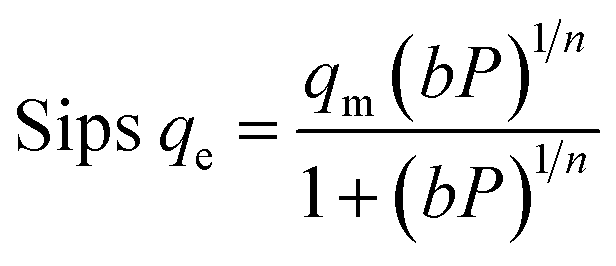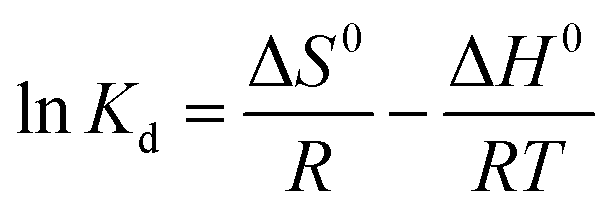DOI:
10.1039/D1RA06781H
(Paper)
RSC Adv., 2021,
11, 36125-36142
Biomass derived hierarchical porous carbon for high-performance O2/N2 adsorption; a new green self-activation approach
Received
9th September 2021
, Accepted 28th October 2021
First published on 10th November 2021
Abstract
Biomass-derived porous carbons are the most common adsorbent materials for O2/N2 adsorption because of their excellent textural properties, high surface area, and low expense. A new synthesis method based on a self-activation technique was developed for a new green porous carbon adsorbent. This ecofriendly system was used for the synthesis of hierarchical porous carbons from walnut-shell precursors. The sorbent was successfully synthesized by facile one-step carbonization, with the activating reagents being gases released during the activation. The sample morphology and structure were characterized by field emission scanning electron microscopy, high-resolution transmission electron microscopy, Raman, Fourier transform infrared spectra, X-ray photoelectron spectroscopy, X-ray powder diffraction, thermogravimetric, and differential thermal analysis. The optimal porous carbons were synthesized at 1000 °C, providing a surface area as high as 2042.4 (m2 g−1) and micropore volume of about 0.499 (m3 g−1). At 298 °K under 9.5 bar pressure, the potential for O2/N2 separation using porous carbon samples was studied, and the sips isotherms with the highest adsorption potential were determined to be 2.94 (mmol g−1) and 2.67 (mmol g−1), respectively. The sample exhibited stable O2/N2 separation over ten cycles, showing high reusability for air separation. Finally, the technology described presents a promising strategy for producing eco-friendly porous carbon from a variety of biomass on an industrial scale.
1 Introduction
Gas separation process technologies play an influential role in various industries including medical, food, chemical, petrochemical, and power generation industries.1,2 Pressure swing adsorption (PSA) is one of the most cost-effective solutions for air separation applications in response to industrial requirements for nitrogen and oxygen processing.3,4 In the 1950s, air separation systems based on a pressure swing were established.5 One of the special applications of air separation technology in the power plant industry is oxyfuel, which is one of the solutions to reduce carbon dioxide.6,7 The PSA technology is a cyclic adsorption process in gas separation using different adsorbents and adsorption capacity rates.8,9 Molecular adsorbents are used to separate and purify various gas species from a gas mixture under pressure using the PSA technique. This process usually operates at low pressure and ambient temperatures.2,10 Various adsorbents such as porous carbon, zeolites, carbon molecular baskets (CMB), and carbon molecular sieves (CMS) are used to adsorb the subject gas molecules at pressure swing.11
2 Literature review
For air separation, both physisorption and chemisorption processes are utilized, but the physisorption mechanism has become more common due to the low bond energy between gas and solid adsorbent, as well as the low-cost energy of sorbent regeneration.11,12 Recently, various physisorption-adsorbent such as zeolites are used in the O2/N2 adsorption as porous crystalline aluminosilicates.13 They have open crystal lattices that contain pores of molecular size, and gas molecules can diffuse them. The selective gas separation by zeolites requires control of the structure and composition of the framework, shape, and distribution of cations within the cavities.14 The breathtaking adsorption of zeolites for air separation is used in the continuous development of nitrogen adsorbents from the air. At room temperature, the ability of zeolites to separate air has revealed that nitrogen uptake capacity is greater than oxygen adsorption capacity.15,16 Metal–organic frameworks (MOFs) are composed of organic ligands or “struts” coordinated to metal-based nodes to form a three-dimensional extended network.17 Much research has been done on the MOFs with tunable pore size, high surface area, ability to functionalize the walls of cavities, and particularly, open metal sites for gas separation and storage.17–24 Experiments at high pressures and extremely low temperatures have been reported, therefore these points impose operational constraints. Carbon-based adsorbents are suitable for gas adsorption and are divided into different categories based on morphology, porosity, and structure, such as carbon nanotubes, graphene, and fullerenes.18,25–29 Despite their high adsorption efficiency, these adsorbents have drawbacks such as high synthesis costs, environmental impacts, and the use of toxic chemicals during synthesis.22,23 Porous carbon, unlike carbon-based adsorbents, does not have the aforementioned drawbacks.
Porous carbon has a wide range of uses, including catalysts, heavy metals adsorbents, separation of various gases, energy storage, and electrochemical materials, due to its unique characteristics.30 The porous carbon media is structurally complex and the total volume, size, and distribution of the pores determine how the molecules of a gas are adsorbed on the porous carbon, which these pores have strong van der Waals forces for the adsorption process.31 Natural precursors biomass can be used for porous carbon synthesis such as rice straw, tobacco, coffee, pine cone, palm shell, etc. due to renewable, cost-effective, and eco-friendly.32–34 The broad range, the abundance of nature, and the renewability of precursors used in the synthesis process distinguish porous carbon fabrication from that of other sorbents.35 Since biomass-derived porous carbon has a large surface area (up to 3072 m2 g−1), its density and morphology are influenced by the synthesis process.36 In recent years the traditional methods such as chemical and physical activation have commonly been used to activate/pyrolyze a variety of carbonaceous biomass. Porous carbons can be pyrolyzed/activated by physical (with CO2, N2, steam, O2, and NH3) or chemical (H3PO4, HCl, KOH, NaOH, and ZnCl2) activation of the carbon precursors that may occur in a one or two-steps process.37,38 The type of precursor, activating agent, activation temperature, and the ratio of activating agent to precursor are the key parameters that impact porosity composition, pore size distribution, and pore volume.29,39,40 The synthesis and carbonization method to prepare porous carbon is one of the influential features in the characteristics of porous carbon materials.41 In the physical activation method, the mentioned gases are used at a temperature below 1000 °C, while in the chemical activation method, acidic and alkaline agents are used to impregnate.42 Chemical activation is favored due to the resulting material's tendency to be mesoporous and tunable porosities, even though physical activation is less expensive and has a lesser environmental effect. Chemical activation, on the other side, is frequently afflicted by toxic gases released during pyrolysis, process complexity, and device corrosion, and is not environmentally friendly.43–45
The direct pyrolysis method is a step forward in the production of porous carbons since it removes the need for post-synthesis activation. A novel self-activation approach for porous carbon synthesis has been developed in recent years as a result of the aforementioned limitations. In recent research, intrinsic hydroxyapatites were employed as natural templates to produce N-doped mesopore-dominant carbon materials utilizing a one-step self-activation method under N2 atmosphere. As discussed, this method is not considered self-activation due to the use of inert gases and templates during the process.46–48
The present work proposed a green, low-cost and facile method to prepare green porous carbon with mesopore-dominant from walnut-shell as precursors using a one-step self-activating system. The proposed approach is based on a closed-loop carbonization system that uses gases decomposition from biomass as the reagent. To better understand the role of the self-activating system, the pyrolysis process was performed with and without argon gas to determine the effect of this system on the structure. In the following, the present report proposed the high-performance green porous carbon adsorbent for O2/N2 adsorption. Adsorption equilibrium and kinetics experiments of O2/N2 were also performed to obtain the adsorption capacity and kinetics. The morphology and structure, as well as chemical constitution of the resulting porous carbon, were characterized by field emission scanning electron and high-resolution transmission electron microscopy, Raman and Fourier-transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), powder X-ray diffraction (XRD), thermogravimetry-differential thermal analysis (DTA-TG) and N2 adsorption–desorption analysis, in which the influence of the treatment method on the morphology and porous texture of the material was also demonstrated. By use of a virtual representation of a large surface area (up to 2042.4 m2 g−1) and availability, the resulting porous carbons show great potential as functional sorbents for gas storage and separation.
3 Experimental
3.1 Materials
In the current study, all chemicals and reagents were analytical grade and were utilized without additional purification as supplied. Walnut-shell biomass was collected from walnut trees on the campus area of Iran University and Science Technology (Tehran, Iran). This biomass was rinsed multiple times before being treated to eliminate visible impurities. Hydrochloric acid (37%) was purchased form was purchased from Dr Mojallali™ Company (Tehran, Iran), and, O2 (99.999%), N2 (99.999%), Ar (99.999%) were supplied by Arman Gas Company (Tehran, Iran).
3.2 Self-activating system for preparation of porous carbon
A sealed chamber, a tubular electric furnace, an air pump, and eventually a condenser are all part of the self-activating synthesis system that is connected by pipes. Both ends of the quartz tube were sealed with rubber. During the synthesis process, no gas is released into the atmosphere since the system is sealed. An aluminum combusting boat holding a specific amount of different types of biomass was put in the hot zone (up to 1373 K) during the pyrolysis process. With the assistance of an air pump positioned at the system, the gases produced throughout the pyrolysis of the feeding biomass are cycled in a closed-loop channel. With the help of these gases, the activation process is carried out. As a result, no further activating agent is required. A condenser is also placed along the gas's path, collecting and converting a part of the exhaust gases to liquid.
To prepare activated carbon, walnut-shell powder was directly pyrolyzed in a tubular electric furnace with a quartz tube under an argon atmosphere and without it at 1000 °C (heating rate: 5 °C min−1). The samples were held at the maximum temperature for 3 h. After cooling to room temperature, the obtained product was washed and stirred with 1 M HCl and ultrapure water to remove impurities. Ultimately, filtration was applied until neutral pH and the residue was thoroughly washed with deionized water and dried at 95 °C for 12 h. The corresponding products were identified as W1000 (self-activated after pickling in 1000 °C), W1000-B (self-activated before pickling), W1000-Ar (self-activated under argon, after pickling in 1000 °C), and W1000-Ar-B (self-activated under argon, before pickling in 1000 °C). Fig. 1 shows a schematic view of the self-activating used in the present study.
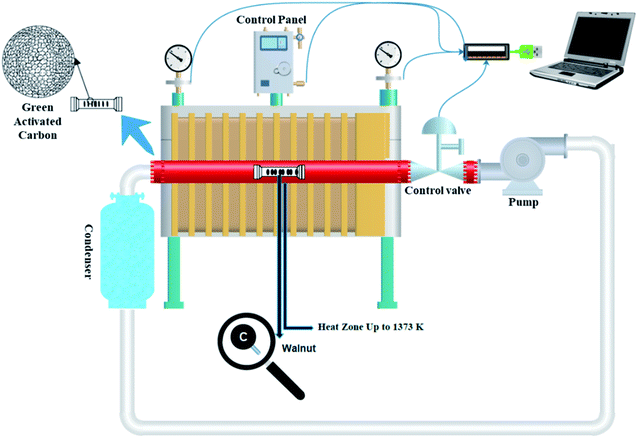 |
| | Fig. 1 Schematic of the self-activating synthesis setup. | |
3.3 Characterization
Micromeritics ASAP2020 (US) adsorption analyzers was employed to measure the N2 adsorption–desorption isotherms at 77 K. Before performing the adsorption–desorption analyses, samples were degassed under dynamic vacuum conditions to constant weight at a temperature of 150 °C for 6 h. FTIR spectroscopy was accomplished on a PerkinElmer Spectrometer in the range of 500–4000 cm−1 with KBr pallets. Raman spectroscopy was conducted on a Takram micro-Raman spectrometer (Teksan™, Iran). X-ray photoelectron spectroscopy (XPS) measurements were carried out on an Al Ka source (XPS Spectrometer Kratos AXIS Supra). High-resolution transmission electron micrographs were obtained on a 300 kV FEI (US) TITAN microscope (HR-TEM). Thermogravimetric and differential thermal analyses (DTA/TG) in argon and air atmosphere were conducted on a Q600 (US) TA. Field emission scanning electron microscopy (FESEM) was observed on a Nanosem 450 microscope.
3.4 O2/N2 Adsorption–desorption measurement
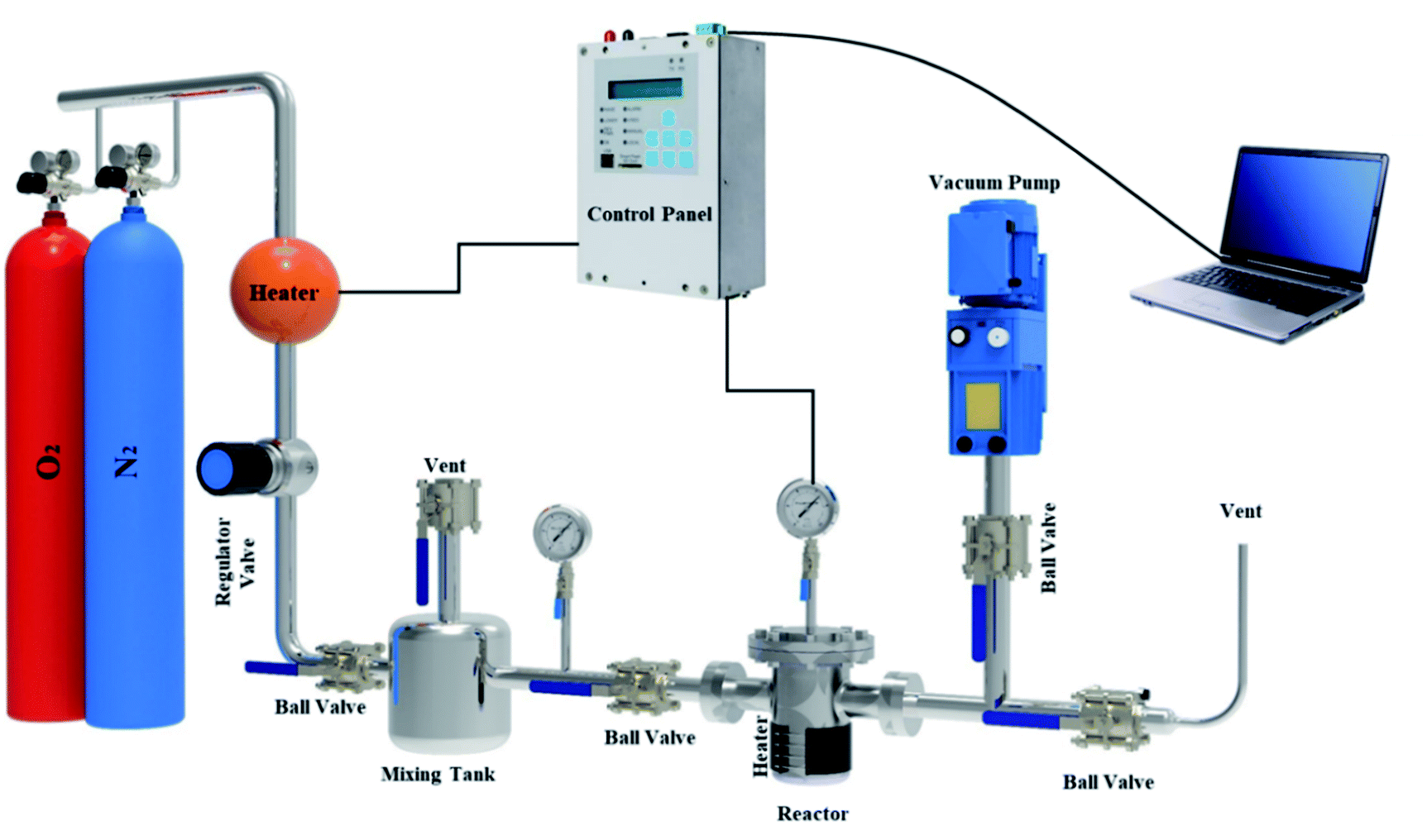
A fixed bed adsorption reactor was built as shown in Fig. 2 to test the adsorption/desorption capacity of sorbent using the volumetric approach. For experiments, pure O2 and N2 were used as a feed to study the adsorption capacity on the sorbent surface. Each experiment by 1 g sorbent was tested in a fixed bed reactor sealed of the experimental set-up. Before the start-up test, the N2 gas purging at 380 °K for 30 min in the experimental set-up was used. The experimental tests were performed at 298, 308, 318, and 328 °K temperatures under pressures rating 2 to 9.5 bar for 60 min. Due to the mixing tank in the passage, the CO2 pressure and temperature were stabilized, and the stable gas was then fed to the adsorbent reactor. The pressure difference recorded at fixed temperature using pressure-temperature sensors and computer panel during the time was used to assess the adsorption parameters. The electrical heat tracing for the temperature stabilizer was used.
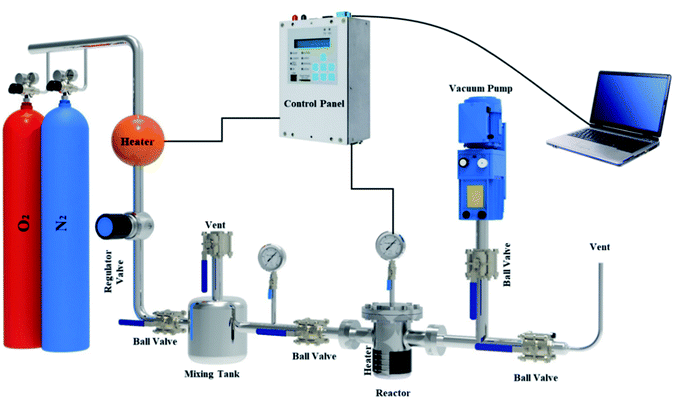 |
| | Fig. 2 The experimental set-up in the schematic view. | |
4 Results and discussions
4.1 Characterization analysis
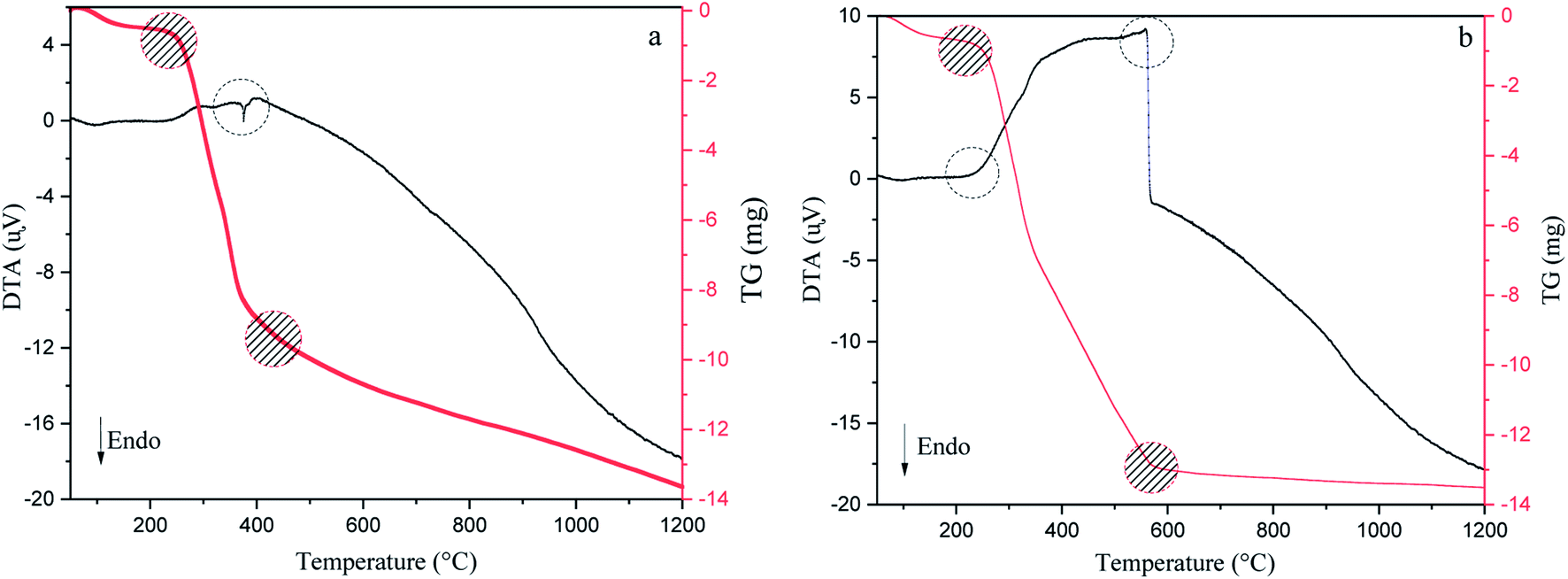
DTA/TG analysis method has been accomplished in argon and air atmospheres to reveal the stability of the precursor, exothermic or endothermic reactions during pyrolysis, the amount of moisture and volatile matter lost from 50 °C to 1200 °C (Fig. 3).12,44 As can be seen in Fig. 3(a and b), in the first step, both conditions' TG curves show a ∼7.5–10.5% mass loss up to 200 °C due to the removal of adsorbed water molecules. Additionally, DTA curves show an endothermic reaction owing to the evaporation of moisture found in walnut-shell.49 The next step begins at the first to the second turning point of the plots (220–400 °C for argon and 220–600 °C for air) has the highest weight loss, which represents the thermal elimination of organic components, hemicellulose, cellulose, and lignin. The third stage is the burning of lignin leftovers, which takes place at elevated temperatures. In the air atmosphere, the weight of the precursor decreases gradually with a temperature up to 580 °C. Conversely, the weight loss process decreases slightly above this temperature (580–850 °C) and reaches a steady pace at high temperatures due to oxygen consumption. In an argon atmosphere, weight loss is fast up to 380 °C, while at high temperatures is moderate. Moreover, DTA curves of both atmospheres show an exothermic reaction that represents the thermal decomposition of the walnut-shell.49,50 As mentioned exothermic peaks, burning the precursor produces, confirm CO2 production, indicating that walnut-shell combustion takes place in both atmospheres. This implies there is enough oxygen in the walnut-shell components for the combustion process to take place.33
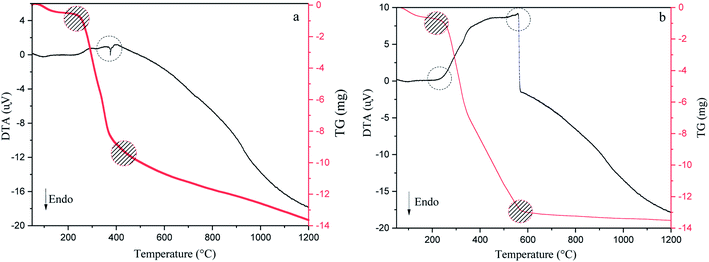 |
| | Fig. 3 Thermogravimetric and differential thermal analysis results of walnut-shell in argon (a) and air (b) atmosphere from 50 °C to 1200 °C. | |
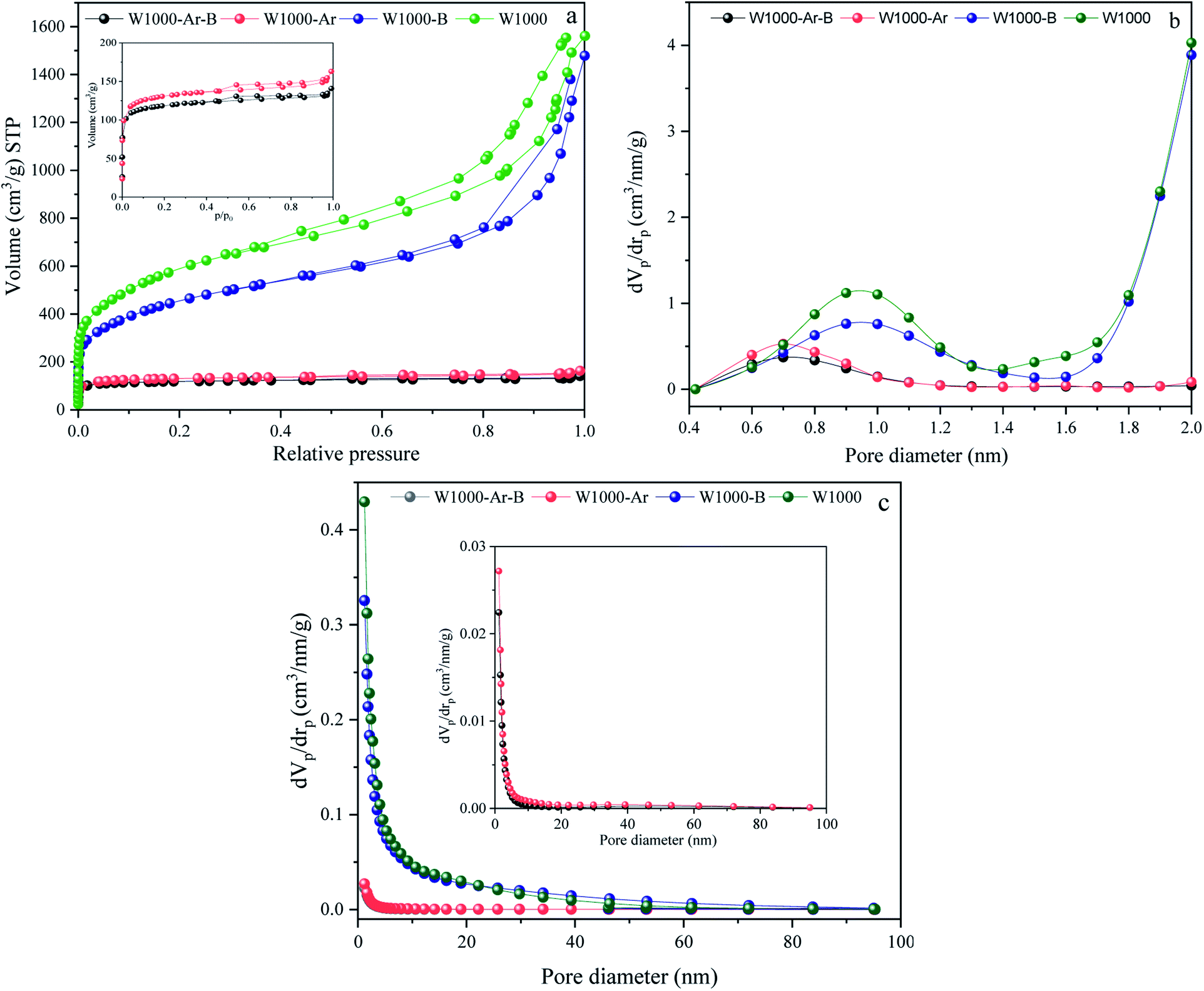
The activation strength of the introduced self-activating system is demonstrated by the nitrogen adsorption–desorption analysis results of the pyrolyzed walnut-shell in Fig. 4. N2 adsorption–desorption, micropores-plots (MP plot), and the pore size distribution (PSD) of various green porous carbons can be seen in Fig. 4(a–c). Moreover, the detailed textural properties of green porous carbons can be observed in Table 1. The specific surface area, mean pore diameter and, total pore volume of the walnut-shell pyrolyzed under argon atmosphere are 488.2 m2 g−1, 2.05 nm, and 0.24 cm3 g−1 which confirms that a self-activating system can form porous carbon under argon atmosphere. Conversely, the self-activating system without argon circulate the released gases such as CO2, CO, H2, and H2O, which shows the increment of mesopore volumes as seen by the much higher N2 adsorption. With the aid of this system, the specific surface area of W1000 has been greatly increased (2042.4 m2 g−1) compared to the W1000-Ar, which shows the capability of a self-activation synthesis system. The volume increased steadily at low relative pressure (P/P0 > 0.1) in the N2 sorption plot, indicating the presence of micropores.51,52 The H1-type hysteresis loop at P/P0 > 0.5 for all samples is the capillary condensation phenomenon, showing the affluence meso–micro porous characteristics which confirm the results of the table. Based on MP plot results, W1000 has 0.83 cm3 g−1 and 1.85 cm3 g−1 mesopores and micropores volume which is much larger than the sample synthesized under argon atmosphere. It shows that more elevated temperatures are favorable for micropore formation. The concurrent rise in micropore and mesopore volume indicates that new pore formation and pore widening occurred at the same time, and the production of micropores did not compromise the W1000 mesopore structure. The PSD presents the hierarchical porous structure of the self-activated instances, composed of micropores (0.5–2 nm), small mesopores (2–10 nm), and large mesopores/macropores (10–60 nm) with large size mesopores and abundant micropores.53–55 As summarized in Table 1, the acid-pickled samples provide a larger specific surface area (488.21–2042.4 m2 g−1) and a pore volume (0.28–2.68 cm3 g−1) than the others, due to removing impurities during pickling. Pickling of porous carbon with acidic solutions (HCl) resulted in the removal of the inorganic compounds that mainly generate mesopore structures. By disappearing these compounds from the surface, voids appear, which firstly increase the volume of porosity and secondly increase the specific surface area.56,57
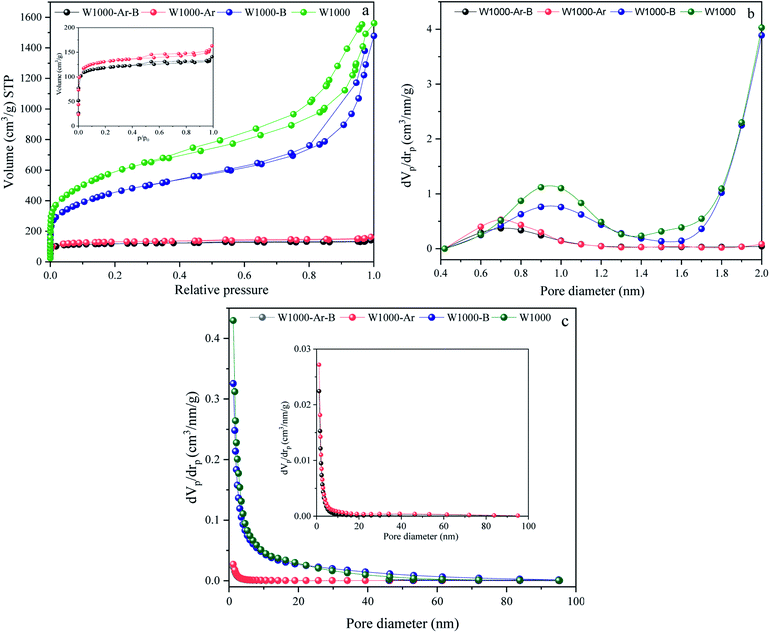 |
| | Fig. 4 N2 adsorption/desorption isotherms, (a), MP plots (pores smaller than 2 nm) (b), and BJH pore size distributions curves (c) of porous carbons. | |
Table 1 Detailed textural properties of green porous carbons
| Sample ID |
Specific surface area (m2 g−1) |
Average pore diameter (nm) |
Total pore volume (cm3 g−1) |
Mesoporous volume (cm3 g−1) |
Microporous volume (cm3 g−1) |
| W1000-Ar-B |
450.94 |
1.9241 |
0.24 |
0.2 |
0.04 |
| W1000-Ar |
488.21 |
2.0542 |
0.28 |
0.22 |
0.06 |
| W1000-B |
1580.1 |
5.4821 |
2.33 |
0.56 |
1.77 |
| W1000 |
2042.4 |
4.6401 |
2.68 |
0.83 |
1.85 |
According to the results of the textural properties and DTA/TG, the general mechanism of self-activation can be achieved, which is expressed in the following reactions:
| | |
C(s) + H2O(g) → CO(g) + H2(g)
| (2) |
| | |
2C(s) + O2(g) → 2CO(g)
| (3) |
| | |
CO(g) + H2O(g) → CO2(g) + H2(g)
| (4) |
| | |
C(s) + CO2(g) → 2CO(g)
| (5) |
At low temperatures, the moisture is gradually removed and converted to vapor. Activation begins slowly at temperatures above 700 °C, and the evolved H2, H2O, CO, and CO2 gases during the pyrolysis process with continuous rotation in the circulatory system, carry out a key role in the self-activation process.58–60 When the activation temperature increases (≥700 °C) the standard Gibbs free energy change of the Boudouard reaction (5) becomes negative. As a result, CO2 and steam (H2O), which are produced by the decomposition gases of cellulose, may react with carbon atoms and can penetrate into the interior of the carbon material to increase the porosity. Both the reactions of carbon with CO2 and H2O are endothermic which are consistent with the DTA results. Pyrolyzation at ≥850 °C is high enough to effectively initiate the reaction (4). As the activation temperature rises, the equilibrium constant decreases. This indicates the Boudouard reaction is faster in favor of CO production and solid carbon consumption, as seen by the TG's weight loss. Through the Boudouard reaction, carbon atoms were separately stripped from their surfaces to create carbon monoxide and hydrogen in the gas phase. As well, as demonstrated in reaction (4), the exothermic water-gas shift reaction between steam and carbon monoxide may occur, resulting in CO2 and H2 as catalyzed by the carbon surface.48,61,62 The pore-forming process in this system has two parts, with temperatures ranging from 200 to 600 °C and 650 to 1000 °C, respectively. Initially, the channel is created by a huge amount of pyrolysis gases escaping from biomass. Secondly, the pyrolysis gases were forced into the channel of char, causing the collapse of adjacent pores. It led to creating micro–meso pores by gasification reactions with carbon.63,64
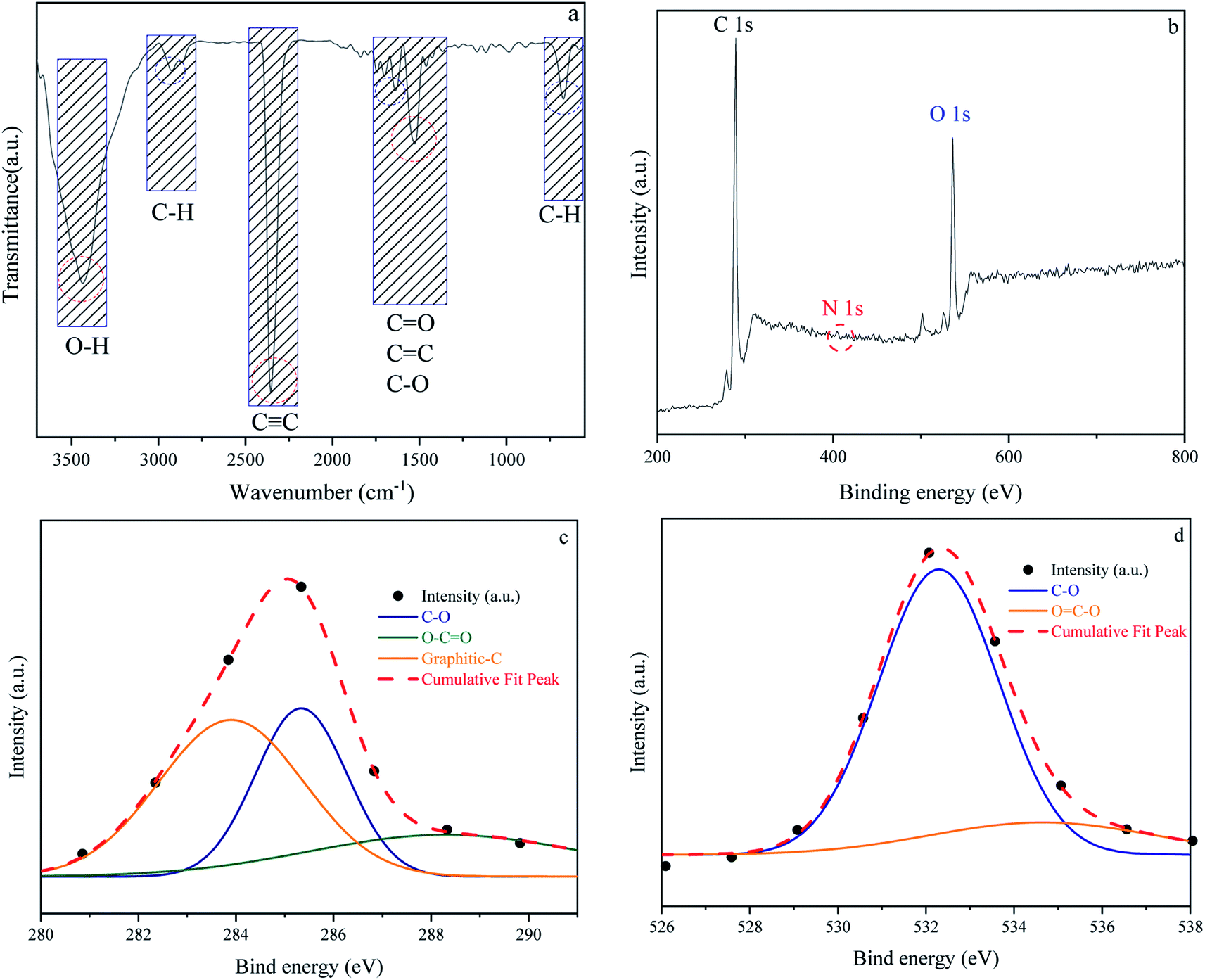
In addition to carbon atoms, self-incorporated heteroatoms such as N- and/or O- dopants, which are typically components of the precursors' original structure, can improve the inherent characteristics of the porous carbon, resulting in enriched surface-active sites.65–67 Furthermore, functional groups comprising the aforementioned heteroatoms can have a significant impact on the final product's behavior in a variety of applications, including gas adsorption. Acidity, electronegativity, and oxyfunctionalized molecules determine the surface chemistry of porous carbons.68–70 Fig. 5 depicts the surface chemical properties of the W1000. As can be seen, the FTIR spectra of chemical bond vibration and specific functional groups have been shown in Fig. 5(a). The mentioned figure demonstrates a peak at approximately 3500 (cm−1), which is typically assigned to O–H stretching vibration of hydroxyl bands. At lower wavenumbers, the location and asymmetry of this band suggest the presence of strong hydrogen bonding. The peak in the 2350 (cm−1) range is related to R–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H bonds and assigns the presence of CC (alkynes) stretching vibration. Moreover, the band at 1732 (cm−1) indicates the presence of the C
C–H bonds and assigns the presence of CC (alkynes) stretching vibration. Moreover, the band at 1732 (cm−1) indicates the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of the carbonyl group in acidic oxygen surface groups; the bands at 1500–1700 (cm−1) display the CC vibrations (alkenes) in the aromatic group. The bands at 1460–1350 (cm−1) depict the C–O vibrations in the carboxylate group. Eventually, a weak bond at around 700 (cm−1) is associated with the structure of aromatic C–H out-of-plane bending vibration (benzene ring).68,71–74 Carboxyl functional bands show the weak intensity of peaks due to the high temperature of porous carbon synthesis. After activation at high temperature, the intensity of CO was apparently decreased, implying that the carboxylic groups were converted to –OH groups. This confirms the role of steam in the self-activating system.75,76
O vibration of the carbonyl group in acidic oxygen surface groups; the bands at 1500–1700 (cm−1) display the CC vibrations (alkenes) in the aromatic group. The bands at 1460–1350 (cm−1) depict the C–O vibrations in the carboxylate group. Eventually, a weak bond at around 700 (cm−1) is associated with the structure of aromatic C–H out-of-plane bending vibration (benzene ring).68,71–74 Carboxyl functional bands show the weak intensity of peaks due to the high temperature of porous carbon synthesis. After activation at high temperature, the intensity of CO was apparently decreased, implying that the carboxylic groups were converted to –OH groups. This confirms the role of steam in the self-activating system.75,76
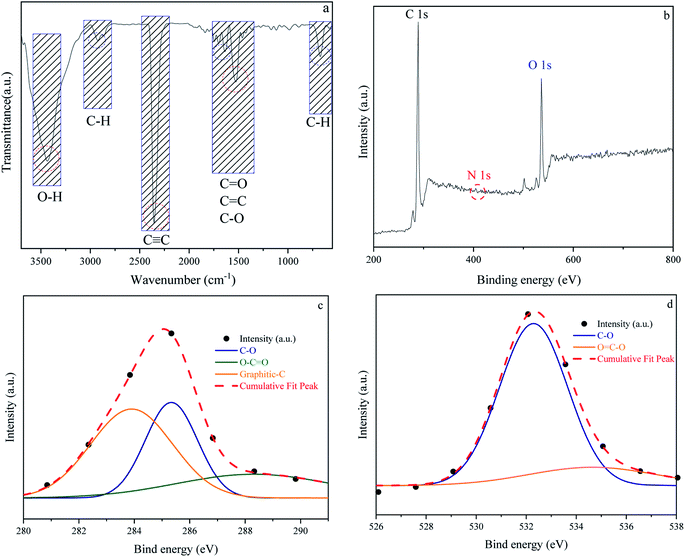 |
| | Fig. 5 Fourier transform infrared spectroscopy results of W1000 (a), XPS survey spectrum of W1000 showing the presence of C, N and O elements in the structure (b), C 1s (c) and O 1s (d), peak deconvolution. | |
The surface elemental composition and chemical bond configuration of W1000 were characterized by XPS measurement. In the survey spectrum carbon, oxygen and nitrogen can be identified by photoelectrons of their 1s orbitals. As shown in Fig. 5(b)–(d) as expected, the surface composition of the sample is composed of carbon species (68%) and a rich distribution of oxygen (31.4%) on the surface. Furthermore, N 1s (0.6%) peak can also be observed at around 401.5 eV on the surface. The C 1s (sp3-boned) spectrum was deconvoluted into three peaks representing sp2 hybridized graphitic-C (284 eV), phenolic C–O (285.9 eV), and O–CO (288.1 eV) groups. O 1s spectrum was fitted by two peaks, located at 532 and 534 eV, which could be belonged to C–O and OC–O groups.43,58,77 The high temperature of the synthesis leads to an increase in the amount of oxygen and nitrogen increasingly disappear, which is consistent with the results of FTIR. These abundant oxygen functional groups on the surface of porous carbon couple with the high specific surface area offer a strong tendency to gas adsorption.78,79
The morphology and crystallinity of the W1000 have been demonstrated in Fig. 6. The development of porosity and changes in the surface morphology was shown by using a FESEM. As shown in Fig. 6(a)–(c), the sample revealed a cauliflower-like structure and a highly irregular porous. It can be seen rougher textures with heterogeneous surfaces and porous with many cavities. Furthermore, it can be seen that the size distribution of the pores, as critical factors determining its applicability, occurred randomly at the sample surface. When synthesis occurs with a gradual increase in temperature, cellulose is changed to porous solid carbon. These changes led to the opening of pores and the spaces between the crystallites. Owing to the thermal degradation of porous carbon occurring at high temperatures, the part of the micropores structure collapses and became mesoporous, confirming the results of BJH. Ultimately, the increase of the pore development in the ACs has a key role due to these pores acting as the active sites for gas adsorption, during the pyrolysis.50,51,80–83
 |
| | Fig. 6 FESEM micrographs showing cauliflower structure at 1000 °C (a)–(c), HRTEM micrographs showing the turbostratic carbon matrix with clear graphite crystallites (d)–(f), Raman (g) and XRD (h) of W1000. | |
The fabrication of the W1000 can be verified by the HRTEM images, showing the development of crystallinity and graphite plates of the porous structure. Fig. 6(d)–(f) illustrates the disordered carbon graphite layers with an approximate short-range lattice spacing (0.07 nm). As it can be seen, the sample consists of disordered mesostructured typical of amorphous carbon with some defects alongside low crystallization. Furthermore, Fig. 6(d) shows an interconnected hierarchical porous structure, including porosity with different dimensions of wormhole-like pores (meso–micro) which comprise large amounts of mesoporous.36,84–86
Raman spectroscopy analyses (Fig. 6(g)) were used to evaluate the degree of structural graphitization of the W1000. Raman spectra display two peaks at 1340 cm−1 (D band) and 1593 cm−1 (G band). D band is attributed to the double-resonance effect of disordered and defects in biomass-based porous carbon (sp3-bond) and G band is ascribed to the in-plane stretching of sp2-bond and graphite in-plane vibrations with E2g symmetry. Moreover, the appearance of a 2D-band about 2700 cm−1 indicates a higher degree of graphitization and is linked to the association between graphite plates, which is the cause of graphite formation in the carbon structure.87–89 The integral intensity ratio of D to G bands (Id/Ig = 1.36) is higher than 1.00. This point indicated that a number of defects present on the carbon surface, poor graphitization, and the tendency of the structure to be amorphous as to be seen in HRTEM Fig. 6(e) and (f). Although higher temperatures usually cause graphite to increase, sometimes destroys the graphite structure, which can be a reason for the irregularity of the synthesized sample at 1000 °C.87,90
As can be observed in Fig. 6(h), X-ray diffraction patterns clearly show the development of turbostratic carbon with structural ordering intermediate between amorphous carbon and crystalline graphite. As shown a broad and weak diffraction peak was observed at about 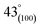 (turbostratic structure) indicating the presence of amorphous graphitic carbon and lower ordering. Besides, the increase in pores at high temperatures due to the higher orientation freedom of the graphite crystallites has led to a decrease in the severity of reflection and expansion at peaks. The absence of a peak at
(turbostratic structure) indicating the presence of amorphous graphitic carbon and lower ordering. Besides, the increase in pores at high temperatures due to the higher orientation freedom of the graphite crystallites has led to a decrease in the severity of reflection and expansion at peaks. The absence of a peak at 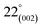 (oriented aromatic carbon) is a sign of the structure's strong tendency to formation of highly disordered and randomly oriented graphitic carbon layers that generate higher specific surface areas, consistent with the observation of Raman and BJH's results. Raman results likewise revealed a more disordered structure, while XRD correlated to lower graphitization degrees.51,80,91–93
(oriented aromatic carbon) is a sign of the structure's strong tendency to formation of highly disordered and randomly oriented graphitic carbon layers that generate higher specific surface areas, consistent with the observation of Raman and BJH's results. Raman results likewise revealed a more disordered structure, while XRD correlated to lower graphitization degrees.51,80,91–93
4.2 Adsorption kinetic and equilibrium isotherms
The gas adsorption processes by porous material is depending on micropores, mesoporous, surface area of adsorbent, and type of different kinetics. Review and analysis of the uptake kinetics reveal that the adsorbents' physical and chemical nature also affects the mechanism. We applied several theoretical kinetic models such as the pseudo-first-order, pseudo-second order, Elovich, and fractional-order kinetic models (eqn (6)–(9)) for kinetic models study.94| | |
qt = qe(1 − e−kft) pseudo-first-order
| (6) |
| | |
qt = (qe2kst)/[1 + qekst] pseudo-second-order
| (7) |
| |
qt = (1/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(αβ)) + (1/β × lnt) Elovich ln(αβ)) + (1/β × lnt) Elovich
| (8) |
| | |
qt = qe − (qe(1 − n) + ((n − 1)/m)kntm)(1/(1 − n)) fractional-order kinetic model
| (9) |
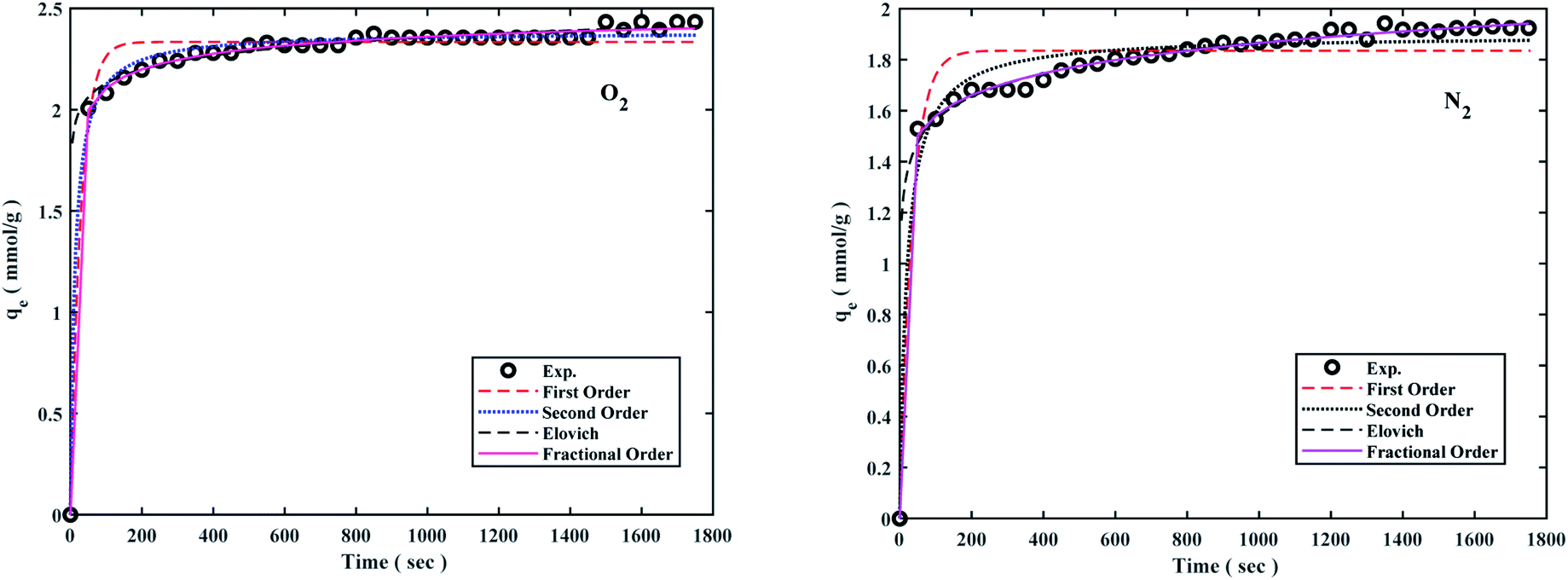
The pseudo-first-order model shows the reversible adsorption with the equilibrium between the adsorbent surface and the pseudo-second-order kinetic model based on the chemisorption process as the adsorption controlling factor. The fractional-order kinetic model shows simultaneous physical and chemical adsorption. Due to the complexity of predicting kinetic parameters, a common approach involves adapting experimental data to a set of mentioned models and selecting one of the best options. The kinetic parameters are presented in Table 2 and the best model is based on correlation coefficient values (R2) in the range of 0.8766 to 0.9987 at 298 °K, 308 °K, 318 °K, and 6 bar were fitted. The results of the models were also plotted to the experiment data to describe the best kinetic model for O2/N2 adsorption, and the kinetic curves of the sorbent are shown in Fig. 7. Fig. 7 shows that the experimental data did not fit the first-order kinetic model, although fractional-order and second-order models were more proportionate.26,95,96
Table 2 Calculated parameters of O2/N2 adsorption kinetic models at 6 bar for the W1000
| Kinetic models |
Parameters |
298 °K |
308 °K |
318 °K |
| O2 |
N2 |
O2 |
N2 |
O2 |
N2 |
| Pseudo-first-order |
qe (mmol g−1) |
2.334 |
1.835 |
1.285 |
1.08 |
0.961 |
0.676 |
| kf (min−1) |
0.035 |
0.029 |
0.016 |
0.023 |
0.029 |
0.017 |
| R2 (%) |
0.9849 |
0.9598 |
0.9859 |
0.9668 |
0.9105 |
0.8766 |
| Pseudo-second-order |
qe (mmol g−1) |
2.384 |
1.896 |
1.340 |
1.118 |
0.965 |
0.721 |
| ks (min−1) |
0.0339 |
0.0274 |
0.0228 |
0.0398 |
0.1544 |
0.0314 |
| R2 (%) |
0.9961 |
0.9848 |
0.9977 |
0.9879 |
0.8865 |
0.9356 |
| Elovich |
(mmol g−1 min−1) α |
2.3 × 108 |
1.1 × 104 |
5.5 × 104 |
6.4 × 104 |
9.1 × 105 |
30.37 |
| β (g mmol−1) |
0.099 |
0.132 |
0.084 |
0.071 |
0.041 |
0.088 |
| R2 (%) |
0.9565 |
0.9858 |
0.8898 |
0.9191 |
0.8942 |
0.9126 |
| Fractional-order |
qe (mmol g−1) |
2.662 |
1.376 |
1.306 |
1.376 |
1.637 |
0.711 |
| kn (min−1) |
0.0685 |
0.055 |
0.0651 |
0.055 |
0.0036 |
0.0103 |
| n |
3.0041 |
6.5344 |
0.949 |
6.5344 |
19.035 |
0.4638 |
| m |
0.5458 |
1.2209 |
0.5273 |
1.2209 |
0.0001 |
0.1353 |
| R2 (%) |
0.9987 |
0.9933 |
0.9982 |
0.9933 |
0.8977 |
0.9782 |
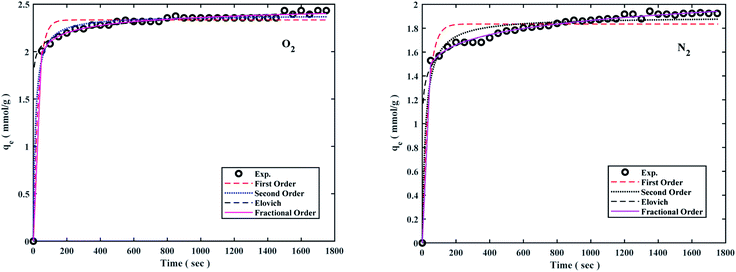 |
| | Fig. 7 O2/N2 adsorption capacities of the W1000 and corresponding fit desirable kinetic models at 298 °K and 6 bar. | |
The kinetic parameters derived from Table 2 revealed that the fractional-order kinetic model was better appropriate for explaining adsorption kinetics. Furthermore, R2 coefficient values for the fractional model ranged from 0.8977 to 0.9987, indicating that the fractional model was well fitted at 298 °K and 6 bar for O2/N2.
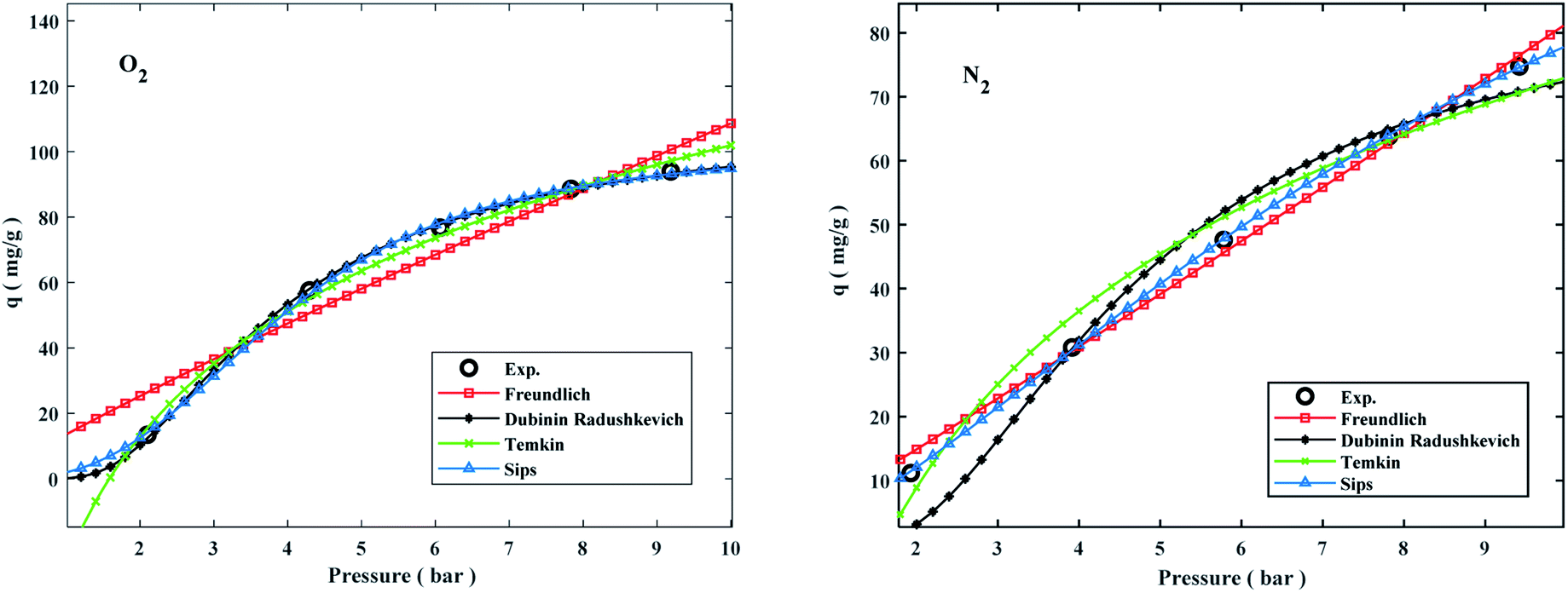
Adsorption isotherms by the following Freundlich, Dubinin–Radushkevich (D–R), Temkin, and sips models were plotted at 298 °K in the range of pressures 2 to 9 bar in Fig. 8. The experimental data and related correlation coefficient of R2 for all coefficients of isotherm parameters models were obtained in Table 3. The decrease in Kf value revealed the physical adsorption behavior, while the increase in qm value revealed the pressure and temperature-dependent O2/N2 adsorption behavior.94
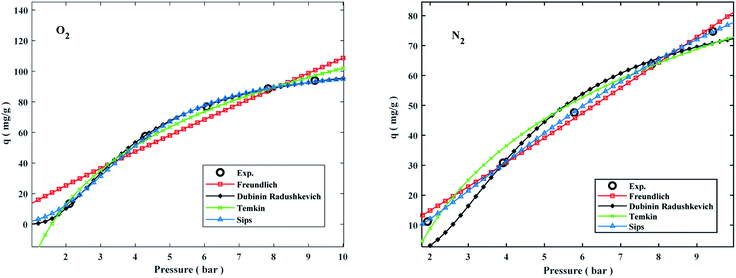 |
| | Fig. 8 Comparison of isotherms and O2/N2 experimental on the W1000 at 298 °K. | |
Table 3 Isotherm models of O2/N2 adsorption for the sorbent at 6 bara
| Models |
Parameters |
298 °K |
308 °K |
318 °K |
| O2 |
N2 |
O2 |
N2 |
O2 |
N2 |
| Where qe is the value of O2/N2 adsorption capacity (mmol g−1), qm is the max adsorption value of O2/N2 (mmol g−1), P is the equilibrium pressure (bar), KF is the Freundlich model constants [(mmol g−1). (bar−1)1/n)] and n is Freundlich isotherm constant. λ is (D–R) model constant (mol2 J−2), ω is Polanyi potential (J mol−1), A is the Temkin model constant (L mol−1), and B is the first virial coefficient (B = RT × bT−1);bT−1 (J mol−1). |
| Freundlich qe = kFPe1/n |
kF |
13.547 |
7.145 |
7.764 |
6.322 |
6.112 |
2.808 |
| n |
1.106 |
0.946 |
0.983 |
1.007 |
0.951 |
0.860 |
| R2 |
0.9633 |
0.9968 |
0.9898 |
0.9929 |
0.9955 |
0.9998 |
| Dubinin–Radushkevich qe = qme−λω2 |
qm |
108.607 |
86.996 |
85.399 |
68.661 |
73.536 |
46.474 |
| λ |
2.409 |
3.398 |
3.253 |
3.420 |
3.304 |
4.337 |
| ω |
0.456 |
0.384 |
0.392 |
0.382 |
0.389 |
0.340 |
| R2 |
0.9997 |
0.9867 |
0.9910 |
0.9910 |
0.9851 |
0.9835 |
| Temkin qe = B × log(A) + B × log(c) |
A |
0.630 |
0.624 |
0.599 |
0.628 |
0.597 |
0.604 |
| B |
55.402 |
39.913 |
41.138 |
31.265 |
35.101 |
19.837 |
| R2 |
0.9967 |
0.9870 |
0.9953 |
0.9923 |
0.9901 |
0.9694 |
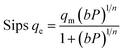 |
qm |
1.966 |
4.345 |
2.928 |
2.845 |
3.481 |
2.543 |
| bP |
2.858 |
1.601 |
1.985 |
1.820 |
1.651 |
1.272 |
| n |
0.0193 |
0.0307 |
0.0285 |
0.0328 |
0.0302 |
0.0094 |
| R2 |
0.9995 |
0.9999 |
1.0000 |
1.0000 |
0.9995 |
1.0000 |
The qe values decreased as the temperature was increased, indicating an exothermic O2/N2 adsorption behavior. All of the isotherm models good agreed with the experimental data but the Sips isotherm model has gained the highest correlation coefficient (R2) values fitted with experimental data. The Sips model proposed a similar equation to the Freundlich model for distinguishing the problem of the ongoing increase in the value adsorbed at increasing in limited pressure for the Freundlich model.97 Furthermore, the beneficial data assigned to the energy parameters is provided by two isotherm models developed by D–R and Temkin, where ω is the mean adsorption free energy and bT is the heat of adsorption. The normal physisorption of O2/N2 adsorption is shown by average λ values in the 3–4 (kJ mol−1) range. The Freundlich constant, n, in the range of 1 to 2, illustrates the desirability of physisorption based on the findings of Table 3. The adsorption process is multi-layer, according to the findings, with O2/N2 adsorbed and penetrated in the surface and inner layers of the sorbent. According to the Sips model, more efficient contact sites are prioritized, meaning that the sorbent surface is heterogeneous and multilayer O2/N2 adsorption is induced by a nonhomogeneous distribution of energy inactive sites.94,98,99 This point in the Sips model represents the physical adsorption process. Furthermore, the isotherm parameters derived from Table 3 revealed that the Sips isotherm model was better appropriate for explaining adsorption isotherm. The coefficient values of the Sips model ranged from 0.9995 to 1, indicating that the Sips model was well fitted from range 298 °K to 308 °K and 6 bar for O2/N2 adsorption. According to the correlation coefficient (R2) values derived in Table 3, the strength of the indicated isotherms models in the description and prediction of adsorption behavior was Sips > Freundlich > D–R > Temkin.94
4.3 The effect of temperature and pressure on O2/N2 adsorption
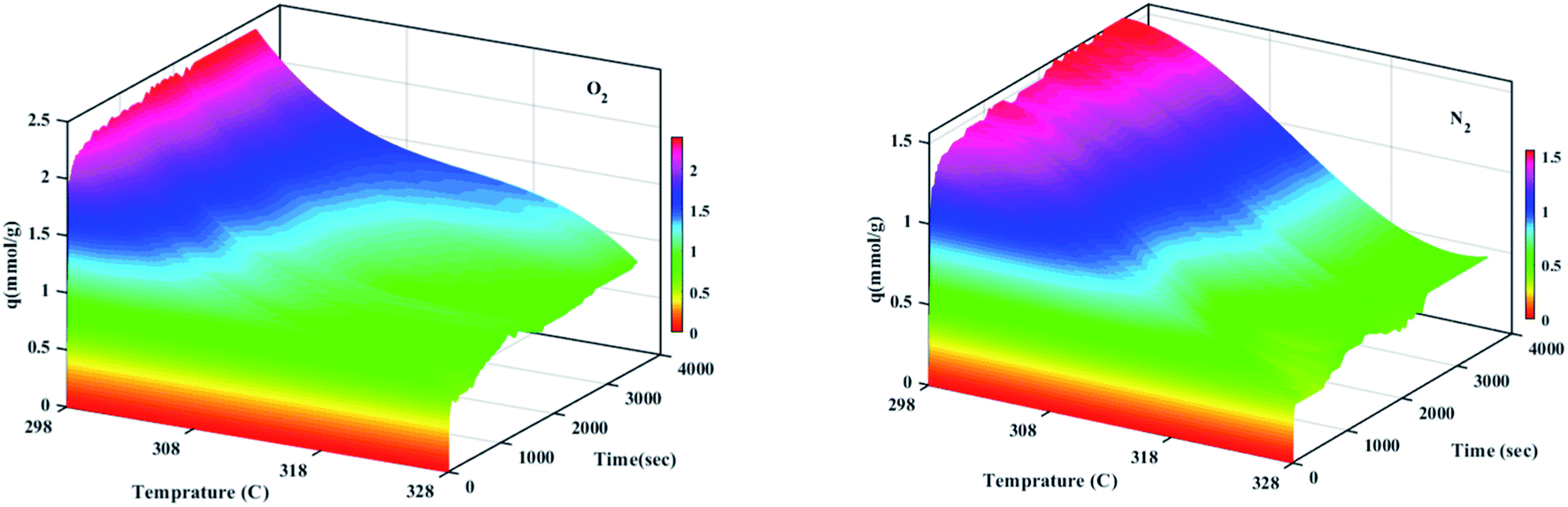
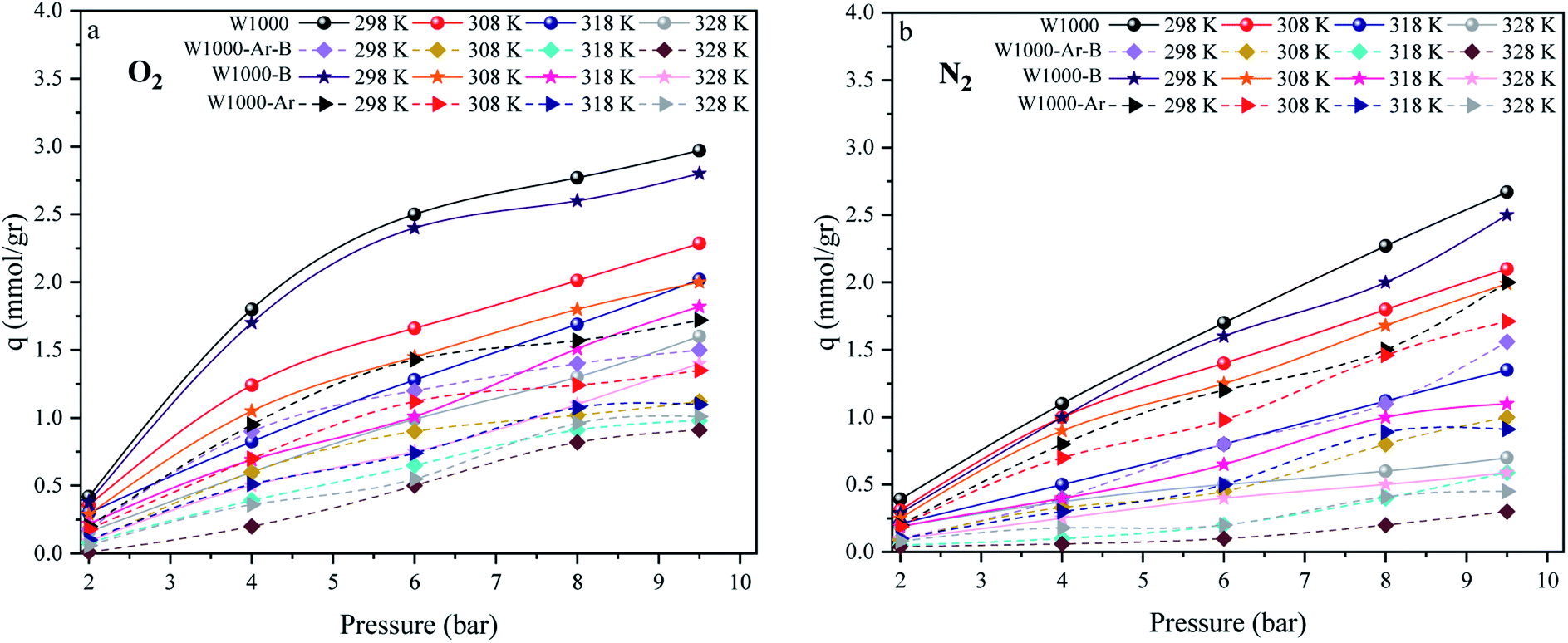
The effect of different temperatures at 6 bar for 60 min on the W1000 for review of O2/N2 adsorption capacity by adsorption setup is shown in Fig. 9. As can be seen, the increase of temperature from 298 °K to 398 °K, reduced the O2/N2 adsorption capacity for the W1000, and the maximum adsorption capacity of both gases was observed at 298 °K. The first thing to cite about the results in Fig. 9 is that denotes the relatively low quantity of N2 adsorbed, resulting in the very interesting selectivity values recorded for W1000. The second thing to cite these results is that the O2 is more quickly than N2 adsorbed at the same time, resulting again in selectivity values recorded for W1000.100 Furthermore, the results show that the O2/N2 adsorption process is exothermic, and O2/N2 adsorption capacity decreases by increasing the temperature. The effect of equilibrium pressure in the range of 298 °K to 398 °K on the W1000, W1000-B, W1000-Ar, and W1000-Ar-B sample sorbents for adsorption capacities of O2/N2 is plotted in Fig. 10. The results show that the W1000 sample is higher efficiency in the adsorption capacity of O2 and N2. Increased pressure has a significant impact on O2/N2 adsorption capability, according to the findings. At 9.2 bar and 298 °K, the maximum adsorption capacities of O2/N2 on the sorbent are 2.94 (mmol g−1) and 2.67 (mmol g−1), respectively.
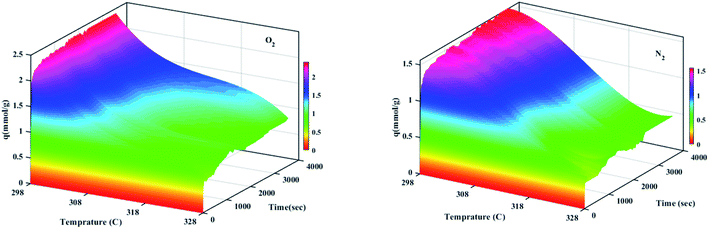 |
| | Fig. 9 Temperature effect on the O2/N2 adsorption capacity of the W1000 at 6 bar for 60 min. | |
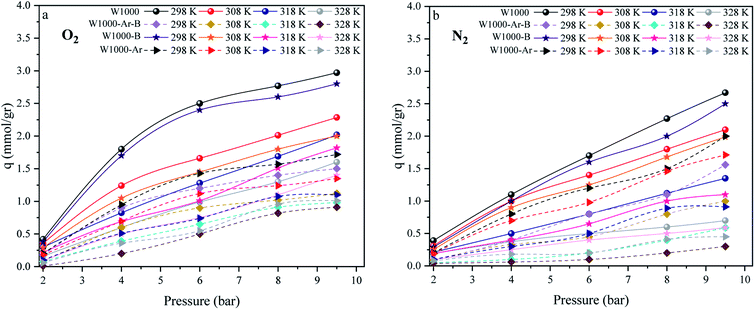 |
| | Fig. 10 The O2/N2 adsorption equilibrium isotherms (a) O2, (b) N2 of the W1000, W1000-B, W1000-Ar-B, and W1000-Ar. | |
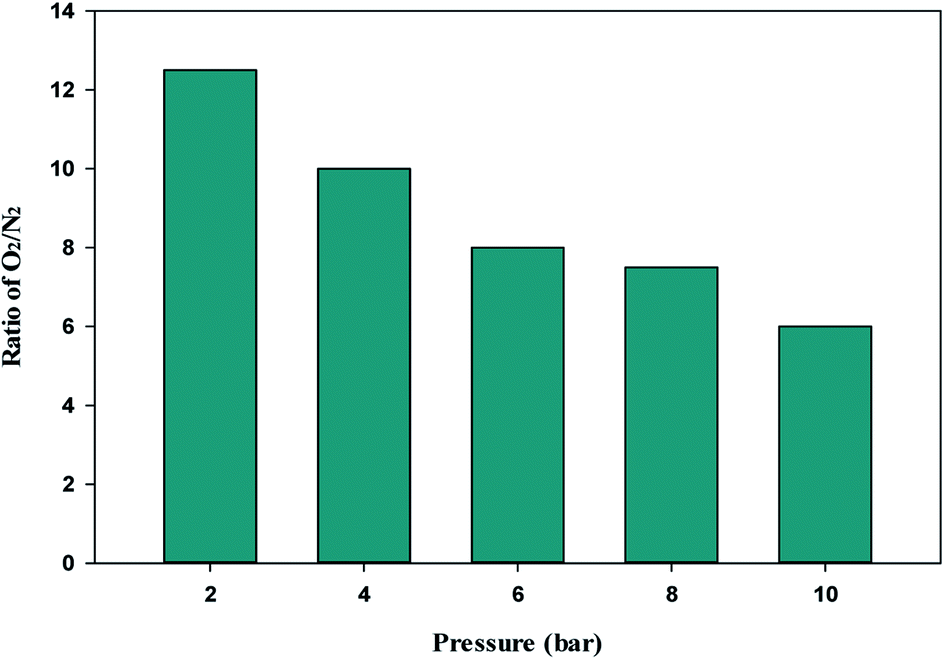
The single-component isotherm is a technique for determining the selectivity of adsorbent for O2 over N2. The equation was used to calculate the adsorption selectivity of O2 over N2.101
VO2 and
VN2 are the volumes of O
2 and N
2 gas adsorbed at a given pressure
P and temperature
T, respectively. The W1000 separation performance was evaluated using the reported equation. At various pressures, the selectivity of O
2 over N
2 was determined, and the results are presented in
Fig. 11. As can be observed from the results, the W1000 performed better in O
2 adsorption than N
2, implying that it has more potential for selective adsorption.
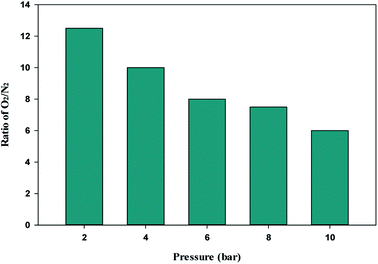 |
| | Fig. 11 O2/N2 adsorption selectivity on W1000 at 298 K and different pressures. | |
4.4 Comparison of O2/N2 uptake
The high adsorption values of these two gases indicate the principal role of mesopores and oxyfunctionalized groups in the sorption and compensation of nitrogen deficiency in the structure. In a selective adsorption application, the pore size and shape are the most significant characteristics. The void areas between highly irregular graphitic plates provide porous carbons pore structure. Moreover, the electrical characteristics of the adsorbate, such as the dipole/quadrupole moment and/or polarizability, influence the rate of sorption. Otherwise, the kinetic diameter, molecular size, and the structure of N2/O2, affect the rate of adsorption. As shown in Fig. 10, at a specified temperature and pressure, porous carbon tends to adsorb more oxygen than nitrogen. Charge transfer from micro graphite to adsorbed oxygen occurs as adsorbed oxygen molecules penetrate smaller pores (microporosities). Owing to the abundance of micropores, this indicates a greater tendency of these pores to adsorb more oxygen. The kinetic diameter of a molecule in a gas is proportional to its mean free path, which indicates the size of the molecule as a target. As a result, the kinetic diameter differs from the atomic diameter, measured in terms of the atom's electron shell size, which is generally significantly smaller. The kinetic diameters of O2 and N2 are 3.46 and 3.64 Å, respectively. These values are so near that separating them using a size exclusion or molecular sieve technique is difficult.94,102,103 O2 has a somewhat lower effective kinetic diameter within porous materials than N2 from a kinetic viewpoint. As a result, O2 diffusion via sorbent meso–micro pores can be quicker than N2 diffusion, particularly when the adsorbent contains reasonably uniform pores with diameters near the kinetic diameter of N2.104 Furthermore, nitrogen and oxygen have a shape factor that is likely to affect (adverse) adsorption kinetics since these molecules must pass through the selective porosity longitudinally, resulting in a loss of rotational flexibility.105 Because activation of porous carbon and high temperatures enlarge pore diameters, it is predicted that N2 contact with pore walls will be reduced more than O2 interaction.106 The quadrupole moment of N2 (−4.9 × 10−26 cm2) is approximately four times that of O2, resulting in increased N2 adsorption, as is well known. Thus, N2 can have a stronger interaction with the sorbent's electrical field gradients than O2. However, by observing the obtained results, it can be understood that this factor does not play a role in the adsorption rate of gases on porous carbon.100 Because of the diamagnetic nature of nitrogen molecules, the dipolar field of paramagnetic oxygen molecules shows better adsorption at very high pressures. The reason for this is that the electron spin resonance does not change for nitrogen molecules at very high pressures.107
4.5 Adsorption thermodynamic analysis
The standard free energy (ΔG0), enthalpy (ΔH0), and entropy (ΔS0) are thermodynamic parameters and calculated using the following equations:| | |
ΔG0 = −RTlnKd
| (12) |
| |
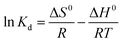 | (13) |
| | |
Kd = (Pi − Pe) × (V/W)
| (14) |
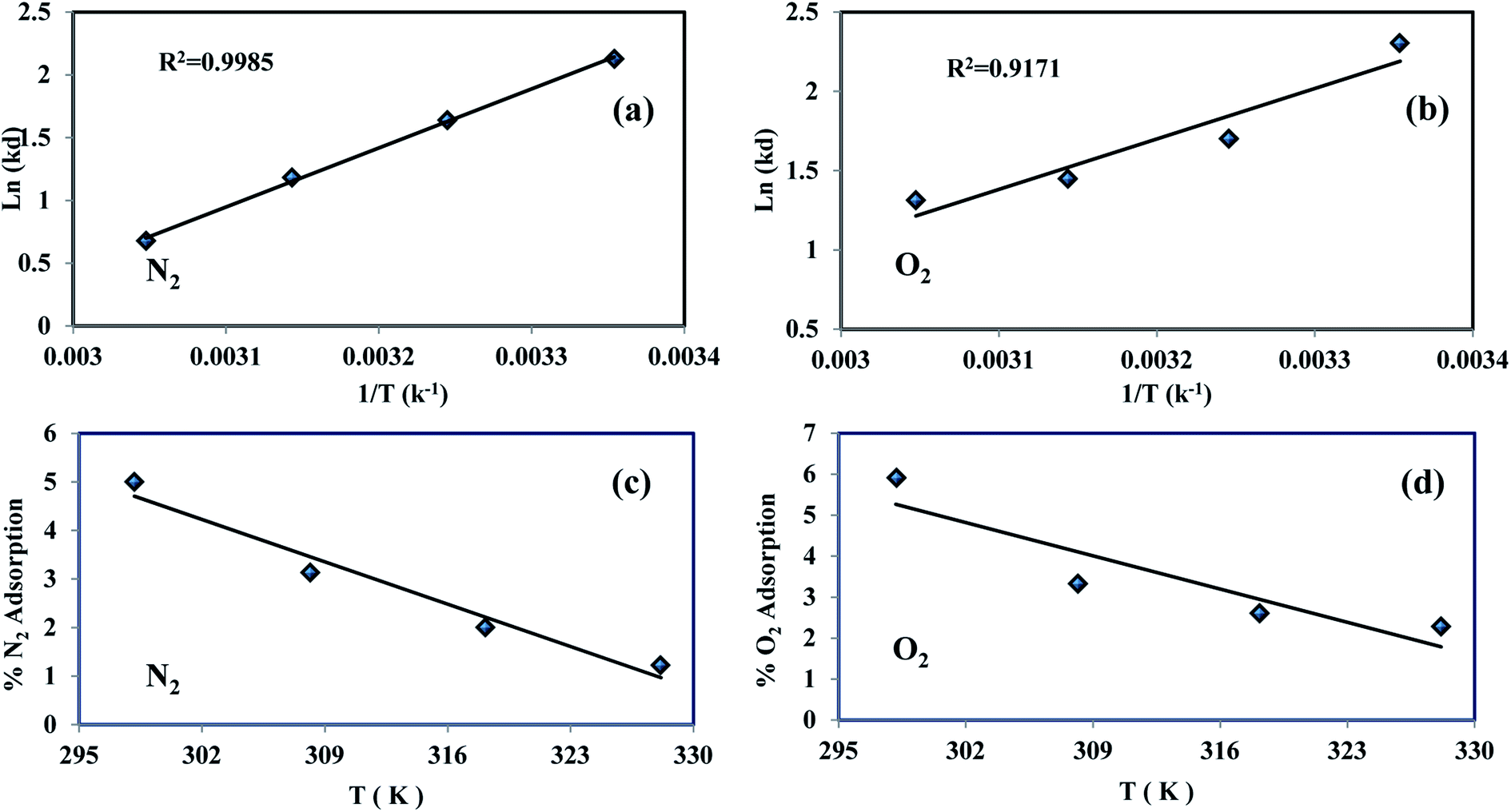
The values of (ΔH0) and (ΔS0) were calculated from the slope and cut off the plot of LnKd versus 1 × T−1. The standard Gibbs free energy change of sorption (ΔG0) was calculated using equations eqn (11) and (12). The Van't Hoff equation was used through equations eqn (11) and (14). In the mentioned equations, R is gas constant (8.314 (J mol−1 K−1)).14 As can be seen in Fig. 12(a) and (b) the values (ΔH0) and (ΔS0) were calculated from the slope and traverse of the plot of LnKd unto 1 × T−1. The results of calculated thermodynamic parameters of O2/N2 were tabulated in Table 4. The value of ΔH0 < 20 (kJ mol−1) represented the physisorption process and if the value of ΔH0 > 40 (kJ mol−1), represents the chemisorption. The ΔS0 is represented the randomized and organized interfaces of gas and solid, which in the case of ΔS0 > 0, it was more random, and in the case of ΔS0 < 0 it was less random.108,109 The Van't Hoff plot of the equilibrium constant of the sorbent which was used for the estimate of ΔS0 and ΔH0 in the range of temperature from 298 °K to 328 °K was plotted in Fig. 12(a and b). The negative values of Gibbs free energy change (ΔG0) and (ΔS0), indicating that the adsorption process is exothermic and spontaneous.
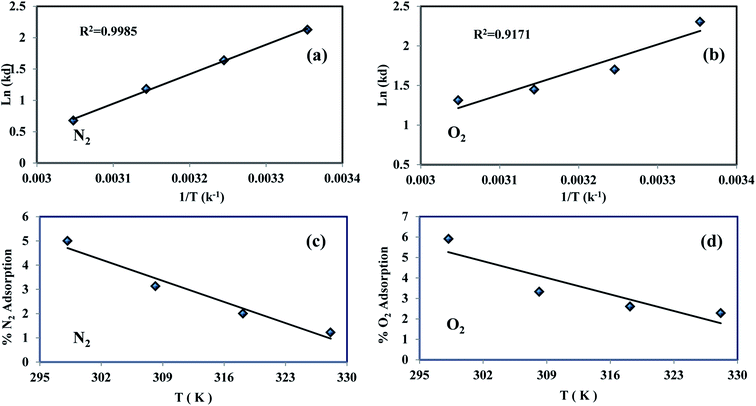 |
| | Fig. 12 O2/N2 experimental Van't Hoff plot (a and b) and adsorption variations percentage vs. temperature (c and d). | |
Table 4 Thermodynamic parameters of the W1000 in O2/N2 adsorption at 6 bar
| Gas |
ΔH (kJ mol−1) |
ΔS (kJ mol K) |
ΔG (kJ mol−1) |
| 298 °K |
308 °K |
318 °K |
328 °K |
| O2 |
−26.449 |
−0.071 |
−5.428 |
−4.723 |
−4.018 |
−3.312 |
| N2 |
−39.079 |
−0.113 |
−5.310 |
−4.178 |
−3.045 |
−1.912 |
The percentage variations of O2/N2 adsorption into temperature were shown in Fig. 12(c and d). The percentage of adsorption capacity for both gases increases with decreasing temperature, which confirms this physisorption process.
4.6 Comparison of various porous carbon
The recent experimental data on biomass-derived precursors for porous carbon adsorbents are summarized in Table 5. The current derived porous carbon fine sample has a considerably greater porosity volume capacity than previous experimental studies due to its high surface area and hierarchical porosity. The high surface area (2042.4 m2 g−1), accessible micro–meso pores, and a reasonable percentage of micropores in W1000 contribute to its excellent capacitance, as seen in Table 1.
Table 5 Experimental parameter ranges of various biomass vs. the present study.110
| Precursors (−) |
Pyrolysis temperature (°C) |
Specific surface area (m2 g−1) |
Porosity volume (cm3 g−1) |
| Lignin waste |
600–700 |
1820–2038 |
0.91–1 |
| Crab shell |
500–650 |
192–1387 |
0.09–0.5 |
| Peanut shell |
680–780 |
1713–1871 |
0.73–0.8 |
| Empty fruit bunch |
600–800 |
1080–1120 |
0.17–0.43 |
| Olive stone |
800 |
1063 |
0.49 |
| Bambo |
873–973 |
1840–1990 |
0.7–0.8 |
| Pollen |
800 |
937 |
0.4 |
| Popcorn |
800 |
867–1489 |
0.40–0.70 |
| Lotus leaf |
450–500 |
792–1924 |
0.29–0.79 |
| Vine shoots |
700–800 |
538–1439 |
0.23–0.67 |
| Chitosan |
800–1000 |
1389–2025 |
0.68–1.15 |
| Poplar wood |
600 |
511 |
0.22 |
| Waste wool |
600 |
1010–1420 |
0.57–0.86 |
| Chrysanthemum tea |
1000 |
1840 |
1.16 |
| Cork |
700 |
2010 |
0.82 |
| Coffee ground |
700–800 |
1476–1692 |
0.61–0.71 |
| Garlic peel |
600–800 |
967–1262 |
0.51–0.7 |
| Wheat |
700 |
1438 |
0.65 |
| Pineapple |
700 |
1076 |
0.08 |
| Coconut shell |
600 |
1172 |
0.43 |
| Paulownia sawdust |
600–900 |
782 |
0.41–1.181 |
| Walnut-shell |
500 |
811 |
0.34 |
| Peanut shell-C |
973 |
956 |
0.43 |
| Sunflower seed shell |
973 |
1790 |
0.77 |
| Catkin |
700–900 |
1005–1455 |
0.4–0.78 |
| Tar |
800 |
1829 |
1.2 |
| Sugarcane bagasse |
750 |
622 |
0.38 |
| Tea seed shell |
700 |
1064 |
0.47 |
| This work (walnut-shell) |
1000 |
2042.4 |
2.68 |
Table 6 compares the adsorption capacities of the porous carbon used in this study to a variety of other adsorbents previously used for O2/N2 adsorption. As shown, the W1000 has much higher adsorption capacities than many other previously reported adsorbents. The findings of this research can be used to develop a new porous carbon green synthesis adsorbent that is both effective and high performance for O2/N2 adsorption.
Table 6 Comparison of the O2/N2 adsorption capacities of the porous carbon derived from walnut-shell with other works
| Material (manufacture) |
Test method |
Surface area (m2 g−1) |
O2/N2 adsorption (mmol g−1) |
Pressure (bar) |
Temperature (K) |
Ref. |
| 5A/3A MSC (Takeda) |
VSM |
— |
0–1.76/0–1.75 |
0–8 |
273–323 |
111 |
| ACF (Kurare) |
ESR |
2000 |
0.181/0.064 |
0–0.02 |
298 |
107 |
| CMS 3A (Takeda) |
Volumetric |
— |
0.63/0.62 |
0–2.2 |
293 |
112 |
| 0.35/0.36 |
323 |
| 0.21/0.23 |
353 |
| CMS-T3A (Takeda) |
Volumetric |
522.59 |
2.2/1.2 |
0–5 |
293 |
113 |
| 1.8/0.85 |
303 |
| 1.7/0.75 |
313 |
| AC (AX-21); (coconut shells based using KOH impregnated) |
Volumetric |
3000 |
9/— |
2.5 |
118 |
98 |
| 2.5/— |
10 |
298 |
| 2.1/— |
10 |
313 |
| CMS 3A (Takeda) |
Gravimetric |
523 |
0.07/0.22 |
0.69/0.68 |
293 |
97 |
| 0.07/0.16 |
0.69/0.64 |
303 |
| 0.06/0.13 |
0.72/0.65 |
313 |
| CMB (palm shell based using PEI impregnated) |
Micrometrics |
1052 |
0.4/0.29 |
0–1 |
298 |
35 |
| AC (SorboNorit B3) |
Volumetric |
1012 |
1.75/— |
9.96 |
303 |
114 |
| 2.63/— |
19.02 |
| 3.39/— |
31.5 |
| AC |
Volumetric |
1306.4 |
1.519/— |
10.1 |
323 |
94 |
| CMS |
436.8 |
1.18/1.06 |
9.56 |
323 |
| AC |
1.86/1.24 |
10.26 |
308 |
| CMS |
1.39/— |
9.78 |
308 |
| AC |
2.23/— |
10.17 |
293 |
| CMS (Kuraray) |
1.66/1.45 |
10.1 |
293 |
| CMS GC-AP (air products and chemicals) |
Volumetric |
— |
0.88/0.78 |
7 |
298 |
99 |
| Porous carbon (walnut-shell based using self-activation method) |
Volumetric |
2042.4 |
2.4/1.7 |
6 |
298 |
This work |
| Volumetric |
2042.4 |
2.94/2.67 |
9.2 |
298 |
This work |
4.7 Regeneration performance of W1000 studies
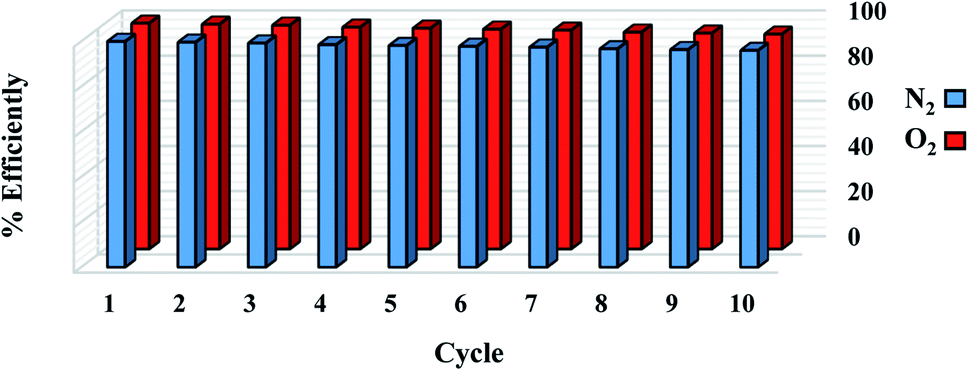
The reuse of sorbent for economic reasons is one of the most significant considerations. For 1 g sorbent, the regeneration process was measured at a pressure of less than 2 bar. For the O2/N2 adsorption operation, 10 adsorption/desorption cycles were reported at 298 °K at 6 bar and regenerated at 450 °K in a vacuum oven for 6 hours. As seen in Fig. 13, there is no significant change in adsorbent potential after each loop. After 10 cycles, the adsorption performance of the sorbent was reduced from 100% to 96%. According to the effects of the regeneration process, the sorbent can be used in industrial applications as a low-cost and cost-effective adsorbent. The steps involved in the processing and synthesizing the sorbent are depicted in Fig. 14.
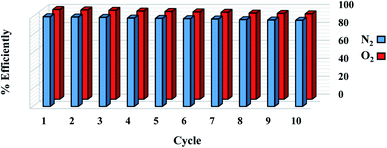 |
| | Fig. 13 Recycling performance of the W1000 for O2/N2 adsorption. | |
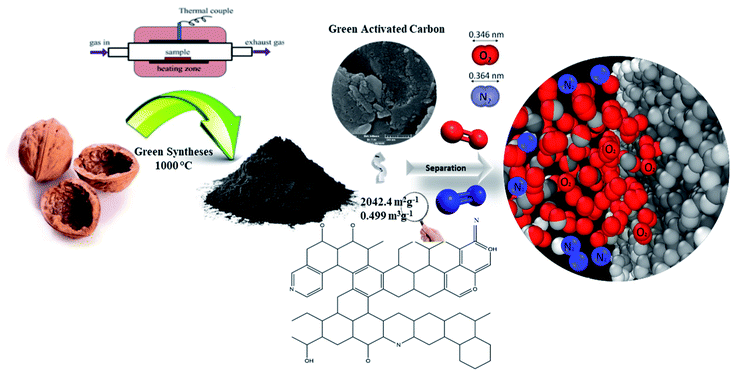 |
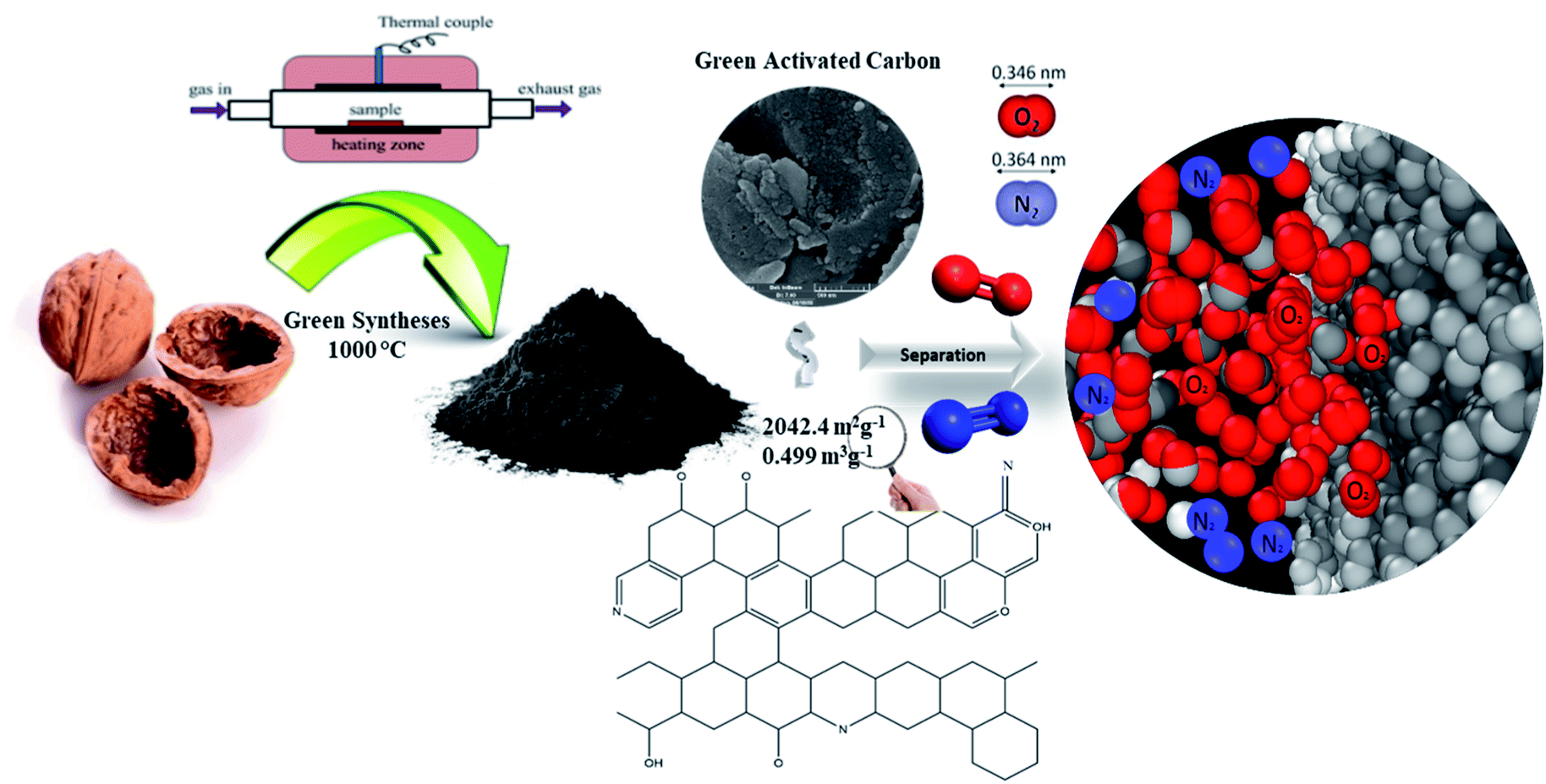
| | Fig. 14 Schematic representation of the porous carbon synthesis for O2/N2 adsorption. | |
5 Conclusions
In this research, a high-performance adsorbent for O2/N2 adsorption is developed using a new green synthesis of porous carbon adsorbent based on the green self-activation process. In summary, a mesopore-dominant porous carbon was synthesized with a high surface area via direct carbonization of walnut-shell. The optimal porous carbon possesses the specific surface area and the pore volume up to 2042 (m2 g−1) and 2.68 (cm3 g−1) at 1000 °C, respectively. During the characterization of the optimal sample, the open porosity with the formation of hierarchically interconnected porous, and turbostratic structures was identified. The mechanism of the self-activating system confirms the effective role of gases emitted from the carbon precursor (such as CO and CO2) in the activation of porous carbon. The green porous carbon preparation confirms an important perspective on the separation efficiency of O2 and N2 gases. Adsorption experiments in the temperature range of 298 °K to 328 °K under pressures up to 9.5 bar were conducted. The kinetics of O2/N2 adsorption on the sorbent has followed the fractional-order kinetic model well and thermodynamic analysis indicated that physisorption process is govern. The sips, fractional-order, and pseudo-second-order isotherm models were well equipped with adsorption isotherm and kinetic adsorption models, respectively, after comparing experimental adsorption effects by models. It was found that the factors such as the kinetic diameter of gases, the dipole/quadrupole moment, polarizability, and the abundance of micropores play a crucial role in better oxygen uptake than nitrogen. The maximal adsorption potential for O2/N2 was found to be 2.94 (mmol g−1) and 2.67 (mmol g−1) at 298 °K under 9.5 bar pressure, respectively. After ten cycles, the adsorption efficiency of the sorbent was reduced from 100% to 96%. Compared to the other carbon-based adsorbents studied, due to the high-performance efficiency and high-efficiency regeneration, the prepared sorbent can be promising for industrial air separation technologies based on the advanced adsorbents. In semi-scale and industrial processes, the new approach reflects a promising technique for preparing the green porous carbon from biomass wastes.
Nomenclature
| A | Temkin model constant (L mol−1) |
| B | First virial coefficient |
| C | Second virial coefficient |
| f | Subscripts refer to final condition |
| i | Subscripts refer to initial condition |
| kL | Langmuir model constant (bar−1) |
| kf | Rate constant of adsorption for the pseudo-first-order (min−1) |
| kF | Freundlich model constants [(mmol g−1) (bar−1)1/n)] |
| ks | Rate constant of adsorption for the first-order model (g mmol−1 min−1) |
| kn | Rate constant of adsorption for the fractional-order model (min−1) |
| Kd | Distribution coefficient |
| m | Mass of gas adsorbed (mg) |
| Mw | Molecular weight of gas (g mol−1) |
| P | Pressure (bar) |
| P0 | Saturated vapor pressure (bar) |
| Pe | Equilibrium pressure (bar) |
| q | Adsorption capacity (mmol g−1), (mg g−1) |
| qe | Equilibrium adsorption capacity (mmol g−1) |
| qm | Maximum adsorption (mmol g−1) |
| qt | Adsorption capacity at specified time t (mmol g−1) |
| R | Universal gas constant (8.314 J mol−1 K−1) |
| R2 | Correlation coefficient (%) |
| T | Temperature (°K), (°C) |
| t | Time (s) |
| w | Mass of adsorbent (g) |
| Z | Compressibility factor |
Greek symbols
| α | Parameter value of Elovich model (mmol g−1 min) |
| β | Parameter value of Elovich model (g mmol−1) |
| λ | Dubinin–Radushkevich (D–R) model constant (mol2 J−2) |
| ω | Polanyi potential (J mol−1) |
Acronyms
| AC | Activated carbon |
| ACF | Activated carbon fiber |
| CMS | Carbon molecular sieve |
| CMB | Carbon molecular basket |
| DTA | Differential thermal analysis |
| ESR | Electron spin resonance |
| FESEM | Field emission scanning electron microscopy |
| FTIR | Fourier transform infra-red |
| HRTEM | High-resolution transmission electron microscopy |
| MSC | Molecular sieve carbon |
| MOFs | Metal–organic frameworks |
| PEI | Poly ethylene imine |
| PSA | Pressure swing adsorption |
| PSD | Pore size distribution |
| TGA | Thermo gravimetric analysis |
| VSM | Vacancy solution model |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- D. Ferreira, P. Barcia, R. D. Whitley and A. l. Mendes, Ind. Eng. Chem. Res., 2015, 54, 9591–9604 CrossRef CAS.
- A. Marcinek, J. Guderian and D. Bathen, Adsorption, 2021, 1–16 Search PubMed.
- S. P. Reynolds, A. D. Ebner and J. A. Ritter, Ind. Eng. Chem. Res., 2006, 45, 3256–3264 CrossRef CAS.
- J. Santos, P. Cruz, T. Regala, F. Magalhaes and A. Mendes, Ind. Eng. Chem. Res., 2007, 46, 591–599 CrossRef CAS.
- M. L. Zanota, N. Heymans, F. Gilles, B. L. Su, M. Frère and G. De Weireld, J. Chem. Eng. Data, 2010, 55, 448–458 CrossRef CAS.
- T. Burdyny and H. Struchtrup, Energy, 2010, 35, 1884–1897 CrossRef CAS.
- N. Zhang and N. Lior, Energy, 2008, 33, 340–351 CrossRef CAS.
- G. Bello, R. García, R. Arriagada, A. Sepulveda-Escribano and F. Rodrıguez-Reinoso, Microporous Mesoporous Mater., 2002, 56, 139–145 CrossRef CAS.
- F. E. Epiepang, X. Yang, J. Li, Y. Liu and R. T. Yang, AIChE J., 2018, 64, 406–415 CrossRef CAS.
- M. J. Kirschner, A. Alekseev, S. Dowy, M. Grahl, L. Jansson, P. Keil, G. Lauermann, M. Meilinger, W. Schmehl and H. Weckler, Ullmann's encyclopedia of industrial chemistry, 2000, pp. 1–32 Search PubMed.
- X. Li, L. Zhang, Z. Yang, P. Wang, Y. Yan and J. Ran, Sep. Purif. Technol., 2020, 235, 116213 CrossRef CAS.
- D. M. Ruthven, Principles of adsorption and adsorption processes, John Wiley & Sons, 1984 Search PubMed.
- P. Li and F. H. Tezel, Microporous Mesoporous Mater., 2007, 98, 94–101 CrossRef CAS.
- Y. Fu, Y. Liu, X. Yang, Z. Li, L. Jiang, C. Zhang, H. Wang and R. T. Yang, Appl. Surf. Sci., 2019, 480, 868–875 CrossRef CAS.
- X. Yang, F. E. Epiepang, J. Li, Y. Wei, Y. Liu and R. T. Yang, Chem. Eng. J., 2019, 362, 482–486 CrossRef CAS.
- A. Jayaraman, R. T. Yang, S.-H. Cho, T. S. Bhat and V. N. Choudary, Adsorption, 2002, 8, 271–278 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- D. A. Reed, D. J. Xiao, M. I. Gonzalez, L. E. Darago, Z. R. Herm, F. Grandjean and J. R. Long, J. Am. Chem. Soc., 2016, 138, 5594–5602 CrossRef CAS PubMed.
- L. Melag, M. M. Sadiq, S. J. Smith, K. Konstas, K. Suzuki and M. R. Hill, J. Mater. Chem. A, 2019, 7, 3790–3796 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- H. Demir, S. J. Stoneburner, W. Jeong, D. Ray, X. Zhang, O. K. Farha, C. J. Cramer, J. I. Siepmann and L. Gagliardi, J. Phys. Chem. C, 2019, 123, 12935–12946 CrossRef CAS.
- Q. M. Wang, D. Shen, M. Bülow, M. L. Lau, S. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55, 217–230 CrossRef CAS.
- J. B. DeCoste, M. H. Weston, P. E. Fuller, T. M. Tovar, G. W. Peterson, M. D. LeVan and O. K. Farha, Angew. Chem., 2014, 126, 14316–14319 CrossRef.
- D. A. Reed, D. J. Xiao, H. Z. Jiang, K. Chakarawet, J. Oktawiec and J. R. Long, Chem. Sci., 2020, 11, 1698–1702 RSC.
- G. Arora and S. I. Sandler, J. Chem. Phys., 2006, 124, 084702 CrossRef.
- A. T. Nasrabadi and M. Foroutan, Comput. Mater. Sci., 2012, 61, 134–139 CrossRef CAS.
- F. Vallejos-Burgos, F.-X. Coudert and K. Kaneko, Nat. Commun., 2018, 9, 1–9 CrossRef CAS.
- E. D. Bloch, W. L. Queen, M. R. Hudson, J. A. Mason, D. J. Xiao, L. J. Murray, R. Flacau, C. M. Brown and J. R. Long, Angew. Chem., 2016, 128, 8747–8751 CrossRef.
- P. Giannozzi, R. Car and G. Scoles, J. Chem. Phys., 2003, 118, 1003–1006 CrossRef CAS.
- S. Chen, Z. Wang, Y. Xia, B. Zhang, H. Chen, G. Chen and S. Tang, RSC Adv., 2019, 9, 25480–25487 RSC.
- L. Zhu, D. Shen and K. H. Luo, J. Hazard. Mater., 2020, 389, 122102 CrossRef CAS PubMed.
- J. E. Sosa, C. Malheiro, R. P. Ribeiro, P. J. Castro, M. M. Piñeiro, J. M. Araújo, F. Plantier, J. P. Mota and A. B. Pereiro, J. Chem. Technol. Biotechnol., 2020, 95, 1892–1905 CrossRef CAS.
- P. González-García, Renewable Sustainable Energy Rev., 2018, 82, 1393–1414 CrossRef.
- J. Feng, R. Tang, X. Wang and T. Meng, ACS Appl. Energy Mater., 2021, 4, 5230–5236 CrossRef CAS.
- M. K. Aroua, W. M. A. W. Daud, C. Y. Yin and D. Adinata, Sep. Purif. Technol., 2008, 62, 609–613 CrossRef CAS.
- H. Sun, B. Yang and A. Li, Chem. Eng. J., 2019, 372, 65–73 CrossRef CAS.
- H. B. M. Emrooz, M. Maleki, A. Rashidi and M. Shokouhimehr, Biomass Convers. Biorefin., 2020, 1–12 Search PubMed.
- J. Maciá-Agulló, B. Moore, D. Cazorla-Amorós and A. Linares-Solano, Microporous Mesoporous Mater., 2007, 101, 397–405 CrossRef.
- M. Xu, D. Li, Y. Yan, T. Guo, H. Pang and H. Xue, RSC Adv., 2017, 7, 43780–43788 RSC.
- R. Acosta, V. Fierro, A. M. De Yuso, D. Nabarlatz and A. Celzard, Chemosphere, 2016, 149, 168–176 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- E. Pérez-Mayoral, I. Matos, M. Bernardo and I. M. Fonseca, Catalysts, 2019, 9, 133 CrossRef.
- J. He, D. Zhang, M. Han, X. Liu, Y. Wang, Y. Li, X. Zhang, K. Wang, H. Feng and Y. Wang, Journal of Energy Storage, 2019, 21, 94–104 CrossRef.
- F. Rodriguez-Reinoso and M. Molina-Sabio, Carbon, 1992, 30, 1111–1118 CrossRef CAS.
- R. I. Kosheleva, A. C. Mitropoulos and G. Z. Kyzas, Environ. Chem. Lett., 2019, 17, 429–438 CrossRef CAS.
- H. Wang, C. Sheng, T. Cai, C. Jin, Q. Sun and C. Wang, RSC Adv., 2018, 8, 12204–12210 RSC.
- J. Dai, S. Tian, Y. Jiang, Z. Chang, A. Xie, R. Zhang and Y. Yan, J. Alloys Compd., 2018, 732, 222–232 CrossRef CAS.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Small Methods, 2020, 4, 1900853 CrossRef CAS.
- G. Asimakopoulos, M. Baikousi, C. Salmas, A. B. Bourlinos, R. Zboril and M. A. Karakassides, J. Hazard. Mater., 2021, 405, 124274 CrossRef PubMed.
- J.-W. Kim, M.-H. Sohn, D.-S. Kim, S.-M. Sohn and Y.-S. Kwon, J. Hazard. Mater., 2001, 85, 301–315 CrossRef CAS PubMed.
- X.-L. Zhu, P.-Y. Wang, C. Peng, J. Yang and X.-B. Yan, Chin. Chem. Lett., 2014, 25, 929–932 CrossRef CAS.
- G. Gou, F. Huang, M. Jiang, J. Li and Z. Zhou, Renewable Energy, 2020, 149, 208–216 CrossRef CAS.
- S. Yu, N. Sun, L. Hu, L. Wang, Q. Zhu, Y. Guan and B. Xu, J. Power Sources, 2018, 405, 132–141 CrossRef CAS.
- G. Wang, D. Wang, S. Kuang, W. Xing and S. Zhuo, Renewable energy, 2014, 63, 708–714 CrossRef CAS.
- S. Sun, Q. Yu, M. Li, H. Zhao and C. Wu, Renewable energy, 2019, 142, 11–19 CrossRef CAS.
- S. Wang and G. M. Lu, Carbon, 1998, 36, 283–292 CrossRef CAS.
- J. Ou, Y. Zhang, L. Chen, Q. Zhao, Y. Meng, Y. Guo and D. Xiao, J. Mater. Chem. A, 2015, 3, 6534–6541 RSC.
- J. Niu, R. Shao, M. Liu, J. Liang, Z. Zhang, M. Dou, Y. Huang and F. Wang, Energy Storage Materials, 2018, 12, 145–152 CrossRef.
- G. Singh, K. S. Lakhi, S. Sil, S. V. Bhosale, I. Kim, K. Albahily and A. Vinu, Carbon, 2019, 148, 164–186 CrossRef CAS.
- C. Bommier, R. Xu, W. Wang, X. Wang, D. Wen, J. Lu and X. Ji, Nano Energy, 2015, 13, 709–717 CrossRef CAS.
- J. Hunt, A. Ferrari, A. Lita, M. Crosswhite, B. Ashley and A. Stiegman, J. Phys. Chem. C, 2013, 117, 26871–26880 CrossRef CAS.
- C. Jiang, G. A. Yakaboylu, T. Yumak, J. W. Zondlo, E. M. Sabolsky and J. Wang, Renewable Energy, 2020, 155, 38–52 CrossRef CAS.
- K. Sun, C.-y. Leng, J.-c. Jiang, Q. Bu, G.-f. Lin, X.-c. Lu and G.-z. Zhu, N. Carbon Mater., 2017, 32, 451–459 CrossRef CAS.
- J. Du, L. Liu, Z. Hu, Y. Yu, Y. Qin and A. Chen, Adv. Funct. Mater., 2018, 28, 1802332 CrossRef.
- L. Yin, M. Park, I. Jeon, J. H. Hwang, J. P. Kim, H. W. Lee, M. Park, S. Y. Jeong and C.-R. Cho, Electrochim. Acta, 2021, 368, 137630 CrossRef CAS.
- B. Kim, J. Park, S. Baik and J. W. Lee, J. Porous Mater., 2020, 27, 451–463 CrossRef CAS.
- X. Hu, X. Sun, Q. Song, Y. Zhu, Y. Long and Z. Dong, Green Chem., 2020, 22, 742–752 RSC.
- E. M. Mistar, T. Alfatah and M. D. Supardan, J. Mater. Res. Technol., 2020, 9, 6278–6286 CrossRef CAS.
- X. Ma, C. Su, B. Liu, Q. Wu, K. Zhou, Z. Zeng and L. Li, Sep. Purif. Technol., 2021, 259, 118065 CrossRef CAS.
- M. S. Ansari, T. N. J. I. Edison and Y. R. Lee, Sustainable Energy Fuels, 2020, 4, 5697–5708 RSC.
- P. J. Wibawa, M. Nur, M. Asy'ari and H. Nur, Heliyon, 2020, 6, e03546 CrossRef PubMed.
- M. Doğan, P. Sabaz, Z. Biçil, B. K. Kizilduman and Y. Turhan, J. Energy Inst., 2020, 93, 2176–2185 CrossRef.
- H. Xu, Y. Liu, H. Liang, C. Gao, J. Qin, L. You, R. Wang, J. Li and S. Yang, Sci. Total Environ., 2021, 759, 143457 CrossRef CAS PubMed.
- J. Yu, X. Li, Z. Cui, D. Chen, X. Pang, Q. Zhang, F. Shao, H. Dong, L. Yu and L. Dong, Renewable Energy, 2021, 163, 375–385 CrossRef CAS.
- C. Zhang, K. B. Hatzell, M. Boota, B. Dyatkin, M. Beidaghi, D. Long, W. Qiao, E. C. Kumbur and Y. Gogotsi, Carbon, 2014, 77, 155–164 CrossRef CAS.
- A. Ganesan and M. M. Shaijumon, Microporous Mesoporous Mater., 2016, 220, 21–27 CrossRef CAS.
- G. Singh, K. S. Lakhi, K. Ramadass, S. Kim, D. Stockdale and A. Vinu, Microporous Mesoporous Mater., 2018, 271, 23–32 CrossRef CAS.
- P. Hao, Z. Zhao, Y. Leng, J. Tian, Y. Sang, R. I. Boughton, C. Wong, H. Liu and B. Yang, Nano Energy, 2015, 15, 9–23 CrossRef CAS.
- F. Kapteijn, J. Moulijn, S. Matzner and H.-P. Boehm, Carbon, 1999, 37, 1143–1150 CrossRef CAS.
- B. Sun, Y. Yuan, H. Li, X. Li, C. Zhang, F. Guo, X. Liu, K. Wang and X. Zhao, Chem. Eng. J., 2019, 371, 55–63 CrossRef CAS.
- N. A. Rashidi, S. Yusup, M. M. Ahmad, N. M. Mohamed and B. H. Hameed, APCBEE Proc., 2012, 3, 84–92 CrossRef CAS.
- M. L. Martinez, M. M. Torres, C. A. Guzman and D. Maestri, Ind. Crops Prod., 2006, 23, 23–28 CrossRef CAS.
- C. Arnas, C. Dominique, P. Roubin, C. Martin, C. Laffon, P. Parent, C. Brosset and B. Pegourie, J. Nucl. Mater., 2005, 337, 69–73 CrossRef.
- A. Sahu, S. Sen and S. C. Mishra, Diamond Relat. Mater., 2020, 108, 107931 CrossRef CAS.
- A. Wang, K. Sun, R. Xu, Y. Sun and J. Jiang, J. Cleaner Prod., 2021, 283, 125385 CrossRef CAS.
- T. Liang, C. Chen, X. Li and J. Zhang, Langmuir, 2016, 32, 8042–8049 CrossRef CAS PubMed.
- H. B. M. Emrooz, M. Maleki and A. Rahmani, J. Taiwan Inst. Chem. Eng., 2018, 91, 281–290 CrossRef.
- J. Deng, T. Xiong, F. Xu, M. Li, C. Han, Y. Gong, H. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC.
- J. Hao, Y. Wang, C. Chi, J. Wang, Q. Guo, Y. Yang, Y. Li, X. Liu and J. Zhao, Sustainable Energy Fuels, 2018, 2, 2358–2365 RSC.
- J. Liu, L. Zhang, C. Yang and S. Tao, J. Mater. Chem. A, 2019, 7, 21168–21175 RSC.
- D. B. Altuntaş, V. Nevruzoğlu, M. Dokumacı and Ş. Cam, Carbon Letters, 2020, 30, 307–313 CrossRef.
- M. O. Prakash, G. Raghavendra, S. Ojha and M. Panchal, Mater. Today: Proc., 2021, 39, 1476–1481 Search PubMed.
- C. Wang, H. Wang, B. Dang, Z. Wang, X. Shen, C. Li and Q. Sun, Renewable Energy, 2020, 156, 370–376 CrossRef CAS.
- D. Park, Y. Ju, J.-H. Kim, H. Ahn and C.-H. Lee, Sep. Purif. Technol., 2019, 223, 63–80 CrossRef CAS.
- V. M. Matsis and H. P. Grigoropoulou, Chem. Eng. Sci., 2008, 63, 609–621 CrossRef CAS.
- A. Ansón, S. M. Kuznicki, T. Kuznicki, T. Haastrup, Y. Wang, C. C. Lin, J. A. Sawada, E. M. Eyring and D. Hunter, Microporous Mesoporous Mater., 2008, 109, 577–580 CrossRef.
- Y.-J. Park, S.-J. Lee, J.-H. Moon, D.-K. Choi and C.-H. Lee, J. Chem. Eng. Data, 2006, 51, 1001–1008 CrossRef CAS.
- Y. Zhou, L. Wei, J. Yang, Y. Sun and L. Zhou, J. Chem. Eng. Data, 2005, 50, 1068–1072 CrossRef CAS.
- M. Andrade, S. C. Rodrigues and A. Mendes, Microporous Mesoporous Mater., 2020, 296, 109989 CrossRef CAS.
- J. V. Nabais, P. Carrott, M. R. Carrott, A. Padre-Eterno, J. Menéndez, A. Dominguez and A. Ortiz, Carbon, 2006, 44, 1158–1165 CrossRef CAS.
- R. R. Pawar, H. A. Patel, G. Sethia and H. C. Bajaj, Appl. Clay Sci., 2009, 46, 109–113 CrossRef CAS.
- D. Saha and M. J. Kienbaum, Microporous Mesoporous Mater., 2019, 287, 29–55 CrossRef CAS.
- B. H. Kim, B. R. Kim and Y. G. Seo, Adsorption, 2005, 11, 207–212 CrossRef.
- J. J. Manyà, B. González, M. Azuara and G. Arner, Chem. Eng. J., 2018, 345, 631–639 CrossRef.
- C. Reid, I. O'koy and K. Thomas, Langmuir, 1998, 14, 2415–2425 CrossRef CAS.
- P. Ehrburger, O. Mahajan and P. Walker Jr, J. Colloid Interface Sci., 1976, 57, 233–239 CrossRef CAS.
- N. Kobayashi, T. Enoki, C. Ishii, K. Kaneko and M. Endo, J. Chem. Phys., 1998, 109, 1983–1990 CrossRef CAS.
- S. M. Wilson and F. H. Tezel, Sep. Purif. Technol., 2021, 258, 117659 CrossRef CAS.
- F. Raganati, M. Alfe, V. Gargiulo, R. Chirone and P. Ammendola, Chem. Eng. Res. Des., 2018, 134, 540–552 CrossRef CAS.
- H. Mashhadimoslem, M. Vafaeinia, M. Safarzadeh, A. Ghaemi, F. Fathalian and A. Maleki, Ind. Eng. Chem. Res., 2021, 60, 13950–13966 CrossRef CAS.
- H. M. Yi, S. Weiruo, B. Maruti, W. Jinqu and G. W. Miller, Sep. Technol., 1991, 1, 90–98 CrossRef.
- S. Rutherford and J. Coons, J. Colloid Interface Sci., 2005, 284, 432–439 CrossRef CAS PubMed.
- Y.-S. Bae and C.-H. Lee, Carbon, 2005, 43, 95–107 CrossRef CAS.
- R. Bazan, M. Bastos-Neto, A. Moeller, F. Dreisbach and R. Staudt, Adsorption, 2011, 17, 371–383 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Mobin Safarzadehb,
Ahad Ghaemi
a,
Mobin Safarzadehb,
Ahad Ghaemi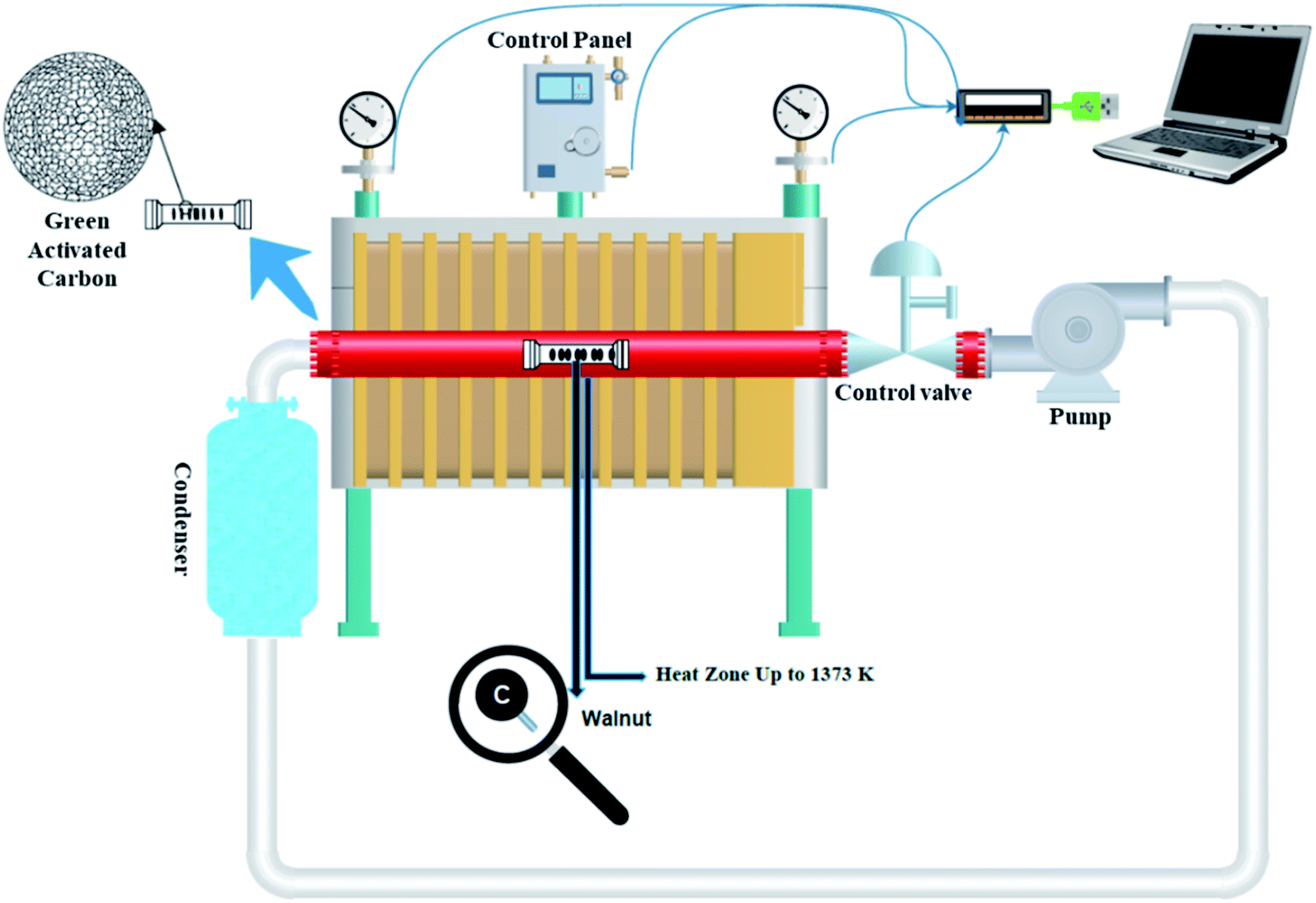

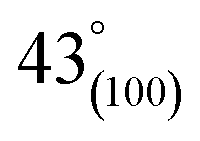 (turbostratic structure) indicating the presence of amorphous graphitic carbon and lower ordering. Besides, the increase in pores at high temperatures due to the higher orientation freedom of the graphite crystallites has led to a decrease in the severity of reflection and expansion at peaks. The absence of a peak at
(turbostratic structure) indicating the presence of amorphous graphitic carbon and lower ordering. Besides, the increase in pores at high temperatures due to the higher orientation freedom of the graphite crystallites has led to a decrease in the severity of reflection and expansion at peaks. The absence of a peak at 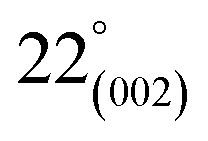 (oriented aromatic carbon) is a sign of the structure's strong tendency to formation of highly disordered and randomly oriented graphitic carbon layers that generate higher specific surface areas, consistent with the observation of Raman and BJH's results. Raman results likewise revealed a more disordered structure, while XRD correlated to lower graphitization degrees.51,80,91–93
(oriented aromatic carbon) is a sign of the structure's strong tendency to formation of highly disordered and randomly oriented graphitic carbon layers that generate higher specific surface areas, consistent with the observation of Raman and BJH's results. Raman results likewise revealed a more disordered structure, while XRD correlated to lower graphitization degrees.51,80,91–93