot, H. A. Burris, S. Stanzel, M. Patre and P. A. Ellis, BMC Cancer, 2019, 19, 517 CrossRef PubMed.
- S.-B. Kim, H. Wildiers, I. E. Krop, M. Smitt, R. Yu, S. Lysbet de Haas and A. Gonzalez-Martin, Int. J. Cancer, 2016, 139, 2336–2342 CrossRef CAS PubMed.
- J. Baselga, G. D. Lewis Phillips, S. Verma, J. Ro, J. Huober, A. E. Guardino, M. K. Samant, S. Olsen, S. L. de Haas and M. D. Pegram, Clin. Cancer Res., 2016, 22, 3755–3763 CrossRef CAS PubMed.
- A. F. Labrijn, M. L. Janmaat, J. M. Reichert and P. W. H. I. Parren, Nat. Rev. Drug Discovery, 2019, 18, 585–608 CrossRef CAS PubMed.
- R. Yuste, Nat. Methods, 2005, 2, 902–904 CrossRef CAS PubMed.
- M. R. Bell, M. J. Engleka, A. Malik and J. E. Strickler, Protein Sci., 2013, 22, 1466–1477 CrossRef CAS PubMed.
- M. D. Witte, J. J. Cragnolini, S. K. Dougan, N. C. Yoder, M. W. Popp and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11993–11998 CrossRef CAS PubMed.
- B. S. Lee, Y. Lee, J. Park, B. S. Jeong, M. Jo, S. T. Jung and T. H. Yoo, J. Biol. Eng., 2019, 13, 56 CrossRef PubMed.
- M. J. Glennie, H. M. McBride, A. T. Worth and G. T. Stevenson, J. Immunol. Res., 1987, 139, 2367–2375 CAS.
- J. M. Scheer, W. Sandoval, J. M. Elliott, L. Shao, E. Luis, S.-C. Lewin-Koh, G. Schaefer and R. Vandlen, PLoS One, 2012, 7, e51817 CrossRef CAS PubMed.
- M. M. Lorenzo, C. G. Decker, M. U. Kahveci, S. J. Paluck and H. D. Maynard, Macromolecules, 2016, 49, 30–37 CrossRef CAS PubMed.
- A. Natarajan, W. Du, C.-Y. Xiong, G. L. DeNardo, S. J. DeNardo and J. Gervay-Hague, Chem. Commun., 2007, 695–697 RSC.
- N. S. Hatzakis, H. Engelkamp, K. Velonia, J. Hofkens, P. C. M. Christianen, A. Svendsen, S. A. Patkar, J. Vind, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Chem. Commun., 2006, 2012–2014 RSC.
- C. J. White and J. W. Bode, ACS Cent. Sci., 2018, 4, 197–206 CrossRef CAS PubMed.
- M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2018, 140, 7065–7069 CrossRef CAS PubMed.
- K. K.-Y. Kung, H.-M. Ko, J.-F. Cui, H.-C. Chong, Y.-C. Leung and M.-K. Wong, Chem. Commun., 2014, 50, 11899–11902 RSC.
- M. A. Waddington, X. Zheng, J. M. Stauber, E. Hakim Moully, H. R. Montgomery, L. M. A. Saleh, P. Král and A. M. Spokoyny, J. Am. Chem. Soc., 2021, 143, 8661–8668 CrossRef CAS PubMed.
- K. Hanaya, J. Ohata, M. K. Miller, A. E. Mangubat-Medina, M. J. Swierczynski, D. C. Yang, R. M. Rosenthal, B. V. Popp and Z. T. Ball, Chem. Commun., 2019, 55, 2841–2844 RSC.
- H. H. Dhanjee, A. Saebi, I. Buslov, A. R. Loftis, S. L. Buchwald and B. L. Pentelute, J. Am. Chem. Soc., 2020, 142, 9124–9129 CrossRef CAS PubMed.
- J. S. Foot, F. E. Lui and R. Kluger, Chem. Commun., 2009, 7315–7317 RSC.
- Y. Yang and R. Kluger, Chem. Commun., 2010, 46, 7557–7559 RSC.
- R. J. Lee, Q. Fang, P. A. Davol, Y. Gu, R. E. Sievers, R. C. Grabert, J. M. Gall, E. Tsang, M. S. Yee, H. Fok, N. F. Huang, J. F. Padbury, J. W. Larrick and L. G. Lum, Stem Cells, 2007, 25, 712–717 CrossRef CAS PubMed.
- M. Yankelevich, S. V. Kondadasula, A. Thakur, S. Buck, N.-K. V. Cheung and L. G. Lum, Pediatr. Blood Cancer, 2012, 59, 1198–1205 CrossRef PubMed.
- J. Liu, L. Cai, W. Sun, R. Cheng, N. Wang, L. Jin, S. Rozovsky, I. B. Seiple and L. Wang, Angew. Chem., Int. Ed., 2019, 58, 18839–18843 CrossRef CAS PubMed.
- H. Khalili, A. Godwin, J.-w Choi, R. Lever, P. T. Khaw and S. Brocchini, Bioconjugate Chem., 2013, 24, 1870–1882 CrossRef CAS PubMed.
- E. A. Hull, M. Livanos, E. Miranda, M. E. B. Smith, K. A. Chester and J. R. Baker, Bioconjugate Chem., 2014, 25, 1395–1401 CrossRef CAS PubMed.
- N. Forte, M. Livanos, E. Miranda, M. Morais, X. Yang, V. S. Rajkumar, K. A. Chester, V. Chudasama and J. R. Baker, Bioconjugate Chem., 2018, 29, 486–492 CrossRef CAS PubMed.
- J. T. Patterson, J. Isaacson, L. Kerwin, G. Atassi, R. Duggal, D. Bresson, T. Zhu, H. Zhou, Y. Fu and G. F. Kaufmann, Bioorg. Med. Chem. Lett., 2017, 27, 5490–5495 CrossRef CAS PubMed.
- A. L. Baumann, S. Schwagerus, K. Broi, K. Kemnitz-Hassanin, C. E. Stieger, N. Trieloff, P. Schmieder and C. P. R. Hackenberger, J. Am. Chem. Soc., 2020, 142, 9544–9552 CrossRef CAS PubMed.
- R. Lonsdale and R. A. Ward, Chem. Soc. Rev., 2018, 47, 3816–3830 RSC.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- B. R. Lanning, L. R. Whitby, M. M. Dix, J. Douhan, A. M. Gilbert, E. C. Hett, T. O. Johnson, C. Joslyn, J. C. Kath, S. Niessen, L. R. Roberts, M. E. Schnute, C. Wang, J. J. Hulce, B. Wei, L. O. Whiteley, M. M. Hayward and B. F. Cravatt, Nat. Chem. Biol., 2014, 10, 760–767 CrossRef CAS PubMed.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- M. E. Abbasov, M. E. Kavanagh, T.-A. Ichu, M. R. Lazear, Y. Tao, V. M. Crowley, C. W. am Ende, S. M. Hacker, J. Ho, M. M. Dix, R. Suciu, M. M. Hayward, L. L. Kiessling and B. F. Cravatt, Nat. Chem., 2021, 13, 1081–1092 CrossRef CAS PubMed.
- E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne Jr., C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef CAS PubMed.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- N. N. Gushwa, S. Kang, J. Chen and J. Taunton, J. Am. Chem. Soc., 2012, 134, 20214–20217 CrossRef CAS PubMed.
- T. Yang, A. Cuesta, X. Wan, G. B. Craven, B. Hirakawa, P. Khamphavong, J. R. May, J. C. Kath, J. D. Lapek, S. Niessen, A. L. Burlingame, J. D. Carelli and J. Taunton, Nat. Chem. Biol., 2022, 18, 934–941 CrossRef CAS PubMed.
- R. Molla, P. N. Joshi, N. C. Reddy, D. Biswas and V. Rai, Org. Lett., 2023, 25, 6385–6390 CrossRef CAS PubMed.
- S. L. Scinto, T. R. Reagle and J. M. Fox, Angew. Chem., Int. Ed., 2022, 61, e202207661 CrossRef CAS PubMed.
- M. M. Zegota, T. Wang, C. Seidler, D. Y. Wah Ng, S. L. Kuan and T. Weil, Bioconjugate Chem., 2018, 29, 2665–2670 CrossRef CAS PubMed.
- C. B. Rosen, R. L. Kwant, J. I. MacDonald, M. Rao and M. B. Francis, Angew. Chem., Int. Ed., 2016, 55, 8585–8589 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |