
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rotaxane PtII-complexes: mechanical bonding for chemically robust luminophores and stimuli responsive behaviour†
Zhihui
Zhang
a,
Graham J.
Tizzard
a,
J. A. Gareth
Williams
 *b and
Stephen M.
Goldup
*a
*b and
Stephen M.
Goldup
*a
aChemistry, University of Southampton, Southampton, SO51 5PG, UK. E-mail: s.goldup@soton.ac.uk
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
First published on 2nd January 2020
Abstract
We report an approach to rotaxanes in which the metal ion of a cyclometallated PtII luminophore is embedded in the space created by the mechanical bond. Our results show that the interlocked ligand environment stabilises a normally labile PtII–triazole bond against displacement by competing ligands and that the crowded environment of the mechanical bond retards oxidation of the PtII centre, without perturbing the photophysical properties of the complex. When an additional pyridyl binding site is included in the axle, the luminescence of the PtII centre is quenched, an effect that can be selectively reversed by the binding of AgI. Our results suggest that readily available interlocked metal-based phosphors can be designed to be stimuli responsive and have advantages as stabilised triplet harvesting dopants for device applications.
Introduction
Many mechanically chelated1 metal complexes, in which a metal ion bridges mechanically bonded covalent sub-components,2 have been synthesised since Sauvage's first report of the CuI templated synthesis of catenanes.3 These complexes are typically reported as intermediates in the synthesis of interlocked molecules,4 as opposed to being objects of study in their own right.5,6 This is despite the potential benefits that embedding the metal ion within the crowded environment of the mechanical bond might bring in terms of the kinetic stabilisation and electronic, magnetic and catalytic properties. Indeed, Sauvage and co-workers highlighted the kinetic stabilisation of a bound CuI ion due to a “catenand effect”.3,7 Recently, we demonstrated that the mechanical bond allows access to complexes of which the non-interlocked equivalents are inaccessible,8 including examples with highly distorted coordination geometries reminiscent of the entatic state of enzyme active sites.9,10 These reports suggest that interlocked metal complexes in which the mechanical bond alters the stability or properties of the ion are an untapped resource in the development of coordination complexes for a range of applications, including catalysis, metallo-pharmaceuticals and light harvesting.Metal-based luminophores have been at the forefront of many recent developments in light-emitting organic electronic materials for various applications.10,11 Iridium(III)-based phosphors are now widely incorporated into commercial organic light-emitting devices (OLEDs) to allow the harvesting of triplet excitons that would otherwise be wasted.12 The strong spin–orbit coupling associated with the heavy metal ion promotes the T1 → S0 phosphorescence process, which is strongly forbidden in purely organic materials.13 Complexes of other 2nd and 3rd row metal ions are also widely investigated in this context, particularly those of platinum(II).14
To date, only a limited number of mechanically chelated heavy-metal complexes have been reported. This is perhaps not surprising as most metal-directed passive template syntheses rely on early transition metals,5 due to their more favourable ligand exchange kinetics. Furthermore, the small number of passive templates based on PdII,15 RuII,16 and AuI (ref.17) reported are not luminescent.18,19 Thus, the limited examples of interlocked molecules containing metal-based luminophores incorporate the metal complex as a substituent of the interlocked scaffold, or as a structural unit. In 2012, Terao and co-workers demonstrated that emissive PtII–acetylide units embedded in the axle of a rotaxane were insulated from the local environment, leading to no change in emission between solution and solid state.20 More recently, Ma and co-workers demonstrated that the emission of a PtII–porphyrin complex appended to a rotaxane could be modulated by mechanical motion.21
Here we describe the synthesis and properties of rotaxane-based PtII complexes in which the metal ion is embedded within the mechanical bond. We show that the mechanical bond does not significantly perturb the photophysical properties of the metal ion but, pleasingly, the chemical stability of the complexes is greatly enhanced relative to non-interlocked analogues. Furthermore, while studying this stabilising influence, we serendipitously identified a metallorotaxane in which the phosphorescence shows a dramatic and selective enhancement in the presence of AgI, through the inhibition of a quenching pathway arising from a pyridyl unit incorporated in the structure.
Results and discussion
Synthesis of mechanically chelated PtII complexes
Our study was in part inspired by a cyclometallated catenane-based PdII complex reported in early work by Sauvage and co-workers.22 Rotaxane 1.H (Scheme 1) was synthesised in excellent yield from readily available starting materials23 using our small macrocycle modification24 of the active template25 Cu-mediated alkyne–azide cycloaddition reaction (AT–CuAAC)26 (see ESI† for details). Treatment of 1.H with PtCl2(DMSO)2 under mild conditions failed to give the target cyclometallated complex [Pt(1)]+ directly, but led instead to trans-Pt(1.H)(DMSO)Cl2, in which the Pt centre is coordinated only by the triazole of the rotaxane axle (Scheme 1a). Subsequent heating of Pt(1.H)(DMSO)Cl2 in AcOH led to [Pt(1)]BF4 in good yield after purification.27 Using these more forcing conditions, it was also possible to obtain this complex directly from 1.H and K2PtCl4 in reasonable yield. The formation of the corresponding non-interlocked complex [Pt(2)(3)]BF4 required a two-step procedure involving initial formation of Pt(2)Cl followed by AgI-mediated substitution of the chloride ligand by the triazole (Scheme 1b).28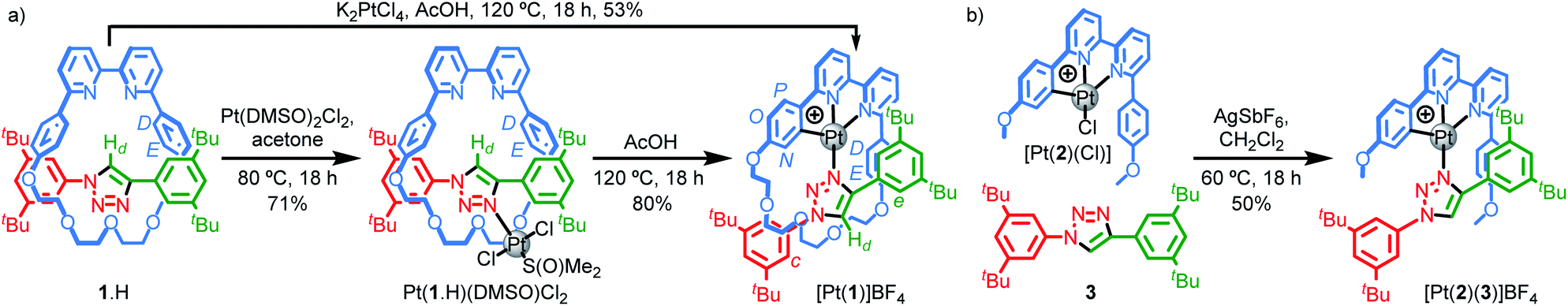 | ||
| Scheme 1 (a) Synthesis of complexes [Pt(1)]BF4. (b) Synthesis of complex [Pt(2)(3)]BF4. | ||
All novel compounds and intermediates were characterised by NMR spectroscopy and MS (see ESI†). The 1H NMR spectrum of [Pt(1)]BF4 clearly shows the expected desymmetrisation of the metallated aromatic ring; individual signals are observed for HN, HO and HP, the first of which is significantly shielded (5.03 ppm). Perhaps counterintuitively, coordination of Pt to the triazole N leads to an upfield shift of Hd as metal coordination interrupts a non-classical H-bonding interaction with the bipyridine Ns. Unusually, whereas HD and HE each appear as 4H doublets in the 1H NMR spectra of 1.H and Pt(1.H)(DMSO)Cl2, indicating free rotation of the flanking aromatic ring, HD and HE of [Pt(1)]BF4 each appear as two 1H doublets suggesting this motion is slow on the 1H NMR timescale, presumably because of the crowded nature of the macrocycle cavity with the large PtII ion included.
Pt(1.H)(DMSO)Cl2 (Table S1†) and [Pt(1)]BF4 (Fig. 1) were further characterised in the solid-state by single-crystal X-ray diffraction (SCXRD). The solid-state structure of [Pt(1)]BF4 clearly demonstrates the mechanically chelating environment of the metal ion which bridges N3 of the triazole axle and the N^N^C chelate provided by the macrocycle. As is typically observed with small, sterically crowded interlocked molecules,24 several short intercomponent contacts are present in addition to metal–ligand interactions (Fig. 1a), including a CH⋯Pt contact. Viewing the solid-state structure in space-filling representation (Fig. 1b) underlines the sterically crowded nature of the mechanical bond; the metal ion is embedded in the cavity of the ring and largely isolated from the local environment.
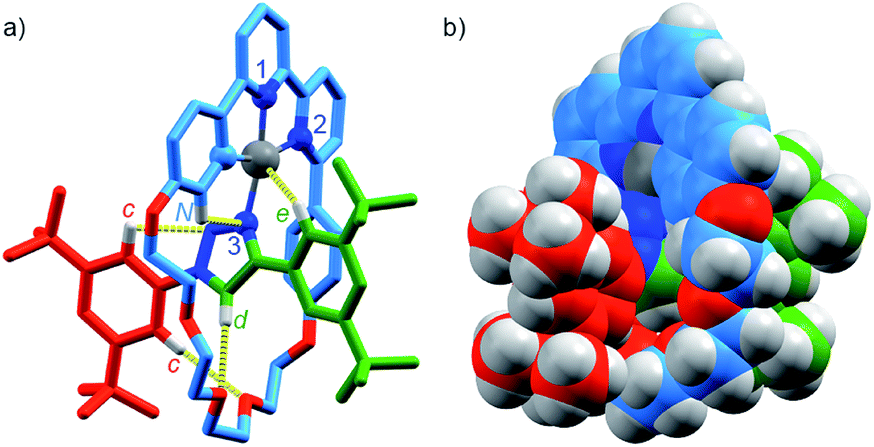 | ||
| Fig. 1 Solid-state structure of rotaxane [Pt(1)]BF4 displayed in (a) partial sticks (rotaxane framework)/ball-and-stick (metal and ligand sphere) representation (anions and majority of hydrogen atoms omitted for clarity) and (b) space-filling representation (anions omitted for clarity). Colours: H white, C as in Scheme 1, N dark blue, O red, Pt dark grey. Selected bond lengths (Å) and angles (°): Pt–C 1.99, Pt–N1 1.97, Pt1–N2 2.17, Pt1–N3 2.05, Hc–N 2.49, Hc–O 2.59, Hd–O 2.53, He–Pt 2.74, HN–N 2.56, C–Pt–N2 160.4, N1–Pt–N3 171.7, C–Pt–N1 81.9, N1–Pt–N2 78.5, N2–Pt–N3 108.4, N3–Pt–C 90.8. | ||
Photophysical properties of metallorotaxane [Pt(1)]BF4 and [Pt(2)(3)]BF4
The photophysical properties of the interlocked PtII complex [Pt(1)]BF4 and its non-interlocked analogue, [Pt(2)(3)]BF4, are remarkably similar (Table 1 and Fig. 2), demonstrating that the mechanical bond does not significantly perturb the pertinent electronic excited states. Both complexes display UV-visible absorption properties typical of cyclometallated PtII complexes of arylpyridine ligands,29 with intense intra-ligand bands in the far-UV and a somewhat less intense band at lower energy (350–380 nm). Bands in the latter region are typically associated with charge–transfer transitions of mixed orbital parentage, that are introduced upon cyclometallation. Notably, there is no such band for the non-cyclometallated Pt(1.H)(DMSO)Cl2 complex (Fig. S59†).
orbital parentage, that are introduced upon cyclometallation. Notably, there is no such band for the non-cyclometallated Pt(1.H)(DMSO)Cl2 complex (Fig. S59†).
| [Pt(1)]BF4 | [Pt(2)(3)]BF4 | |
|---|---|---|
| a Quantum yield in deoxygenated solution measured using [Ru(bpy)3]Cl2(aq) as the standard. b Luminescence lifetimes in deoxygenated solution; values in parenthesis refer to air-equilibrated solution. c Radiative kr and non-radiative Σknr rate constants estimated using the approximation that the emissive state is formed with unitary efficiency and thus kr = Φ/τ and Σknr = (1 − Φ)/τ.32 | ||
| 298 K in deoxygenated CH 2 Cl 2 | ||
| λ abs max/nm (ε/M−1 cm−1) | 264 (58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900), 358 (16400), 441 sh (946) 900), 358 (16400), 441 sh (946) |
262 (48700), 356 (14700), 448 sh (1120) |
| λ em max/nm | 575 | 582 |
| Φ lum × 102a | 2.7 | 1.9 |
| τ/μs | 2.1 [0.46]b | 2.2 [0.39]b |
| k r/103 s−1c | 13 | 8.6 |
| Σknr/105 s−1c | 4.6 | 4.5 |
|
||
77 K in frozen EPA (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2:1 v/v Et2O–iC5H12–EtOH) :2:1 v/v Et2O–iC5H12–EtOH) |
||
| λ em max/nm | 536, 576, 623 | 521, 559, 603 |
| τ/μs | 15.9 | 19 |
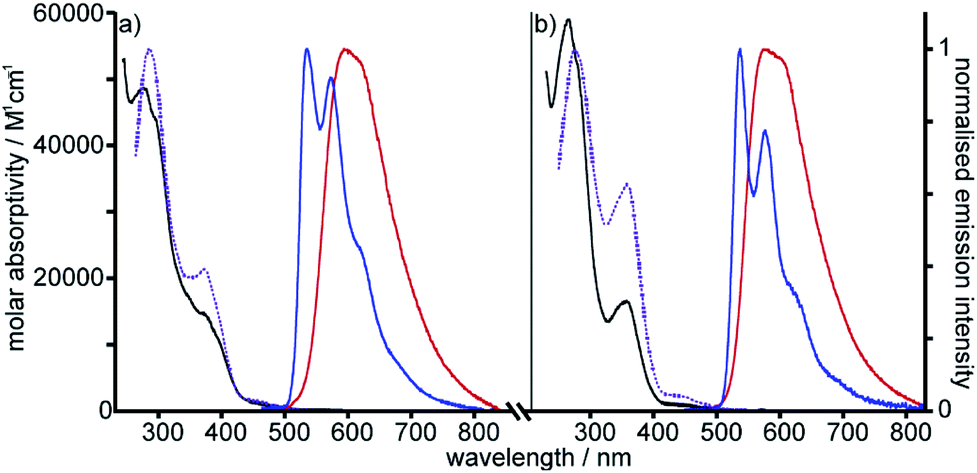 | ||
| Fig. 2 Absorption and excitation spectra in CH2Cl2 at 298 K (black and dotted purple lines respectively), and emission spectra in CH2Cl2 at 298 K and in EPA at 77 K (red and blue lines respectively) for (a) Pt[(1)]BF4 and (b) [Pt(2)(3)]BF4. (EPA is defined in Table 1). | ||
Excitation of both cyclometallated complexes at 360 nm in degassed solution leads to an emission band centred around 575 nm. The corresponding luminescence lifetimes of around 2 μs in deoxygenated solution are indicative of a formally spin-forbidden phosphorescence process, promoted by the spin–orbit coupling associated with the metal ion. Such phosphorescence is typical of Pt(II) complexes of 6-phenylbipyridine-based ligands, of the form Pt(N^N^C)X or [Pt(N^N^C)L]+, and assigned to a 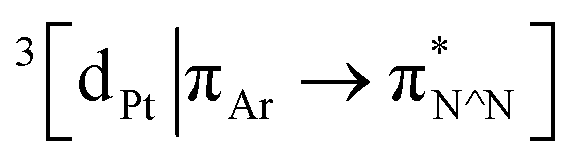 state.30 The measured quantum yields of around 2–3% under these conditions are also quite typical of such complexes. The phosphorescence is modestly quenched by dissolved molecular oxygen, as evident from the shorter lifetimes observed in air-equilibrated solution. At 77 K, vibrational structure becomes clearly resolved, with a progression of around 1300 cm−1, quite typical of complexes featuring aromatic ligands. The complexes are also luminescent in the solid state, with similar quantum yields and lifetimes (Table S8†).31
state.30 The measured quantum yields of around 2–3% under these conditions are also quite typical of such complexes. The phosphorescence is modestly quenched by dissolved molecular oxygen, as evident from the shorter lifetimes observed in air-equilibrated solution. At 77 K, vibrational structure becomes clearly resolved, with a progression of around 1300 cm−1, quite typical of complexes featuring aromatic ligands. The complexes are also luminescent in the solid state, with similar quantum yields and lifetimes (Table S8†).31
Chemical stability of interlocked PtII complexes
Having confirmed that metallorotaxane [Pt(1)]BF4 retains the emissive properties typical of Pt(N^N^C) complexes, we turned to the effect of the mechanical bond on the stability of the metal complex. Triazole ligands are only weakly coordinating33 and indeed, treatment of the non-interlocked complex [Pt(2)(3)]BF4 with NBu4Cl led to decomposition to produce [Pt(2)Cl] and axle 3 through exchange of the monodentate ligand (Scheme 2). In contrast, the interlocked analogue [Pt(1)]BF4 is stable over at least 5 days under the same conditions. Similarly, treatment of [Pt(2)(3)]BF4 with tBuO2H led to rapid decomposition to a complex mixture of products (Scheme 2). The same experiment with [Pt(1)]BF4 led to a slow reaction to give an oxidised PtIV complex as the major product, which was identified by ESI-MS as [Pt(1)Cl2]BF4. The Cl ligands in this compound are presumably provided through oxidative decomposition of CH2Cl2 over the extended reaction time (Scheme 2).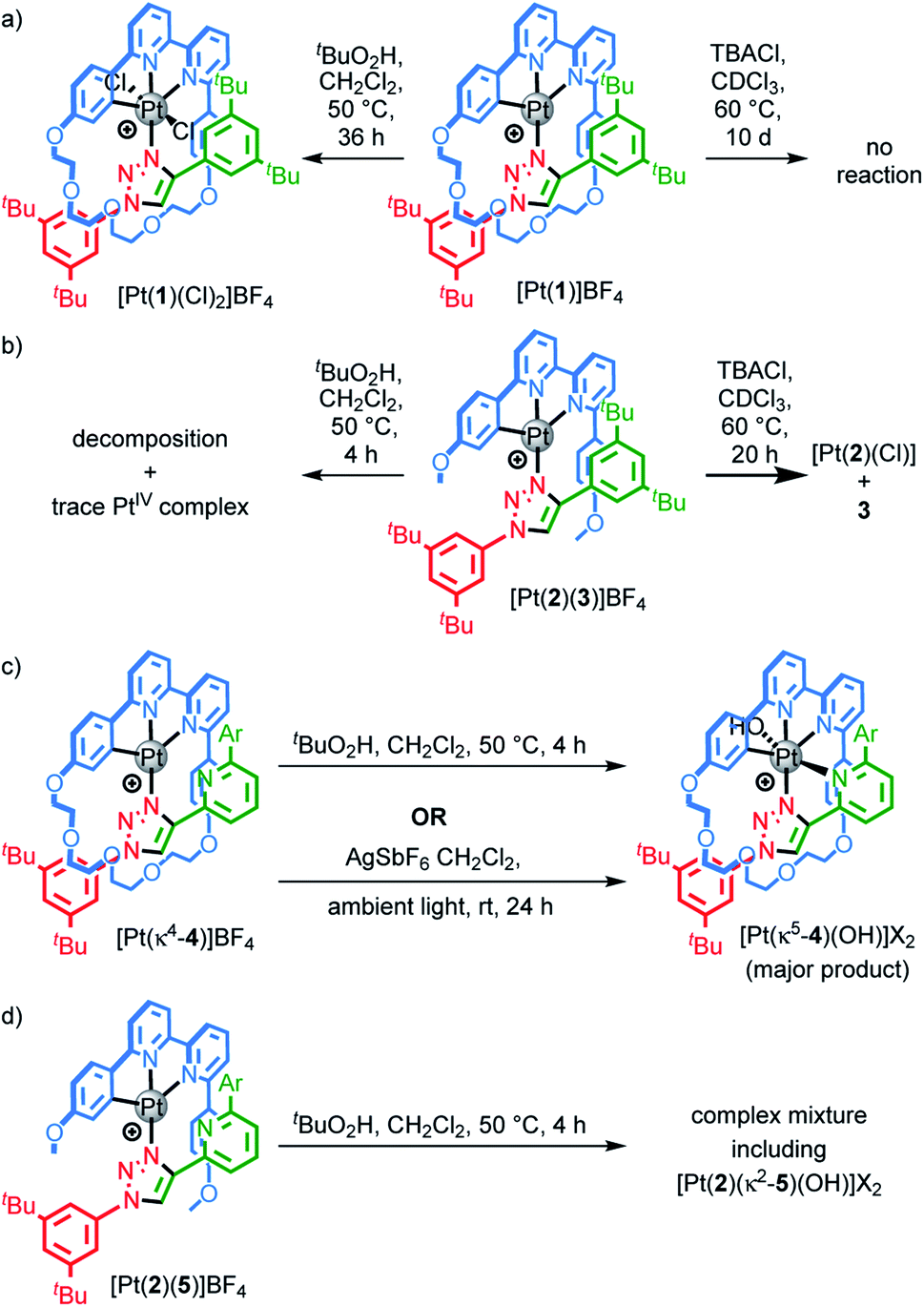 | ||
| Scheme 2 Reactions of (a) [Pt(1)]BF4, (b) [Pt(2)(3)]BF4, (c) [Pt(κ4-4)]BF4 and (d) [Pt(2)(5)]BF4. Ar = 3,5-di-tBu-C6H3. | ||
These results suggest that the mechanical bond significantly retards displacement of the weakly coordinating triazole ligand, presumably due to steric repulsion of the incoming Cl nucleophile and destabilisation of the expected 5-coordinate intermediate of the presumed associative pathway. The same steric hindrance would also thermodynamically destabilise the product of the reaction with [Pt(1)]BF4 as the macrocycle cannot readily accommodate both a Pt–Cl moiety and the uncoordinated axle. The mechanical bond also appears to kinetically disfavour oxidation of the PtII centre, presumably for similar reasons, as the change from PtII to PtIV requires the inclusion of two additional ligands within the metal primary coordination sphere.
To examine if the latter effect could be overcome by providing an additional donor atom in the rotaxane framework, we synthesised complex [Pt(κ4-4)]BF4 (see ESI†) and examined its stability to oxidation. As predicted, when [PtII(κ4-4)]BF4 was treated with tBuO2H, 1H NMR analysis (Fig. S55†) revealed that a rapid reaction (<2 h) took place to give a mixture in which the species assigned as [PtIV(κ5-4)(OH)](BF4)2 (m/z = 616.3 corresponding to M2+) was the major product. Replacing tBuO2H with H2O2 led to a cleaner reaction (Fig. S56†) in which the species assigned as [PtIV(κ5-4)(OH)](BF4)2 was the only significant product. This assignment was supported by SCXRD analysis of crystals grown from the crude product mixture which, although of low quality, confirmed the expected connectivity (Table S5†).
Serendipitously (vide infra), when [Pt(κ4-4)]BF4 was treated with AgSbF6 without protection from light (Scheme 2c), higher quality crystals were produced and found to correspond to [Pt(κ5-4)(OH)](SbF6)2 in which the same coordination environment was also observed (Fig. 3b). The structures of [Pt(κ4-4)]BF4 and [Pt(κ5-4)(OH)](SbF6)2 (Fig. 3) clearly demonstrate the change from a square-planar PtII unit to a pseudo-octahedral PtIV complex in which the rotaxane framework provides five of the six donor atoms required to complete the PtIV coordination sphere (three from the macrocycle and now two from the axle).
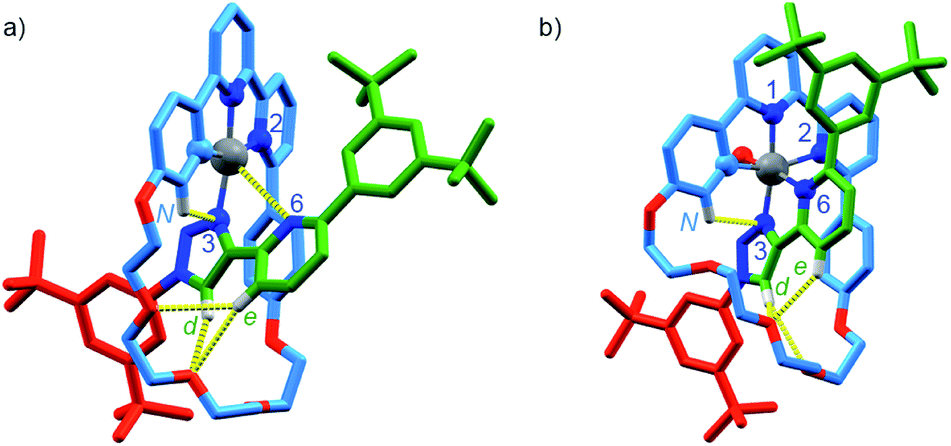 | ||
| Fig. 3 Solid-state structures of (a) [Pt(κ4-4)]BF4 and (b) [Pt(κ5-4)(OH)](SbF6)2 in partial sticks (rotaxane framework)/ball-and-stick (metals and ligand sphere) representation (anions and majority of hydrogen atoms omitted for clarity). Colours: H white, C as in Scheme 2, N dark blue, O red, Pt dark grey. [Pt(κ4-4)]BF4 selected distances (Å) and angles (°): Pt–C 1.97, Pt–N1 1.95, Pt–N2 2.17, Pt–N3 2.03, Pt–N6 3.39, Hd–O 2.25, He–O 2.79, He–O 2.80, HN–N 2.55, C–Pt–N2 160.7, N1–Pt–N3 172.0, C–Pt–N1 82.1, N1–Pt–N2 78.8, N2–Pt–N3 107.3, N3–Pt–C 91.6. [Pt(κ4-4)(OH)](SbF6)2 selected distances (Å) and angles (°): Pt–C 2.01, Pt–N1 1.99, Pt–N2 2.22, Pt–N3 2.01, Pt–N6 2.13, Pt–O 1.97, Hd–O 2.49, Hd–O 2.76, He–O 2.55, HN–N3 2.54, C–Pt–N2 160.3, N1–Pt–N3 172.6, N6–Pt–O 171.6, C–Pt–N1 82.4, N1–Pt–N2 77.9, N2–Pt–N3 108.2, N3–Pt–C 91.4. | ||
The corresponding non-interlocked complex, [Pt(2)(5)]BF4, also reacted rapidly with tBuO2H to give a mixture of products (Fig. S57†), including uncoordinated axle 5, and a species that was assigned as PtIV complex by ESI-MS analysis (m/z = 551.2 corresponding to M2+). Replacing tBuO2H with H2O2 simplified the product mixture at short reaction times (Fig. S58†). However, whereas [PtIV(κ5-4)(OH)](BF4)2 proved stable over extended reaction times, treatment of [Pt(2)(5)]BF4 with H2O2 for 24 h led to decomposition to produce significant quantities of non-interlocked axle (Fig. S58†), suggesting that even in the PtIV oxidation state, the Pt–triazole bond remains labile.
Photophysical properties of [PtII(κ4-4)]BF4 – response to AgI
The pyridine appended rotaxane complex [Pt(κ4-4)]BF4 displays a similar phosphorescence spectrum to that of [Pt(1)]BF4. However, its emission quantum yield in solution is dramatically reduced by around 20-fold, and the phosphorescence lifetime is an order of magnitude shorter (Table 2vs.Table 1). A similar difference was observed between the corresponding non-interlocked complexes [Pt(2)(5)]BF4 and [Pt(2)(3)]BF4. Estimation of the radiative and non-radiative rate constants, kr and Σknr respectively, from the quantum yields and lifetimes suggests that the effect is due primarily to the introduction of an additional non-radiative decay pathway that increases knr by an order of magnitude when the additional pyridine ring is present. Interestingly, the quenching effect is apparently eliminated in the solid state (Table S9†), under which conditions the quantum yields and lifetimes of all of the complexes are similar to one another, suggesting that the quenching process in the pyridine-functionalised structures requires some degree of intramolecular reorganisation that is inhibited in the rigid environment of a solid matrix.34| [Pt(4)]BF4 | [Pt(2)(5)]BF4 | |
|---|---|---|
| a Quantum yield in deoxygenated solution measured using [Ru(bpy)3]Cl2(aq) as the standard. b Luminescence lifetimes in deoxygenated solution; values in parenthesis refer to air-equilibrated solution. c Radiative kr and non-radiative Σknr rate constants estimated using the approximation that the emissive state is formed with unitary efficiency and thus kr = Φ/τ and Σknr = (1 − Φ)/τ. | ||
| 298 K in deoxygenated CH 2 Cl 2 | ||
| λ abs max/nm (ε/M−1 cm−1) | 268 (48500), 359 (12900), 445 sh (698) |
266 (47000), 352 (12900), 443 (1120) |
| λ em max/nm | 591 | 595 |
| Φ lum × 102a | 0.12 | 0.22 |
| τ/μsb | 0.22 [0.18] | 0.10 [0.07] |
| k r/103 s−1c | 5.5 | 22 |
| Σknr/105 s−1c | 45 | 99 |
|
||
| 298 K in deoxygenated CH 2 Cl 2 with Ag I (3 equiv.) | ||
| λ em max/nm | 581 | 578 |
| Φ lum × 102a | 10 | 6.0 |
| τ/μsb | 4.3 [0.95] | 5.4 [1.0] |
| k r/103 s−1c | 23 | 11 |
| Σknr/105 s−1c | 2.1 | 1.7 |
|
||
|
77 K in frozen EPA (2:2:1 v/v Et2O–iC5H12–EtOH) |
||
| λ em max/nm | 534, 574, 622 | 529, 555 |
| τ/μs | 19 | 18 |
Examining the solid state structure of [Pt(κ4-4)]BF4, a striking feature is that the lone pair of the axle pyridine moiety is projected near to the Pt centre (3.39 Å) in the ground state. Thus, one explanation for the quenching of the emission is through exciplex formation, where a conformational rearrangement takes place to bring the pyridine lone pair closer to the PtII centre, which becomes more electrophilic in the 3MLCT excited state. Indeed, it has been noted that PtII complexes with “dangling” nucleophilic groups are often non-emissive (or only weakly so), ostensibly due to such axial interactions, whilst similar reasoning has been invoked to explain the frequently lower quantum yields of PtII complexes in solvents incorporating potential donor atoms like DMF.35 McMillin and co-workers have highlighted the correlation between the Gutmann donor number36 and the rate constant for intermolecular quenching of the luminescence of PtII complexes by donors such as pyridine.37
To probe the role of the axle pyridine in [Pt(κ4-4)]BF4, we attempted to protonate it, in order to determine if its elimination as a potential donor group would enhance the luminescence. However, no change was observed by 1H NMR spectroscopy (Fig. S61†) when [Pt(κ4-4)]BF4 was treated with trifluoroacetic acid. The apparent lack of basic behaviour may be due to charge–charge repulsion between the formally cationic PtII centre and the protonated pyridine moiety that destablises the protonated state. We therefore examined instead the binding of the soft, monovalent cations CuI, AgI, and AuI in the hope that this charge–charge repulsion might be compensated for by attractive metal–metal interactions. Pleasingly, when [Pt(κ4-4)]BF4 was titrated with AgSbF6 or [Cu(MeCN)4]PF6, significant changes were observed by 1H NMR (Fig. 4a), suggesting that AgI or CuI may bind to the pyridine ring of the axle. No significant changes were observed upon addition of [AuCl(SMe2)]. Similarly, divalent cations ZnII and CdII failed to elicit any observable change by 1H NMR (Fig. S61†).
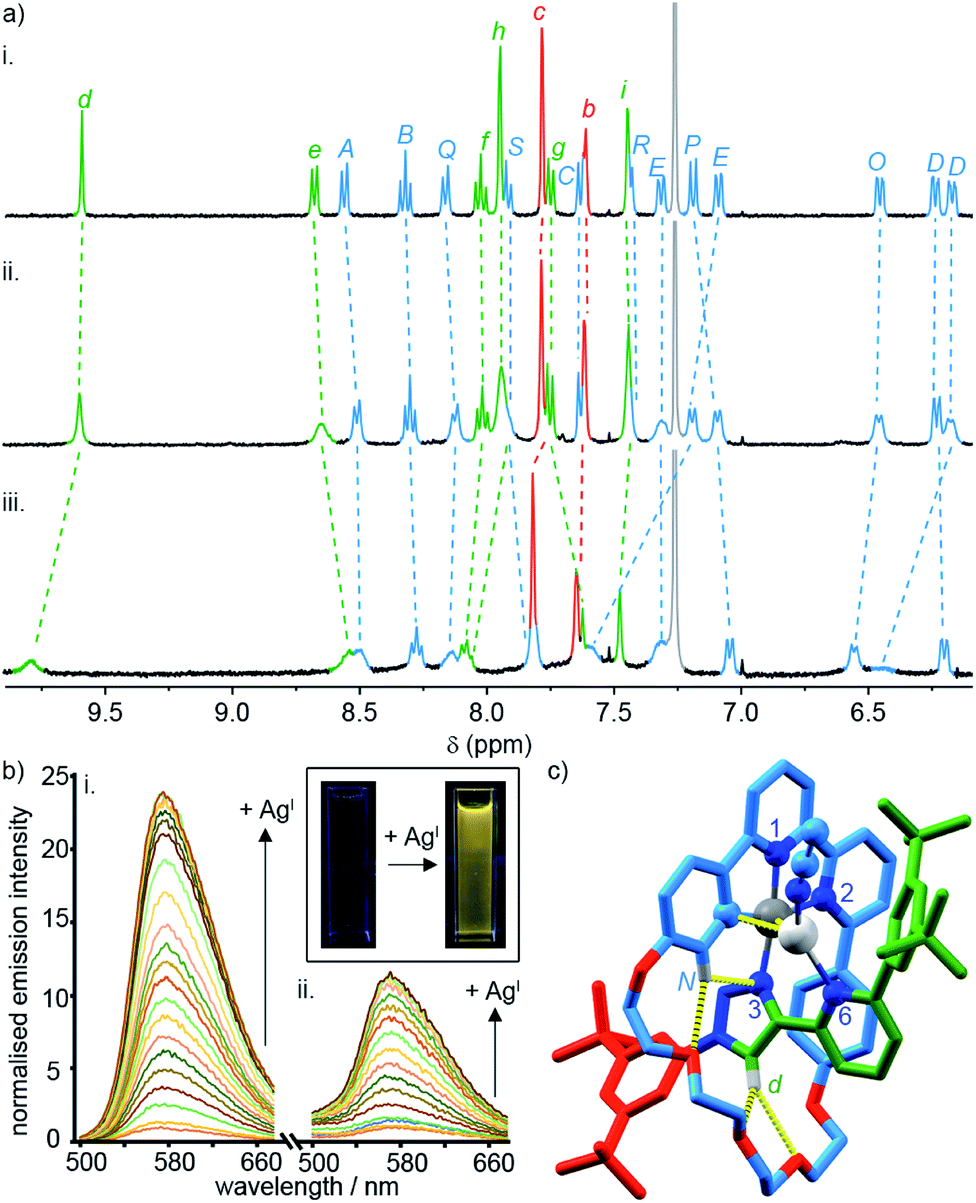 | ||
| Fig. 4 (a) Partial 1H NMR of (i) [Pt(κ4-4)]BF4, (ii) [Pt(κ4-4)]BF4 + 1 equiv. AgSbF6, (iii) [Pt(κ4-4)]BF4 + 1 equiv. of [Cu(MeCN)4]PF6. (b) Phosphorescence response of (i) [Pt(κ4-4)]BF4 and (ii) [Pt(2)(5)]BF4 to the portion-wise addition of AgSbF6 (inset: visual demonstration of the luminescent switch-on of [Pt(κ4-4)]BF4 in the presence of 10 equiv. of AgI). (c) Solid-state structure of [Pt(κ4-4)Ag(MeCN)]BF4 in partial sticks (rotaxane framework)/ball-and-stick (metals and ligand sphere) representation (anions and majority of Hs omitted for clarity). Colours: H white, C as in Scheme 2, N dark blue, O red, Pt dark grey, Ag light grey. Selected distances (Å) and angles (°): Pt–C 1.98, Pt–N1 1.95, Pt–N2 2.17, Pt–N3 2.02, Ag–Pt 2.83, Ag–C 2.54, Hd–O 2.28, Hd–O 2.60, HN–N3 2.51, HN–O 2.59, C–Pt–N2 161.3, N1–Pt–N3 172.1, C–Pt–N1 82.2, N1–Pt–N2 79.1, N2–Pt–N3 108.4, N3–Pt–C 90.3. | ||
Titration of [Pt(κ4-4)]BF4 with AgI and CuI monitored by UV-vis or luminescence spectroscopy revealed significant changes with incremental addition of metal salt (Fig. S61–S70†). Most strikingly, a switch-on of the emission was observed as a result of AgI binding but not in the case of CuI; in the presence of 3 equiv. of AgI, the emission intensity increased by ∼25-fold at λmax upon excitation at 400 nm (Fig. 4b). The quantum yield and lifetime of the emission increased by two orders of magnitude, and the corresponding estimated rate constants (Table 2) suggest that the dramatic effect of silver is due to the combined effect of suppressed non-radiative decay and an increase in kr. The consequent increase in luminescence is strikingly visible to the naked eye (Fig. 4b inset).
SCXRD analysis of crystals obtained from a solution of [Pt(κ4-4)]BF4 and AgSbF6 in the dark confirmed that the isolated product incorporates a AgI ion coordinated to the pyridine ring of the axle (Fig. 4c). This heterodinuclear complex appears to be stabilised both by a metal–metal interaction between the PtII and AgI centres (2.83 Å) and by an η2 π-interaction between the AgI centre and the bipyridine unit (2.54 Å), although it should be noted that in crystals grown under different conditions, only the Ag–Pt contact is maintained (Table S7†). Thus, we tentatively assign the observed increase in the luminescence quantum yield of [Pt(κ4-4)]BF4 in the presence of AgI to inhibition of the proposed quenching mechanism involving the pyridine-N⋯Pt interaction in the excited state, thus decreasing Σknr. The increase in kr, albeit a modest one, may reflect more efficient spin–orbit coupling pathways in the dinuclear system, in line with other recent reports of hetero-dinuclear38 and polynuclear39 complexes incorporating PtII, although these pathways remain poorly understood.
Titration of [Pt(2)(5)]BF4 with AgI revealed an increase in luminescence efficiency, albeit with a lower switch-on effect (∼12 fold). The difference in behaviour between [Pt(κ4-4)]BF4 and the corresponding non-interlocked complex may reflect the lower rigidity of the non-interlocked system; the pyridine moiety is sterically constrained in [Pt(κ4-4)]BF4 to project towards the PtII centre, whereas the non-interlocked complex has more conformational freedom which may entropically disfavour the PtII⋯N interaction and thus lower the efficiency of the proposed quenching mechanism.
Conclusions
In conclusion, we have demonstrated that interlocked cyclometallated PtII complexes – which are readily synthesised using an active template strategy – retain the photophysical properties of the parent non-interlocked complexes. The mechanical bond sterically stabilises the metal centre towards ligand displacement and – when no additional donors are present in the structure – towards oxidation, suggesting that this approach may have significant benefits for the construction of more stable emitters (e.g., as required in applications such as OLEDs, where stability and longevity are crucial). In particular, it allows one or more weakly coordinating ligands to be considered for the optimisation of the luminescence properties, since the chemical integrity of the luminophore is maintained by the mechanical bond rather than relying on the intrinsic strength of the metal–ligand interaction.33 In addition, we serendipitously identified a luminescent sensory system for Ag+ with >20 fold switch-on, in which the preorganisation provided by the mechanical bond appears to lead to enhanced performance compared with the non-interlocked analogue.More generally, we have demonstrated that interlocked, mechanically chelated late transition metal complexes can be stabilised by taking advantage of the catenand effect, something that has previously, to our knowledge, only been observed in the case of CuI.7 Just taking the example of platinum alone, such complexes are not only of interest for their photophysical properties but also in cancer chemotherapy – where both PtII and PtIV species are known to be effective40,41 – and in catalysis.42 Our results indicate that it may be possible to use the mechanical bond to augment the stability of key reaction intermediates or even divert the system down an alternative pathway43 in such chemical applications.
Finally, it is also noteworthy that all of the interlocked Pt complexes reported here are examples of mechanically planar chiral rotaxanes – systems in which the mechanical bond acts as a stereogenic unit.44 Indeed, 1H NMR analysis of [Pt(1)]BF4 in the presence of the chiral anion “trisphat” revealed the appearance of two sets of signals corresponding to the diastereomeric ion pairs of Rmp-[Pt(1)]Δ-trisphat and Smp-[Pt(1)]Δ-trisphat (Fig. S75†). [Pt(κ5-4)(OH)](BF4)2 also contains a stereogenic PtIV centre, the configuration of which is dictated by the mechanical stereochemistry of the precursor. Although all of these compounds were formed as racemic mixtures in the present study, we have recently developed methodology to stereoselectively access mechanically planar chiral rotaxanes45 and related topologically chiral catenanes.46 These results add further perspectives to the potential applications of interlocked heavy metal complexes in, for example, asymmetric catalysis47 and circularly polarised luminescence.48
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the China Scholarship Council (studentship to ZZ) and the European Research Council (Consolidator Grant to SMG, agreement no. 724987). The authors thank Dr J. E. M. Lewis (Imperial College) for assistance with SCXRD and helpful discussions.Notes and references
- By this we mean complexes in which a metal ion bridges donor atoms provided by two or more interlocked covalent subcomponents: J. E. M. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2017, 53, 298–312 RSC.
- It is an open question whether an interlocked molecule in which the components are bridged in this manner are true rotaxanes or if the metal–ligand interactions constitute a covalent link, rendering them entangled but not mechanically bonded, strictly speaking. Sauvage proposed the term “catenate” to denote such complexes in the context of catenanes. The equivalent noun “rotaxanate” is more commonly used as a verb meaning “to make a rotaxane”. Here we use the term “metallorotaxane”, which has seen some use to denote the mechanically chelated complex.5a.
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- Reviews: (a) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; (b) J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC.
- Reviews: (a) C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, 2016 CrossRef; (b) N. H. Evans and P. D. Beer, Chem. Soc. Rev., 2014, 43, 4658–4683 RSC; (c) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- Selected examples: (a) D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Angew. Chem., Int. Ed., 2005, 44, 4557–4564 CrossRef CAS PubMed; (b) D. A. Leigh, P. J. Lusby, R. T. McBurney, A. Morelli, A. M. Z. Slawin, A. R. Thomson and D. B. Walker, J. Am. Chem. Soc., 2009, 131, 3762–3771 CrossRef CAS PubMed; (c) A. Noor, W. K. C. C. Lo, S. C. Moratti and J. D. Crowley, Chem. Commun., 2014, 50, 7044–7047 RSC; (d) G. Baggi and S. J. Loeb, Angew. Chem., Int. Ed., 2016, 55, 12533–12537 CrossRef CAS PubMed; (e) G. Baggi and S. J. Loeb, Chem.–Eur. J., 2017, 23, 14163–14166 CrossRef CAS.
- A. M. Albrecht-Gary, Z. Saad, C. O. Dietrich-Buchecker and J. P. Sauvage, J. Am. Chem. Soc., 1985, 107, 3205–3209 CrossRef CAS.
- M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, J. Am. Chem. Soc., 2019, 141, 879–889 CrossRef CAS.
- (a) R. J. P. Williams, J. Mol. Catal., 1985, 30, 1–26 CrossRef CAS; (b) R. J. P. Williams, Eur. J. Biochem., 1995, 234, 363–381 CrossRef CAS PubMed.
- For selected examples and reviews of synthetic complexes designed to display “entatic” behaviour see: (a) P. Comba, Coord. Chem. Rev., 2000, 200–202, 217–245 CrossRef CAS; (b) G. Chaka, J. L. Sonnenberg, H. B. Schlegel, M. J. Heeg, G. Jaeger, T. J. Nelson, L. A. Ochrymowycz and D. B. Rorabacher, J. Am. Chem. Soc., 2007, 129, 5217–5227 CrossRef CAS PubMed; (c) A. Hoffmann, S. Binder, A. Jesser, R. Haase, U. Flörke, M. Gnida, M. Salomone Stagni, W. Meyer-Klaucke, B. Lebsanft, L. E. Grünig, S. Schneider, M. Hashemi, A. Goos, A. Wetzel, M. Rübhausen and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2014, 53, 299–304 CrossRef CAS PubMed; (d) L. Garcia, F. Cisnetti, N. Gillet, R. Guillot, M. Aumont-Nicaise, J. P. Piquemal, M. Desmadril, F. Lambert and C. Policar, J. Am. Chem. Soc., 2015, 137, 1141–1146 CrossRef CAS PubMed; (e) E. W. Dahl and N. K. Szymczak, Angew. Chem., Int. Ed., 2016, 55, 3101–3105 CrossRef CAS; (f) B. Dicke, A. Hoffmann, J. Stanek, M. S. Rampp, B. Grimm-Lebsanft, F. Biebl, D. Rukser, B. Maerz, D. Göries, M. Naumova, M. Biednov, G. Neuber, A. Wetzel, S. M. Hofmann, P. Roedig, A. Meents, J. Bielecki, J. Andreasson, K. R. Beyerlein, H. N. Chapman, C. Bressler, W. Zinth, M. Rübhausen and S. Herres-Pawlis, Nat. Chem., 2018, 10, 355–362 CrossRef CAS PubMed.
- (a) Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC; (b) Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC; (c) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
- (a) Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley, 2007 Search PubMed; (b) Y. Chi and P. T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC; (c) L. F. Gildea and J. A. G. Williams, in Organic Light-Emitting Diodes (OLEDs): Materials, devices and applications, Woodhead, 2013, pp. 77–113 Search PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- (a) W. Y. Wong and C. L. Ho, Chem. Soc. Rev., 2009, 253, 1709 CAS; (b) J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS; (c) C. M. Che, C. C. Kwok, S. W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Y. Zhu and H. Yersin, Chem.–Eur. J., 2010, 16, 233 CrossRef CAS PubMed; (d) C. Cebrian, M. Mauro, D. Kourkoulos, P. Mercandelli, D. Hertel, K. Meerholz, C. A. Strassert and L. De Cola, Adv. Mater., 2013, 25, 437–442 CrossRef CAS PubMed; (e) S. Q. Huo, J. Carroll and D. A. K. Vezzu, Asian J. Org. Chem., 2015, 4, 1210 CrossRef CAS; (f) M. C. Tang, A. K. W. Chan, M. Y. Chan and V. W. W. Yam, Top. Curr. Chem., 2016, 374, 46 CrossRef PubMed.
- Selected examples: (a) A. M. Fuller, D. A. Leigh, P. J. Lusby, I. D. H. Oswald, S. Parsons and D. B. Walker, Angew. Chem., Int. Ed., 2004, 43, 3914–3918 CrossRef CAS PubMed; (b) Y. Furusho, T. Matsuyama, T. Takata, T. Moriuchi and T. Hirao, Tetrahedron Lett., 2004, 45, 9593–9597 CrossRef CAS; (c) A. M. L. Fuller, D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, J. Am. Chem. Soc., 2005, 127, 12612–12619 CrossRef CAS PubMed; (d) A.-M. L. Fuller, D. A. Leigh and P. J. Lusby, Angew. Chem., Int. Ed., 2007, 46, 5015–5019 CrossRef CAS PubMed; (e) A.-M. L. Fuller, D. A. Leigh and P. J. Lusby, J. Am. Chem. Soc., 2010, 132, 4954–4959 CrossRef CAS PubMed.
- (a) J. P. Sauvage and M. Ward, Inorg. Chem., 1991, 30, 3869–3874 CrossRef CAS; (b) P. Mobian, J. M. Kern and J. P. Sauvage, J. Am. Chem. Soc., 2003, 125, 2016–2017 CrossRef CAS PubMed; (c) P. Mobian, J. M. Kern and J. P. Sauvage, Angew. Chem., Int. Ed., 2004, 43, 2392–2395 CrossRef CAS PubMed.
- (a) C. P. McArdle, J. J. Vittal and R. J. Puddephatt, Angew. Chem., Int. Ed., 2000, 39, 3819–3822 CrossRef CAS; (b) C. P. McArdle, S. Van, M. C. Jennings and R. J. Puddephatt, J. Am. Chem. Soc., 2002, 124, 3959–3965 CrossRef CAS; (c) S. M. Goldup, D. a. Leigh, P. J. Lusby, R. T. McBurney and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 6999–7003 CrossRef CAS PubMed; (d) R. J. Puddephatt, J. Organomet. Chem., 2015, 792, 13–24 CrossRef CAS.
- For an example of a cyclometallated pseudorotaxane-based Pt complex see: D. Sooksawat, S. J. Pike, A. M. Z. Slawin and P. J. Lusby, Chem. Commun., 2013, 49, 11077–11079 RSC.
- Examples in which late transition metals are structural components of non-emissive interlocked molecules: (a) M. Fujita, F. Ibukuro, K. Yamaguchi and K. Ogura, J. Am. Chem. Soc., 1995, 117, 4175–4176 CrossRef CAS; (b) C. P. McArdle, J. J. Vittal and R. J. Puddephatt, Angew. Chem., Int. Ed., 2000, 39, 3819–3822 CrossRef CAS; (c) A. Hori, A. Akasaka, K. Biradha, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3269–3272 CrossRef CAS; (d) A. Hori, K. I. Yamashita and M. Fujita, Angew. Chem., Int. Ed., 2004, 43, 5016–5019 CrossRef CAS PubMed; (e) T. J. Burchell, D. J. Eisler and R. J. Puddephatt, Dalton Trans., 2005, 268–272 RSC; (f) N. Weisbach, Z. Baranová, S. Gauthier, J. H. Reibenspies and J. A. Gladysz, Chem. Commun., 2012, 48, 7562 RSC; (g) Z. Baranová, H. Amini, N. Bhuvanesh and J. A. Gladysz, Organometallics, 2014, 33, 6746–6749 CrossRef; (h) W. Wang, L.-J. Chen, X.-Q. Wang, B. Sun, X. Li, Y. Zhang, J. Shi, Y. Yu, L. Zhang, M. Liu and H.-B. Yang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5597–5601 CrossRef CAS PubMed; (i) Y. X. Wang, Q. F. Zhou, L. J. Chen, L. Xu, C. H. Wang, X. Li and H. B. Yang, Chem. Commun., 2018, 54, 2224–2227 RSC; (j) X.-Q. Wang, W. Wang, W.-J. Li, L.-J. Chen, R. Yao, G.-Q. Yin, Y.-X. Wang, Y. Zhang, J. Huang, H. Tan, Y. Yu, X. Li, L. Xu and H.-B. Yang, Nat. Commun., 2018, 9, 3190 CrossRef PubMed.
- (a) H. Masai, J. Terao, S. Makuta, Y. Tachibana, T. Fujihara and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 14714–14717 CrossRef CAS PubMed; (b) H. Masai and J. Terao, Bull. Chem. Soc. Jpn., 2019, 529–539 CrossRef CAS.
- X. Ma, J. Zhang, J. Cao, X. Yao, T. Cao, Y. Gong, C. Zhao and H. Tian, Chem. Sci., 2016, 7, 4582–4588 RSC.
- A. J. Blake, C. O. Dietrich-Buchecker, T. I. Hyde, J. P. Sauvage and M. Schröder, J. Chem. Soc., Chem. Commun., 1989, 1663–1665 RSC.
- J. E. M. Lewis, R. J. Bordoli, M. Denis, C. J. Fletcher, M. Galli, E. A. Neal, E. M. Rochette and S. M. Goldup, Chem. Sci., 2016, 7, 3154–3161 RSC.
- (a) H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 CrossRef CAS; (b) E. A. Neal and S. M. Goldup, Chem. Sci., 2015, 6, 2398–2404 RSC; (c) E. A. Neal and S. M. Goldup, Angew. Chem., Int. Ed., 2016, 55, 12488–12493 CrossRef CAS; (d) J. E. M. Lewis, F. Modicom and S. M. Goldup, J. Am. Chem. Soc., 2018, 140, 4787–4791 CrossRef CAS PubMed.
- Reviews: (a) J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC; (b) M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 0061 CrossRef CAS.
- (a) V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS PubMed; (b) V. Aucagne, J. Berna, J. D. Crowley, S. M. Goldup, K. D. Hänni, D. A. Leigh, P. J. Lusby, V. E. Ronaldson, A. M. Z. Slawin, A. Viterisi and D. B. Walker, J. Am. Chem. Soc., 2007, 129, 11950–11963 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- J. H. K. Yip, Suwarno and J. J. Vittal, Inorg. Chem., 2000, 39, 3537–3543 CrossRef CAS.
- (a) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS; (b) S. Fernández, J. Forniés, B. Gil, J. Gómez and E. Lalinde, Dalton Trans., 2003, 2, 822–830 RSC; (c) F. Niedermair, O. Kwon, K. Zojer, S. Kappaun, G. Trimmel, K. Mereiter and C. Slugovc, J. Chem. Soc., Dalton Trans., 2008, 4006–4014 RSC; (d) M. Spencer, A. Santoro, G. R. Freeman, Á. Díez, P. R. Murray, J. Torroba, A. C. Whitwood, L. J. Yellowlees, J. A. G. Williams and D. W. Bruce, Dalton Trans., 2012, 41, 14244–14256 RSC; (e) H. Uesugi, T. Tsukuda, K. Takao and T. Tsubomura, Dalton Trans., 2013, 42, 7396–7403 RSC; (f) J. Moussa, G. R. Freeman, J. A. G. Williams, L. M. Chamoreau, P. Herson and H. Amouri, Eur. J. Inorg. Chem., 2016, 2016, 761–767 CrossRef CAS.
- (a) S. Lai, M. C. Chan, T. Cheung, S. Peng and C. M. Che, Inorg. Chem., 1999, 38, 4046–4055 CrossRef CAS; (b) T. Cheung and C. Che, J. Chem. Soc., Dalton Trans., 1996, 2, 1645–1651 RSC.
- Non-cyclometallated complex Pt(1.H)(DMSO)Cl2 displays weak ligand-centred fluorescence and no detectable phosphorescence at room temperature, but phosphoresces in the red region at 77 K (Fig. S59†). The long lifetime of 140 μs is consistent with a more ligand-centred formulation to the emissive excited state, with little participation of the metal, reflecting the absence of the Pt–C covalent bond.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006, p. 9 Search PubMed.
- (a) G. Aromí, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485–546 CrossRef; (b) R. A. S. Vasdev, D. Preston and J. D. Crowley, Dalton Trans., 2017, 46, 2402–2414 RSC.
- For related examples see: (a) M. L. Clarke, Polyhedron, 2001, 20, 151–164 CrossRef CAS; (b) M. Ghedini, A. Golemme, I. Aiello, N. Godbert, R. Termine, A. Crispini, M. La Deda, F. Lelj, M. Amati and S. Belviso, J. Mater. Chem., 2011, 21, 13434–13444 RSC; (c) S. Liu, H. Sun, Y. Ma, S. Ye, X. Liu, X. Zhou, X. Mou, L. Wang, Q. Zhao and W. Huang, J. Mater. Chem., 2012, 22, 22167–22173 RSC.
- S. Chatterjee, J. A. Krause, K. Madduma-Liyanage and W. B. Connick, Inorg. Chem., 2012, 51, 4572–4587 CrossRef CAS PubMed.
- V. Gutmann, Chimia, 1977, 31, 1–7 CAS.
- D. K. Crites Tears and D. R. McMillin, Coord. Chem. Rev., 2001, 211, 195–205 CrossRef CAS.
- G. Turnbull, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2017, 53, 2729–2732 RSC.
- (a) E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS; (b) S. Culham, P.-H. Lanoë, V. L. Whittle, M. C. Durrant, J. A. G. Williams and V. N. Kozhevnikov, Inorg. Chem., 2013, 52, 10992–11003 CrossRef CAS PubMed; (c) M. Z. Shafikov, R. Daniels, P. Pander, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, ACS Appl. Mater. Interfaces, 2019, 11, 8182–8193 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- (a) M. D. Hall and T. W. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS; (b) S. J. Berners-Price, Angew. Chem., Int. Ed., 2011, 50, 804–805 CrossRef CAS PubMed.
- (a) M. L. Clarke, Polyhedron, 2001, 20, 151–164 CrossRef CAS; (b) M. L. Clarke, Polyhedron, 2001, 20, 151–164 CrossRef CAS.
- F. Modicom, E. M. G. Jamieson, E. Rochette and S. M. Goldup, Angew. Chem., Int. Ed., 2019, 58, 3875–3879 CrossRef CAS PubMed.
- (a) N. H. Evans, Chem.–Eur. J., 2017, 1–13 CAS; (b) N. Pairault and J. Niemeyer, Synlett, 2018, 29, 689–698 CrossRef CAS; (c) E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- (a) R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817–4820 CrossRef CAS PubMed; (b) M. A. Jinks, A. de Juan, M. Denis, C. J. Fletcher, M. Galli, E. M. G. Jamieson, F. Modicom, Z. Zhang and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 14806–14810 CrossRef CAS PubMed.
- M. Denis, J. E. M. Lewis, F. Modicom and S. M. Goldup, Chem, 2019, 5, 1512–1520 CAS.
- (a) H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L. A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS; (b) C. Wang, L. A. Chen, H. Huo, X. Shen, K. Harms, L. Gong and E. Meggers, Chem. Sci., 2015, 6, 1094–1100 RSC; (c) Y. Zheng, Y. Tan, K. Harms, M. Marsch, R. Riedel, L. Zhang and E. Meggers, J. Am. Chem. Soc., 2017, 139, 4322–4325 CrossRef CAS; (d) N. Hu, H. Jung, Y. Zheng, J. Lee, L. Zhang, Z. Ullah, X. Xie, K. Harms, M. H. Baik and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 6242–6246 CrossRef CAS PubMed; (e) C. Zhang, S. Chen, C.-X. Ye, K. Harms, L. Zhang, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2019, 58, 14462–14466 CrossRef CAS PubMed.
- (a) C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem. Sci., 2014, 5, 1915–1927 RSC; (b) J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation data for all novel compounds. CCDC 1961263–1961269. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05507j |
| This journal is © The Royal Society of Chemistry 2020 |