
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An anti-poisoning defective catalyst without metal active sites for NH3-SCR via in situ stabilization†
Ge
Li
a,
Baodong
Wang
 *ab,
Ziran
Ma
*a,
Jing
Ma
a,
Hongyan
Wang
a,
Jiali
Zhou
a,
Shengpan
Peng
a,
Jessica Jein
White
c,
Yonglong
Li
a,
Jingyun
Chen
a,
Zhihua
Han
a,
Hui
Wei
a,
Chuang
Peng
d,
Yujie
Xiong
*e and
Yun
Wang
*c
*ab,
Ziran
Ma
*a,
Jing
Ma
a,
Hongyan
Wang
a,
Jiali
Zhou
a,
Shengpan
Peng
a,
Jessica Jein
White
c,
Yonglong
Li
a,
Jingyun
Chen
a,
Zhihua
Han
a,
Hui
Wei
a,
Chuang
Peng
d,
Yujie
Xiong
*e and
Yun
Wang
*c
aNational Institute of Clean-and-Low-Carbon Energy, Beijing 102211, China. E-mail: baodong.wang.d@chnenergy.com.cn; ziran.ma@chnenergy.com.cn
bNICE Europe Research GmbH, Stockholmer Platz 1, Stuttgart 70173, Germany
cCentre for Catalysis and Clean Energy, School of Environment and Science, Gold Coast Campus, Griffith University, Queensland 4222, Australia. E-mail: yun.wang@griffith.edu.au
dSchool of Resource and Environmental Sciences and Hubei International Scientific and Technological Cooperation Base of Sustainable Resource and Energy, Wuhan University, Wuhan 430072, China
eSchool of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: yjxiong@ustc.edu.cn
First published on 17th January 2023
Abstract
NOx emission can be controlled through selective catalytic reduction (SCR) by ammonia in industry. However, the SCR catalysts are sensitive to contaminants. Searching for anti-poisoning catalysts has become a perpetual quest for large-scale SCR. Here, we report that hydrogenated titanium dioxide particles containing oxygen vacancies undergo in situ N-doping during NH3-SCR reaction. The N-doped hydrogenated TiO2−x exhibits high denitrification activity and selectivity, long-term stability, H2O and SO2 tolerance, and high poisoning resistance. The DRIFT spectra combined with density functional theory computations demonstrate that the N-dopant as the catalytic active site can enhance O2 and NO adsorption, which can be reduced by NH3via the Eley–Rideal mechanism. This is greatly different from traditional catalysts with metal active sites for NH3 adsorption. The high anti-poisoning performance can be ascribed to the weak interaction between N and toxic reactants. This discovery creates a new concept that non-metal active sites can replace conventional precious/transition metals to avoid poisoning, while being stabilized by in situ doping with reactants.
Broader contextNOx emission can be controlled through selective catalytic reduction (SCR) by ammonia in industry. However, the SCR catalysts are sensitive to contaminants. Searching for anti-poisoning catalysts has become a perpetual quest for large-scale SCR. We report that hydrogenated titanium dioxide particles containing oxygen vacancies undergo in situ N doping during NH3-SCR reaction. The N-doped hydrogenated TiO2-x exhibits high denitrification activity and selectivity, long-term stability, H2O and SO2 tolerance, and high poisoning resistance. The DRIFT spectra combined with density functional theory computations demonstrate that the N-dopant as the catalytic active site can enhance O2 and NO adsorption, which can be reduced by NH3via the Eley–Rideal mechanism. This is greatly different from traditional catalysts with metal active sites for NH3 adsorption. The high anti-poisoning performance can be ascribed to the weak interaction between N and toxic reactants. This discovery creates a new concept that non-metal active sites can replace conventional precious/transition metals. Moreover, the results of this work provide a better understanding of the role of defect engineering for the design of highly poison-resistant, stable catalysts for a wide variety of reactions. |
Introduction
Nitrogen oxides (NOx) are one of the main air pollutants, and the source of acid rain, ozone, photochemical smog, regional ultrafine particle (PM2.5) and haze, which affect human health and sustainable economic development. Selective catalytic reduction (SCR) is the most efficient and mature denitrification technology in stationary and mobile sources. Its success is determined by the development of NH3-SCR catalysts with high activity, selectivity and long-term stability. The active components are often made of precious/transition metals that account for 50–70% of the total cost of the catalyst. Moreover, in the real denitrification process, alkali/alkaline metals (Na, K, Ca etc.), heavy metals (As, Pd, etc.), P, SO2 and water vapor could deactivate the catalysts severely. These poisons can block, directly combine with or react with the active components of the catalysts, leading to limited useable lifespan of the catalysts and high economic cost due to frequent catalyst replacement.1–6 To this end, the anti-poisoning performance of denitrification catalysts is not only an important indicator affecting the cost of the catalysts, but also an aspect of the sustainable development of NH3-SCR.Various additives including sacrificial agents or acidic/basic additives have been used to modify the denitrification catalyst support structures. Other approaches have involved controlling the catalyst synthesis to tune the crystalline structure, morphology, distribution of active components and interaction of the metal with the support to improve the anti-poisoning characteristics.2–6 However, these methods only reduce poisoning agents without completely eliminating them. Therefore, the development of low-cost, poisoning-resistant denitrification catalysts is an ultimate goal. One promising strategy for the design of anti-poisoning catalysts is through defect engineering.7–25 The distortion of the crystal structure can modify the Fermi energy level to promote the electron transitions necessary for the catalytic effect.7–9 In addition, the defects can act as the active components for reactant adsorption, facilitate electron interaction and transfer, and reduce intrinsically wide band gaps. More importantly, the precious/transition metals are no longer required to act as active components because the defects can act as the active sites with unsaturated coordination numbers. As such, the poisoning of precious/transition metals would be mitigated, while the expense of the catalysts can be greatly reduced. However, some defects are very unstable, forming a major obstacle for practical applications. For example, the oxygen vacancies can be re-oxidized during prolonged storage in the atmosphere.26–29 Therefore, such oxygen vacancy-based catalysts need to be stored in isolation from oxygen, which increases the cost and restricts industrial applications. To address this issue, it is desirable to design poison-resistant defective catalysts with outstanding stability, but this is challenging.
Here, we demonstrated that the defect sites designated for NH3-SCR reaction can be stabilized by their interaction with reactants (NH3 and NO), overcoming the dilemma between catalyst activity and stability. To eliminate the use of precious/transition metals, oxygen vacancies were created in TiO2 crystals using a hydrogenation method. The hydrogenated TiO2−x (H-TiO2−x) was used as a catalyst for the NH3-SCR, which leads to in situ N-doping on the defective H-TiO2−x catalyst (N-H-TiO2−x). Our results reveal that N-H-TiO2−x exhibits superior denitrification activity. More importantly, it has long-term stability because of its tolerance of H2O and SO2 and significant poisoning resistance, such as SO2, H2O, P, and alkali metals. The results of this work provide a better understanding of the role of defect engineering for the design of highly poison-resistant, stable catalysts for a wide variety of reactions.
Results and discussion
NH3-SCR performance of the hydrogenated TiO2−x catalyst
Fig. 1 shows the NH3-SCR activity of the H-TiO2−x catalyst compared with those of pristine TiO2 and commercial VW/Ti catalysts. From Fig. 1a the denitrification efficiency of the pristine TiO2 was only 7.3–12.8% at 250–400 °C. However, following hydrogenation, the denitrification activity was dramatically increased, to more than 90% at 300–400 °C, representing an increase of approximately 80%, almost reaching the efficiency of the commercial VW/Ti catalyst. The selectivity for N2 was essentially unchanged after hydrogenation, remaining above 87.5% (Fig. S1, ESI†). The stability and resistance to H2O, SO2, K2O and P2O5 of the H-TiO2−x catalyst were also tested. As shown in Fig. 1b, a 500 h stability test was carried out to investigate the stability of the catalyst at 350 °C. The denitrification efficiency was found to slightly decrease by 4.86%, while the N2 selectivity dropped by only 3.29% for 200 h with NH3, NO and O2 blowing in. The resistance of the H-TiO2−x catalyst to H2O and SO2 at 350 °C is followed, conducted as shown in Fig. 1b, which indicates that the denitrification efficiency dropped sharply (by 31.3%) after 8% H2O was introduced into the reaction system for 30 h. Water molecules are believed to have covered the oxygen vacancies on the surface of the catalyst or to have competed with NO and/or NH3 for adsorption on the catalyst surface, resulting in a decrease in denitrification activity. Interestingly, when 800 ppm SO2 was also introduced into the reaction system, the denitrification activity slowly increased and was maintained at 77–79% for 220 h. These results demonstrate that hydrogenated TiO2−x showed good water and sulfur resistance and suitable long-term stability. The trial using 8% H2O and 800 ppm SO2 at 350 °C provided a N2 selectivity of greater than 98% (Fig. 1b). This performance can possibly be ascribed to the formation of more acidic sites on the surface of the catalyst following the introduction of SO2, leading to an increase in the activity and the N2 selectivity.30,31 When the supply of SO2 was cut off, the NOx conversion remained relatively constant, while after the H2O flow was stopped, the NOx conversion increased sharply and returned to its initial level. These results establish that the effects of water on the catalyst were reversible. To better evaluate the anti-poisoning performance of the hydrogenated TiO2−x catalyst, a conventional SCR catalyst (1V8W/TiO2) was prepared for comparison and simulated poisoning experiments were conducted using an impregnation method. Fig. 1c demonstrates that, under the same temperature and reaction conditions, the denitrification activity of the 1V8W/TiO2 catalyst was decreased by 7.6% and 22.67% with K and P poisoning, respectively. Surprisingly, the performances of the hydrogenated TiO2−x catalysts poisoned by K and P were almost unchanged.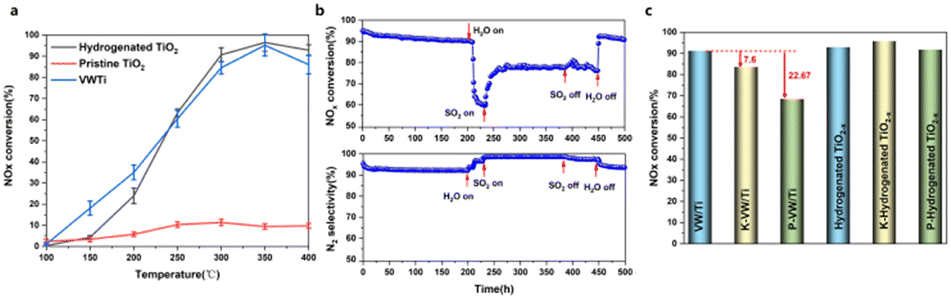 | ||
| Fig. 1 NH3-SCR denitrification activity of the catalyst. (a) The denitrification performances of pristine TiO2, H-TiO2−x and VW/Ti catalysts. (b) Data from 500 h durability trials with the H-TiO2−x catalyst at 350 °C. (c) Tolerance of K2O and P2O5 of the H-TiO2−x catalyst and of commercial VW/Ti catalysts at 350 °C. | ||
Reaction mechanism
The HRTEM and XRD results shown in Fig. S2 and S3 (ESI†) demonstrated that the phase composition of the H-TiO2−x was primarily anatase. The Raman, EPR, 1H NMR and photoluminescence spectra combined with the O2-TPD and H2-TPD shown in Fig. S4–S9 (ESI†) further demonstrate the existence of oxygen vacancies. The in situ X-ray photoelectron spectroscopy (XPS; Fig. S10–S12, ESI†) results confirm that oxygen vacancies and Ti3+/Ti4+ were produced on the surface of the H-TiO2−x catalyst. The in situ XPS data obtained during the NH3-SCR reaction are presented in Fig. 2 and Fig. S13–S15 (ESI†). Fig. 2a presents the N 1s spectra of the material. The spectra obtained over the range of 150–400 °C each contain a O–Ti–N peak related to interstitial doping at approximately 399 eV. The transition from a mixture of Ti3+ and Ti4+ to solely Ti4+ after N-doping is shown in Fig. S14 (ESI†). Because the electronegativity and radius of the N ions (3.04 and 0.171 nm, respectively) are similar to those of O ions (3.44 and 0.140 nm, respectively), N-doping of the H-TiO2−x crystal lattice is expected. The N-doping into the H-TiO2−x catalyst might have come from NH3 or NOx in the reaction gas. Thus, XPS spectra were obtained from the H-TiO2−x after trials in which only NH3 or NO + O2 were present. Fig. S16 (ESI†) demonstrates that N-doping of the H-TiO2−x catalyst surface occurred under both conditions. Previous studies have shown that the nitrogen-doping of TiO2 can proceed under an NH3 atmosphere at 500 °C as N from NH3 partially replaces the lattice oxygen of TiO2, forming O–Ti–N and oxygen vacancies. The present hydrogenation process generated oxygen vacancies in the TiO2 in advance, making nitrogen-doping more likely to occur at lower temperatures (150–400 °C), which is a unique phenomenon in the deNOx reaction. Based on the NIST database,32 the peaks near 406–407 eV were attributed to N–H bonds in NH4NO3. As shown in Fig. 2b, the N–H peak gradually decreased with increasing temperature, while the O–Ti–N peak gradually increased, suggesting that elevated temperature promotes N-doping. Comparing these data to the results shown in Fig. 1a demonstrates that the degree of N-doping was proportional to the denitrification activity. The results from 200 h stability trials with only NH3, NO and O2 blowing in indicated that in situ N-doping improved the stability of the catalyst.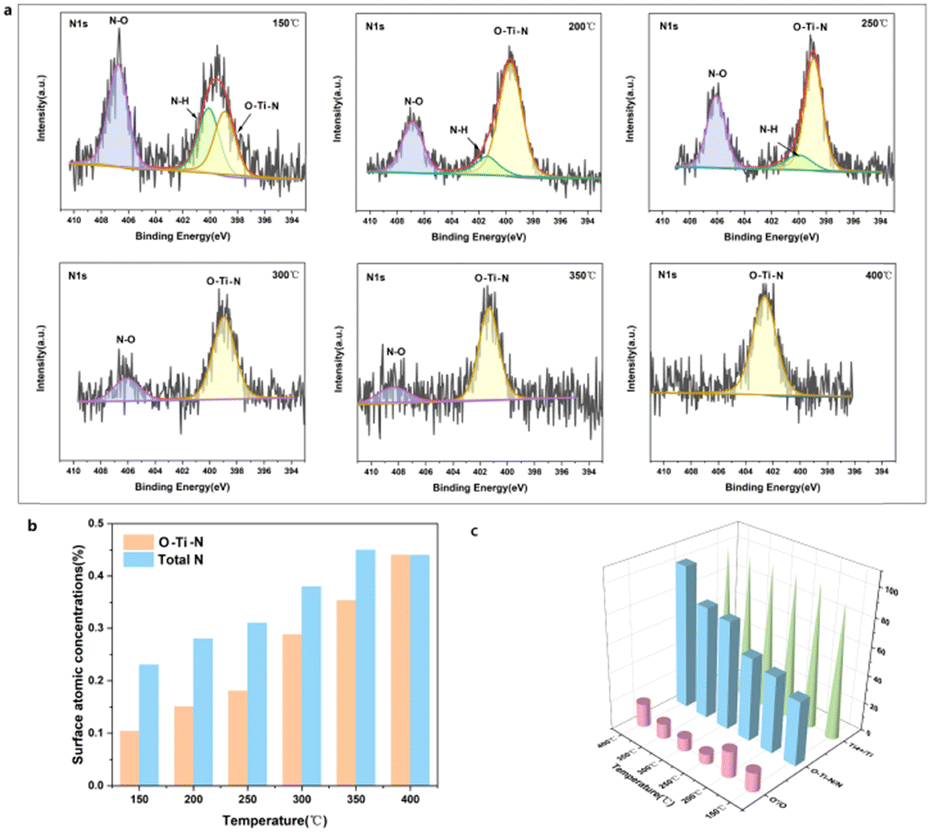 | ||
| Fig. 2 The in situ XPS spectra obtained from the H-TiO2−x during the NH3-SCR process at 150–400 °C. (a) N1s. (b) Surface N atom concentrations and O–Ti–N concentrations at various temperatures. (c) Percentage content of the active components O, N and Ti (O′ = Oβ + Oγ). | ||
The local atomic structure and cation coordination in the catalyst before and after N-doping were ascertained using XAFS (Fig. 3). The Ti K-edge X-ray absorption near-edge structure (XANES) spectra of hydrogenated TiO2−x and the N-hydrogenated TiO2−x catalyst possessed three typical pre-edge peaks associated with the anatase phase (denoted as A1, A2, and A3 at 4968, 4971, and 4974 eV, respectively), corresponding to quadruple-allowed 1s to 3d transitions.19 Furthermore, it has recently been suggested that most of the contribution to pre-edge splitting is from the edge and corner sharing of Ti ions in the TiO6 octahedra with oxygen ions, which lead to non-local, intersite hybrid excitations. Therefore, the variation in pre-edge peak area during sample etching can be interpreted as the change in the Ti 3d unoccupied states due to the interaction of Ti 4p, O 2p, and Ti 3d states. The total area of the pre-peaks was associated with the number of unoccupied next-nearest-neighbour Ti 3d states. As clearly shown in Fig. 3, for both hydrogenated TiO2−x and the N-hydrogenated TiO2−x catalyst, the area of the near-side peaks was significantly larger than that of the TiO2 standard sample (anatase), which was due to oxygen vacancies. For the hydrogenated TiO2−x catalyst and N-hydrogenated TiO2−x catalyst, the A1, A2, and A3 peaks were all observed to shift to a higher energy compared with the TiO2 standard sample (anatase). This was attributed to the higher oxidation state of the core metal atom (Ti), as induced by oxygen vacancies in the coordination sphere. The intensity of the A2 peak increased with increasing site distortion in the order TiO2 < hydrogenated TiO2−x ≈ N-hydrogenated TiO2−x. The introduction of oxygen vacancies made the crystal lattice more disordered. From Fig. 3a, after N-doping, the A2 peak of the catalyst was shifted to a slightly lower energy, supporting that nitrogen filled the oxygen vacancies. Fig. 3b and c show the EXAFS oscillations for the standard, hydrogenated and N-hydrogenated TiO2−x, respectively. Fig. 3d clearly demonstrates that the Ti–O coordination environment was only minimally changed after N-doping, while the Ti–Ti coordination environment changed greatly. These changes confirm the transition from a mixture of Ti3+ and Ti4+ to solely Ti4+ after N-doping.
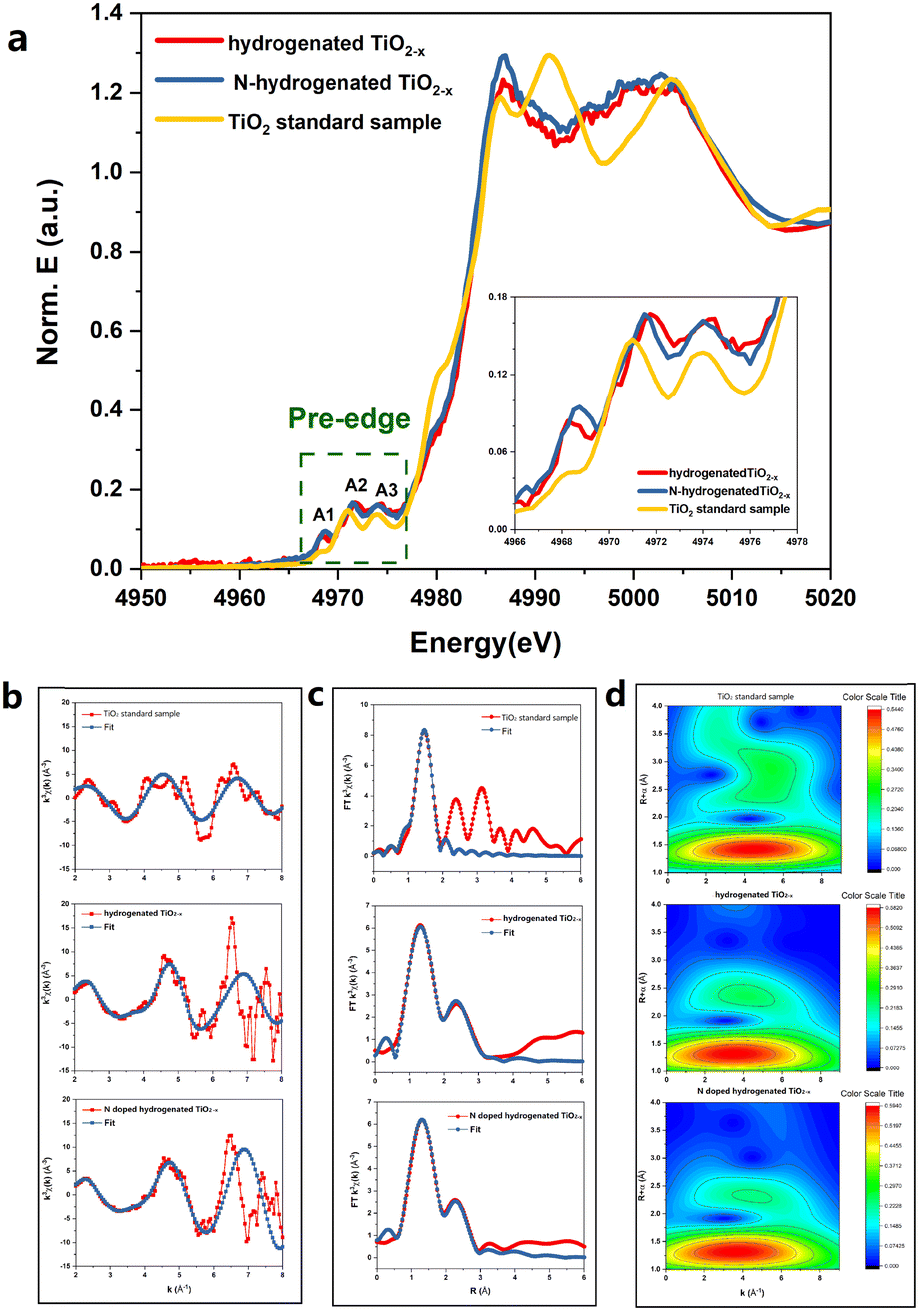 | ||
| Fig. 3 The XANES spectra of the catalyst samples before and after the SCR reaction. (a) The Ti K-edge, (b) extended region fitting using ARTEMIS in R-space, (c) fitted magnitude of k3-weighted Ti K-edge EXAFS spectra, and (d) wavelet transform images obtained for the TiO2 standard sample (anatase), hydrogenated TiO2−x and N-hydrogenated TiO2−x. | ||
The distributions of formal valences in the catalyst before and after N-doping were probed using EELS-STEM, while simultaneously acquiring Ti–L, N–K and O–K edge data (Fig. 4). The Ti–L edge provided a fingerprint of the Ti4+ and Ti3+ states and the O–K edge was sensitive to the presence of oxygen vacancies. In the case of the H-TiO2−x catalyst, the EELS spectra were collected close to (0.9, 1.8, 2.7 and 3.6 nm) and far from (6.3 nm) the interface and the results are presented in Fig. 4a. These data indicate that the Ti-L2 peak was shifted by 0.6 eV from the crystalline side to the amorphous side, demonstrating the presence of Ti3+ in the amorphous layer, at the interface and on the crystalline side close to the interface (within 2.7 nm). In Fig. 4b, the N–K peak appeared at 0.9 nm, showing that the surface nitrogen-doping occurred primarily in the amorphous layer. Furthermore, no Ti–L peak shifts were observed, further demonstrating that Ti4+ was the primary state in the N-hydrogenated TiO2−x.
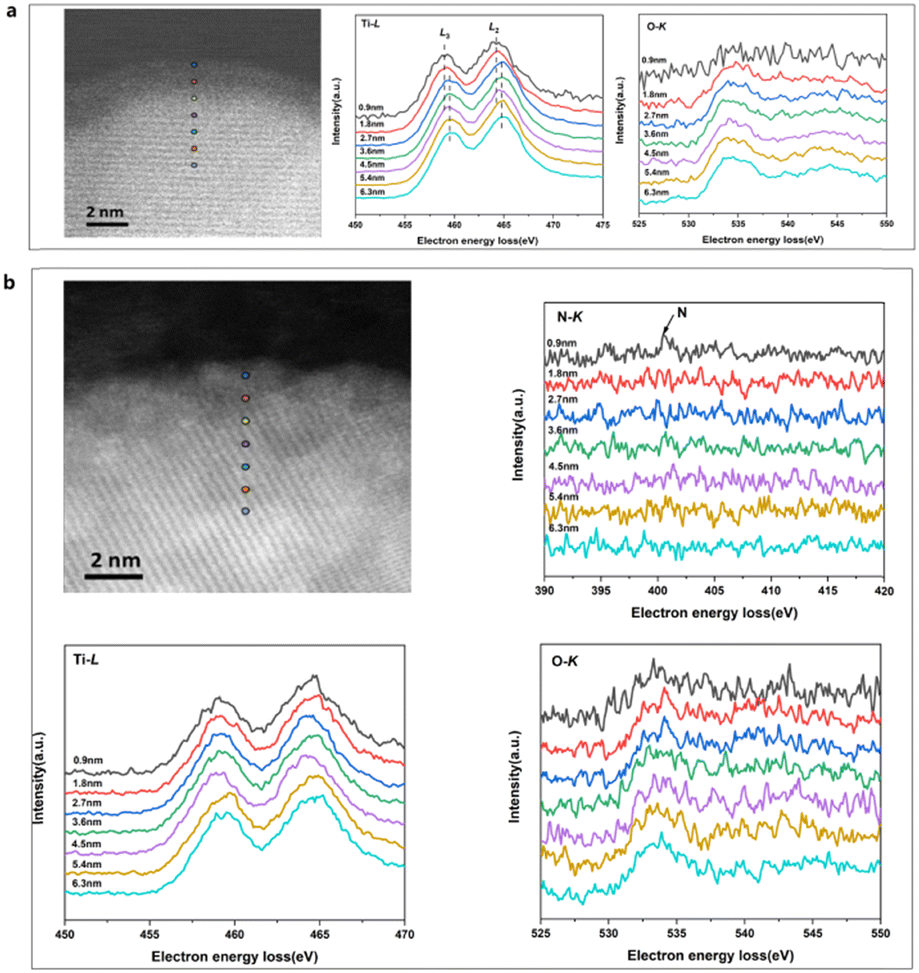 | ||
| Fig. 4 The EELS-STEM spectra of the catalysts. (a) The H-TiO2−x catalyst and (b) the N-H-TiO2−x catalyst. | ||
Fig. 5 and Fig. S17 (ESI†) provide the in situ infrared spectra of H-TiO2−x and pristine TiO2. These data indicate that very little NO or O2 was adsorbed on the pristine TiO2 (Fig. 5a), while bridged bidentate nitrate (at 1610 cm−1), chelated bidentate nitrate (1578 and 1238 cm−1) and monodentate nitrite (1350 cm−1)30 were formed on the hydrogenated TiO2−x. These results are in agreement with the poor denitrification activity of this material. This difference is attributed to the abundant oxygen vacancies present on the H-TiO2−x surface, which are able to oxidize NO to NO2 and generate a series of nitrates and nitrites. Exposure to NH3 did not modify the surface of the pristine TiO2 although the chelated bidentate nitrate on the H-TiO2−x disappeared and no NH3 adsorption peak was obtained, indicating that gaseous NH3 reacted with nitrate on the catalyst surface. Following the surface adsorption of NH3 (Fig. 5b), the pristine TiO2 generated an NH3 adsorption peak at the L acid position at 1103 cm−1. The results obtained from NH3-temperature programmed desorption (Fig. S18, ESI†) and pyridine adsorption infrared spectroscopy (Fig. S19, ESI†) show that the pristine TiO2 was somewhat acidic and was also able to adsorb NH3. In contrast, the surface adsorption of NH3 on the H-TiO2−x generated NH4+ at B acid sites (1633 cm−1) together with NH2 (1322 and 1492 cm−1) and NH3 at L acid sites (1197 cm−1). Exposure to NO and O2 had little effect on the surface of the pristine TiO2. However, the H-TiO2−x formed bridged bidentate nitrate (1606 cm−1), chelated bidentate nitrate (1578 and 1236 cm−1), monodentate nitrite (1517 cm−1) and ammonium nitrate (1346 cm−1). The NH3 adsorption peaks at L acid and B acid sites also disappeared, indicating that NOx had reacted with adsorbed NH3. These results confirmed that the adsorption of NH3 and gaseous NOx proceeded according to the Eley–Rideal mechanism.33–35
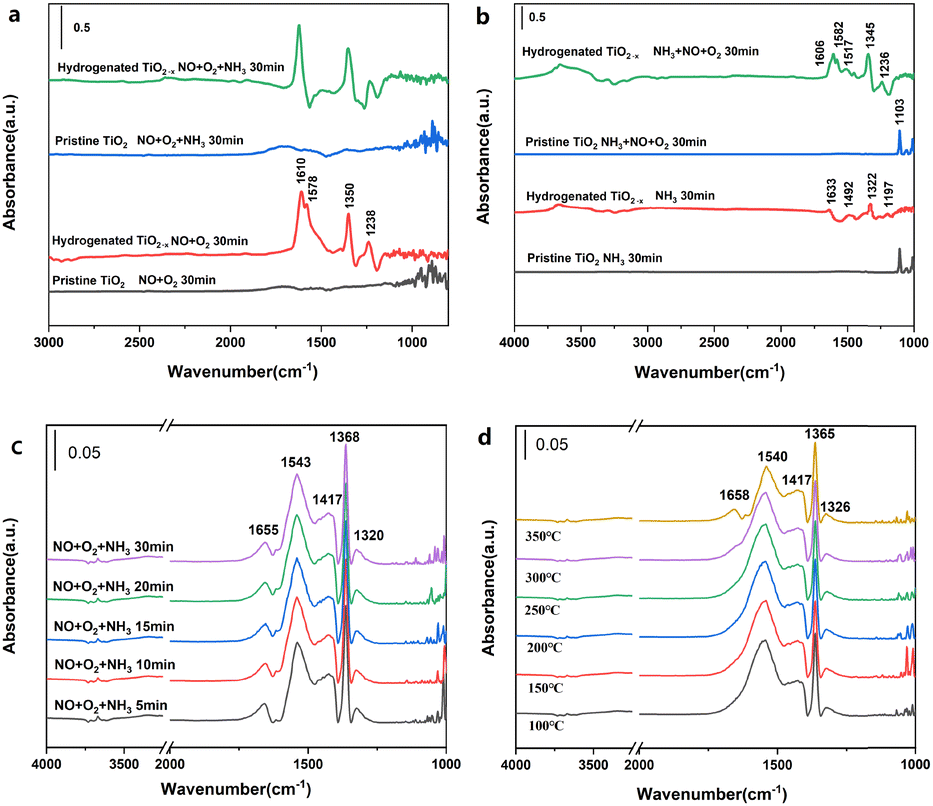 | ||
| Fig. 5 The DRIFT spectra obtained from the hydrogenated TiO2−x and pristine TiO2. (a) NO and O2 followed by NH3 at 300 °C. (b) NH3 followed by NO + O2 at 300 °C. (c) NH3 + NO + O2 for 30 min at 300 °C. (d) NH3 + NO + O2 with a temperature gradient. | ||
Fig. 5c shows the in situ infrared spectra obtained from the N-H-TiO2−x catalyst at 300 °C following a simultaneous exposure to NO, O2 and NH3. When the three gases were introduced for 5 min, peaks related to bridged bidentate nitrate (1655 cm−1), monodentate nitrate (1543 cm−1) and monodentate nitrite (1368, and 1320 cm−1) appeared.36 With prolonged exposure to the three gases simultaneously, the intensity of these peaks changed only minimally, indicating that these compounds were stable on the catalyst surface. No NH3 adsorption peak was observed, which suggests that NH3 is the primary participant in the gas phase NH3-SCR reaction. Fig. 5d provides the steady state in situ infrared spectra acquired from the N-H-TiO2−x catalyst surface at different temperatures in conjunction with the simultaneous introduction of NH3, NO and O2 during the SCR reaction. At 100 °C, a series of peaks attributed to NOx species adsorbed on the catalyst surface appeared. The peaks at 1658 and 1540 cm−1 represent bridged and chelated bidentate nitrates, respectively, while those at 1365 and 1326 cm−1 indicate monodentate nitrite. As the temperature was gradually increased, the intensity of the chelated bidentate nitrate peak decreased significantly, due to desorption from the catalyst surface. Again, peaks related to the adsorbed NH3 were not observed. It is apparent that NH3 and chelated bidentate nitrate were active on the catalyst surface and participated in the SCR reaction through the Eley–Rideal mechanism. We will carry out kinetic experiments in the subsequent research and quantify the proportion of E–R mechanism according to the kinetic calculation. It is worthy of note that the assignment of the band at 1417 cm−1 still needed further identification. The peaks in some studies37–40 are close to 1417 cm−1, but their attribution of this peak is various. Moreover, we have not found literature with exactly the same peak as ours. This may be because other studies are all on catalysts with metal active sites and the functional groups on the catalyst surface are different from those of our catalysts without metal active sites. The peak of 1417 cm−1 in the in situ infrared spectrum is unique to our catalyst. As such, the ascription of the strong IR absorption peak at 1417 cm−1 could be based on other characterizations and DFT simulation in our subsequent research.
DFT calculations
Density functional theory (DFT) calculations were performed to better understand the role of lattice defects in the catalytic process.37–39 The adsorption properties of specific small molecules on the pristine TiO2 (101), H-TiO2−x, and N-H-TiO2−x surfaces are presented in Fig. 6a–c. The Raman, EPR, 1H NMR and photoluminescence spectra combined with the O2-TPD and H2-TPD shown in Fig. S4–S9 (ESI†) demonstrate the existence of oxygen vacancies in H-TiO2−x. As a result, one surface oxygen vacancy was introduced here to model H-TiO2−x. The oxygen vacancy was highlighted by a blue circle shown in Fig. 6b. The adsorption energies indicate that the pristine TiO2 has a weak interaction with O2 and NO. As a comparison, the adsorption of NH3 on this surface was stronger. The surface Ti4+ as L acid sites can adsorb a significant quantity of NH3. On H-TiO2−x, the O2 and NO can tightly bind with the vacancy. NH3 can still adsorb on L acid sites with an enhanced interaction strength. The adsorption energies of reactants match the NH3-TPD and Py-IR spectra shown in Fig. S18 and S19 (ESI†). However, the strong adsorption of NO and NH3 disagrees with DRIFT data with the Eley–Rideal mechanism because NH3 needs to directly interact with the adsorbed reactants. This indicates that the oxygen vacancy is not the active site during the reactions. Interestingly, the NO and O2 can tightly adsorb through the interaction with the N-dopant after in situ N-doping. This indicates that the surface N-dopant can act as the catalytic active site. In particular, the NO adsorption energy on N via a N–N–O configuration is −2.88 eV (see Fig. 6c), which is 1.04 and 1.80 eV lower than those of O2 and NH3. The formation of a N–N bond weakens the N–O bond in NO, as evidenced by the enlarged N–O bond length from 1.17 in NO gas to 1.32 Å after adsorption. More importantly, the NH3 can dissociatively adsorb on the pre-adsorbed NO to form *OH and *N–NH2 as shown in Fig. 6d. The dissociative adsorption of NH3 on the –N–N–O site is because N in NO carries a positive charge and, therefore unoccupied orbitals. On the other hand, the N in NH3 has lone-pair electrons. As a result, the N in NH3 can strongly bind with N in adsorbed NO through dative covalent bonding. At the same time, the NH3 was dissociated after the adsorption because of the strong O–H bonding between the negatively charged O in NO and positively charged H in NH3. The adsorption energy is −1.24 eV, which is 26.5% lower than that of the direct adsorption of NH3 on the Ti4+ L acid site. Moreover, there is no steric effect in the interaction between NH3 and NO. Consequently, the Eley–Rideal mechanism derived from the DRIFT spectra is supported by the DFT results. The novel SCR reaction route through the adsorption of NO on the N active sites in the N-H-TiO2−x catalyst is illustrated in Fig. 6e, which is compared with the mechanism through the adsorption of NH3 on metal active sites in the traditional catalysts shown in Fig. 6f.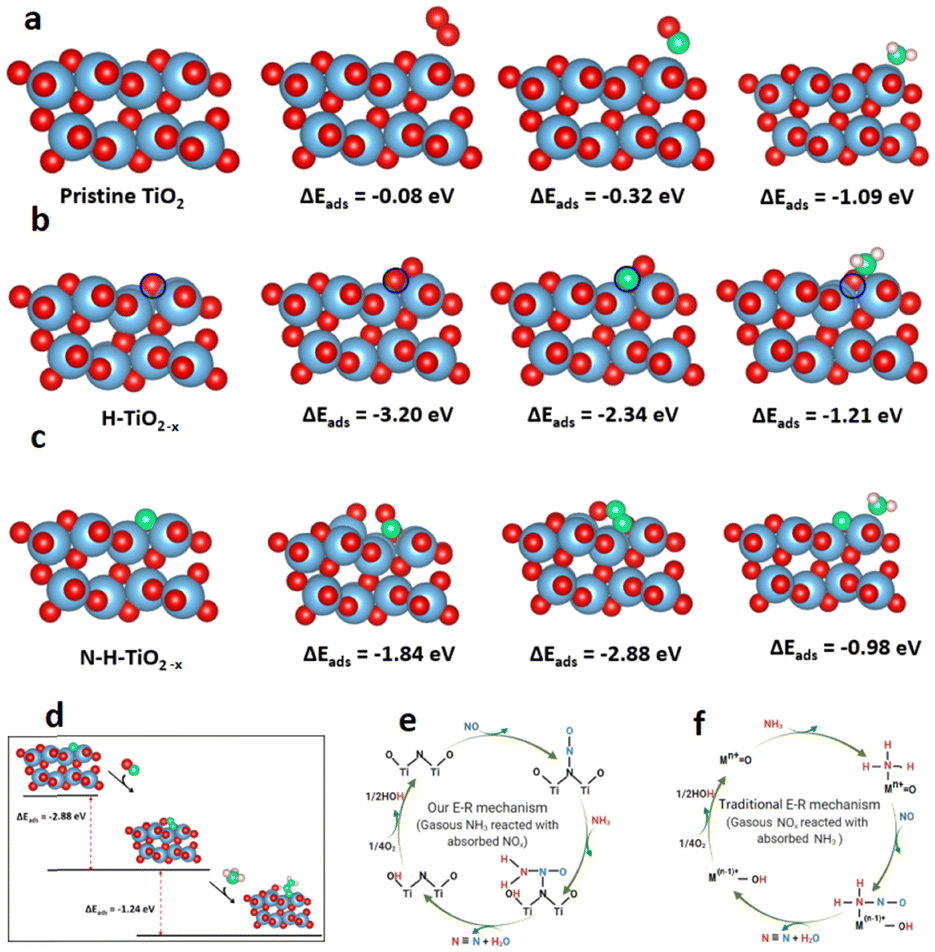 | ||
| Fig. 6 (a) Adsorption of O2, NO and NH3 on pristine TiO2 (101), (b) adsorption of O2, NO and NH3 on H-TiO2−x, (c) adsorption of O2, NO and NH3 on N-H-TiO2−x, (d) reaction coordinates of NH3-SCR via the Eley–Rideal mechanism, (e) SCR reaction routes at the N active sites in the N-H-TiO2−x catalyst, and (f) SCR reaction routes at the metal active sites in the traditional catalysts. Legend: red = O; blue = Ti; pink = H; and green = N; blue circle = oxygen vacancy. | ||
The DFT computations combined with in situ DRIFTS experiments were also used to explain the resistance of the N-H-TiO2−x catalyst to H2O, SO2 and K poisoning. Fig. 7a shows that the water can adsorb at the bridge site between surface Ti4+ and N atoms on N-H-TiO2−xvia hydrogen bonding. As a result, the N active sites can be blocked by adsorbed H2O. This explains why the activity drops after the exposure of water, as shown in Fig. 1b. However, as the interaction strength of −0.87 eV is moderate via hydrogen bonding, the impact of H2O adsorption on the electronic properties of N-H-TiO2−x is limited. As such, the selectivity only changes slightly. After the further exposure of SO2, the adsorbed water can interact with SO2 and O2 to form H2SO4.40,41 The formation and desorption of H2SO4 is an exothermal process with the reaction energy of −1.32 eV. This indicates that the adsorbed water can be removed after the exposure of SO2 and O2, which can reactivate N active sites for denitrification, as shown in Fig. 1b, so that the activity of the N-H-TiO2−x catalysts recovers after the exposure of SO2 to the catalyst with pre-adsorbed water. The DFT calculations also reveal that K atoms prefer not to adsorb on the oxygen vacancy, even when the K atom was initially placed at the vacancy site. The most stable configuration is shown in Fig. 7b. This is reasonable because the system becomes more stable when the K atom is bonded with more surface oxygen atoms. This means that the vacancy for the formation of active sites via in situ N-doping will not be poisoned by K. More interestingly, the NO adsorption can be strengthened when K is next to the N active site since K can extract O from NO, which leads to the formation of N2 at the active site. This result explains why the denitrification efficiency increases slightly after K poisoning, as shown in Fig. 1c. When the metal acts as the active site in traditional NH3-SCR catalysts, e.g., VW/Ti, the metal as L acid sites can accommodate the lone pair electrons of O and S in toxic substances, which is the origin of the poisoning of these catalysts. Our results, therefore, demonstrate the unique anti-poisoning advantage of the nonmetal active site for NH3-SCR.
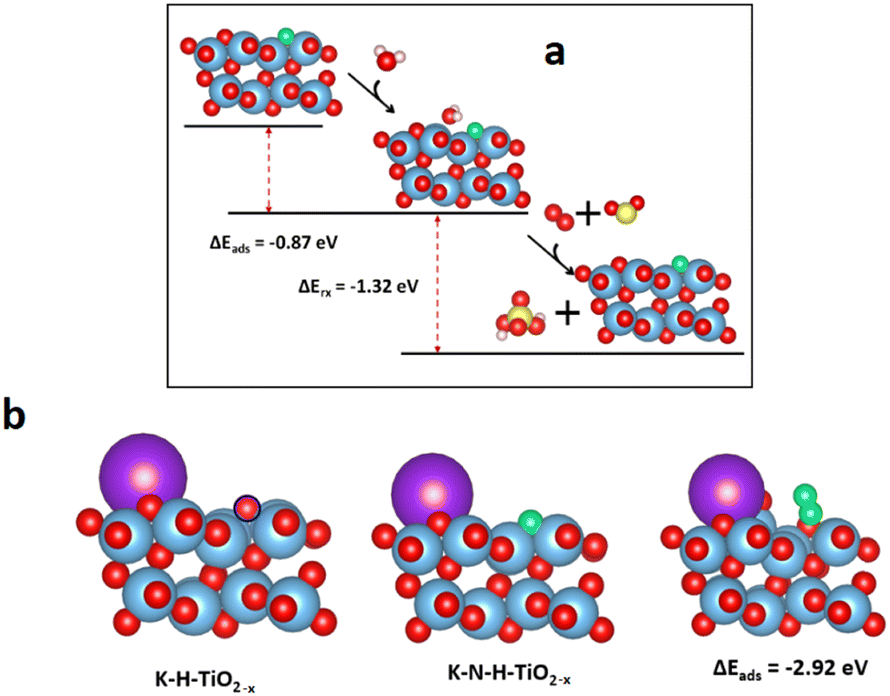 | ||
| Fig. 7 (a) Reaction coordinates of adsorption of water followed by SO2 on N-H-TiO2−x, and (b) adsorption of K on H-TiO2−x and N-H-TiO2−x and NO adsorption on K–N-H-TiO2−x. Legend: red = O; blue = Ti; pink = H; green = N; yellow = S; purple = K; and blue circle = oxygen vacancy. | ||
Experimental
Synthesis of hydrogenated TiO2−x catalysts
Commercial titanium dioxide (Denox Advanced Material Co., Ltd in China, TiO2 97%, BET 323.36 m2 g−1, rutile and anatase) powder was placed into a fixed bed reactor. Hydrogenated TiO2−x catalysts with Ti3+ were prepared by a hydrogen thermal reduction treatment method by heating commercial titanium dioxide powders in 99.99% H2 at 450 °C (heating rate 5 °C min−1) under ambient pressure for 12 h. The H2 flow velocity (100 mL min−1) was controlled using a gas flow controller during hydrogen reduction treatment until cooling to room temperature. The hydrogenated TiO2−x powder is placed in a vacuum dryer to prevent the oxygen in the air from further oxidizing the catalyst. Hydrogenated TiO2−x powder was obtained as shown in eqn (1) and (2).| TiO2 + xH2(g) → TiO2−x + xH2O(g) | (1) |
| O2− (s) + H2(g) → H2O(g) + VO2− | (2) |
A typical impregnation method was applied to prepare the alkali and P-poisoned catalysts. According to the literature, the mass ratios of K and P are 0.6 and 1 wt%, respectively. Therefore, 1 g of catalyst, 50 mL of deionized water, and a corresponding amount of KCl or/and NH4H2PO4 were stirred together, dried, and calcined at 550 °C for 4 h in N2. For comparison, commercial 1%V8%W/Ti catalyst poisoning was also prepared according to the above method. The poisoned samples were denoted as K-hydrogenated TiO2−x, P-hydrogenated TiO2−x, K-VW/Ti and P-VW/Ti.
Characterization of the materials
X-Ray diffraction (XRD) was performed using a D8 ADVANCE diffractometer (Bruker, Germany), using Cu Kα radiation and a step size of 0.02°. Data were collected from 5° to 90° at a scanning rate of 3° min−1.High-resolution transmission electron microscopy (HRTEM) was performed using a JEOL JEM ARM200F instrument (Japan).
The electron paramagnetic resonance (EPR) spectra of the samples were acquired using a Bruker A320 instrument. The samples (0.12 g) were loaded into quartz tubes. EPR experiments of titanium dioxide powders and hydrogenated TiO2−x samples were conducted under normal temperature and pressure conditions. The reactor was placed in a heating furnace and heated to 500 °C at a heating rate of 10 °C min−1 and pretreated with 2% O2 for 1 h. After pretreatment, the gas supply was stopped and the reactor was sealed and cooled. Upon reaching room temperature, the EPR test was repeated. The magnetic field was swept from 1500 to 5000 G.
The Raman spectra were obtained using a LabRam HR-800 spectrometer (Horiba Jobin Yvon, France) with laser excitation at 532 nm.
X-Ray photoelectron spectroscopy (XPS) was performed using an ESCALAB 250xi instrument (Thermo Scientific, UK) using monochromatic Al Kα radiation (1486.6 eV) at 25 W. The samples were outgassed overnight at room temperature in an ultrahigh-vacuum chamber (<5 × 10−7 Pa). All binding energies were referenced to the C 1s peak at 284.6 eV. Experimental errors were within ±0.1 eV.
Solid-state NMR experiments were conducted on a Bruker Avance III HD/89 mm spectrometer with a 4 mm triple-resonance magic angle spinning probe.
O2-temperature-programmed desorption (TPD) experiments were performed to determine the adsorption/desorption characteristics of each species over the catalyst. Each catalyst (200 mg) was loaded into the reactor, pretreated with N2 (50 mL min−1) at 100 °C for 30 min, and then cooled to room temperature in the same stream. The pretreated sample was then exposed to O2 (3%) at a flow rate of 70 mL min−1 for 30 min. Physisorbed O2 was removed by flushing the catalyst with He at a flow rate of 30 mL min−1 for 30 min at 120 °C prior to TPD. The samples were subjected to TPD in He (30 mL min−1) from 100 to 600 °C at a heating rate of 10 °C min−1.
H2-temperature-programmed reduction (TPR) experiments were performed with an AutoChem II 2920 instrument (USA). The catalyst (0.3 g) was pretreated with Ar flow (50 mL min−1) for 30 min at 500 °C to remove water and other impurities. As the samples cooled, the Ar flow was replaced with a reductive mixture of 10.0% H2 in Ar and the reactor temperature was increased to 800 °C at a heating rate of 10 °C min−1.
NH3-TPD experiments were performed using an automatic physical and chemical adsorption instrument (AutoChem II 2920, Micromeritics). Before NH3 adsorption at 373 K, the samples were heated at 773 K under He flow. The amount of NH3 desorbed between 373 and 873 K at a heating rate of 10 K min−1 was determined using an on-line gas chromatograph equipped with a thermal conductivity detector.
The diffuse reflectance infrared Fourier transform (DRIFT) spectra were recorded using an IR Prestige-21 instrument (Shimadzu) at a resolution of 4 cm−1 and averaged over 500 scans. These experiments were performed by heating precalcined powder samples in situ from room temperature to 673 K at a heating rate of 5 K min−1 under a pure N2 flow (40 mL min−1). The samples were kept at 673 K for 3 h and then cooled to 323 K. Pyridine vapor (20 μL) was then introduced under a N2 flow. The IR spectra were recorded at various stages of pyridine desorption, which was maintained by evacuation at progressively higher temperatures (323–473 K). A resolution of 4 cm−1 was achieved after averaging 500 scans for all the IR spectra recorded.
The in situ X-ray photoelectron spectroscopy (in situ XPS) measurements were conducted on a near ambient pressure (up to 2.5 kPa) X-ray photoelectron spectroscopy system (SPECS) equipped with a monochromatized Al Kα source (hν = 1486.6 eV), gas atmosphere XPS analysis chamber (up to 2.5 kPa, 1000 K), electron analyzer (Phoibos 150), three-differential pumping stage that separates the analysis chamber from the electron energy analyzer, and sample preparation chamber (up to 3.0 MPa and 873 K). The X-ray source was set at an acceleration voltage of 15 kV and an irradiation power of 80 W. A preparation chamber was used for catalyst activation. Afterwards, the chamber was evacuated, and the sample was directly transferred into the analysis chamber under vacuum to avoid exposure to air. Firstly, the temperature was raised to 150 °C and then NO (1% NO, N2, and 2 L h−1) and O2 (99.99%, 2 L h−1) were blown in simultaneously. NH3 (1% NH3, N2, and 2 L h−1) is then introduced one hour later. After the catalyst adsorption equilibrium, the Ti spectra (including the valence band spectra), O spectra and N spectra of the catalyst at 150 °C, 200 °C, 250 °C, 300 °C, 350 °C and 400 °C were collected, respectively.
The distribution of Brønsted (B) and Lewis (L) acids on the synthesized catalyst was characterized using the pyridine infrared adsorption (Py-IR) method (IRPrestige-21, Shimadzu, Japan). Catalyst powder (20–30 mg) was weighed onto a sheet with a diameter of 13 mm, which was then fixed in an infrared cell. The temperature was increased to 500 °C for 60 min, then decreased to 350, 200, 100, and 30 °C, holding at each temperature for 5 min to measure the background value. Pyridine was adsorbed at 30 °C for 1 h, then purged for 30 min, and the adsorption spectrum was obtained. The temperature was then increased to 100, 200, and 350 °C, holding at each temperature for 5 min to measure the desorption spectra. The infrared spectra of the catalyst were recorded in the region of 1700–1400 cm−1.
Photoluminescence spectroscopy (PL) was used to investigate oxygen vacancies on the catalyst surface (LabRam HR-800, Horiba Jobin Yvon, France). The light-induced spectral curve was measured at room temperature, and He–Cd was used as the laser source (λ = 325 nm).
Thermogravimetric analysis, conducted on an STA 449 F3/QMS 403C instrument (Netzsch, Germany), was used to assess catalyst loss.
The catalyst electronic structure was determined using electron energy loss spectroscopy (EELS). The energy resolution of EELS with a monochromator was 0.15 eV measured at the full width at half-maximum of the zero-loss peak under vacuum, with the highest energy dispersion of 0.025 eV per channel. To obtain surface structures through direct observation of the atomic arrangement, an aberration-corrected dedicated FEI Titan Themis 60–300 instrument was used, achieving atomic resolution (70 pm) in the high-angle annular dark-field imaging (HAADF) mode.
The X-ray absorption fine structure (XAFS) Ti K-edge spectra were collected at the BL07A1 beamline of the National Synchrotron Radiation Research Center (NSRRC). Data were collected in fluorescence mode using a Lytle detector, while the corresponding reference samples were collected in transmission mode. Samples were ground and uniformly coated on a special adhesive tape. The as-obtained XAFS data were processed in Athena (version 0.9.26) for background, pre-edge line, and post-edge line calibrations. Fourier transform fitting was then conducted in Artemis (version 0.9.26). The k3 weighting, k range of 1–6 Å−1, and R range of 1–4 Å were used for the fitting. The four parameters, namely, coordination number, bond length, Debye–Waller factor, and E0 shift (CN, R, σ2, and ΔE0), were fitted without any being fixed, constrained, or correlated. For wavelet transform analysis, χ(k) exported from Athena was imported into the Hama Fortran code. The parameters were as follows: R range, 1–4 Å; k range, 0–6 Å−1; and k weight, 2; the Morlet function with κ = 10 and σ = 1 was used as the mother wavelet to provide the overall distribution.
NH3-SCR activity measurements
The SCR reaction was evaluated in a fixed-bed reactor. The samples (0.3 g) were placed in reaction tubes, which were then placed in simulated flue gas for 2 h. The gas mixture contained NO (300 ppm), NH3 (300 ppm), and O2 (3%), with N2 as the balancing gas. The gas hourly space velocity was ∼30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The catalytic activity was determined by analyzing the inlet and outlet gases using a flue gas analyzer (MultiGas 6030, MKS) at temperatures between 100 and 400 °C. At each temperature (100, 150, 200, 250, 300, 350, and 400 °C), the catalyst activity was determined after a reaction time of 1 h. The NOx conversion and N2 selectivity were calculated as follows:
000 h−1. The catalytic activity was determined by analyzing the inlet and outlet gases using a flue gas analyzer (MultiGas 6030, MKS) at temperatures between 100 and 400 °C. At each temperature (100, 150, 200, 250, 300, 350, and 400 °C), the catalyst activity was determined after a reaction time of 1 h. The NOx conversion and N2 selectivity were calculated as follows:| NOx conversion = [([NOx]in − [NOx]out)/[NOx]in] × 100% | (3) |
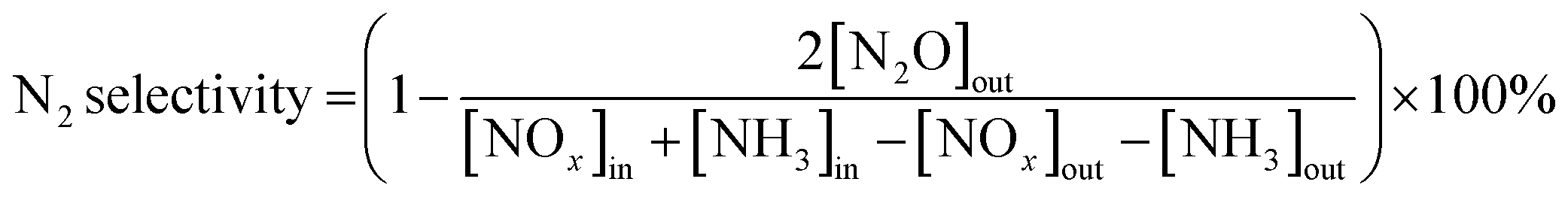 | (4) |
For H2O and combined SO2 + H2O resistance experiments, the SCR reaction was allowed to stabilize for 200 h at 350 °C with only 300 ppm NO + 3% O2 + 300 ppm NH3 blew in, generating steady-state conditions prior to introducing 8% H2O for 40 h and a 500 ppm SO2 + 8% H2O combination for 340 h at 350 °C into the flue gas, respectively. Finally, SO2 and H2O were successively cut off.
DFT calculations
The Vienna ab initio package (VASP) was used to perform all density functional theory (DFT) calculations within the generalized gradient approximation (GGA) using the PBE formulation. The spin-polarization was considered in all computations. The projected augmented wave (PAW) potentials were selected to describe the ionic cores and account for valence electrons using a plane wave basis set with a kinetic energy cutoff of 500 eV. Partial occupancies of the Kohn–Sham orbitals were allowed using the Gaussian smearing method and a width of 0.05 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−5 eV. A geometry optimization was considered convergent when the force change was smaller than 0.01 eV Å−1. On-site corrections (DFT+U) were applied to the 3d electrons of Ti atoms (Ueff = 4.5 eV) using the approach of Dudarev et al.The equilibrium lattice constants of the anatase TiO2 unit cell were optimized to a = b = 3.858 Å, c = 9.652 Å using a 10 × 10 × 4 Monkhorst-Pack k-point grid for Brillouin zone sampling. These constants were then used to construct a TiO2(101) surface model with p(1 × 3) periodicity in the x and y directions and two stoichiometric layers in the z direction with a vacuum depth of 15 Å to separate the surface slab from its periodic duplicates. This TiO2(101) surface model contained 24 Ti and 48 O atoms. The TiO2(101) surface with one oxygen vacancy was used to represent the hydrogenated TiO2 (H-TiO2−x). The N-doped hydrogenated TiO2 (N-H-TiO2−x) was simulated by using one N atom to replace the surface O atoms in TiO2(101). During structural optimization, a (2 × 2 × 1) k-point grid in the Brillouin zone was used for k-point sampling and the bottom stoichiometric layer was fixed, while the remaining atoms were allowed to fully relax.
The adsorption energy (Eads) of adsorbate A was defined as follows:
| ΔEads = EA/surf − Esurf − EA(g) | (5) |
Conclusions
This work shows that N-H-TiO2−x constructed in conjunction with lattice defect engineering exhibited good NH3-SCR performance. The denitrification efficiency of N-H-TiO2−x was greater than 90% at 300–400 °C. More importantly, this catalyst exhibited long-term stability, resistance to H2O and SO2 and good poisoning tolerance. The results of this study confirmed that the in situ N-doping on oxygen vacancies in hydrogenated TiO2−x is the key step. The DFT calculations combined with the DRIFT spectra data demonstrate that the N-dopant acts as the catalytic active site for the NH3-SCR denitrification process via the Eley–Rideal mechanism. The introduction of nonmetal active sites greatly reduces the impact of toxic substances. This is because precious/transition metals are no longer required to act as active components. As such, the poisoning of precious/transition metals would be mitigated, while the expense of the catalysts can be greatly reduced. We anticipate that this research will assist in the future design of anti-poisoning catalysts with non-metal active sites through lattice defect engineering, which can be used in wide industrial catalysis applications.Author contributions
G. L. carried out all the relevant experiments, data analysis and wrote the first draft of the manuscript. B. D. W. led the research program and supervised this project. Y. J. X. provided suggestions for the project development. B. D. W., Y. W., Y. J. X. and C. P. advised and revised the manuscript. J. J. W. and Y. W. performed all the DFT calculations and analyzed the catalytic mechanisms. Y. L. L. and J. Y. C. did the in situ XPS experiments of hydrogenation of TiO2−x. Z. H. H. did the EELS-STEM measurements. H. W. did some catalyst characterization measurements. H. Y. W. drew some figures. S. P. P., Z. R. M., J. M. and J. L. Z. did some NH3-SCR measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge W. F. Tu for the assistance during the in situ XPS experiments and data analysis. The in situ XPS experiments were performed at the School of Chemical Engineering and Energy, Zhengzhou University, China. This work was supported by the National Key Research and Development Program of China (grant no. 2019YFC1907500) and by the National Energy Group Project (grant no. ST930021011C and no. ST930022006C). This research was undertaken on the supercomputers in the National Computational Infrastructure (NCI) in Canberra, Australia, which is supported by the Australian Commonwealth Government and the Pawsey Supercomputing Centre in Perth with funding from the Australian Government and the Government of Western Australia.Notes and references
- Catalyst Deactivation, in Advances in Chemistry, ed. J. B. Butt, American Chemical Society, Washington, DC, 1972, vol. 109, pp. 259–518 Search PubMed.
- Y. Peng, J. H. Li, W. Z. Si, J. M. Luo, Y. Wang, J. Fu, X. Li, J. Crittenden and J. M. Hao, Appl. Catal., B, 2015, 168–169, 195–202 CrossRef CAS.
- Z. W. Huang, H. Li, J. Y. Gao, X. Gu, L. Zheng, P. P. Hu, Y. Xin, J. X. Chen, Y. X. Chen, Z. L. Zhang, J. M. Chen and X. F. Tang, Environ. Sci. Technol., 2015, 49, 14460–14465 CrossRef CAS PubMed.
- F. Tang, B. Xu, H. Shi, J. Qiu and Y. Fan, Appl. Catal., B, 2010, 94, 71–76 CrossRef CAS.
- J. X. Liu, H. F. Cheng, H. L. Zheng, L. Zhang, B. Liu, W. Y. Song, J. Liu, W. S. Zhu, H. M. Li and Z. Zhao, ACS Catal., 2021, 11(24), 14727–14739 CrossRef CAS.
- Z. Chen, C. Bian, Y. B. Guo, L. Pang and T. Li, ACS Catal., 2021, 11(21), 12963–12976 CrossRef CAS.
- C. Xie, D. F. Yan, H. Li, S. Q. Du, W. Chen, Y. Y. Wang, Y. Q. Zou, R. Chen and S. Y. Wang, ACS Catal., 2020, 10, 11082–11098 CrossRef CAS.
- C. M. Yang, Y. X. Lu, L. Zhang, Z. J. Kong, T. Y. Yang, L. Tao, Y. Q. Zou and S. Y. Wang, Small Struct., 2021, 2100058, 1–25 Search PubMed.
- Y. Q. Zhang, L. Tao, C. Xie, D. D. Wang, Y. Q. Zou, R. Chen, Y. Y. Wang, C. K. Jia and S. Y. Wang, Adv. Mater., 2020, 1905923, 1–22 Search PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- Y. Liu, L. H. Tian, X. Y. Tan, X. Li and X. B. Chen, Sci. Bull., 2017, 62, 431–441 CrossRef CAS PubMed.
- V. A. Glezakou and R. Rousseau, Nat. Mater., 2018, 17, 856–857 CrossRef CAS PubMed.
- S. G. Ullattil, S. B. Narendranath, S. C. Pillai and P. Periyat, Chem. Eng. J., 2018, 343, 708–736 CrossRef CAS.
- T. S. Rajaraman, S. P. Parikh and V. G. Gandhi, Chem. Eng. J., 2020, 389, 123918 CrossRef CAS.
- H. Y. Hu, Y. Lin and Y. H. Hu, Phys. Lett. A, 2019, 383, 2978–2982 CrossRef CAS.
- S. X. Liu, S. Yuan, Q. Zhang, B. Xu, C. Wang, Z. Ming and O. Teruhisa, J. Catal., 2018, 366, 282–288 CrossRef CAS.
- C. Y. Han, S. L. Zhang, L. N. Guo, Y. Q. Zeng, X. H. Li, Z. C. Shi, Y. Zhang, B. Q. Zhang and Q. Zhong, Chem. Eng. Res. Des., 2018, 136, 219–229 CrossRef CAS.
- Y. Q. Zeng, Y. N. Wang, S. L. Zhang and Q. Zhong, Phys. Chem. Chem. Phys., 2018, 20, 22744–22752 RSC.
- H. Q. Tan, Z. Zhao, M. Niu, C. Y. Mao, D. P. Cao, D. J. Cheng, P. Y. Feng and Z. C. Sun, Nanoscale, 2014, 6, 10216–10223 RSC.
- Y. Lu, W. J. Yin, K. L. Peng, K. Wang, Q. Hu, A. Selloni, F. R. Chen, L. M. Liu and M. L. Sui, Nat. Commun., 2018, 9, 2752 CrossRef PubMed.
- K. Lan, R. C. Wang, Q. L. Wei, Y. X. Wang, A. Hong, P. Y. Feng and D. Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 17676–17683 CrossRef CAS PubMed.
- T. Q. Lin, C. Y. Yang, Z. Wang, H. Yin, X. J. Lü, F. Q. Huang, J. H. Lin, X. M. Xie and M. H. Jiang, Energ. Environ. Sci., 2014, 7, 967 RSC.
- P. Pichat, Cataly. Today, 2020, 340, 26–33 CrossRef CAS.
- L. Pan, M. H. Ai, C. Y. Huang, L. Yin, X. Liu, R. R. Zhang, S. B. Wang, Z. Jiang, X. W. Zhang, J. J. Zou and W. B. Mi, Nat. Commun., 2020, 11, 418 CrossRef CAS PubMed.
- F. Li, W. H. Huang and X. Q. Gong, Chin. Chem. Lett., 2018, 29, 765–768 CrossRef CAS.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. D. Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- M. I. Nandasiri, V. Shutthanandan, S. Manandhar, A. M. Schwarz, L. Oxenford, J. V. Kennedy, S. Thevuthasan and M. A. Henderson, J. Phys. Chem. Lett., 2015, 6, 4627–4632 CrossRef CAS PubMed.
- R. Schaub, E. Wahlström, A. Rønnau, E. Lægsgaard, I. Stensgaard and F. Besenbacher, Science, 2003, 299, 377–379 CrossRef CAS PubMed.
- D. N. Pei, L. Gong, A. Y. Zhang, X. Zhang, J. J. Chen, Y. Mu and H. Q. Yu, Nat. Commun., 2015, 6, 8696 CrossRef CAS PubMed.
- D. Pietrogiacomi, D. Sannino, A. Magliano, P. Ciambelli, S. Tuti and V. Indovina, Appl. Catal., B, 2002, 36, 217–230 CrossRef CAS.
- L. Ma, J. Li, R. Ke and L. Fu, J. Phys. Chem. C, 2011, 115, 7603–7612 CrossRef CAS.
- U.S. Department of Commerce. The National Institute of Standards and Technology (NIST) X-ray Photoelectron Spectroscopy (XPS) Database Main Search Menu (2003); https://www.nist.gov/.
- N. Y. Topsoe, Science, 1994, 265, 1217–1219 CrossRef CAS PubMed.
- N. Y. Topsoe, J. A. Dumesic and H. Topsoe, J. Catal., 1995, 151, 241–252 CrossRef CAS.
- G. Z. He, Z. H. Lian, Y. B. Yu, Y. Yang, K. Liu, X. Y. Shi, Z. D. Yan, W. P. Shan and H. He, Sci. Adv., 2018, 4, eaau4637 CrossRef CAS PubMed.
- S. J. Yang, Y. F. Guo, H. Z. Chang, L. Ma, Y. Peng, Z. Qu, N. Q. Yan, C. Z. Wang and J. H. Li, Appl. Catal., B, 2013, 136–137, 19–28 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. T. Wilburn and T. L. Wright, PowerPlant Chem., 2004, 6, 295–314 CAS.
- J. M. Burke and K. L. Johnson, Environmental Protection Agency, Washington, DC, United States, 1982 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ey00077f |
| This journal is © The Royal Society of Chemistry 2023 |