
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxanorbornenes: promising new single addition monomers for the metathesis polymerization†
Subhajit
Pal
,
Mahshid
Alizadeh
,
Phally
Kong
and
Andreas F. M.
Kilbinger
 *
*
Department of Chemistry, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: andreas.kilbinger@unifr.ch
First published on 7th April 2021
Abstract
Higher ring-opening metathesis propagation rates of exo-norbornene derivatives over endo derivatives are well established in the literature. Here, we report for the first time that endo-isomers of oxanorbornene derivatives show higher reactivity towards ring-opening metathesis with Grubbs' 3rd generation catalyst (G3) than the corresponding exo-isomers. A very high selectivity for the reaction of G3 with endo over the exo-isomers could be shown. Furthermore, single molecular addition of the endo-isomers with G3 was observed. On the other hand, pure exo-monomers could successfully be homopolymerized. Mixtures of exo- and endo- monomers, however, prevented the homopolymerization of the exo-monomer. Such mixtures could successfully be copolymerized with cycloalkenes, resulting in alternating copolymers. An oxanorbornadiene derivative could be shown to undergo single addition reactions, exploited in the preparation of mono-end functional ROMP polymers. These could be selectively derivatized via endgroup selective thiol-ene click reactions. A thiol and alcohol end functional ROMP polymer was synthesized, and the efficient end functionalization was confirmed by 1H NMR spectroscopy and MALDI-ToF spectrometry.
Introduction
Today, the ring-opening metathesis polymerization (ROMP) has become a routine polymerization technique in the toolbox of polymer chemists. Typically, ruthenium and molybdenum based metal carbene complexes are used for ROMP.1–3 Features such as living polymerization, high functional group tolerance of metathesis catalysts, mild reaction conditions, and short reaction times make ROMP popular in polymer as well as materials chemistry.4–7 Release of ring strain of the cyclic olefins is the driving force for polymerization; as a result, highly strained cyclic olefins like cyclopropene, cyclobutene, cyclooctene, norbornene, etc. are used as monomers in ROMP.8In addition to fast and living polymerizations, control over molecular structure and monomer sequence has been a central goal of polymer chemistry. Several synthetic approaches have been made to tune the polymers' microscopic properties by the precise placement of chemical functionality.9,10 However, a strictly sequential arrangement of individual monomer units in a polymer remains challenging.
Much progress has been made regarding the precise placement of end functional groups in ROMP polymers.6,11 Within the broad field of reactive polymer chain ends, the thiol group has received significant attention. From bioconjugation to transition metal complexation, thiols are often superior over their oxo-derivatives.12,13 One of the most common approaches for ROMP polymer end functionalization is the chain transfer with a symmetrical chain transfer agents (CTA) to install a particular end group.6 However, the synthesis of different CTAs for each different end group can be very laborious. Simultaneously, the intolerance of Ru metal carbene complexes for thiol, makes a thiol end-functionalized ROMP polymer synthesis particularly challenging.14
Over the past few years, ROMP has been used to synthesize sequence defined polymers, mostly alternating copolymers.15–21,66 Several approaches have been made to achieve alternating ring-opening metathesis polymerization (AROMP) either via catalyst modification or through monomer design.22–30 In 2009, Sampson and co-workers reported an AROMP strategy where cyclobutene-1-carboxylic esters undergo ring-opening metathesis with a ruthenium catalyst giving an enoic carbene incapable of homopropagation.31–39
However, alternating copolymerization of cyclobutene-1-carboxylic esters with cycloalkenes lead to a sterically accessible double bond in the polymer backbone, resulting in secondary metathesis (chain transfer to the polymer). Later, Xia and co-workers introduced a creative approach for AROMP using the single molecular addition of sterically hindered cyclopropene derivatives.40–44 However, the preparation of cyclopropenes for AROMP is synthetically challenging.
Monomers like norbornene (NBE) and its derivatives (Fig. 1) are the most common choice in metathesis polymerization due to easy synthetic accessibility. Fast polymerization kinetics, easy functionalization, and absence of irreversible chain-transfer allows for a living polymerization and make NBE superior to many other monomers.45 However, among the two different isomers (exo- and endo-, Fig. 1) of NBE derivatives, exo-isomers are predominantly used.46 It is well-known that in the presence of a suitable metal carbene initiator, exo-isomers undergo rapid polymerization, whereas endo-isomers exhibit much slower propagation kinetics.47–50 The difference in reactivity of the two isomers of NBE derivatives is attributed to a higher ring strain and a more reactive propagating carbene of the exo-derivative.51–56 However, a lower ring strain and a less reactive propagating carbene of the endo-isomer could potentially be advantageous in controlling the polymer microstructure. Unfortunately, the detailed exploration of endo- norbornene derivatives towards sequential incorporation during polymerization is rare.
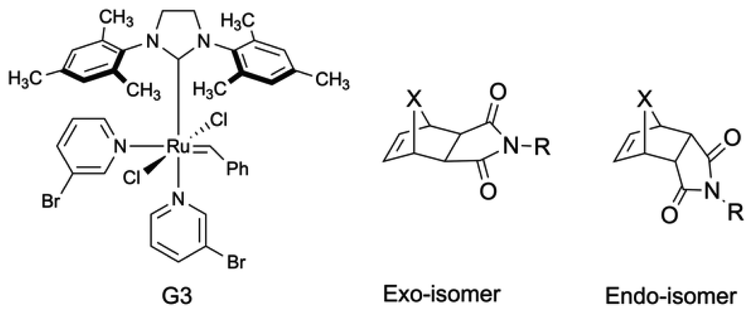 | ||
| Fig. 1 (Left) commercially available Grubbs' third generation (G3) catalyst. (Right) two general representations of exo- and endo- norbornene imide derivatives where X are either C (carba) or O (oxo). | ||
Several approaches have been reported for the controlled incorporation of norbornene derivatives throughout the copolymerization. The higher reactivity of NBE derivatives makes a sequential arrangement challenging. Coughlin and co-workers reported AROMP using oxanorbornene imide derivatives with Grubbs' 1st (G1) and 2nd (G2) generation catalysts.57,58 Recently, our group also reported AROMP with oxanorbornene imide (oxaNBE) derivatives even at elevated temperature using G2.59 However, under similar conditions, the corresponding norbornene derivatives exhibiting comparable ring strain mainly yielded block copolymers.60–63 To the best of our knowledge, a norbornene derivative that undergoes a strict single addition to a metathesis initiator has not yet been reported.
The slower reactivity of oxanorbornene derivatives compared to their norbornene counterparts and a more straightforward synthetic accessibility inspired us to explore such derivatives as single addition monomers. Here, we are reporting for the first time the single monomer addition of an oxanorbornene derivative to Grubbs' 3rd generation catalyst (G3, Fig. 1), which gives access to AROMP and a unique end functionalization method for mono-telechelic ROMP polymers.
Results and discussion
Initiation studies
Faster propagation kinetics of exo-norbornene derivatives compared to endo-derivatives are well established.47–50 However, we are not aware of detailed studies investigating the initiation kinetics of exo- and the corresponding endo-isomers.To understand the initiation preference of G3 (Fig. 1) for either the exo- or the endo-isomer, a control 1H NMR experiment was performed with a mixture of 1-exo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-endo: G3 (1:1:0.4) in the presence of 30 eq. of 3-bromo pyridine. 3-Bromo pyridine was used to slow down the reaction with G3 so that it could be followed by 1H NMR spectroscopy. Surprisingly, preferential initiation of G3 with 1-endo over 1-exo (80% endo carbene, 20% exo carbene) was clearly observed (Fig S2 and S3†). Over the course of the reaction, the concentration of the endo carbene increased even in the presence of 1-exo. To generalize whether the observed preference is also valid for (7-carba) norbornene derivatives, another control 1H NMR experiment was performed with a 1:1 mixture of endo and exo N-phenylnorbornene imide in the presence of G3 (1:1:0.5) and 30 eq. of 3-bromo pyridine. Interestingly, in this case preferential initiation with the exo-isomer was observed (Fig. S4†). Initially, G3 reacted mainly with the exo isomer (30% endo-carbene, 70% exo-carbene) and over time (60 min) converted exclusively to the endo carbene after complete consumption of the exo-derivative.
:1-endo: G3 (1:1:0.4) in the presence of 30 eq. of 3-bromo pyridine. 3-Bromo pyridine was used to slow down the reaction with G3 so that it could be followed by 1H NMR spectroscopy. Surprisingly, preferential initiation of G3 with 1-endo over 1-exo (80% endo carbene, 20% exo carbene) was clearly observed (Fig S2 and S3†). Over the course of the reaction, the concentration of the endo carbene increased even in the presence of 1-exo. To generalize whether the observed preference is also valid for (7-carba) norbornene derivatives, another control 1H NMR experiment was performed with a 1:1 mixture of endo and exo N-phenylnorbornene imide in the presence of G3 (1:1:0.5) and 30 eq. of 3-bromo pyridine. Interestingly, in this case preferential initiation with the exo-isomer was observed (Fig. S4†). Initially, G3 reacted mainly with the exo isomer (30% endo-carbene, 70% exo-carbene) and over time (60 min) converted exclusively to the endo carbene after complete consumption of the exo-derivative.
The steric demand of both, 7-carba and 7-oxanorbornene derivatives is very similar and it is not immediately apparent to us what causes the observed difference in initiation kinetics. Ab initio calculations are currently being carrying out in an attempt to answer this question.
As far as the propagation rate is concerned, oxaNBEimides follow a similar trend to NBEimide derivatives. We believe this is due to the lower reactivity of the propagating endo carbene compared to the propagating exo-carbene. The lower reactivity of propagating endo-carbenes of NBEimide derivatives is well established in the literature.51–56 For geometrical reasons, the ring-opening of any endo norbornene or 7-oxanorbornene imide derivative results in the imide group pointing towards the Ru metal center. This results in a sterically more crowded metal coordination site and, likely, a coordination between the carbonyl group of the imide with the Ru metal center. We believe this to be the reason for the observed lower reaction rates for such carbene complexes.
In order to achieve a true single monomer addition the monomer should react only once with an initiator and should not undergo homopolymerization. The fast initiation and slow homopropagation kinetics of the endo oxanorbornene derivatives appeared suitable to be considered as single addition monomers. Although, 1-endo has a low reactivity it still undergoes homopolymerization.59 The lower reactivity of bridgehead substituted oxanorbornene imide derivatives towards ROMP had been reported previously.64 The presence of steric bulk at the bridgehead position makes the monomer less accessible for coordination to alkylidene catalysts and thus results in slower polymerization.
Slower monomers design
Therefore, the unique reactivity of oxaNBEimide derivatives was explored more carefully by synthesizing sterically more demanding and hence less reactive bridgehead substituted derivatives of monomer 1 (monomers 2, 3, 4 and 5Scheme 1E).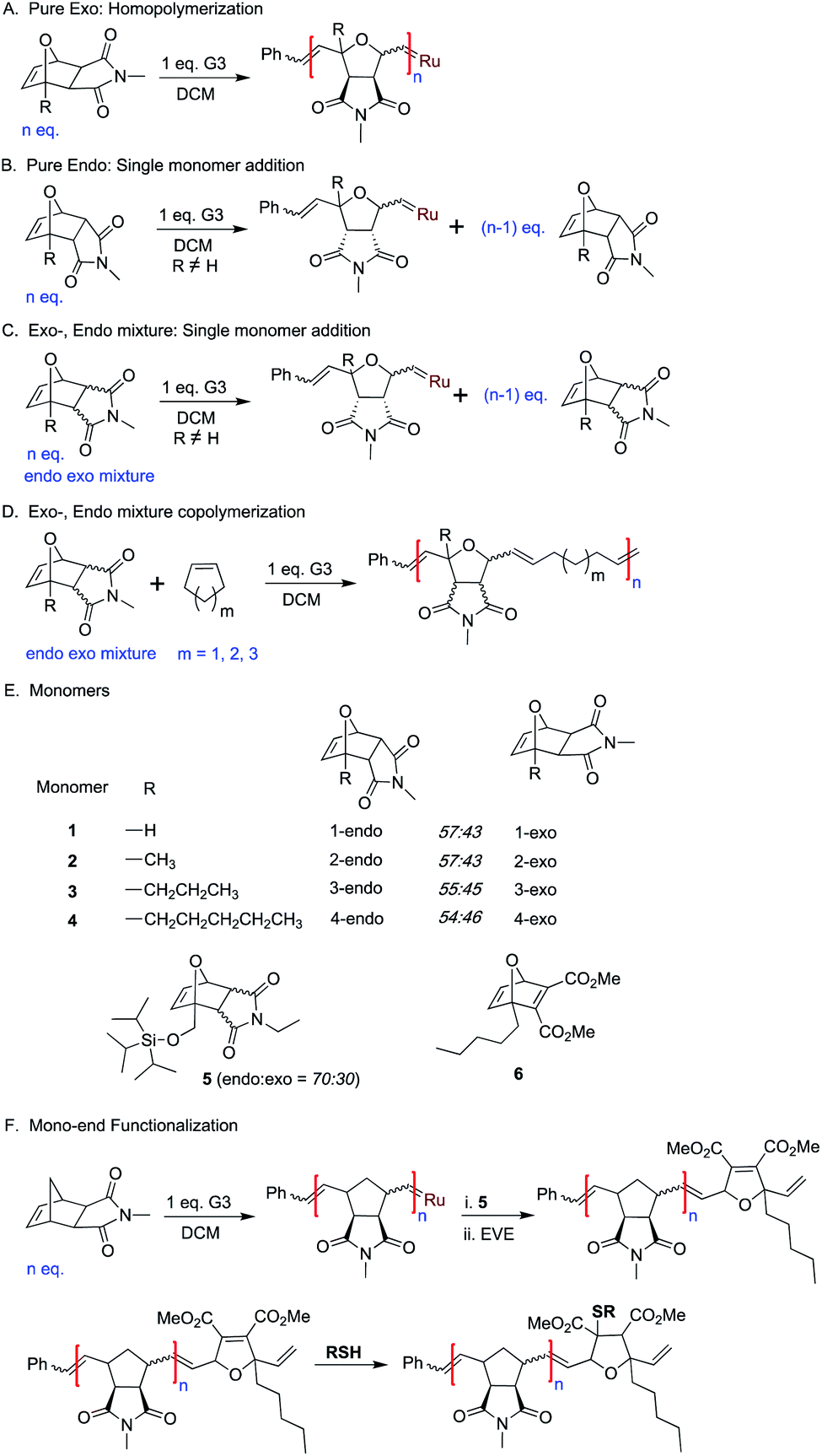 | ||
| Scheme 1 General representation of exo and endo-oxanorbornene reactivity. | ||
To investigate the initiation selectivity of G3 and the newly synthesized monomers, control 1H NMR experiments were performed with mixtures of exo- and endo-monomers 2–4 and G3 (1:1:0.4) in dichloromethane-d2 (Fig. 2). The lower reactivity of monomers 2–4 allows careful 1H NMR analysis even in the absence of 3-bromo pyridine. Interestingly, more than 94% endo:exo carbene selectivity was observed for monomer 2 (40 min, Fig. 2A, B, S5 and S6†), whereas 99.9% endo:exo selectivity was observed in the case of monomer 4 (55 min, Fig. 2C, D, S7 and S8†).
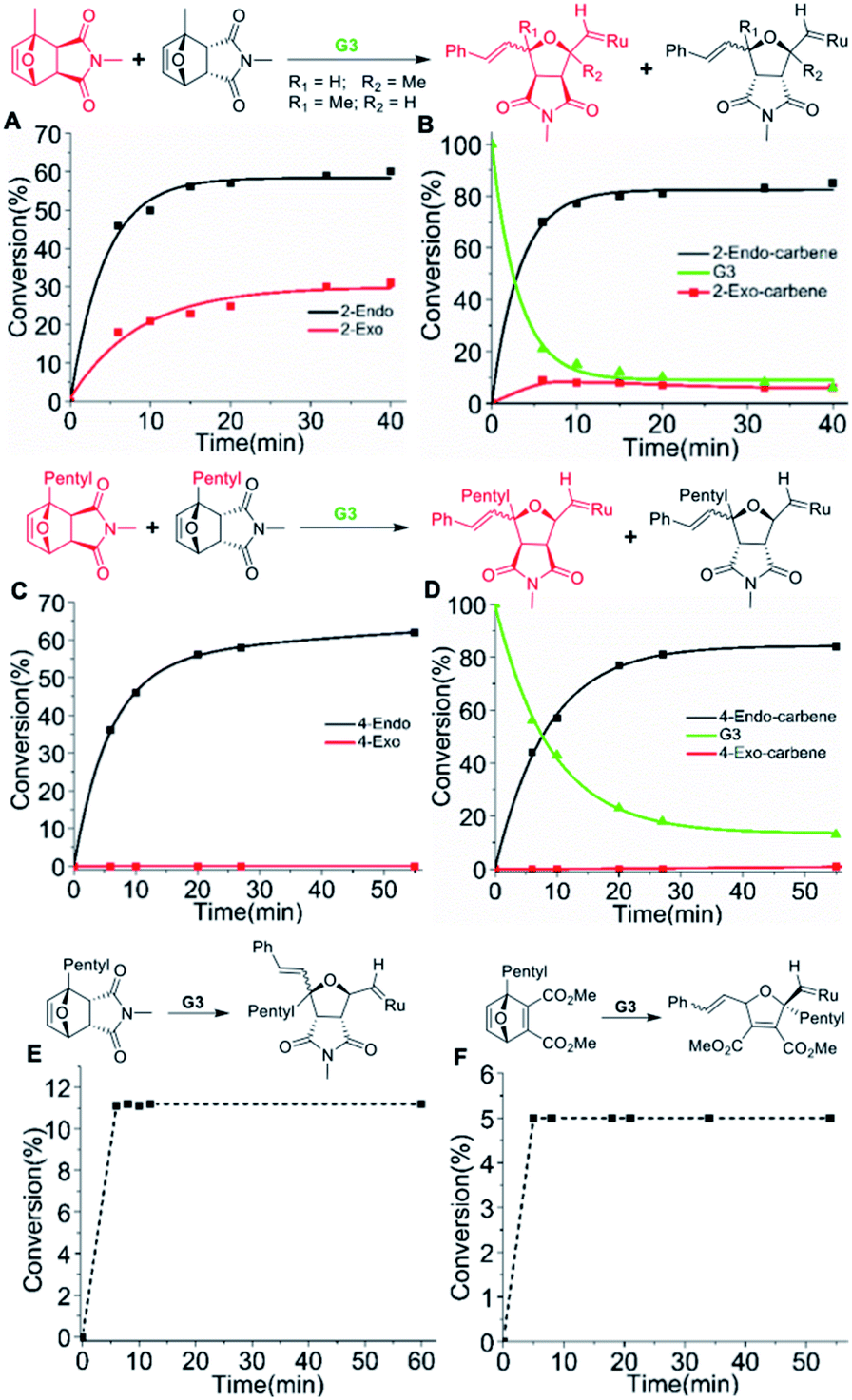 | ||
| Fig. 2 Plot of monomer and initiator conversion determined by 1H NMR spectra (CD2Cl2, 400 MHz) of the reactions: (A and B) mixture of 2-endo:2-exo (1:1) with 0.4 eq. G3. (A) Conversion of 2-endo, 2-exovs. time showing preference for 2-endo (B) Conversion to endo carbene and exo carbene vs. time showing preference for endo carbene. (C and D) Mixture of 4-endo:4-exo (1:1) with 0.4 eq. G3. (C) Conversion of 4-endo, 4-exovs. time showing selective reaction with 4-endo (D) conversion to endo carbene and exo carbene vs. time showing exclusive formation of endo carbene. (E) Conversion of monomer 4-endovs. time showing single molecular addition of 4-endo to G3 (11 mol%). (F) Conversion of monomer 6vs. time showing single molecular addition of 6 to G3 (5 mol%). | ||
Next, the endo and exo-isomers of monomers 2 and 4 were isolated by column chromatography (see ESI†) and the homopropagation of the individual isomers was followed by 1H NMR spectroscopy. The effect of increasing steric bulk towards slowing down the polymerization was confirmed by homopolymerization experiments of monomers 2-exo and 4-exo (Scheme 1A). The 1H NMR analysis of the homopolymerization of monomer 2-exo in the presence of 3 mol% of G3, shows complete consumption of the monomer within 5 hours whereas monomer 4-exo was consumed only 75% in 24 hours (Fig. S9 and S10†).
Then, homopolymerizations of 2-endo, 3-endo, and 4-endo with 3 mol% of G3 were attempted. Surprisingly, no homopolymer was obtained in any of the experiments. To understand the “inertness” of the endo monomers towards homopolymerization, a control 1H NMR experiment was performed with 4-endo and 10 mol% G3 in dichloromethane-d2 (Scheme 1B, Fig. 2E and S11†). Surprisingly, only a single monomer addition to G3 was observed immediately and the 1H-NMR spectrum remained unchanged even after 13 hours. A similar observation was also made when 2-endo was reacted with 10 mol% of G3 (Scheme 1B, Fig S12†).
Thereafter, homopolymerization studies were performed with mixtures of the corresponding exo and endo isomers of monomer 2 and 4 using 6.6 mol% of G3 (Scheme 1C, Fig S13 and S14†). Even in these cases, only a single addition of the endo-monomer but no homopolymerization of the exo isomer was observed (Fig S13 and S14†).
Copolymerizations
This clearly shows that G3 reacts fast and selectively with the endo-isomers of monomers 2, 3 and 4 and that the resulting carbene complexes cannot further propagate neither with the endo nor the exo isomer due to increased steric bulk. Consequently, in the absence of homopolymerization of the exo-, endo- mixture of monomers 2–4, the determination of propagation preference of G3 becomes challenging. However, a bulky monomer that does not homopolymerize could still undergo copolymerization with sterically less bulky comonomers. A set of monomers that undergo single monomer addition but do not homopolymerize is most useful for sequence-controlled polymer synthesis. One of the simplest examples for sequence control is the alternating copolymerization. If two monomers cannot homopolymerize individually but form a copolymer when mixed together, the resulting copolymer must necessarily be a strictly alternating copolymer.Next, the potential of monomers 2–4 towards alternating copolymerization was studied. Alternating copolymerization of monomers 2–4 will allow the determination of the propagation preference of G3 among the exo, endo isomers. Exhibiting little or no ring strain, small cycloalkenes such as cyclohexene (Chex), cyclopentene (Cpen), or cycloheptene (Chep) are popular comonomers in alternating ROMP polymer synthesis.31,32,40,41 We hypothesized that, after fast initiation of G3 with a bulky strained monomer (such as 2–4, see above), the formed alkylidene might still react with a less bulky cycloalkene monomer. This would lead to a sterically less hindered alkylidene which could react once more with a new strained but bulky monomer leading, eventually, to an alternating sequence.
To verify our hypothesis, a control 1H NMR copolymerization experiment was performed using an endo/exo-mixture (57:43) of monomer 2 (16 eq.) cyclohexene (Chex) (320 eq.) and 6.6 mol% of G3 (1 eq.) in the presence of an internal standard (1,3,5-trimethoxybenzene) in dichloromethane-d2 (Scheme 1D). Interestingly, under these conditions the consumption of both, endo (2-endo) and exo monomer (2-exo), was observed over the course of the reaction. A faster consumption of the endo isomer was clearly observed, which confirmed our earlier observations (see above and Fig. 3A). The fact that the exo monomer (2-exo) is also consumed over time shows that the ruthenium alkylidene complex formed after ring opening of cyclohexene is less selective than the G3 benzylidene complex: the sterically bulky and less reactive metal carbene complex of 2-endo cannot homopolymerize. After reaction with cyclohexene (Chex), an alkylidene is formed that is sterically less hindered and more reactive.65 This carbene is less selective than the initial benzylidene complex. It still results in a faster endo monomer (2-endo) consumption but as the concentration of the endo-monomer (2-endo) decreases, so does its rate of reaction, and thus the consumption of the exo-monomer (2-exo) increases over time.
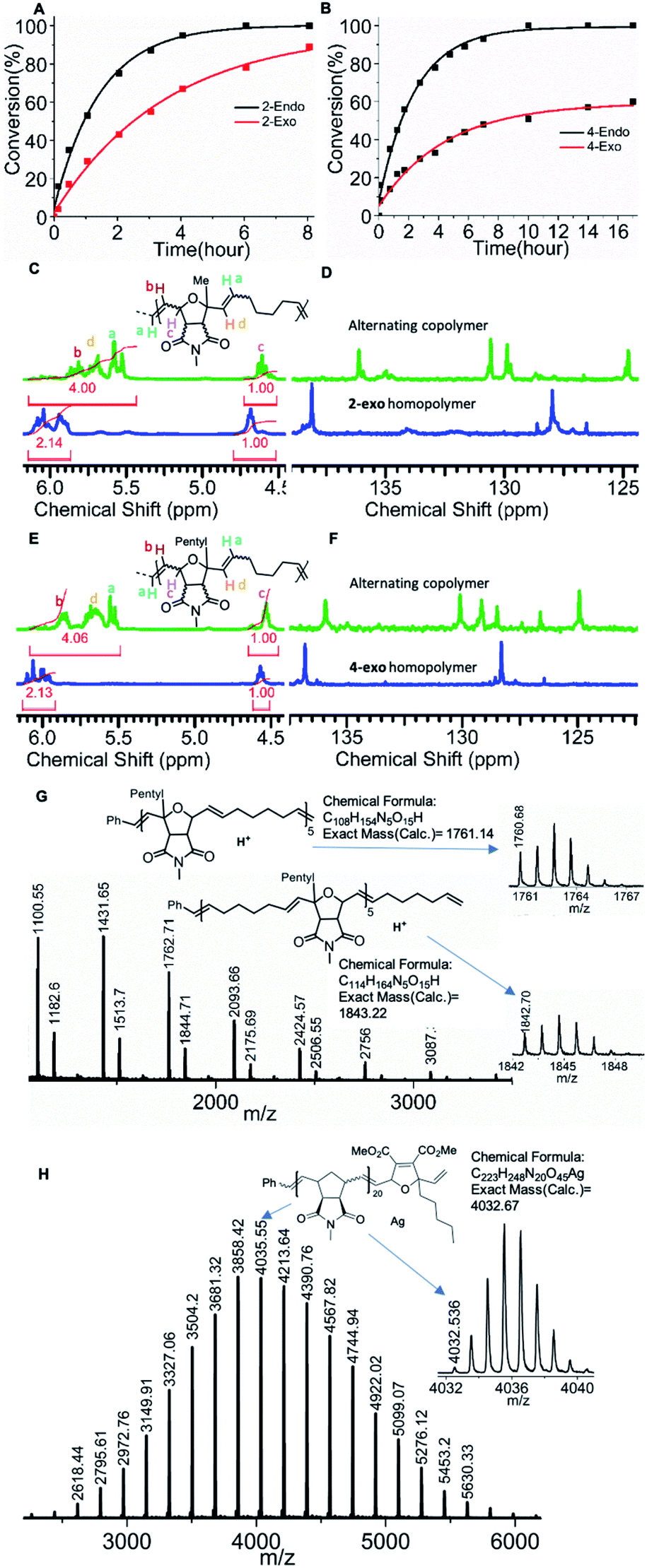 | ||
| Fig. 3 (A) Plot of 2-endo, 2-exo (57:43, 16 eq.) conversion vs. time in the copolymerization with cyclohexene (320 eq.) and 6.6 mol% G3 (1 eq.). (B) Plot of 4-exo, 4-endo (54:46, 16 eq.) conversion in the copolymerization with cyclohexene (320 eq.) and 6.6 mol% G3 (1 eq.). (C and D) Stacking of 1H and 13C NMR spectrum of the alternating copolymer synthesized from endo/exo mixture (2 + cyclohexene) and homopolymer of 2-exo. (E and F) Stacking of 1H and 13C NMR spectrum of the alternating copolymer synthesized from endo/exo mixture (4 + cyclohexene) and homopolymer of 2-exo. (G) MALDI-ToF mass spectrum of alternating copolymer synthesized from 4-endo and cyclohexene (P14). (H) MALDI-ToF mass spectrum of single end capped polymer P18. | ||
Next, the degree of alternation in the polymer backbone was analyzed by NMR spectroscopy. As the endo/exo monomer mixture of 2 and cyclohexene (Chex) cannot undergo homopolymerization, a high degree of alternation was expected. A comparison of 1H and 13C NMR signals of the resulting copolymer P1 with homopolymer of 2-exo (Fig. 3C, D, see ESI†) shows a characteristic shift in the olefinic signals, which confirms an excellent alternating nature of the copolymer (Fig. 3C, D, see ESI,† 90% alternating diads).57,59 The molecular mass of the polymer P1 obtained by size exclusion chromatography (SEC, THF, Mn = 4.1 kDa.; Đ = 1.2, Table 1) was close to the molecular mass determined by the [2]:[G3] ratio. Another polymerization using monomer 2 (endo/exo-mixture 57:43, 33 eq.) cyclohexene (660 eq.) and 3 mol% G3 (1 eq.) targeting a higher molecular weight confirmed good molecular weight control (SEC, THF, P2, Mn = 10.6 kDa.; Đ = 1.5, Table 1).
| Polymer | Monomers | Monomer/G3 | M n(theo) | M n(GPC) | Đ |
|---|---|---|---|---|---|
| a All copolymers were analysed by GPC (THF) except (b) which were measured in CHCl3. | |||||
| P1 |
2:Chex |
16 | 4.4 | 4.1 | 1.2 |
| P2 |
2:Chex |
33 | 9 | 10.6 | 1.5 |
| P3 |
2:Chep |
16 | 4.6 | 4.2 | 1.2 |
| P4 |
2:Chep |
33 | 9.5 | 10.9 | 1.6 |
| P5 |
2:Cpen |
16 | 4.2 | 3.9 | 1.2 |
| P6 |
3:Chex |
16 | 4.8 | 5.3 | 1.2 |
| P7 |
3:Chex |
60 | 18 | 13 | 1.3b |
| P8 |
3:Cpen |
16 | 4.6 | 3.7 | 1.2 |
| P9 |
3:Chep |
16 | 5.1 | 5 | 1.2 |
| P10 |
4:Chex |
16 | 5.3 | 5.6 | 1.1 |
| P11 |
4:Chex |
60 | 19.8 | 14.1 | 1.4b |
| P12 |
4:Cpen |
16 | 5.1 | 4.4 | 1.2 |
| P13 |
4:Chep |
16 | 5.5 | 5 | 1.2 |
| P14 |
4-endo:Chex |
16 | 5.3 | 5.8 | 1.1 |
| P15 |
5:Chex |
16 | 7.3 | 10.6 | 1.3b |
| P16 |
5:Chex |
30 | 13.8 | 17.8 | 1.3b |
| P17 |
5:Chex |
60 | 27.6 | 24.9 | 1.3b |
Thereafter, copolymerizations with either cycloheptene or cyclopentene were explored with the endo/exo mixture (57:43) of monomer 2. Copolymerization of monomer 2 with an equivalent amount of cycloheptene (Chep) with 6.6 and 3 mol% G3 yielded polymers P3 (SEC, THF, Mn = 4.2 kDa.; Đ = 1.2) and P4 (SEC, THF, Mn = 10.9 kDa.; Đ = 1.6). Monomer 2 was also copolymerized with cyclopentene (Cpen) in the presence of 6.6 mol% G3 to obtain polymer P5 (SEC, THF, Mn = 3.9 kDa.; Đ = 1.2). The good control over molecular weight and excellent degree of alternation of all copolymers obtained was confirmed by SEC and NMR spectroscopy (see ESI, Table 1,† 92% alternating diads for P3–P5).
The copolymerizations of endo/exo mixture (55:45) of monomer 3 with cyclohexene (Chex), cyclopentene (Chep), and cycloheptene (Chep) were also carried out. Monomer 3 was copolymerized with cyclohexene in the presence of 6.6 mol% and 1.6 mol% G3 to yield polymer P6 (SEC, THF, Mn = 5.3 kDa.; Đ = 1.2) and P7 (SEC, CHCl3, Mn = 13 kDa.; Đ = 1.3). Then, monomer 3 was polymerized with an equivalent amount of cyclopentene and 6.6 mol% of G3 to obtain polymer P8 (SEC, THF, Mn = 3.7 kDa.; Đ = 1.2). Polymer P9 (SEC, THF, Mn = 5 kDa.; Đ = 1.2) was obtained by polymerizing monomer 3 with cycloheptene in the presence of 6.6 mol% G3(see ESI, Table 1).
Another control 1H NMR copolymerization experiment was performed using an endo/exo mixture (54:46) of monomer 4 (16 eq.) cyclohexene (320 eq.) and 6.6 mol% of G3 in the presence of an internal standard (1,3,5-trimethoxybenzene) in dichloromethane-d2.
Similar to the copolymerization of monomer 2, faster endo conversion was observed, which supports our previous observation (Fig. 3B). The polymer P10 (SEC, THF, Mn = 5.6 kDa.; Đ = 1.1) obtained by this copolymerization was analyzed by SEC and NMR spectroscopy (Fig. 3E, F, see ESI, Table 1, 96% alternating diads). A high molecular weight copolymer P11 (SEC, CHCl3, Mn = 14.1 kDa.; Đ = 1.4) was also synthesized with monomer 4 (60 eq.), cyclohexene (1200 eq.) and 1.6 mol% G3 (1 eq.). Good control over the molecular weight even at a higher monomer to initiator ratio and the high degree of alternation of the synthesized copolymers confirmed the versatility of the copolymerization.
Copolymerization of endo/exo mixture (54:46) of monomer 4 with an equivalent amount of cyclopentene and 6.6 mol% G3 yielded polymer P12 (SEC, THF, Mn = 4.4 kDa.; Đ = 1.2, 96% altering diads). The endo/exo mixture (54:46) of monomer 4 was also copolymerized with cycloheptene in the presence of 6.6 mol% G3 to obtain polymer P13 (SEC, THF, Mn = 5 kDa.; Đ = 1.2, 94% alternating diads). The SEC and NMR analysis confirmed good molecular weight control as well as an excellent degree of alternation of the resulting polymers (see ESI, Table 1).
A copolymerization was also performed with pure 4-endo (15 eq.) and cyclohexene (300 eq.) in the presence of 6.6 mol% G3 (1 eq.) to obtain polymer P14 (SEC, THF, Mn = 5.8 kDa.; Đ = 1.1). Neither of the two monomers has the possibility to form a homopolymer, yet a polymer (P14) was formed from the mixture. This by itself could be regarded as proof for strict alternation, however, the 1H, 13C NMR, and MALDI-ToF analysis confirmed the strictly alternating character of the polymer (Fig. 3G, S65, S91–92, S117–118). In particular, the MALDI-ToF mass spectrum shows a pattern in which two peaks (the higher intensity one at lower mass and the lower intensity one at higher mass) repeat. The higher intensity peak can be assigned to polymers with equal numbers of 4-endo and cyclohexene units, whereas the lower intensity peak is formed by polymers containing one unit of cyclohexene more than 4-endo. Other combinations of these two monomers could not be assigned which is a strong indication for a strictly alternating polymerization.
Next, a bridgehead functional monomer 5 (Scheme 1E) was tested under alternating copolymerization conditions. A control 1H NMR copolymerization experiment was performed with the endo/exo mixture of monomer 5 (16 eq.), cyclohexene (300 eq.) and 6.6 mol% G3 (1 eq.) in the presence of an internal standard (1,3,5-trimethoxybenzene) in dichloromethane-d2. Similar to monomers 2–4, faster consumption of 5-endo was observed over 5-exo during the course of the copolymerization (Fig S18,† S19). Slower copolymerization kinetics of monomer 5 are attributed to the increased steric bulk at the bridgehead position. The obtained polymer P15 (SEC, CHCl3, Mn = 10.6 kDa.; Đ = 1.3) was analyzed by SEC and NMR, which confirmed good control over the molecular weight and a high degree of alternation of the copolymer. Two high molecular weight copolymers P16 and P17 were also synthesized with the endo/exo mixture of monomer 5, cyclohexene and G3 (3.3 and 1.6 mol%). The SEC analysis of P16 (SEC, CHCl3, Mn = 17.8 kDa.; Đ = 1.3) and P17 (SEC, CHCl3, Mn = 24.9 kDa.; Đ = 1.3) confirmed good control over the molecular weight (see ESI†). Furthermore, NMR analysis confirmed a high degree of alternation of the resulting copolymers (see ESI†) (Table 1).
End-functionalization
Furthermore, we studied the use of single monomer addition towards the end functionalization of ROMP polymers. A monomer that undergoes single molecular addition while containing a highly reactive group could be very useful for the installation of functional end groups via post-polymerization reactions. The wide commercial availability of functional thiol derivatives and the efficient thiol-ene click reaction prompted us to design monomer 6. We assumed that the planar geometry of the carbonyl groups in the ring-opened form of this 7-oxanorbornadiene derivative would be efficient for chelation with the metal carbene and lead to a single monomer addition.To prove our hypothesis, a control 1H NMR experiment was performed with 6 and 10 mol% G3 in dichloromethane-d2 (Fig. 2F, S22†). As proposed, a single monomer addition of 6 to G3 was observed immediately and the 1H-NMR spectrum remained unchanged even after 9 hours.
Next, a monomer 6 terminated ROMP polymer was synthesized. Exo N-methylnorbornene imide (MNI, 10 eq.) was polymerized for 10 min with 3.3 mol% of G3 followed by the addition of monomer 6 (Scheme 1F). The polymerization was quenched with an excess of ethyl vinyl ether 10 min after monomer 6 addition (see ESI†) to obtain P18 (SEC, CHCl3, Mn = 6.1 kDa.; Đ = 1.08). A sample was collected after addition of each reagent and analysed by 1H NMR spectroscopy confirming complete carbene conversion (Fig S23†). 1H NMR and MALDI-ToF MS spectroscopy confirmed the precise single molecular addition of monomer 6 at the polymer (P18) chain end (Fig. 3H, S66, S103†).
The presence of an activated maleic ester in monomer 6 makes it a very good reaction partner for thiol ene click derivatization (Scheme 1F). Hence, different end functional polymers can be achieved from one single parent polymer simply by using commercial functional thiols.
To prove our hypothesis, two samples of polymer P18 were treated separately with 10 eq. of thioethanol (P19) or pentanedithiol (P20) (Scheme 1F, see ESI†). The 1H NMR and MALDI-ToF analysis of both polymers confirmed the presence of polymers with either an alcohol or thiol end group with a high degree of end functionalization (>98% for the alcohol end group by 1H-NMR spectroscopy, Fig S67, S68, S105;† unfortunately, the percentage of thiol end groups could not be determined by 1H-NMR spectroscopy.).
Conclusions
In conclusion, we have successfully developed an oxanorbornene type single addition monomer. We observed that in 7-oxanorbornene derivatives the endo isomer initiates faster with G3 than the exo isomer. Together with a slow propagation reaction of the endo-isomer, this allowed us to use endo 7-oxanorbornenes as single addition monomers. The observed selective initiation of G3 with the endo isomer prompted us to synthesize a set of bridgehead substituted 7-oxanorbornene monomers where no homopolymerization could be observed at all. The absence of homopolymerization and successful copolymerization with cycloalkenes allowed us to prepare alternating copolymers. The synthesis of a strictly alternating copolymer with 4-endo and cyclohexene confirmed the single molecular addition in the presence of G3. End functionalization of ROMP polymers with reactive single addition monomer 6 allowed us to prepare a polymer carrying a reactive end group for post-polymerization functionalization. Two different end functional polymers were obtained from a single parent polymer with commercial thiol derivatives, one yielding an alcohol and the other a thiol end group. We believe that the detailed understanding of the reactivity of oxaNBE derivatives combined with their straightforward synthesis will allow easy access to sequence-controlled and end functional ROMP polymers. These polymers could be exploited as a building block for the preparation of well-defined architectures, new functional materials, and in biomedical applications.Author contributions
P. K. synthesized the exo and endo N-phenylnorborneneimides, M. A. carried out several polymerizations and helped in editing the manuscript, S. P. synthesized all remaining monomers, polymers and carried out all analyses. S. P. and A. F. M. K. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Fabio Zobi for helpful discussions and the National Center of Competence in Research (NCCR Bioinspired Materials) and the Fribourg Center for Nanomaterials (FriMat) for support.References
- Y. Chen, M. M. Abdellatif and K. Nomura, Tetrahedron, 2018, 74, 619 CrossRef CAS.
- R. R. Schrock, in Handbook of Metathesis, ed. R. H. Grubbs and A. G. Wenzel, Wiley, 2nd edn, 2015, vol. 1–27. G. C Search PubMed.
- M. R. Buchmeiser, Chem.–Eur. J., 2018, 24, 14295 CrossRef CAS PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1 CrossRef CAS.
- S. Kress and S. Blechert, Chem. Soc. Rev., 2012, 41, 4389 RSC.
- S. Hilf and A. F. M. Kilbinger, Nat. Chem., 2009, 1, 537 CrossRef CAS PubMed.
- R. H. Grubbs, in Handbook of Metathesis, Wiley-VCH, Weinheim, 2003 Search PubMed.
- S. Sutthasupa, M. Shiotsuki and F. Sanda, Polym. J., 2010, 42, 905 CrossRef CAS.
- J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- A. Anastasaki, B. Oschmann, J. Willenbacher, A. Melker, M. H. C. Van Son, N. P. Truong, M. W. Schulze, E. H. Discekici, A. J. McGrath, T. P. Davis, C. M. Bates and C. J. Hawker, Angew. Chem., Int. Ed., 2017, 56, 14483 CrossRef CAS PubMed.
- S. Pal, F. Lucarini, A. Ruggi and A. F. M. Kilbinger, J. Am. Chem. Soc., 2018, 140, 3181 CrossRef CAS PubMed.
- D. S. Everhart, in Handbook Appl. Surf. Colloid Chem. 2002, 2, p. 99 Search PubMed.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062 CrossRef CAS PubMed.
- S. Hilf and A. F. M. Kilbinger, Macromolecules, 2009, 42, 4127 CrossRef CAS.
- A. K. Pearce, J. C. Foster and R. K. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3020 CrossRef.
- S. Al Samak, A. G. Carvill, J. G. Hamilton, J. J. Rooney and J. M. Thompson, Chem. Commun., 1997, 2057 RSC.
- Z. Wu and R. H. Grubbs, Macromolecules, 1995, 28, 3502 CrossRef CAS.
- J. G. Hamilton, K. J. Ivin, J. J. Rooney and L. C. Waring, Chem. Commun., 1983, 159 RSC.
- V. Amir-Ebrahimi and J. J. Rooney, J. Mol. Catal. A: Chem., 2004, 208, 115 CrossRef CAS.
- M. R. Buchmeiser, I. Ahmad, V. Gurram and P. S. Kumar, Macromolecules, 2011, 44, 4098 CrossRef CAS.
- K. Vehlow, D. Wang, M. R. Buchmeiser and S. Blechert, Angew. Chem., Int. Ed., 2008, 120, 2655 CrossRef.
- T.-L. Choi, I. M. Rutenberg and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 114, 3995 CrossRef.
- H. Jeong, J. M. John and R. R. Schrock, Organometallics, 2015, 34, 5136 CrossRef CAS.
- H. Jeong, J. M. John, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2015, 137, 2239 CrossRef CAS PubMed.
- V. Paradiso and F. Grisi, Adv. Synth. Catal., 2019, 361, 4133 CrossRef CAS.
- S. Torker, A. Müller and P. Chen, Angew. Chem., Int. Ed., 2010, 49, 3762 CrossRef CAS PubMed.
- M. Lichtenheldt, D. Wang, K. Vehlow, I. Reinhardt, C. Kühnel, U. Decker, S. Blechert and M. R. Buchmeiser, Chem.–Eur. J., 2009, 15, 9451 CrossRef CAS PubMed.
- E. S. Jang, J. M. John and R. R. Schrock, J. Am. Chem. Soc., 2017, 139, 5043 CrossRef CAS PubMed.
- H. K. Lee, K. T. Bang, A. Hess, R. H. Grubbs and T. L. Choi, J. Am. Chem. Soc., 2015, 137, 9262 CrossRef CAS PubMed.
- K. Vehlow, D. Wang, M. R. Buchmeiser and S. Blechert, Angew. Chem., Int. Ed., 2008, 47, 2615 CrossRef CAS PubMed.
- A. Song, K. A. Parker and N. S. Sampson, J. Am. Chem. Soc., 2009, 131, 3444 CrossRef CAS PubMed.
- K. A. Parker and N. S. Sampson, Acc. Chem. Res., 2016, 49, 408 CrossRef CAS PubMed.
- G. Li and N. S. Sampson, Macromolecules, 2018, 51, 3932 CrossRef CAS PubMed.
- J. Romulus, L. Tan, M. Weck and N. S. Sampson, ACS Macro Lett., 2013, 2, 749 CrossRef CAS PubMed.
- L. Tan, K. A. Parker and N. S. Sampson, Macromolecules, 2014, 47, 6572 CrossRef CAS PubMed.
- L. Tan, G. Li, K. A. Parker and N. S. Sampson, Macromolecules, 2015, 48, 4793 CrossRef CAS PubMed.
- J. Romulus, L. Tan, M. Weck and N. S. Sampson, ACS Macro Lett., 2013, 2, 749 CrossRef CAS PubMed.
- L. Tan, G. Li, K. A. Parker and N. S. Sampson, Macromolecules, 2015, 48, 4793 CrossRef CAS PubMed.
- L. Tan, K. A. Parker and N. S. Sampson, Macromolecules, 2014, 47, 6572 CrossRef CAS PubMed.
- B. R. Elling and Y. Xia, J. Am. Chem. Soc., 2015, 137, 9922 CrossRef CAS PubMed.
- B. R. Elling, J. K. Su, J. D. Feist and Y. Xia, Chem, 2019, 5, 2691 CAS.
- B. R. Elling, J. K. Su and Y. Xia, ACS Macro Lett., 2010, 9, 180 CrossRef.
- J. K. Su, Z. Jin, R. Zhang, G. Lu, P. Liu and Y. Xia, Angew. Chem., Int. Ed., 2019, 131, 17935 CrossRef.
- B. R. Elling, J. K. Su and Y. Xia, ACS Macro Lett., 2020, 9, 180 CrossRef CAS.
- T.-L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743 CrossRef CAS PubMed.
- K. J. Ivin and J. C. Mol, in Olefin Metathesis and Metathesis Polymerization, Academic Press, San Diego, CA, 1997 Search PubMed.
- J. M. Pollino, L. P. Stubbs and M. Weck, Macromolecules, 2003, 36, 2230 CrossRef CAS.
- N. Seehof, S. Grutke and W. Risse, Macromolecules, 1993, 26, 695 CrossRef CAS.
- D. Moatsou, C. F. Hansell and R. K. O'Reilly, Chem. Sci., 2014, 5, 2246 RSC.
- M. L. Gringolts, M. V. Bermeshev, Yu. V. Rogan, M. V. Moskvicheva, M. P. Filatova, E. S. Finkelshtein and G. N. Bondarenko, Silicon, 2015, 7, 107 CrossRef CAS.
- J. D. Rule and J. S. Moore, Macromolecules, 2002, 35, 7878 CrossRef CAS.
- V. Lapinte, J. C. Brosse and J. C. Fontaine, Macromol. Chem. Phys., 2004, 205, 824 CrossRef CAS.
- Y. Nishihara, Y. Inoue, Y. Nakayama, T. Shiono and K. Takagi, Macromolecules, 2006, 39, 7458 CrossRef CAS.
- A. B. Chang, T. P. Lin, N. B. Thompson, S. X. Luo, A. L. Liberman-Martin, H. Y. Chen, B. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 17683 CrossRef CAS PubMed.
- W. J. Wolf, T.-P. Lin and R. H. Grubbs, J. Am. Chem. Soc., 2019, 141, 17796 CrossRef CAS PubMed.
- M. G. Hyatt, D. J. Walsh, R. L. Lord, J. G. A. Martinez and D. Guironnet, J. Am. Chem. Soc., 2019, 141, 17918 CrossRef CAS PubMed.
- M. F. Ilker and E. B. Coughlin, Macromolecules, 2002, 35, 54 CrossRef CAS.
- C. S. Daeffler and R. H. Grubbs, Macromolecules, 2013, 46, 3288 CrossRef CAS.
- S. Pal, M. Alizadeh and A. F. M. Kilbinger, ACS Macro Lett., 2019, 8, 1396 CrossRef CAS.
- J. Alonso-Villanueva, J. L. Vilas, I. Moreno, J. M. Laza, M. Rodriguez and L. M. Leon, J. Macromol. Sci., Pure Appl. Chem., 2011, 48, 211 CrossRef CAS.
- T. Hino, N. Inoue and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6599 CrossRef CAS.
- D. Liaw, C. Huang and E. Kang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2901 CrossRef CAS.
- L. Zheng, S. Hong, G. Cardoen, E. Burgaz, S. P. Gido and E. B. Coughlin, Macromolecules, 2004, 37, 8606 CrossRef CAS.
- M. Yasir, P. Liu, I. Tennie and A. F. M. Kilbinger, Nat. Chem., 2019, 11, 488 CrossRef CAS PubMed.
- P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100 CrossRef CAS.
- A. B. Chang, G. M. Miyake and R. H. Grubbs, Sequence-Controlled Polymers by Ruthenium-Mediated Ring-Opening Metathesis Polymerization, Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties, ed. J.-F. Lutz, T. Y. Meyer, M. Ouchi and M. Sawamoto, American Chemical Society, Washington, DC, 2014, vol. 1170, pp. 161–188 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00036e |
| This journal is © The Royal Society of Chemistry 2021 |