
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of N-acyl amidines to amidoximes: a convenient synthetic approach to molnupiravir (EIDD-2801) from ribose†
Ajaz Ahmedab,
Qazi Naveed Ahmed ab and
Debaraj Mukherjee*ab
ab and
Debaraj Mukherjee*ab
aNatural Product and Medicinal Chemistry Division, Indian Institute of Integrative Medicine (IIIM), Jammu 180001, India. E-mail: dmukherjee@iiim.ac.in; dmukherje@iiim.res.in
bAcademy of Scientific and Innovative Research (AcSIR-IIIM), Jammu-180001, India
First published on 10th November 2021
Abstract
An efficient method is described for the preparation of molnupiravir (EIDD-2801) an antiviral agent via regioselective conversion of an N-acyl-nucleoside intermediate, generated through stereo and regioselective glycosylation of protected ribose and N4-acetyl cytosine, to an amidoxime. This method avoids use of expensive starting materials, enzymes, complex reagents, and cumbersome purification procedures.
Introduction
Amidoximes, having both hydroxylamino and imino groups at the same carbon atom, and their O-substituted derivatives form a unique class of pharmaceutical products with varied activities such as anti-bacterial, anti-tuberculosis, insecticidal, antiviral etc.1 A number of amidoximes are currently in use as drugs, or are undergoing clinical trials. Molnupiravir, also known as EIDD-2801 and MK-4482, is one such compound being studied in late-stage clinical trials for combating infections caused by COVID-19, and acts by disrupting RNA synthesis.2–5 Its advantages over remdesivir, the other drug in use, are oral bioavailability and structural simplicity allowing its easier manufacture.6The reported synthetic route for molnupiravir disclosed by the original developers consists of a five-step synthesis starting from uridine.7 The method suffers from low yield (ca. 17%) and the use of an inefficient (yield 29%) triazole insertion step (Scheme 1A). In a revised route with an improved yield reported by Kappe, Dallinger, and co-workers, the triazole insertion was introduced in the first step of a modified procedure.8 Snead and co-workers published another route to molnupiravir with 60% yield starting with cytidine and using acetonide protection, esterfication, hydroxyamination and acetonide deprotection.9 The same group also published two different two-step routes from cytidine, where selective enzymatic esterification of the primary alcohol replaced the diol protection/deprotection sequence.10,11 However, the drawbacks include use of expensive oxime ester, need for immobilized enzyme in large amounts, formation of diester (both at O- and NHO-) along with the desired monoester, and the necessity for removal of O-ester during transamination (Scheme 1B).
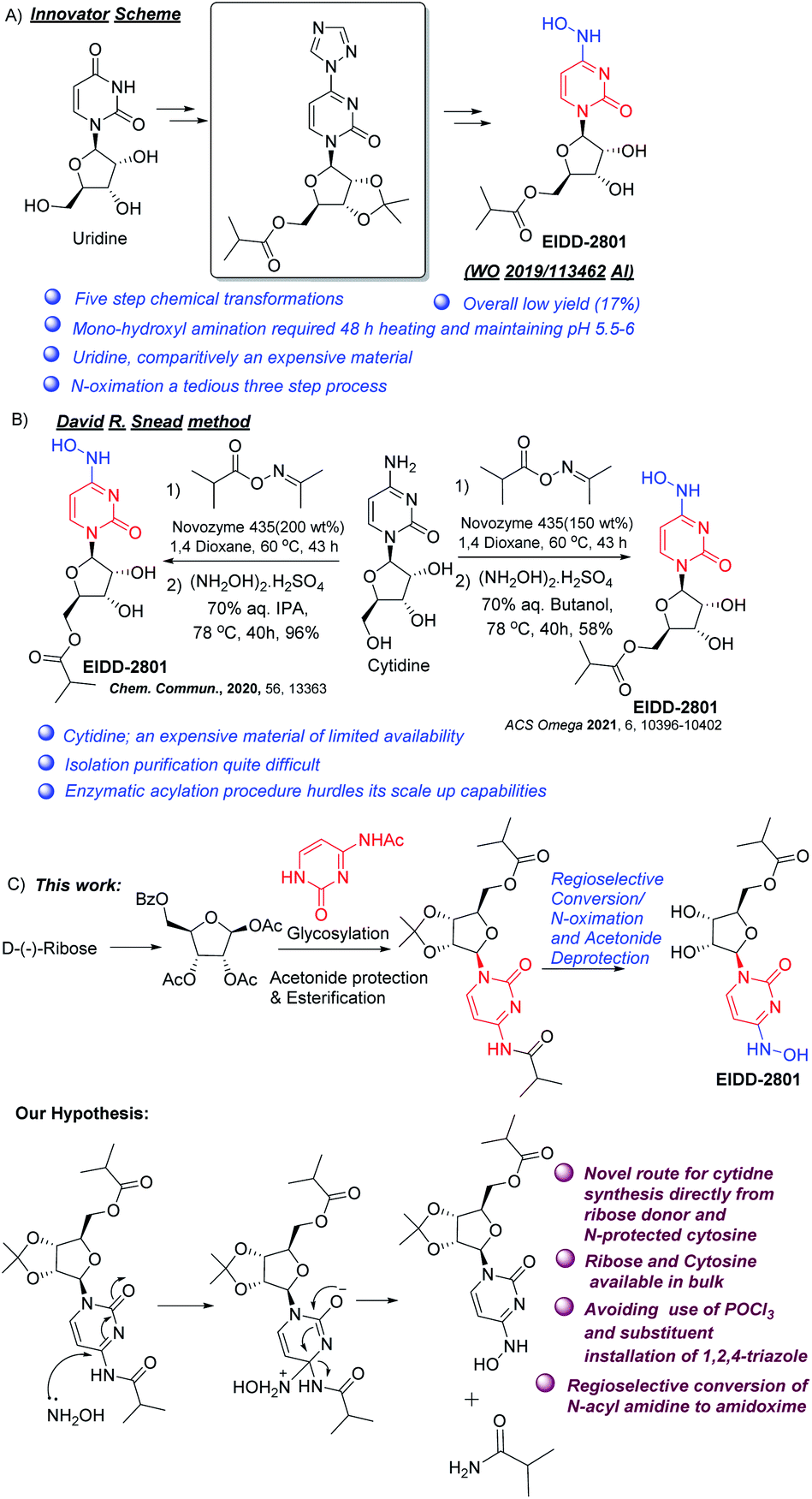 | ||
| Scheme 1 Different approaches to Molnupiravir (A) Innovator synthetic method, (B) David R Snead method, (C) our method and hypothesis. | ||
Merck & Co (USA) carried out an elegant enzymatic synthesis starting from ribose and uracil.12 However it entails the use of cocktails of costly enzymes and buffers used in the glycosylation step, while the high water solubility of the final product makes its isolation tricky. Our interest in nucleoside synthesis13,18 led us to devise an alternative to overcome the shortcomings. We envisaged that the amino group at C-4 might get substituted by hydroxylamine to form 4-hydroxylamino derivative (Scheme 1C). In the case of cytidine, this exchange may be facilitated by the presence of an electron withdrawing group (perhaps an acyl) on the nitrogen at C-4 and thereby N-acyl cytidine can be converted directly to amidoximes under mild conditions by the removal of corresponding amides. For this, our initial task was to synthesize N-acyl protected cytidine from the free base and thereafter attempt to selectively convert it to the 4-hydroxylamine derivative under controlled conditions to avoid deprotection of the ester at the primary hydroxyl group (Scheme 1C). The advantages of the envisaged process will include (a) avoiding chlorinating solvents like POCl3, and (b) better yield in the coupling step as compared to uracil. This would also present an alternative strategy to EIDD-2801, employing ribose and cytosine rather than cytidine. Thus, we focused on the development of an efficient route to access N-acyl cytidine derivative for a regio and stereo selective glycosylation.
Results and discussion
We initiated the process by synthesising the key intermediate 2 from commercially available D-(−)-ribose. It could be locked in its furanoid form employing an HCl mediated acetonide protection procedure,14 followed by benzoylation of the free hydroxyl group of 1 with benzoyl chloride in pyridine at ambient temperature (Scheme 2). Hydrolysis of 2 with trifluoroacetic acid and water (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) resulted in the removal of the acetonide group at 2,3-position and the methyl group from anomeric hydroxy in a single step. Subsequent acetylation with acetic anhydride in pyridine furnished 1,2,3-tri-O-acetyl-5-benzoyl-β-D-ribofuranose 3 in 90% overall yield (Scheme 2).
:1) resulted in the removal of the acetonide group at 2,3-position and the methyl group from anomeric hydroxy in a single step. Subsequent acetylation with acetic anhydride in pyridine furnished 1,2,3-tri-O-acetyl-5-benzoyl-β-D-ribofuranose 3 in 90% overall yield (Scheme 2).
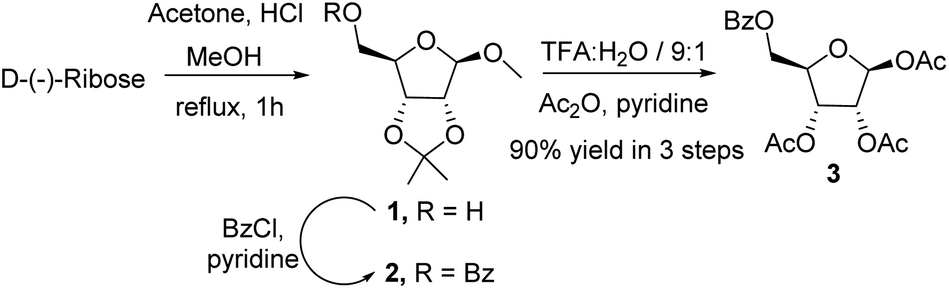 | ||
| Scheme 2 Access of furanoid ribose as donor. | ||
The key step involved in the synthesis of molnupiravir is a stereo and regioselective glycosylation, where coupling of nucleobases 4a–c with 3 is desired. Silylation of nucleobases with hexamethyldisilazane (HMDS) in catalytic amount of TMSOTf prior to coupling with sugar donors is well known Silyl Hilbert–Johnson reaction which is reported mostly for the synthesis of uridine derivatives.15 But, mostly cytidine is obtained from uridine after a series of steps which involves chlorination with POCl3 and substituent installation of 1,2,4-triazole to get amine with the treatment of hydroxylamine16 (Scheme 3A). For direct access of cytidine Raffaele et al. reported microwave accelerated glycosylation approach from N-benzoyl cytosine with 50% yield17 (Scheme 3B), So in order to improve the yield further optimization of reaction was needed. Therefore we firstly treated N-protected cytosine 4a with HMDS and TMSOTf (catalytic amount) in acetonitrile at 80 °C for 1 h, then added 3 and SnCl4 and stirred at the same temperature only to end up with a mixture of regioisomers of cytidine derivatives in low yield (ESI,† Table 1, entry 1).
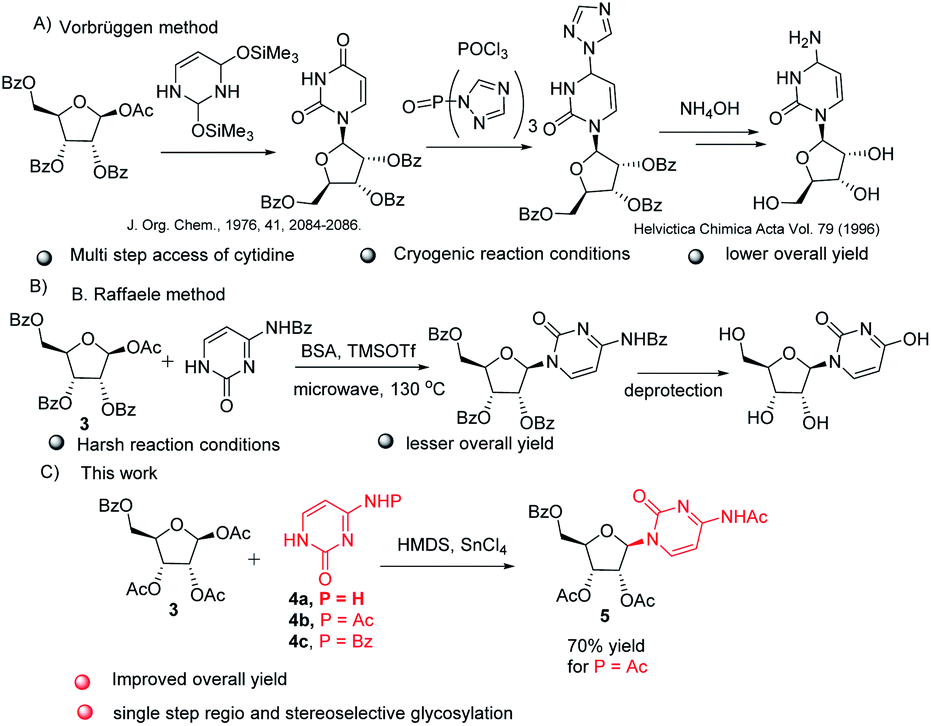 | ||
| Scheme 3 Literature reports for the synthesis of cytidine, (A) Vorbruggen glycosylation method, (B) B. Raffaele microwave assisted method and (C) our method. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cytosine | Promotor (equiv.) | Solvent | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1 equiv. of N-acetyl cytosine 4b, HMDS (1.5 equiv.) and TMSOTf (0.1 equiv.) at 80 °C for 1 h followed by cooling the reaction mixture down to 60 °C before the addition of 0.5 equiv. of 3 and 1 equiv. SnCl4 and allowing the reaction mixture to stir for 3 h.b Isolated yield. | ||||||
| 1 | 4a | SnCl4 (1.0) | ACN | 80 | 12 | 20 |
| 2 | 4a | SnCl4 (1.0) | DCM | 80 | 5 | 28 |
| 3 | 4a | SnCl4 (1.0) | DCE | 80 | 3 | 54 |
| 4 | 4a | SnCl4 (1.0) | DMF | 80 | 3 | 5 |
| 5 | 4a | SnCl4 (1.0) | 1,4 dioxane | 80 | 3 | 15 |
| 6 | 4b | SnCl4 (1.0) | DCE | 80 | 3 | 64 |
| 7 | 4c | SnCl4 (1.0) | DCE | 80 | 3 | 62 |
| 8 | 4b | SnCl4 (1.0) | DCE | 60 | 3 | 70 |
| 9 | 4b | SnCl4 (1.0) | DCE | 40 | 5 | 68 |
| 10 | 4b | TMSOTf (1.0) | DCE | 60 | 3 | 48 |
| 11 | 4b | ZnCl2 (1.0) | DCE | 40 | 3 | 18 |
| 12 | 4b | TiCl4 (1.0) | DCE | 40 | 3 | 11 |
| 13 | 4b | SnCl4 (0.5) | DCE | 60 | 3 | 52 |
| 14 | 4b | SnCl4 (2.0) | DCE | 60 | 3 | 70 |
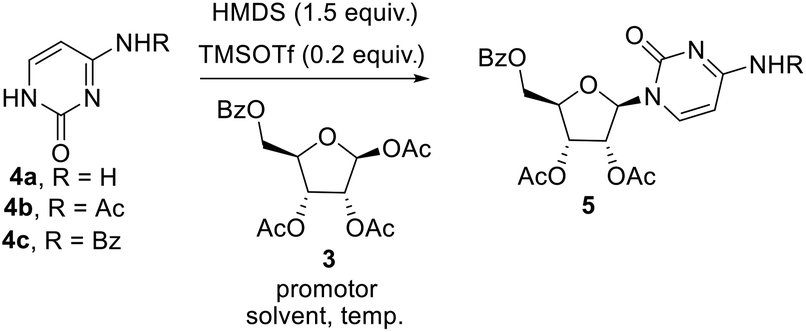
To increase the yield and regioselectivity we screened different solvents like DCM, DCE, DMF, and 1,4-dioxane. This showed that DCE was the best solvent to obtain a high yield of the product but regioselectivity remained a concern (ESI,† Table 1, entries 2–5). In order to improve this, we planned to perform the glycosylation with N-masked cytosine derivatives. To our delight, replacing cytosine with N-acetyl cytosine 4b and N-benzoyl cytosine 4c ensured the desired regioselectivity (ESI,† Table 1, entries 6–7). Decreasing the temperature from 80 °C to 60 °C resulted in still more increase in the selectivity and yield of the product, but further lowering to 40 °C was not helpful (ESI,† Table 1, entries 8–9). Also, using promoters other than SnCl4 gave unsatisfactory results (ESI,† Table 1, entries 10–12). Although decreased proportion of promoter affects the yield of the product, but increase beyond the stoichiometric amount did not improve the yield (ESI,† Table 1, entry 13–14). Consolidating the outcome of our optimization study, it appeared that using 1 equiv. of N-acetyl cytosine 4b with HMDS (1.5 equiv.) and TMSOTf (0.1 equiv.) at 80 °C for 1 h followed by cooling the reaction mixture down to 60 °C before the addition of 0.5 equiv. of 3 and 1 equiv. SnCl4 and allowing the reaction mixture to stir for 3 h at the same temperature delivers the desired nucleoside in good yield (Scheme 3).
In order to remove all the protecting groups, 5 was exposed to ammonia (20%) in methanol at 50 °C which resulted in deprotection of N-acetyl, O-acetyl and O-benzoyl groups within 5 h to deliver cytidine 6 (Scheme 3). To selectively introduce the isobutyl ester at the 5′-hydroxy and 4-amino positions of 6, the other hydroxy groups at 2′ and 3′ needed to be protected first. Using the reported conditions for the acetonide protection (5 mol% H2SO4 in acetone),15 about 20% conversion to the desired acetonide 7 took place. The use of Iodine (5 mol%) in place of H2SO4 did not significantly improve the outcome. However addition of 2 equiv. of 2,2-dimethoxypropane (DMP) showed an increase in conversion. The best results were obtained by changing the solvent from acetone to MeCN, which in combination with the use of DMP smoothly effected a 94% conversion to 7 (Scheme 3). Next, attempting the esterification of both 5′-hydroxy and 4-amino positions of 7 according to the reported method14 revealed that 2 equiv. of isobutyric anhydride produced a mixture of mono and diacylated products. In order to reach full conversion of 7 to 8, use of 3.0 equiv. of anhydride at ambient temperature was found to work best (Scheme 3). The most crucial step involved in the process was conversion of amidine amino to hydroxylamino without affecting the primary O-ester group. To achieve this, we used different hydroxyl amine derivatives in the presence of bases, varying temperature as well as time. Thus heating 8 with 2 molar hydroxylamine solution in the presence of sodium acetate in isopropanol at 50 °C caused hydrolysis of both O-ester and amide groups to form 9b (Table 2, entry 1). Replacement of hydroxylamine solution with hydroxylamine hydrochloride resulted in the formation of the N-hydroxylated product 9a along with 9b (Table 2, entry 2). In order to increase the yield of the desired product, isopropanol was replaced with water to derive satisfactory results (Table 2, entry 3). Yield of the desired product was found better at the boiling point of water instead of 50 °C (Table 2, entry 4). Also time played a critical role on the outcome of the reaction. For instance, lowering of the time period of the reaction from 12 h to 40 min increased the yield of the desired product (Table 2, entry 4–5). To check the role of NaOAc, a reaction in the absence of it was performed only to get 9b in 20% yield (Table 2, entry 6). Replacement of NaOAc with other bases failed to improve the yield (Table 2, entries 7–11). On decreasing the proportion of hydroxylamine hydrochloride, the yield of the product was found to be decreased whereas an increase had little influence (Table 2, entries 12–13). Consolidating these outcomes, employing acetonide diester 8 (1 equiv.), hydroxyl amine hydrochloride (4.0 equiv.), and sodium acetate (2.5 equiv.) in water at 100 °C for 40 min proved to be the best condition for obtaining the desired product 9a (Scheme 3). Deprotection of the acetonide group of 8 to EIDD-2801 has been described to proceed in H2SO4 in combination with iPrOH as solvent. However, we failed to achieve satisfactory conversion under the reported conditions. But screening of different acids like formic acid, acetic acid, and trifluoroacetic acid (TFA) for different time periods revealed that an aqueous solution of TFA worked best, effecting the highest conversion (90%) to EIDD-2801 10 in 1 h at room temperature (Scheme 3). Finally, keeping in mind the requirement of large amount of drug and the availability of cytidine as a cheap raw material, we have carried out the 2-step synthesis of EIDD-2801 from cytidine 6, one of the intermediates in our synthetic route, in 62% overall yield (Scheme 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Hydroxyl-amine deriv. (4 equiv.) | Base (2.5 equiv.) | Solvent | Time (h) | Yieldb 9a:9b |
| a Reaction conditions: 1 equiv. acetonide diester 8, 4.0 equiv. (except entry 12, 13) hydroxyl amine hydrochloride (2 M NH2OH for entry 1), and 2.5 equiv. base in water (iPrOH for entry 1,2) at 100 °C (50 °C for entry 1–3) for specified time.b Isolated yield. | |||||
| 1 | NH2OH (2 M) | NaOAc | iPrOH | 12 | 0:50 |
| 2 | NH2OH·HCl | NaOAc | iPrOH | 12 | 10:30 |
| 3 | NH2OH·HCl | NaOAc | H2O | 12 | 30:20 |
| 4 | NH2OH·HCl | NaOAc | H2O | 2 | 50:15 |
| 5 | NH2OH·HCl | NaOAc | H2O | 40 min | 88:0 |
| 6 | NH2OH·HCl | — | H2O | 40 min | 0:20 |
| 7 | NH2OH·HCl | Na2CO3 | H2O | 1 | 00:15 |
| 8 | NH2OH·HCl | NaHCO3 | H2O | 1 | 00:23 |
| 9 | NH2OH·HCl | Cs2CO3 | H2O | 1 | 00:18 |
| 10 | NH2OH·HCl | KOAc | H2O | 1 | 40:10 |
| 11 | NH2OH·HCl | Ca(OAc)2 | H2O | 1 | 23:14 |
| 12 | NH2OH·HCl (2.0 equiv.) | NaOAc | H2O | 1 | 57:12 |
| 13 | NH2OH·HCl (6.0 equiv.) | NaOAc | H2O | 1 | 75:0 |
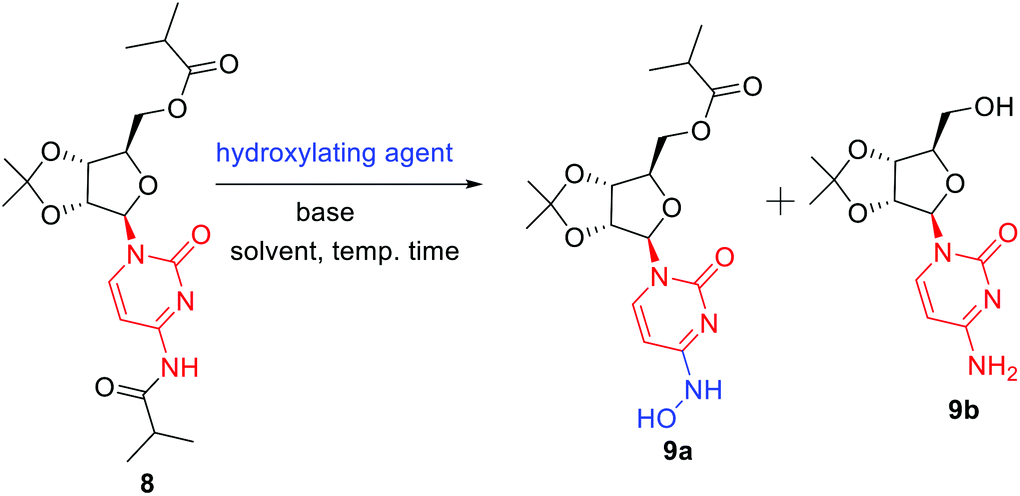
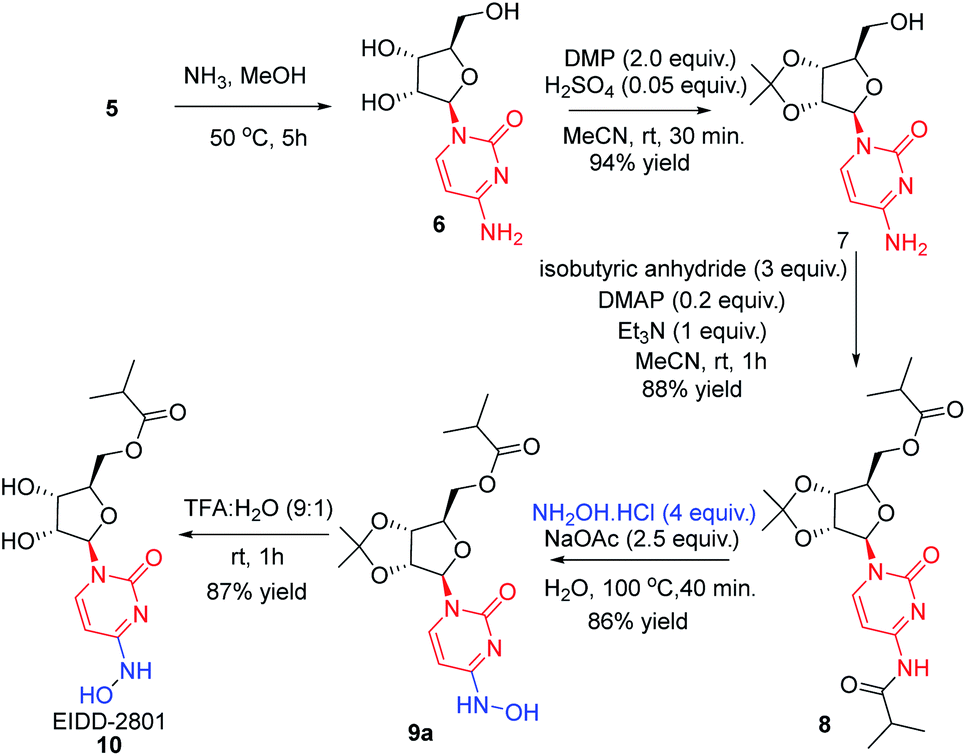 | ||
| Scheme 4 Diester synthesis and regioselective conversion of N-hydroxylaton. | ||
Conclusions
In conclusion, we have developed an efficient process for the synthesis of molnupiravir starting with cheaper commercially available ribose and N4-acetyl cytosine, which ensures stereo and regioselective glycosylation and regioselective conversion of N-acyl to hydroxylamino derivative in good to excellent yields. Other N-hydroxy cytidine derivatives could also be synthesized by employing this method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to CSIR-India (HCP-29), SERB-India (EMR/2016/004710) for funding. A. A. is thankful to CSIR New Delhi for SRF. IIIM Publication No. CSIR-IIIM/IPR/00311.Notes and references
- K. Fylaktakidou, D. Hadjipavlou-Litina, K. Litinas, E. Varella and D. Nicolaides, Curr. Pharm. Des., 2008, 14, 1001–1047 CrossRef CAS PubMed.
- T. P. Sheahan, A. C. Sims, S. Zhou, R. L. Graham, A. J. Pruijssers, M. L. Agostini, S. R. Leist, A. Schäfer, K. H. Dinnon 3rd, L. J. Stevens, J. D. Chappell, X. Lu, T. M. Hughes, A. S. George, C. S. Hill, S. A. Montgomery, A. J. Brown, G. R. Bluemling, M. G. Natchus, M. Saindane, A. A. Kolykhalov, G. Painter, J. Harcourt, A. Tamin, N. J. Thornburg, R. Swanstrom, M. R. Denison and R. S. Baric, Sc. Trans. Med., 2020, 12(541), eabb5883 CrossRef CAS PubMed.
- (a) M. L. Agostini, A. J. Pruijssers, J. D. Chappell, J. Gribble, X. Lu, E. L. Andres, G. R. Bluemling, M. A. Lockwood, T. P. Sheahan and A. C. Sims, J. virol., 2019, 93(24), e01348-19 CrossRef PubMed; (b) N. Urakova, V. Kuznetsova, D. K. Crossman, A. Sokratian, D. B. Guthrie, A. A. Kolykhalov, M. A. Lockwood, M. G. Natchus, M. R. Crowley, G. R. Painter, E. I. Frolova and I. Frolov, J. Virol., 2018, 92(3), e01965-17 CrossRef PubMed.
- Ridgeback Biotherapeutics and Merck Announce Preliminary Findings from a Phase 2a Trial of Investigational COVID-19 Therapeutic Molnupiravir. Merck, March 6, 2021, https://www.merck.com/news/ridgeback-biotherapeutics-and-merck-announce-preliminary-findingsfrom-a-phase-2a-trial-of-investigational-covid-19-therapeuticmolnupiravir/, accessed 2021-04-14 Search PubMed.
- (a) Merck and Ridgeback Bio Announce Closing of Collaboration and Licensing Transaction, July 1, 2020, Merck, https://www.merck.com/news/merck-and-ridgeback-bio-announce-closing-of-collaborationand-licensing-transaction/, accessed 2021-04-14 Search PubMed; (b) Efficacy and Safety of Molnupiravir (MK-4482) in Hospitalized Adult Participants with COVID-19 (MK-4482-001), https://clinicaltrials.gov/ct2/show/NCT04575584, accessed 2021-04-14 Search PubMed.
- D. J. Paymode, N. Vasudevan, S. Ahmad, A. L. Kadam, F. S. P. Cardoso, J. Burns, D. W. Cook, R. W. Stringham and D. R. Snead, Org. Process Res. Dev., 2021, 25, 1822 CrossRef CAS.
- G. R. Painter, G. R. Bluemling, M. G. Natchus and D. Guthrie, N4-Hydroxycytidine and Derivatives and Anti-viral Uses Related Thereto, WO 2016/106050 Al, June 13, 2019.
- A. Steiner, D. Znidar, S. B. Ö. tvös, D. R. Snead, D. Dallinger and C. O. Kappe, Eur. J. Org. Chem., 2020, 2020, 6736 CrossRef CAS PubMed.
- V. Gopalsamuthiram, C. Williams, J. Noble, T. F. Jamison, B. F. Gupton and D. R. Snead, Synlett, 2021, 32, 326 CrossRef CAS.
- N. Vasudevan, G. P. Ahlqvist, C. P. McGeough, D. J. Paymode, F. S. Cardoso, T. Lucas, J.-P. Dietz, T. Opatz, T. F. Jamison, F. B. Gupton and D. R. Snead, Chem. Commun., 2020, 56, 13363–13364 RSC.
- G. P. Ahlqvist, C. P. McGeough, C. Senanayake, J. D. Armstrong, A. Yadaw, S. Roy, S. Ahmad, D. R. Snead and T. F. Jamison, ACS Omega, 2021, 6, 10396–10402 CrossRef CAS PubMed.
- T. Benkovics, J. A. McIntosh, S. M. Silverman, J. Kong, P. Maligres, T. Itoh, H. Yang, M. A. Huffman, D. Verma, W. Pan, H.-I. Ho, J. Vroom, A. Knight, J. Hurtak, W. Morris, N. A. Strotman, G. Murphy, K. M. Maloney and P. S. Fier, Evolving to an Ideal Synthesis of Molnupiravir, an Investigational Treatment for COVID-19, ChemRxiv, 2020 DOI:10.26434/chemrxiv.13472373.v1.
- (a) F. Rasool and D. Mukherjee, Org. Lett., 2017, 19, 4936 CrossRef CAS PubMed; (b) A. Ahmed, F. Rasool, G. Singh, M. Katoch and D. Mukherjee, Eur. J. Org. Chem., 2020, 2020, 4408 CrossRef CAS.
- A. B. Smith, Q. H. Paul, A. S. Breslin and G. K. Beauchamp, Org. Lett., 2005, 7, 5075 CrossRef CAS PubMed.
- U. Niedballa and H. Vorbrüggen, J. Org. Chem., 1976, 41, 2084–2086 CrossRef CAS PubMed.
- A. Amanteal, M. Henz and P. Strazewski, Helv. Chim. Acta, 1996, 79, 244 CrossRef.
- B. C. Bookser and N. B. Raffaele, J. Org. Chem., 2007, 72, 173–179 CrossRef CAS PubMed.
- A. Ahmed, J. S. Banday, Q. N. Ahmed, D. Mukherjee, Non infringing process for the synthesis of N4-hydroxycytidine and its derivatives, IN202111020678, May, 6, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06912h |
| This journal is © The Royal Society of Chemistry 2021 |