
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed asymmetric reductive aryl-allylation of unactivated alkenes†
Zhiyang
Lin‡
,
Youxiang
Jin‡
,
Weitao
Hu
and
Chuan
Wang
 *
*
Hefei National Laboratory for Physical Science at the Microscale, Department of Chemistry, Center for Excellence in Molecular Synthesis, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 20237, P. R. China. E-mail: chuanw@ustc.edu.cn
First published on 9th April 2021
Abstract
Herein we report a nickel-catalyzed asymmetric reductive aryl-allylation of aryl iodide-tethered unactivated alkenes, wherein both acyclic allyl carbonates and cyclic vinyl ethylene carbonates can serve as the coupling partners. Furthermore, the direct use of allylic alcohols as the electrophilic allyl source in this reaction is also viable in the presence of BOC anhydride. Remarkably, this reaction proceeds with high linear/branched-, E/Z- and enantio-selectivity, allowing the synthesis of various chiral indanes and dihydrobenzofurans (50 examples) containing a homoallyl-substituted quaternary stereocenter with high optical purity (90–98% ee). In this reductive reaction, the use of pregenerated organometallics can be circumvented, giving this process good functionality tolerance and high step-economy.
Introduction
In recent years, the rapid development of transition-metal catalyzed asymmetric aryl-carbofunctionalizations of aryl borane- or aryl (pseudo)halide-tethered alkenes allow the efficient synthesis of chiral benzene-annulated cyclic compounds in a highly enantioselective fashion (Scheme 1A).1 The enantiodetermining step of these reactions relies on facially selective intramolecular Heck-type arylmetallation. After ring closure, the generated chiral alkyl metal species can be intercepted by a boronic acid, an organo(pseudo)halide, a terminal alkyne, or carbon monoxide to deliver the cross-coupling products in either a redox-neutral or a reductive pathway. Hitherto, the successfully installed carbo-moieties following this reaction sequence include alkyl,2–4 aryl,4–6 benzyl,7 alkenyl,4,8 alkynyl,9 and carbonyl.10 However, transition-metal catalyzed highly enantioselective olefin carbo-allylation still remains elusive mainly due to the issues in controlling chemo-, regio-, and stereo-selectivity.11,12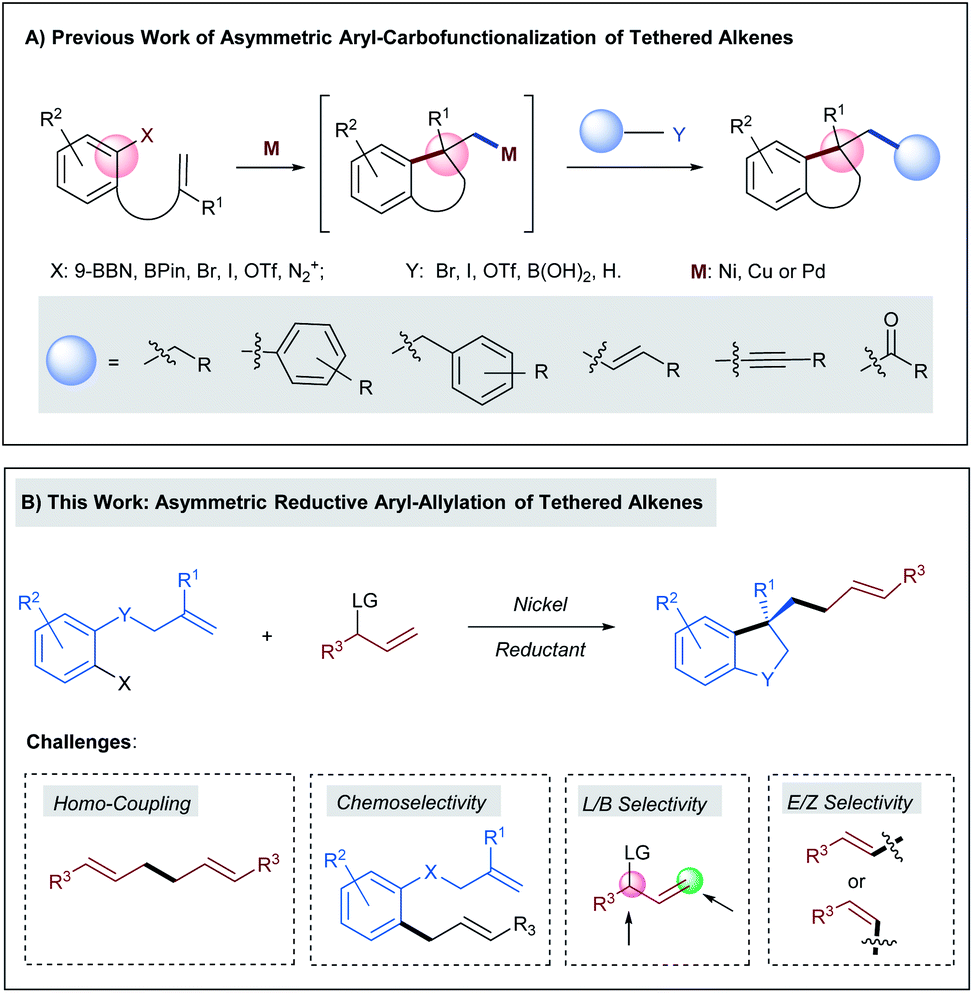 | ||
| Scheme 1 (A) Previous work of asymmetric aryl-carbofunctionalizations of tethered alkenes; (B) asymmetric reductive aryl-allylation of tethered alkenes. | ||
Herein we anticipated that a two-component enantioselective aryl-allylation of aryl-halide-tethered alkenes would be achieved through a reaction with an appropriate electrophilic allylating agent under reductive nickel catalysis13 (Scheme 1B). The challenges of the target reaction lie in the following aspects: (1) allylic electrophiles are highly reactive and thus could easily outcompete aryl halides in the interaction with low-valent nickel, leading to undesired homo-coupling; (2) the direct coupling between aryl halides and allylic electrophiles is also a significant complicating factor; (3) difficulty in discriminating both ends of the generated π-allylic nickel intermediate could result in a mixture of linear and branched products; (4) the newly formed C–C double bond gives rise to a potential issue of controlling E/Z-selectivity.
Results and discussion
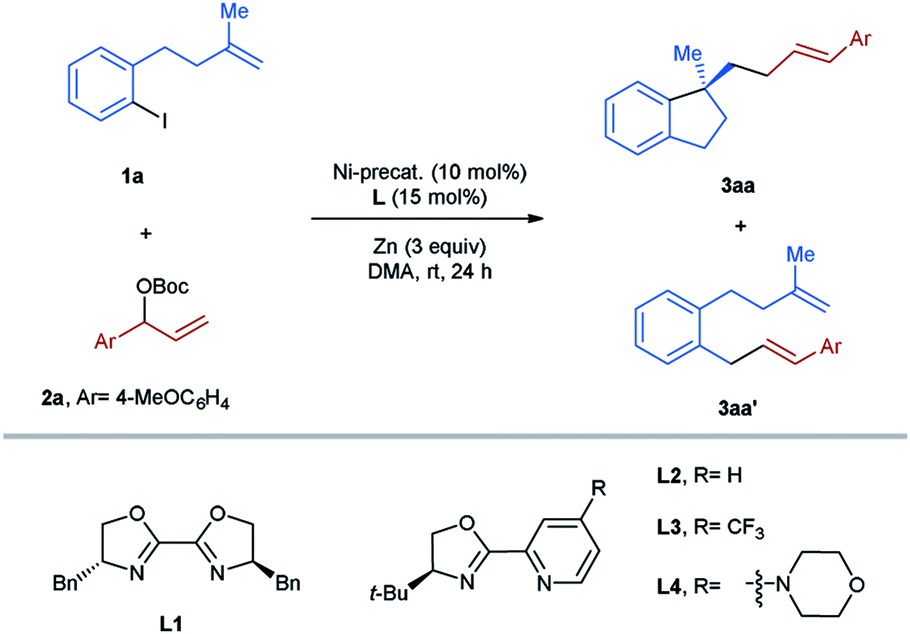
For the optimization of the reaction conditions, the aryl-iodide-tethered alkene 1a and the allylic Boc carbonate 2a (ref. 14) were selected as the standard substrates (Table 1). In our previous work,3,7,15 BiOX L1 and pyrox L2 have proved to be successful ligands for various asymmetric transformations with arylnickelation as the enantiodetermining step. Therefore, these two ligands were first tested for the studied reaction using NiBr2(dme) (10 mol%) as a precatalyst and Zn as a reductant in DMA at room temperature for 24 h. In both cases, the desired aryl-allylation product 3aa was formed in a mixture with the direct cross-coupling product 3aa′, which could not be separated through column chromatography (entries 1 and 2). It is noteworthy that the issue of the competing direct coupling did not exist in our previously reported aryl-alkylation reaction.3 Encouragingly, the cyclization/cross-coupling cascade reaction proceeded with complete regio- and E/Z-selectivity, affording only the linear product with an E-configured olefinic unit with high enantioselectivity. Since a better result concerning both efficiency and asymmetric induction was achieved in the reaction using L2, we continued the optimization sticking to the use of L2 as the ligand. Next, the bromo analog of 1a was employed as the substrate, but only the homo-coupling of 2a occurred, indicating that the reactivity of the allylic carbonate 2a outcompetes the aryl bromide completely (entry 3). We also examined the acetate analog of 2a in the studied reaction. In this case, the formation of 3aa′ turned out to be the major reaction (entry 4). Replacing Zn by Mn as the reducing agent led to the complete shut-down of the desired reaction (entry 5). Performing the reaction in other polar aprotic solvents like DMF and NMP could also provide the aryl-allylation product 3aa, but with less selectively (entries 6 and 7). Subsequently, several nickel salts were screened as the precatalysts (entries 8–11). To our delight, the formation of 3aa′ could be avoided in the case of NiCl2 (entry 10). Raising the reaction temperature to 50 °C improved the yield to 67%, while the enantioselectivity remained excellent (entry 12). Further elevating the temperature to 70 °C showed a detrimental effect on the reaction efficiency (entry 13). When the catalyst loading was increased to 20 mol%, the target product 3aa was obtained in 74% yield and 96%ee (entry 14). We also investigated the substitution effect on the pyridine ring of the pyrox L2. In the case of L3 bearing an electron-withdrawing CF3 group, the aryl-allylation reaction did not occur (entry 15). Moreover, the introduction of an electron-donating amine group also gave rise to a relatively low yield, although the enantiomeric ratio was slightly higher (entry 16). Finally, we tested cinnamyl bromide and chloride as the allylating agents for the studied reaction, but did not observe the formation of the desired product (entry 17).|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Ni-precatalyst | Yield 3aa (%)b | 3aa/3aa'c | ee (%)c |
| a Unless otherwise specified, reactions were performed on a 0.2 mmol scale of the aryl iodide-tethered alkene 1a using 1.5 equiv of the allylic carbonate 2a, 10 mol% Ni-precatalyst, 15 mol% ligand L and 3 equiv of Zn in 1.0 mL DMA at room temperature for 24 h. b Yields of the isolated product obtained through column chromatography. c Determined by HPLC-analysis on a chiral stationary phase. d The bromo analog of 1a was used instead of 1a. e The acetate analog of 2a was used instead of 2a. f Mn was used as the reductant instead of Zn. g Reaction was performed in DMF. h Reaction was performed in NMP. i Reaction was performed at 50 °C. j Reaction was performed at 70 °C. k Reaction was performed using a 20 mol% Ni-precatalyst and 20 mol% ligand. l Cinnamyl chloride or bromide was used instead of 2a. | |||||
| 1 | L1 | NiBr2(dme) | 43 | 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 :4 |
−86 |
| 2 | L2 | NiBr2(dme) | 60 | 90:10 |
94 |
| 3d | L2 | NiBr2(dme) | 0 | — | — |
| 4e | L2 | NiBr2(dme) | 21 | 39:61 |
n.d. |
| 5f | L2 | NiBr2(dme) | 0 | — | — |
| 6g | L2 | NiBr2(dme) | 42 | 58:42 |
92 |
| 7h | L2 | NiBr2(dme) | 51 | 81:19 |
92 |
| 8 | L2 | NiI2 | 24 | 52:48 |
90 |
| 9 | L2 | NiBr2 | 46 | 91:9 |
94 |
| 10 | L2 | NiCl2 | 61 | 98:2 |
96 |
| 11 | L2 | Ni(acac)2 | 0 | — | — |
| 12i | L2 | NiCl2 | 67 | >99:1 |
96 |
| 13j | L2 | NiCl2 | 50 | >99:1 |
95 |
| 14i,k | L2 | NiCl2 | 74 | >99:1 |
96 |
| 15i,k | L3 | NiCl2 | 0 | — | — |
| 16i,k | L4 | NiCl2 | 52 | >99:1 |
97 |
| 17l | L2 | NiCl2 | 0 | — | — |
After establishing the optimal reaction conditions, we started to evaluate the substrate scope of this nickel-catalyzed aryl-allylation reaction. First, we turned our attention to the substrate spectrum of allylic carbonates (Table 2). Various acyclic aryl-substituted secondary allylic carbonates (2b–g) were reacted with the aryl iodide-tethered alkene 1a. In the case of alkoxy-substituted phenyl (2a–d), the reactions proceeded smoothly under the optimized conditions, providing the products 3aa–ad in moderate to good yields and excellent enantioselectivities. When the phenyl ring is substituted by an electron-withdrawing group (2e–g), modifications of the reaction conditions were needed. Under the amended conditions (NiBr2(dme) instead of NiCl2), the corresponding products 3ae–ag were obtained in moderate efficiency with a high level of asymmetric induction. Furthermore, both α- and β-naphthyl substituted carbonates (2h and 2i) also turned out to be competent precursors, affording the coupling products 3ah and 3ai in moderate yields and excellent enantiomeric excesses. When the allylic position of the carbonates is substituted by a tertiary alkyl group (2j–l), the aryl-allylation reaction still occurred, furnishing the products 3aj–al in yields ranging from 51–58% with excellent enantioselectivities. Unfortunately, no desired products were formed in the case of primary or secondary alkyl-substituted allylic carbonates. Of note is that complete linear- and E-selectivities were achieved for all the isolated products mentioned above.
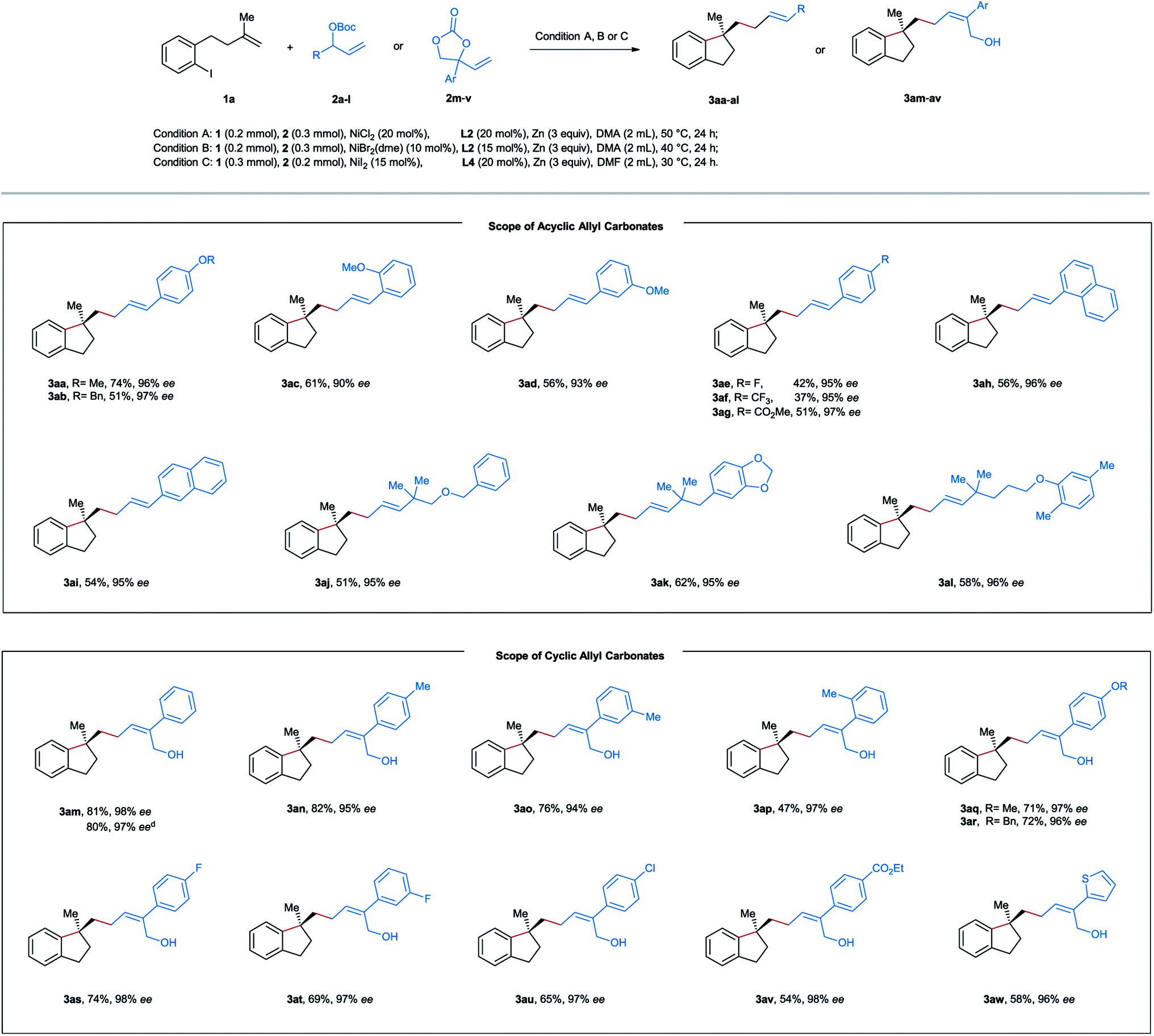
As a specific class of cyclic allyl carbonates, vinyl ethylene carbonates have been widely applied as a substrate with various coupling partners under the catalysis of Pd, Cu, Ir, or Rh.16,17 However, the use of vinyl ethylene carbonates in nickel-catalyzed reductive cross-electrophile coupling is still unknown. After screening various reaction parameters, we reoptimized the reaction conditions for vinyl ethylene carbonates as follows: NiI2 (15 mol%), L4 (20 mol%), and Zn (3 equiv) in DMF at 30 °C for 24 h. Under these conditions, a series of vinyl-substituted 1,3-dioxolan-2-ones 2m–w were successfully utilized as the allyl source in our reductive aryl-allylation, and the corresponding Z-configured chiral allylic alcohols 3am–av and E-configured 3aw were obtained as the only geometry isomer in moderate to good efficiency, complete linear-selectivity and excellent enantioselectivities (Table 2). Notably, the reported reaction could be simply scaled up to 5 mmol in the case of 3am with a similar yield.
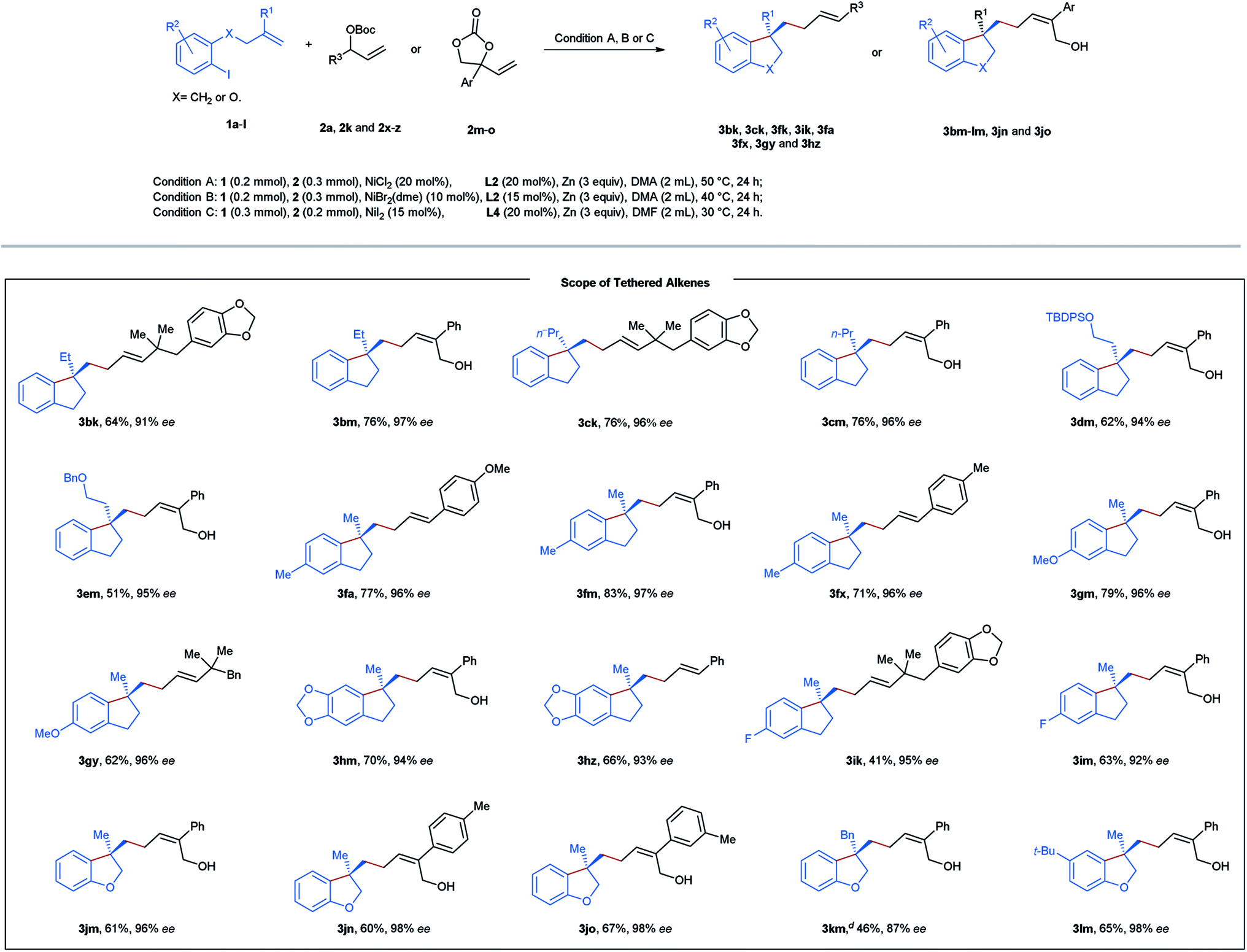
Next, diverse tethered alkenes 1b–l were subjected to the reaction with different acyclic and cyclic carbonates under the optimal reaction conditions (Table 3). Generally, all the reactions provided the desired products with complete linear- and E/Z-selectivities as well as excellent enantioselectivities. Larger geminal substituents of the olefinic unit (1b–e) posed no problem, and moderate to good efficiency could be achieved in these cases. Substituted iodobenzene (1f–i) also turned out to be suitable precursors, producing chiral indanes in yields ranging from 41 to 83%. Moreover, our method is also applicable to the efficient construction of a chiral dihydrobenzofuran scaffold starting from the allyl phenyl ethers 1j–l. In addition, we attempted to construct an indoline and isochroman framework by means of this nickel-catalyzed reaction. Unfortunately, no target cyclization products were formed in these cases.
In the case of 3hz, we utilized the linear cinnamyl acetate 2z′ as the coupling partner instead of its branched analog 2z, and found that the same product was afforded with a similar outcome concerning both yields and selectivities (Scheme 2). This result suggests the presence of a π-allylic nickel intermediate in the reaction, and the linear selectivity could be attributed to a regioselective reductive elimination.
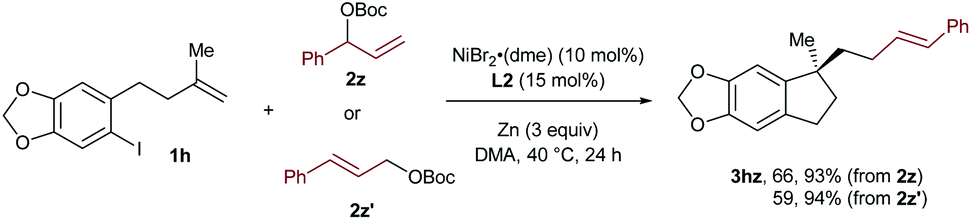 | ||
| Scheme 2 Comparison of the reactions using linear and branched allylic carbonates. | ||
To improve the step economy of our method, we tested the reactions using directly allylic alcohols as the substrates, which were transformed into the corresponding carbonates in situ via a reaction with BOC anhydride (Table 4). Gratifyingly, the target products 3aa and 3ak were yielded with excellent enantioselectivities in the selected reactions, although the efficiency was somewhat lower than those obtained using the corresponding carbonates. During the process of scope evaluation of acyclic allyl carbonates, we noticed that some of them, particularly heteroaryl-substituted secondary allylic carbonates, could not be isolated through column chromatography due to rapid decomposition on silica gel. Therefore, this one-pot procedure provides an entry to prepare aryl-allylation products, which are inaccessible following the reaction employing carbonates. A group of moieties including methoxy-substituted naphthalene (3aaa), pyridine (3aab), pyrrole (3aac), indole (3aad), thiazole (3aae), and thiophene (3gaf) were successfully incorporated into the framework of the aryl-allylation products according to the protocol of the reactions employing allylic alcohols.
| a Unless otherwise specified, reactions were performed on a 0.2 mmol scale of the aryl iodide-tethered alkene 1a or 1g using 2.0 equiv of allylic alcohols 2a, 2k and 2aa–af, 10 mol% NiBr2(dme), 15 mol% ligand L2 and 3 equiv of Zn in 1.0 mL DMA at 40 °C for 24 h. b Yields of the isolated product obtained through column chromatography. c Enantiomeric Excesses were determined by HPLC analysis on a chiral stationary phase. |
|---|
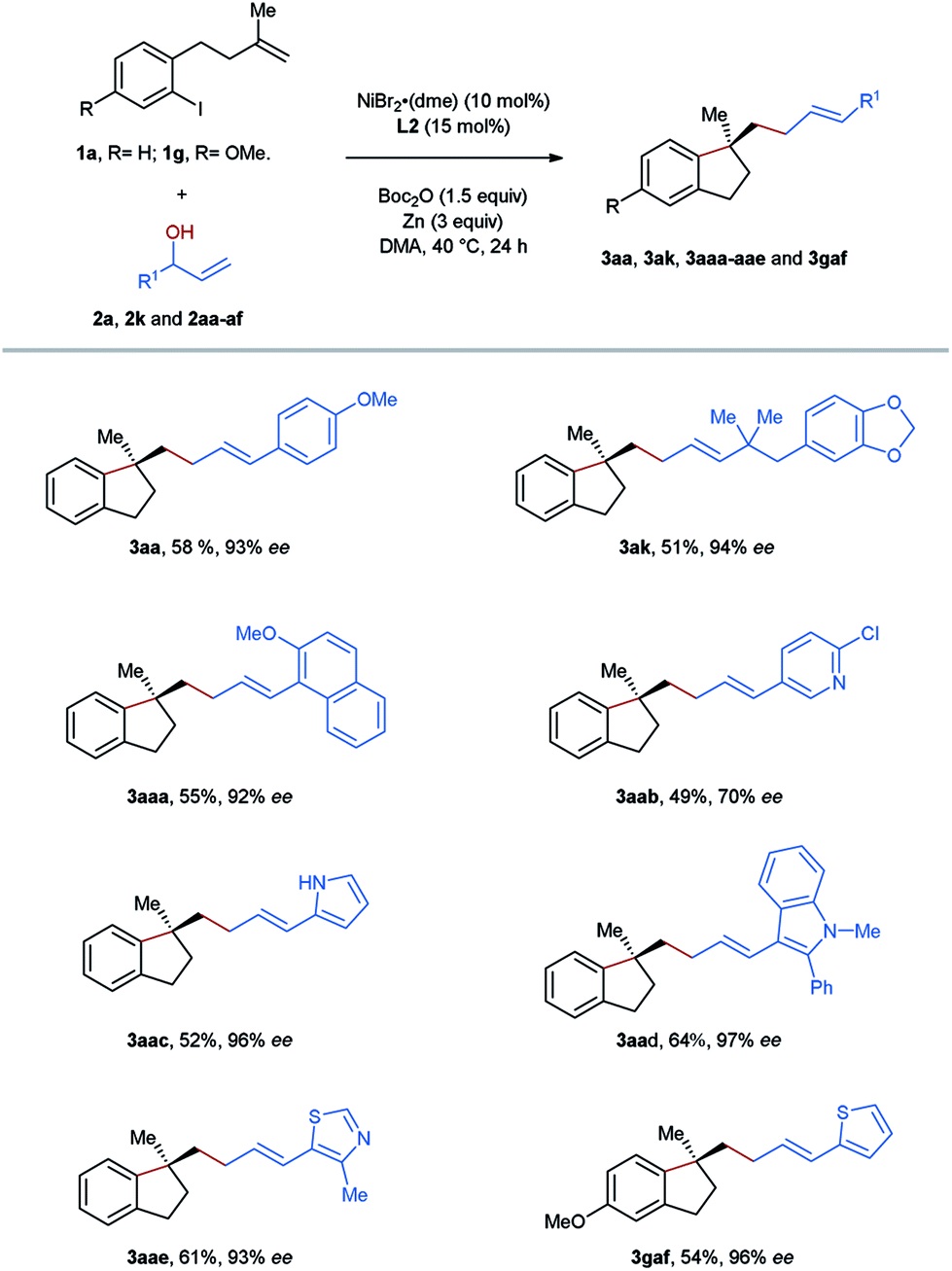
|
Taking advantage of the attached hydroxyl group as a functional handle, various derivatizations of the aryl-allylation product 3am were conducted, and the results are demonstrated in Scheme 3. First, compound 3am was oxidized into an α,β-unsaturated aldehyde 4 in 75% yield by means of Dess–Martin oxidation. Next, PBr3-mediated bromination of 3am followed by nucleophilic substitution with morpholine furnished an allylic amine 5 in 83% yield over two steps. Moreover, 3am was subjected to the Mitsunobu reaction using phenol or phthalimide as the nucleophile, delivering an allyl phenyl ether 6 and an allylic imide 7 in 76% and 87% yields, respectively. Notably, the enantioselectivities of all the derivatization products mentioned above remained high.
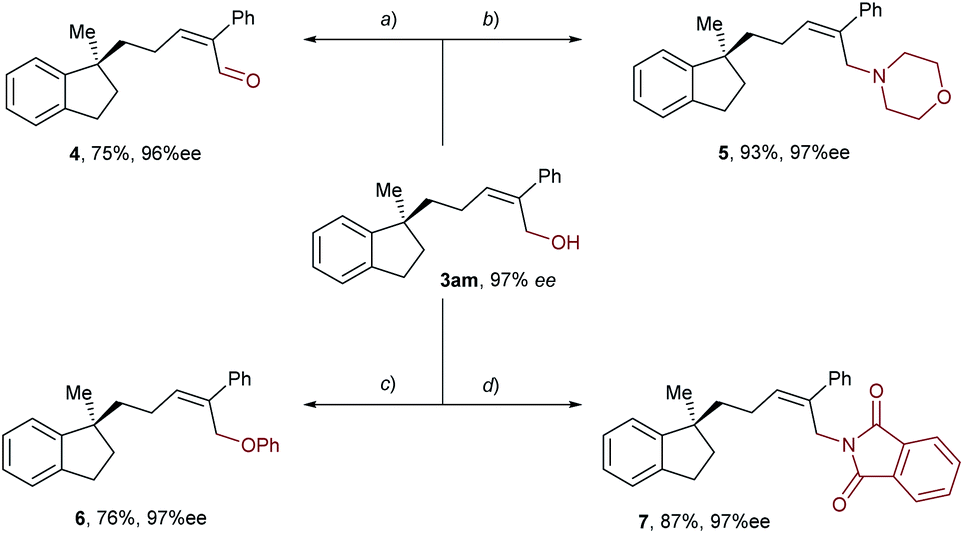 | ||
| Scheme 3 Derivatizations of the aryl-allylation product. | ||
On the basis of previous reports, we proposed a plausible mechanism for this nickel-catalyzed aryl-allylation reaction in Scheme 4. Initially, Ni(0) is formed under reductive conditions and undergoes subsequently oxidative addition with the alkene-tethered aryl iodide 1a. The resultant Ni(II) complex I performs a facially selective intramolecular migratory insertion into the pendant olefinic unit, to complete the construction of the chiral benzene-fused ring framework.6–8 Alternatively, the enantiodiscriminating step could also be facially selective oxidative addition prior to the insertion.18 The following zinc-mediated single-electron reduction affords the Ni(I) intermediate II, to which the allylic carbonates 2a and 2m are oxidatively added. In the case of the cyclic allyl carbonate 2m, decarboxylation succeeds the oxidative addition. Next, terminal selective reductive elimination from the generated π-allylic nickel species III or IV yields the linear aryl-allylation product 3aa or 3am, respectively.14,19 Finally, Zn-mediated reduction of Ni(I) regenerates Ni(0) for the next catalytic cycle.
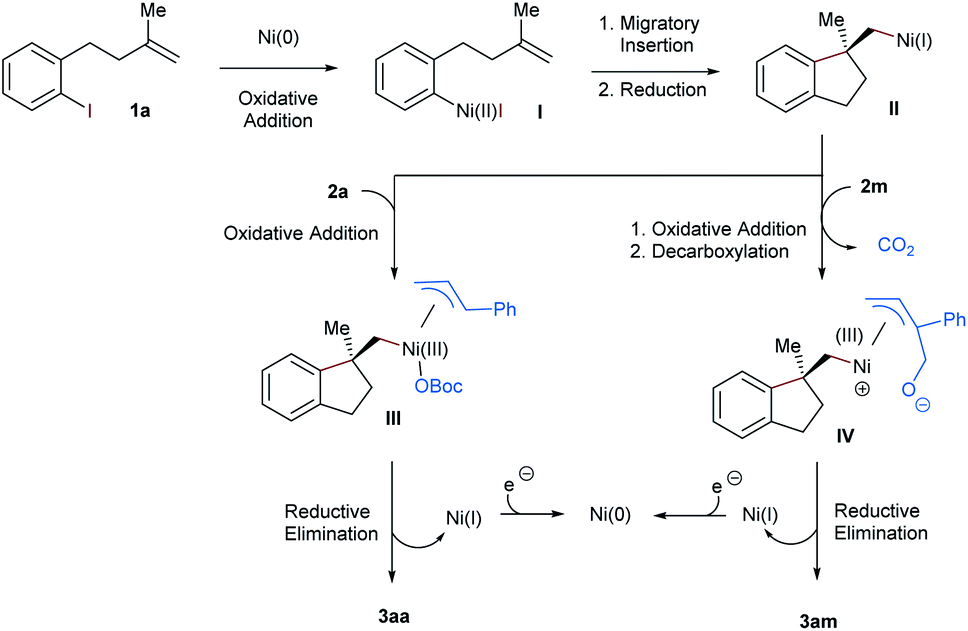 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a highly enantioselective nickel-catalyzed reductive aryl-allylation of aryl iodide-tethered unactivated alkenes with both acyclic and cyclic allyl carbonates as the allylating agent. Moreover, allylic alcohols can be employed directly as the electrophilic allyl source in the presence of BOC anhydride. The enantiodetermining step of the studied reaction relies on facially selective intramolecular Heck-type arylnickelation, while regioselective reductive elimination from the π-allylic nickel intermediates leads to the linear selectivity of the coupling product. Furthermore, the formation of a new C–C double bond proceeds with high E/Z-selectivity. The aforementioned high chemo-, regio- and stereo-control along with mild reductive reaction conditions allow the preparation of diverse highly enantioenriched indanes and dihydrobenzofurans bearing a homoallyl-substituted quaternary stereogenic center with a high tolerance of functionalities.Author contributions
C. W. conceived and designed the experiments. Z. L., Y. J. and W. H. performed the experiments and prepared the Supporting Information. C. W. directed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by “1000-Youth Talents Plan” start up funding, the National Natural Science Foundation of China (21772183, 22071230), the Fundamental Research Funds for the Central Universities (WK2060190086) and the University of Science and Technology of China.Notes and references
- For reviews on transition-metal catalyzed dicarbo-functionalization, see: (a) R. Giri and S. KC, J. Org. Chem., 2018, 83, 3013 CrossRef CAS PubMed; (b) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287 RSC; (c) H.-Y. Tu, S. Zhu, F.-L. Qing and L. Chu, Synthesis, 2020, 52, 1346 CrossRef CAS; (d) X. Qi and T. Diao, ACS Catal., 2020, 10, 8542 CrossRef CAS PubMed; (e) Y.-C. Luo, C. Xu and X. Zhang, Chin. J. Chem., 2020, 38, 1371 CrossRef CAS; (f) Y. Ping and W. Kong, Synthesis, 2020, 52, 979 CrossRef.
- H. Cong and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 3788 CrossRef CAS PubMed.
- Y. Jin and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6722 CrossRef CAS PubMed.
- Z.-M. Zhang, B. Xu, L. Wu, Y. Wu, Y. Qian, L. Zhou, Y. Liu and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14653 CrossRef CAS PubMed.
- W. You and M. K. Brown, J. Am. Chem. Soc., 2015, 137, 14578 CrossRef CAS PubMed.
- K. Wang, Z. Ding, Z. Zhou and W. Kong, J. Am. Chem. Soc., 2018, 140, 12364 CrossRef CAS PubMed.
- Y. Jin, H. Yang and C. Wang, Org. Lett., 2020, 22, 2720 CrossRef PubMed.
- (a) Z.-X. Tian, J.-B. Qiao, G.-L. Xu, X. Pang, L. Qi, W.-Y. Ma, Z.-Z. Zhao, J. Duan, Y. F. Du, P. Su, X.-Y. Liu and X.-Z. Shu, J. Am. Chem. Soc., 2019, 141, 7637 CrossRef CAS PubMed; (b) T. Ma, Y. Chen, Y. Li, Y. Ping and W. Kong, ACS Catal., 2019, 9, 9127 CrossRef CAS.
- (a) X. Bai, C. Wu, S. Ge and Y. Lu, Angew. Chem., Int. Ed., 2020, 59, 2764 CrossRef CAS PubMed; (b) L. Zhou, S. Li, B. Xu, D. Ji, L. Wu, Y. Liu, Z.-M. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 2769 CrossRef CAS PubMed.
- (a) R. C. Carmona, O. D. Köster and C. R. D. Correia, Angew. Chem., Int. Ed., 2018, 57, 12067 CrossRef CAS PubMed; (b) H. Hu, F. Teng, J. Liu, W. Hu, S. Luo and Q. Zhu, Angew. Chem., Int. Ed., 2019, 58, 9225 CrossRef CAS PubMed; (c) C. Cheng, B. Wan, B. Zhou, Y. Gu and Y. Zhang, Chem. Sci., 2019, 10, 9853 RSC; (d) M. Chen, X. Wang, P. Yang, X. Kou, Z.-H. Ren and Z.-H. Guan, Angew. Chem., Int. Ed., 2020, 59, 12199 CrossRef CAS PubMed; (e) Z. Yuan, Y. Zeng, Z. Feng, Z. Guan, A. Lin and H. Yao, Nat. Commun., 2020, 11, 2544 CrossRef CAS PubMed.
- For examples of the racemic version of transition metal-catalyzed olefin carbo-allylation, see: (a) T. Taniguchi and K. Ogasawara, Chem. Commun., 1998, 1531 RSC; (b) K. Semba, N. Ohta, Y. Yano and Y. Nakao, Chem. Commun., 2018, 54, 11463 RSC; (c) K. Semba, N. Ohta and Y. Nakao, Org. Lett., 2019, 21, 4407 CrossRef CAS PubMed; (d) H. Li, M. Zhang, H. Mehfooz, D. Zhu, J. Zhao and Q. Zhang, Org. Chem. Front., 2019, 6, 3387 RSC; (e) V. T. Tran, Z.-Q. Li, T. J. Gallagher, J. Derosa, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7029 CrossRef CAS PubMed.
- During the preparation of our manuscript, Nakao et al. reported an enantioselective example of olefin aryl-allylation with moderate enantiocontrol: K. Semba, N. Ohta, F. Paulus, M. Ohata and Y. Nakao, Chemistry – A European Journal, 2021, 27, 5035 CrossRef CAS PubMed.
- For general reviews on reductive cross-coupling, see: (a) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793 CrossRef CAS PubMed; (b) T. Moragas, A. Correa and R. Martin, Chemistry – A European Journal, 2014, 20, 8242 CrossRef CAS PubMed; (c) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411 RSC; (d) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed; (e) X. Wang, Y. Dai and H. Gong, Top. Curr. Chem., 2016, 374, 43 CrossRef PubMed; (f) E. Richmond and J. Moran, Synthesis, 2018, 50, 499 CrossRef CAS; (g) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237 CrossRef CAS PubMed; (h) Y. Jin and C. Wang, Synlett, 2020, 31, 1843 CrossRef CAS.
- For examples of the use of allylic carbonates in nickel-catalyzed cross-electrophile coupling, see: (a) Y. Dai, F. Wu, Z. Zang, H. You and H. Gong, Chemistry – A European Journal, 2012, 18, 808 CrossRef CAS PubMed; (b) F. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Angew. Chem., Int. Ed., 2017, 56, 13103 CrossRef PubMed; (c) F. Song, F. Wang, L. Guo, X. Feng, Y. Zhang and L. Chu, Angew. Chem., Int. Ed., 2020, 59, 177 CrossRef CAS PubMed.
- F. Yang, Y. Jin and C. Wang, Org. Lett., 2019, 21, 6989 CrossRef CAS PubMed.
- For a review on the use of vinyl ethylene carbonates in transition-metal catalysis, see: W. Guo, J. E. Gómez, À. Cristòfol, J. Xie and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 13735 CrossRef CAS PubMed.
- For examples on the use of vinyl ethylene carbonates in transition-metal catalysis, see: (a) B. M. Trost and G. R. Granja, Tetrahedron Lett., 1991, 32, 2193 CrossRef CAS; (b) B. M. Trost and A. Aponick, J. Am. Chem. Soc., 2006, 128, 3931 CrossRef CAS PubMed; (c) Y. J. Zhang, J. H. Yang, S. H. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 4562 CrossRef CAS PubMed; (d) A. Khan, R. Zheng, Y. Kan, J. Ye, J. Xing and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6439 CrossRef CAS PubMed; (e) A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11257 CrossRef CAS PubMed; (f) W. Guo, L. Martínez-Rodríguez, R. Kuniyil, E. Martin, E. C. Escudero-Adán, F. Maseras and A. W. Kleij, J. Am. Chem. Soc., 2016, 138, 11970 CrossRef CAS PubMed; (g) A. Cai, W. Guo, L. Martínez-Rodríguez and A. W. Kleij, J. Am. Chem. Soc., 2016, 138, 14194 CrossRef CAS PubMed; (h) W. Guo, L. Martínez-Rodríguez, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2016, 55, 11037 CrossRef CAS PubMed; (i) A. Khan, S. Khan, I. Khan, C. Zhao, Y. Mao, Y. Chen and Y. J. Zhang, J. Am. Chem. Soc., 2017, 139, 10733 CrossRef CAS PubMed; (j) L.-C. Yang, Z.-Q. Rong, Y.-N. Wang, Z. Y. Tan, M. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2017, 56, 2927 CrossRef CAS PubMed; (k) H. Wang, M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 6339 CrossRef CAS PubMed; (l) Z.-Q. Rong, L.-C. Yang, S. Liu, Y.-N. Wang, Z. Y. Tan, R.-Z. Huang, Y. Lan and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 15304 CrossRef CAS PubMed; (m) N. Miralles, J. E. Gómez, A. W. Kleij and E. Fernández, Org. Lett., 2017, 19, 6096 CrossRef CAS PubMed; (n) S. Singha, T. Patra, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2018, 140, 3551 CrossRef CAS PubMed; (o) A. Khan, H. Zhao, M. Zhang, S. Khan and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 1340 CrossRef CAS PubMed; (p) Y. Wang, J. Chai, C. You, J. Zhang, X. Mi, L. Zhang and S. Luo, J. Am. Chem. Soc., 2020, 142, 3184 CrossRef CAS PubMed; (q) L. Chen, H. Quan, Z. Xu, H. Wang, Y. Xia, L. Lou and W. Yang, Nat. Commun., 2020, 11, 2151 CrossRef CAS PubMed.
- J.-N. Desrosier, J. Wen, S. Tcyrulnikov, S. Biswas, B. Qu, L. Hie, D. Kurouski, L. Wu, N. Grinberg, N. Haddad, C. A. Busacca, N. K. Yee, J. J. Song, N. K. Garg, X. Zhang, M. C. Kozlowski and C. H. Senanayake, Org. Lett., 2017, 19, 3338 CrossRef PubMed.
- L. S. Hegedus and D. H. P. Thompson, J. Am. Chem. Soc., 1985, 107, 5663 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectra data, NMR data, and HPLC data. See DOI: 10.1039/d1sc01115d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |