
Circularly polarized luminescence reveals interaction between commercial stains and protein matrices used in paintings†
Sibilla Orsini*,
Francesco Zinna*,
Tarita Biver,
Lorenzo Di Bari and
Ilaria Bonaduce
and
Ilaria Bonaduce
Dipartimento di Chimica e Chimica Industriale, Università di Pisa, via Moruzzi 13, I-56126 Pisa, Italy. E-mail: sibilla.orsini@for.unipi.it; francesco.zinna@for.unipi.it
First published on 3rd October 2016
Abstract
Chemistry is at the front line of preservation of work of arts. Reliable but simple protocols for localizing proteins in art samples are necessary to guide conservators in their work. In this article, we investigated the interaction between some commonly used fluorescent stains and protein-based paint binders. Beside fluorescence, we used for the first time circularly polarized luminescence (CPL) as a tool to reveal interactions in complex matrices relevant for the field of cultural heritage. We show that, in such cases, CPL provides unique information compared to more common techniques such as fluorescence and circular dichroism. These findings will contribute to provide a rational base to empirical observations in staining procedures, even in arduous and complex cases.
Introduction
Painters from all periods and across the globe rely on a wide range of organic substances as painting materials, such as oils, saccharide gums, terpenoid resins and proteinaceous materials. The development of specific methods for the stratigraphic localization and identification of these materials in paint samples is one of the most important steps to understand painting techniques, to highlight degradation phenomena, to identify the best conservation conditions and to prevent considerable damage brought on by misguided restoration interventions on painted artworks.1–4Today, the stratigraphic localization of proteinaceous materials in art samples is still relatively arduous.5,6 Following a common technique, the sample is mounted on a cross-section, after embedding it in a synthetic resin, which is subsequently polished in order to expose the sample stratigraphy. Cross-sections can then be analyzed by imaging techniques based on magnetic resonance, mass spectrometry or optical spectroscopies6 such as Raman microscopy.7,8 For proteins, one may take advantage of staining methods using visible or fluorescent dyes. Such methods do not require specific instrumentation or complex sample handling and are very appealing to non-specialized conservation laboratories.6,9,10 These dyes may be coupled to immunochemical assays, for obtaining a selective detection of specific epitopes.11–15 However, proteins can undergo severe changes within the complex matrix of a paint upon (very) long term exposure to the changeable and sometimes harsh environment, where the artwork is displayed or stored.10,16–19 Degradation processes of proteinaceous binders include oxidation and deamidation, partial hydrolysis, crosslinking, formation of aggregates and complexes with other organic binders and inorganic pigments and fillers.20,21 As a consequence, antibodies selected to recognize native proteins may fail to localize degraded proteins or their aggregates in aged paint cross-sections. On the other hand, there is a wide variety of low molecular weight dyes, developed to reveal proteins or peptides non-specifically, by visual inspection, through colour or fluorescence on/off response.
We may distinguish between covalent22–24 and non-covalent dyes,10,25,26 depending on the type of interaction they establish with the protein or the peptide.
For the localization of proteinaceous binders in paint cross sections, one may use common fluorescent protein gel stains, although false negatives and positives are reported.26,27 In order to carry out a preliminary study of the interaction between fluorescent stains with protein-based painting matrices in solution, here we propose to employ circularly polarized luminescence (CPL) to complement more common techniques, such as electronic circular dichroism (ECD) or fluorescence.
CPL is the differential emission of left and right circularly polarized light from chiral molecules or systems. It is therefore the natural complement of fluorescence, much in the same way as ECD is for absorption spectroscopy. In the same way CPL is highly sensitive to the stereochemical environment surrounding the fluorophore.
In general chiroptical spectroscopies are a well known tool to monitor the interaction between a small achiral or racemic molecule and a chiral guest such as proteins or DNA.28 Chiroptical techniques are particularly beneficial when complex systems are involved, thanks to the specificity of the chiroptical response. Most commonly, ECD is used to this purpose,28 because induced ECD can be an extremely powerful way to reveal and follow small molecule–protein binding. On the other hand, CPL is complementary to ECD, because the latter monitors mainly interactions between molecules in their ground state, while CPL responds essentially to excited state perturbations.29
Indeed, some research groups reported and studied CPL-responsive bioprobes30,31 mainly based on lanthanides,32,33 to target proteins34 or other biomolecules.35–39 However, despite its potential, this technique is not of general use, probably because it still relies mostly on home-made instruments.
Moreover, especially where small polarization effects are expected, one should be aware that different artifacts may affect the true CPL signal.40 Such artifacts are mainly due to linear anisotropies and overlap between CPL and ECD bands.41,42 The first problem is usually solved exciting the sample either by using an in-line geometry with non polarized light or by employing a 90° geometry with excitation light linearly polarized along the detection direction.
The aim of this work is to study the interaction of selected fluorescent stains with the proteins contained in real painting matrices.
Our long term goal is to provide a rational approach to the development of successful and simple fluorescent stains, to understand scopes and limitations of protein staining in painting cross-sections and to put forward an improved, reliable and robust methodology for restorers based on fluorescence.
Results and discussion
For the reasons given above, in order to obtain a realistic insight, we had to employ products of the typical grade which would actually be employed in painting practice, together with some commercial fluorescent stains. We screened five widespread stains (Scheme 1), which normally are used to reveal denatured proteins or peptides,25–27,43,44 mainly on gel electrophoresis, and three relevant protein-based substrates, namely dried egg white (EW) based on ovalbumin, casein (CAS) from dried cow milk, and rabbit skin glue (RSG) based on partially hydrolysed collagen. As a standard reference we used a purified protein (chicken egg ovalbumin, OVA).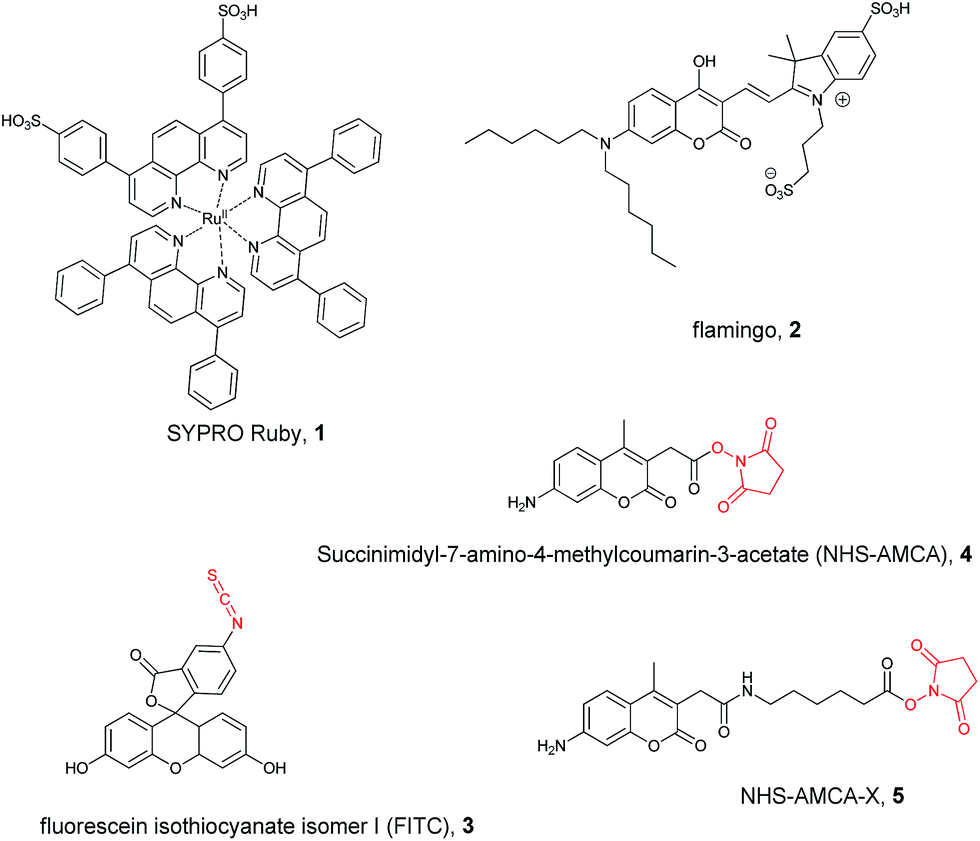 | ||
| Scheme 1 Structures of the fluorescent stains studied in this work. The reactive moieties of the covalent stains are highlighted in red. | ||
In a first screening of cross sections containing our three different model proteinaceous binders (CAS, RSG and OVA) we observed remarkably different responses from the five stains. In Fig. 1, we report the fluorescence microscopy images of cross-sections prepared with the three proteins after staining with the fluorophores reported in Scheme 1. SYPRO Ruby (1) and FITC (3) give CAS and EW-containing cross-sections a characteristic red (in the case of staining agent 1) and green (in the case of 3) fluorescence (Fig. 1), while no staining emission is visible with RSG. NHS-AMCA (4) and NHS-AMCA-X (5) give blue fluorescence with the three proteins, while Flamingo (2) does not give any fluorescence with any of the samples.
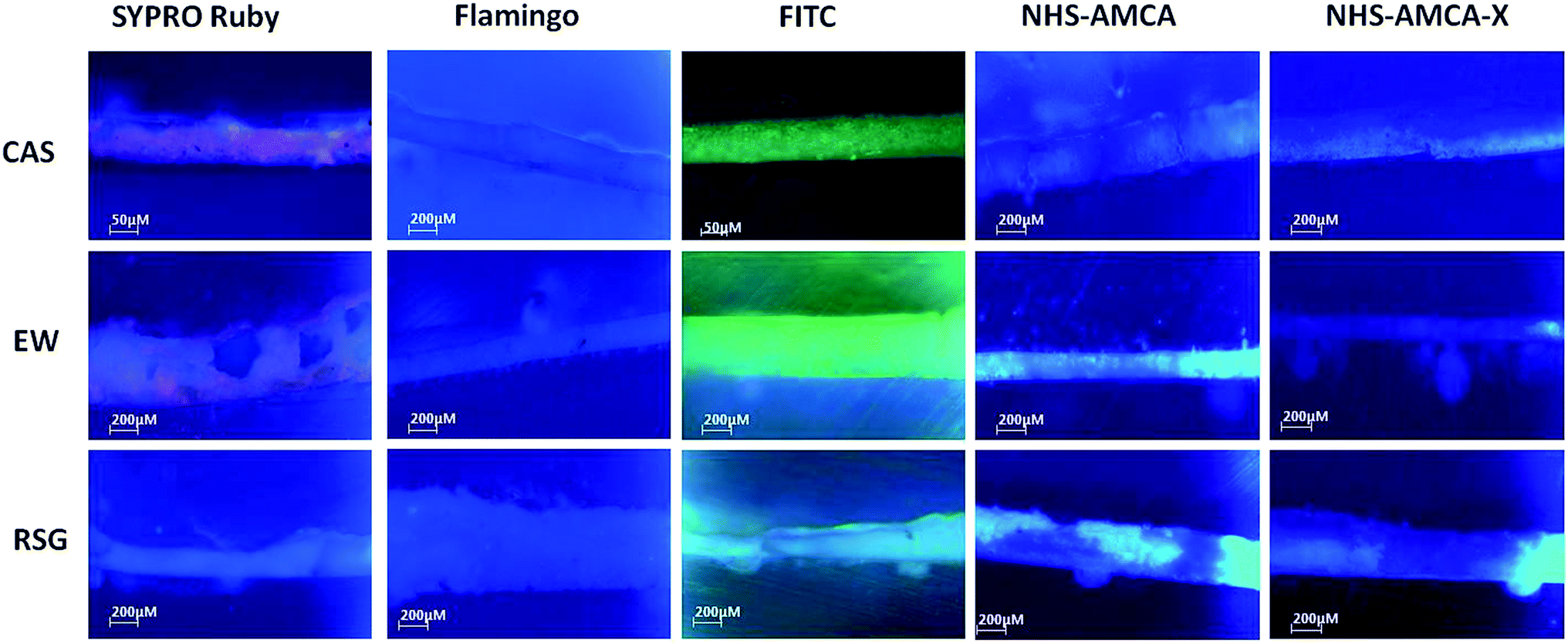 | ||
| Fig. 1 Visible fluorescence microscopy images of cross sections of a CAS matrix (top row), EW (middle row) and a RSG (bottom row) matrix, stained with SYPRO Ruby (first column from the left), Flamingo (second column), FITC (third column), NHS-AMCA (fourth column) and NHS-AMCA-X (fifth column) under UV (λexc = 365 nm) light. | ||
In this work, for the first time we exploited CPL to signal the interaction of some of these commercial achiral (or racemic) fluorophores with common painting grade protein-based matrices. These results are complemented with the affinity constants of the fluorescent stains obtained by fluorescence titrations in aqueous solution.
SYPRO Ruby 1 and Flamingo 2 (Scheme 1) are known to bind denatured proteins on a gel, and were recently proposed to localize proteins in paint cross sections.10,45 Both 1 and 2 are only weakly emissive in aqueous solution (Tris buffer pH 7.2), while they become brighter upon irradiation with 365 nm UV-B light when they interact with certain proteins, peptides or some proteinaceous material.
In the presence of CAS, EW and OVA, 1 yields red luminescence, which allowed us to carry out titration experiments, detailed in the ESI and depicted in Fig. S1, S2a, S3† and Table 1. On the contrary, RSG did not induce any luminescence enhancement at any mole ratio (see ESI, Fig. S2a†). A similar behaviour is also observed for the Flamingo stain (2). 2 shows very weak fluorescence when it is alone in aqueous solution, but the binding with CAS, EW and RSG leads to an increase of the fluorescence intensity upon addition of proteins (see ESI, Fig. S2b†). Moreover, an increase of the protein content generates a shift in the fluorescence emission maximum to lower wavelengths, from 550 nm of 2 alone to 535 nm with CAS, 536 nm with EW and 543 nm with RSG (see ESI, Fig. S4†). These changes further confirm the interaction between 2 and the three proteins.
| Proteinaceous materials | SYPRO Ruby | Flamingo | ||||
|---|---|---|---|---|---|---|
| Emission maximum [nm] (λex 470 nm) | Δφ [M−1] at the saturating level | Kb | Emission maximum [nm] (λex 490 nm) | Δφ [M−1] at the saturating level | Kb | |
| Without proteins | 603 | 550 | ||||
| CAS | 603 | 5.8 × 107 | 3.1 × 105 | 535 | 6.1 × 108 | 1.1 × 105 |
| EW | 603 | 4.1 × 107 | 5.2 × 104 | 536 | 7.1 × 108 | 2.6 × 105 |
| RSG | 603 | 4.8 × 106 | — | 543 | 2.5 × 108 | 9.5 × 103 |
| OVA | 603 | 7.2 × 107 | 2.5 × 105 | |||
In order to compare quantitatively the binding affinity of stains 1 and 2 with the proteins, we determined the association constant values (Kb) by monitoring the changes of emission at the maximum band position with increasing concentration of proteins. In Table 1, we report the constants Kb, determined using the equation
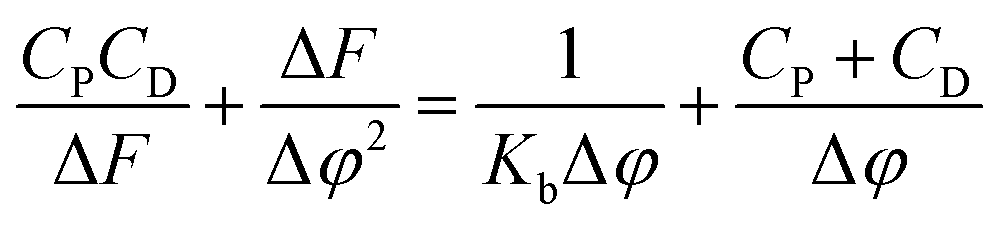 | (1) |
The comparison of the Kb values obtained for 1 and 2 shows that there are very large differences between these two stains: 1 has strong affinity for CAS and OVA and only to a much lesser extent to EW, while it does not bind to RSG. The large differences between EW and the purified protein OVA indicate that the latter may not be a satisfactory model for the complex EW and therefore a study of the real complex matrix is mandatory to understand the interaction of the stains and the proteins in real samples.
On the contrary, 2 prefers EW and CAS over RSG. Interestingly, in cross-sections no fluorescence was visible (Fig. S15†), suggesting that in the solid phase different phenomena might take place, such as quenching of fluorescence, or the binding site of the protein is not available when not in solution.
These quantitative data deserved further insight and we proceeded to chiroptical measurements. In the first place, we resorted to ECD spectroscopy in the visible range, aiming at the detection of induced ECD of the dye-centred absorption bands.
In this context, we should notice that 1 is the racemic mixture of a (tris(phenanthroline)ruthenium(II) disulfonate),46 which stems from the octahedral coordination of Ru(II) ion with the bidentate ligand (phen) enabling two enantiomeric configurations, (Δ) and (Λ).
On the contrary, Flamingo (2) is achiral, as well as all the other dyes (shown in Scheme 1) that we shall discuss below.
For a racemic molecule like 1, ECD may arise only if: (a) one of the two enantiomers interacts more strongly than the other with the protein and at the same time this interaction induces significant changes in the ECD spectrum of the bound species compared to the free one; (b) if both enantiomers bind to a similar extent but in different sites, such that the ECD spectra of the two become different and no longer cancel out (as it would normally occur in the absence of the protein).28
In our case, we could detect no ECD signal in the region of the absorption of 1 (450 nm, see ESI Fig. S11†), therefore we must exclude both possibilities. Moreover, we monitored the protein UV-ECD spectrum as well, but we could not detect significant changes. This reveals that the binding event does not cause any secondary structure change in the protein.47
On account of the large effect on luminescence, we turned to CPL of 1 with proteins in plateau conditions. In the case of CAS, EW and OVA, we observed a relatively intense positive signal whose maximum around 605 nm roughly matches the luminescence spectrum of 1 (Fig. 2). The extent of CPL activity can be quantified by the dissymmetry factor glum, defined as:
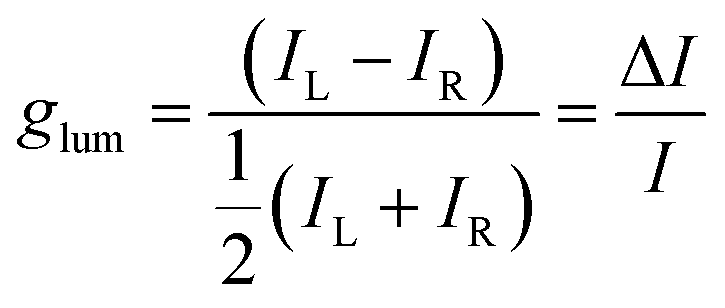 | (2) |
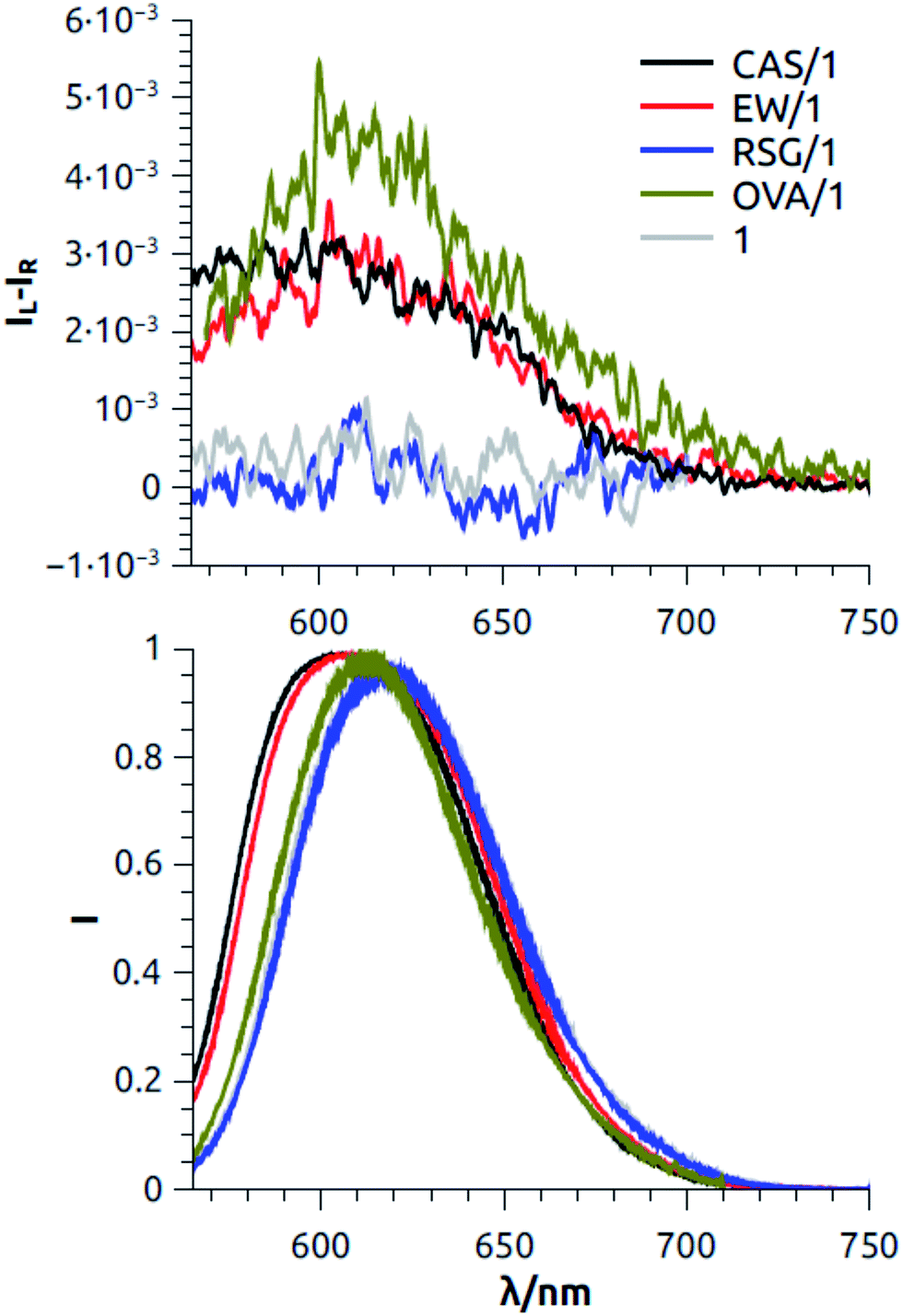 | ||
| Fig. 2 CPL spectra of SYPRO Ruby, 1, without protein and with the three proteins studied (CAS, EW and RSG) and with the model protein OVA. The spectra are normalized to the maximum of the fluorescence intensity, so that the glum values can be directly read on y-axis at the maximum of each curve (Top). Normalized total fluorescence spectra measured simultaneously with CPL spectra (Bottom). The spectra are cut at higher energy where the onset of the excitation source becomes visible. | ||
This reasoning might be proved by repeating the experiments with the two enantiomers resolved which unfortunately are not commercially available.
Moreover, the sign of the band gives information about which enantiomer is preferentially interacting. The absolute configuration of the interacting Ru species would be unambiguously assessed only by comparing the spectra in Fig. 2 with the CPL spectra of the resolved enantiomers of 1. Since to the best of our knowledge there are no CPL data of similar Ru complexes in the literature, the stereochemistry of the interacting species can be supposed employing the following reasoning. If upon protein binding there is no major variation between the geometry of the ground and the emitting state of the dye, the sign of CPL is expected to be concordant with the one of the most red-shifted ECD band. ECD spectra of optically pure (Λ)-Ru(phen)3 found in the literature50 show that the most red shifted signal is a positive couplet around 450 nm, thus in our case the positive CPL bands is consistent with a preferential interaction of the Λ enantiomer.
This holds true for CAS, EW and also for the model protein OVA. Ceteris paribus, the most intense signal was recorded with the model protein OVA, which is in agreement with fluorescence data. Also in agreement with the fluorescence data is the absence of any CPL signal of the solution of 1 with RSG.
In order to confirm the reliability of the signals, we also measured the CPL spectrum of 1 alone in the same conditions and we checked that it gave no signal, as expected. From these observations, we may conclude that CAS and OVA can host 1 in a cavity, where it is shielded from luminescence quenching effects and which is enantioselective, with a preference for Λ-configuration. EW possibly responds similarly to OVA, although with some weakened effect, possibly on account of the fact that OVA is somewhat diluted in EW.
In the case of 2/protein systems, we were not able to measure any significant CPL spectra due to its overall low fluorescence.
Covalent stains (FITC, AMCA and AMCA-X, Scheme 1) open on a different landscape, where again CPL plays a fundamental role. In this case the binding to the protein is irreversible and in principle one can securely wash off the excess stain. This applies when developing electrophoretic gels or working with paint cross-sections, because the protein is immobilized, but it is poorly fit for model studies conducted in solution (unless proceeding to a dialysis step). In contrast with what we required for 1 and 2, which are necessarily in free/bound exchange, here no change in fluorescence between free and bound dye is necessary. As a result one might not be able to gain any information from total fluorescence in solution, while chiroptical methods could be of use if the dye, upon binding, experiences a chiral environment.
Indeed, when a fixed concentration of FITC in aqueous solution of carbonate buffer at pH 9 was titrated with increasing amount of proteins (CAS, EW and RSG, respectively), the fluorescence did not change in a noticeable way (ESI, Fig. S5 and S6†). Similarly, the proteins had no significant effect on fluorescence intensity of AMCA and AMCA-X during the titrations in carbonate buffer (ESI, Fig. S7–S10†).
We tried to record ECD spectra of FTIC (3), but, as in the previous cases, no induced ECD signal was detected corresponding to the absorption band of 3 around 470 nm (ESI, Fig. S12†). On the contrary, taking advantage of the strong fluorescence of FTIC (3), we measured the CPL spectra of the three proteins covalently bonded with 3 (Fig. 3). We were able to measure a weak but significant positive CPL signal (glum ≈ +6 × 10−4) in the case of 3/CAS and 3/EW, while the spectrum measured for 3/RSG was not significantly different from the baseline recorded on a buffer solution containing 3 without any protein. The mere presence of a CPL signal in the two cases is direct evidence of the occurrence of an interaction between achiral fluorophore 3 and the proteins. It was reported before that fluorescein can give rise to a relatively strong CPL signal (but notably without ECD) when it is in a chiral medium such as in a chiral solvent.51 It is therefore possible that, also in our case, the chiral interactions between the proteins and 3 differ significantly in the ground and in the excited emitting state.
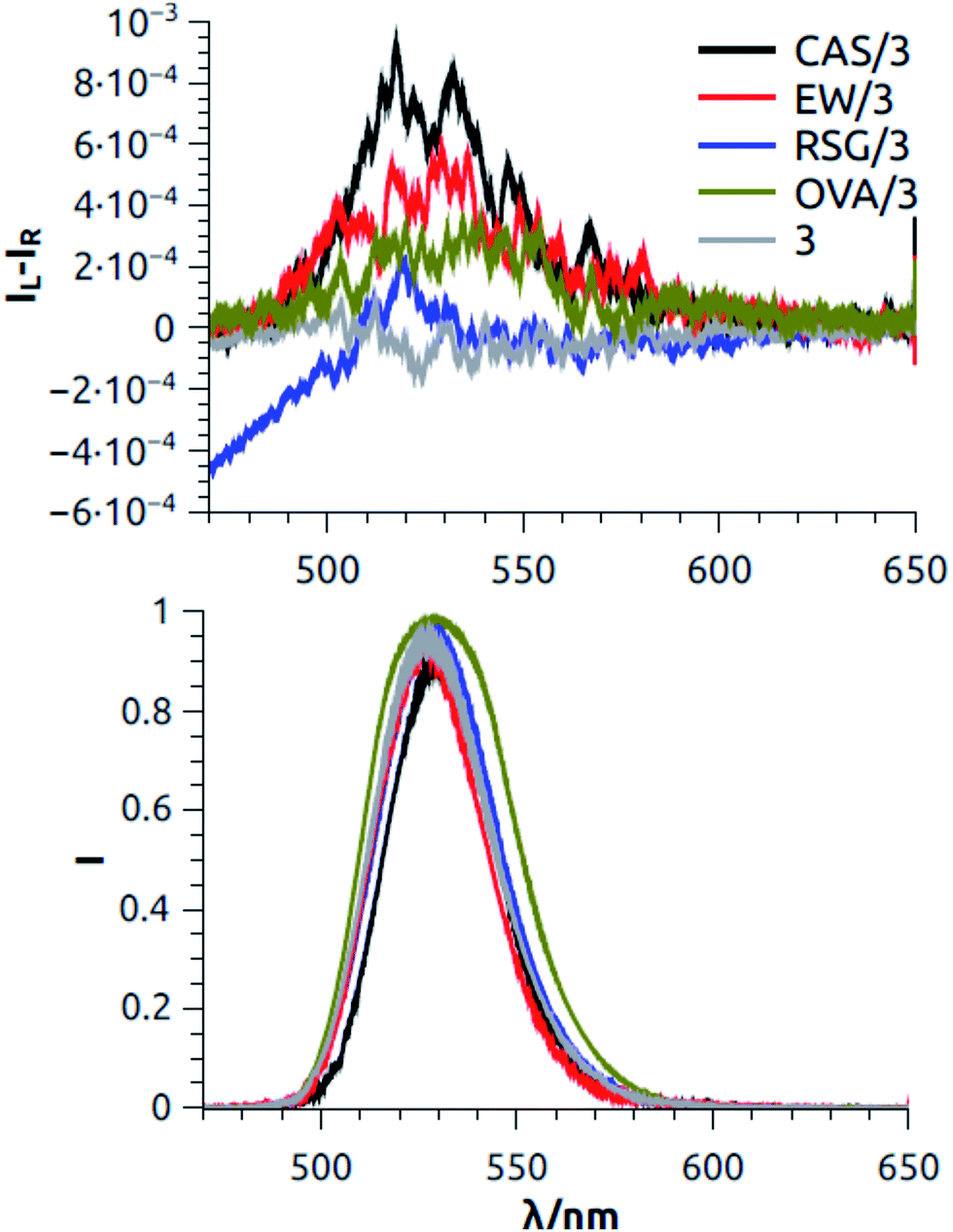 | ||
| Fig. 3 CPL spectra of FTIC without protein and with the three proteins studied (CAS, EW, and RSG). The spectra are normalized to the maximum of the fluorescence intensity, so that the glum values can be directly read on y-axis at the maximum of each curve (Top). Normalized total fluorescence spectra measured simultaneously with CPL spectra (Bottom). | ||
These results are in agreement with the staining tests carried out on cross-sections (Fig. 1). It is therefore apparent that in such a case CPL provides a clear indication of the interaction, while ECD and fluorescence do not.
Unfortunately, the emission region of AMCA and AMCA-X is currently unsuited to our CPL apparatus.
Conclusions
In conclusion, we have shown that CPL can be conveniently employed to shed light on the selective interaction between commercial achiral or racemic fluorophores with proteinaceous materials of interest in the field of cultural heritage. CPL can complement fluorescence data or even give information about the occurrence of an interaction when fluorescence or ECD fail, as in the case of the covalent stain FITC.These data will help rationalizing empirical observations from the use of fluorescent stains to localize proteins in samples from works of art, and will provide a rational way to design stains, which reliably, selectively and/or specifically respond to various types of proteinaceous layers in paint cross-sections. A possible development of this research would be the study of aged samples, although it might reveal very complex, due to the loss of solubility of proteins, which was observed in model and paint samples.52–54 Such a future study could help to ascertain if limitations of labelling with fluorescent stains are related to the chemical changes undergone by the proteins during aging, or if they arise because when embedded in the complex matrix of a solid paint sample, the proteins are less available for the fluorescent ligand.
Acknowledgements
We wish to dedicate this work to the memory of the late Dr Ettore Castiglioni: he gave us constant advice on CPL instrument operation. University of Pisa (PRA 2016, “Functional Materials”) is acknowledged for financial support.Notes and references
- C. Cennini, Il libro dell'arte, Firenze, 1859 Search PubMed
.
- M. P. Colombini and F. Modugno, Organic Mass Spectrometry in Art and Archeology, Pisa, 2009 Search PubMed
- M. Doener, The Materials of the Artist and their use in painting, Orlando, 1984 Search PubMed
- M. D. Gottsegen, The Painter's Handbook, Watson Guptill Publications, New York, 2006 Search PubMed
- M. P. Colombini, A. Andreotti, I. Bonaduce, F. Modugno and E. Ribechini, Acc. Chem. Res., 2010, 43, 715–727 CrossRef CAS PubMed
- S. Dallongeville, N. Garnier, C. Rolando and C. Tokarski, Chem. Rev., 2016, 116, 2–79 CrossRef CAS PubMed
- L. Burgio, R. J. H. Clark and R. R. Hark, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5726 CrossRef CAS PubMed
- R. Clark, Chem. Soc. Rev., 1995, 24.3, 187–196 RSC
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS PubMed
- I. C. A. Sandu, A. C. A. Roque, P. Matteini, S. Schäfer, G. Agati, C. R. Correia and J. F. F. P. Viana, Microsc. Res. Tech., 2012, 75, 316–324 CrossRef
- L. Dolci, G. Sciutto, M. Guardigli, M. Rizzoli, S. Prati, R. Mazzeo and A. Roda, Anal. Bioanal. Chem., 2008, 392, 29–35 CrossRef CAS PubMed
- G. Sciutto, L. S. Dolci, A. Buragina, S. Prati, M. Guardigli, R. Mazzeo and A. Roda, Anal. Bioanal. Chem., 2010, 399, 2889–2897 CrossRef PubMed
- G. Sciutto, L. S. Dolci, M. Guardigli, M. Zangheri, S. Prati, R. Mazzeo and A. Roda, Anal. Bioanal. Chem., 2012, 405, 933–940 CrossRef PubMed
- G. Sciutto, S. Prati, R. Mazzeo, M. Zangheri, A. Roda, L. Bardini, G. Valenti, S. Rapino and M. Marcaccio, Anal. Chim. Acta, 2014, 831, 31–37 CrossRef CAS PubMed
- I. Sela-Culang, V. Kunik and Y. Ofran, Front. Immunol., 2013, 4, 1–13 CAS
- C. Duce, E. Bramanti, L. Ghezzi, L. Bernazzani, I. Bonaduce, M. P. Colombini, A. Spepi, S. Biagi and M. R. Tine, Dalton Trans., 2013, 42, 5975–5984 RSC
- C. Duce, L. Ghezzi, M. Onor, I. Bonaduce, M. Colombini, M. Tine' and E. Bramanti, Anal. Bioanal. Chem., 2012, 402, 2183–2193 CrossRef CAS PubMed
- L. Ghezzi, C. Duce, L. Bernazzani, E. Bramanti, M. P. Colombini, M. R. Tiné and I. Bonaduce, J. Therm. Anal. Calorim., 2015, 122, 315–322 CrossRef CAS
- D. Pellegrini, C. Duce, I. Bonaduce, S. Biagi, L. Ghezzi, M. P. Colombini, M. R. Tinè and E. Bramanti, Microchem. J., 2016, 124, 31–35 CrossRef CAS
- G. Leo, I. Bonaduce, A. Andreotti, G. Marino, P. Pucci, M. P. Colombini and L. Birolo, Anal. Chem., 2011, 83, 2056–2064 CrossRef CAS PubMed
- A. Nevin, D. Anglos, S. Cather and A. Burnstock, Appl. Phys. A, 2008, 92, 69–76 CrossRef CAS
- J. E. Berlier, A. Rothe, G. Buller, J. Bradford, D. R. Gray, B. J. Filanoski, W. G. Telford, S. Yue, J. Liu and C.-Y. Cheung, J. Histochem. Cytochem., 2003, 51, 1699–1712 CrossRef CAS PubMed
- D. Kretschy, G. Koellensperger and S. Hann, Anal. Chim. Acta, 2012, 750, 98–110 CrossRef CAS PubMed
- K. Muramoto, H. Kamiya and H. Kawauchi, Anal. Biochem., 1984, 141, 446–450 CrossRef CAS PubMed
- S. Kuckova, I. C. A. Sandu, M. Crhova, R. Hynek, I. Fogas and S. Schafer, J. Cult. Herit, 2013, 14, 31–37 CrossRef
- D. Magrini, S. Bracci and I. C. A. Sandu, Procedia Chem., 2013, 8, 194–201 CrossRef CAS
- J. M. Messinger II, JAIC, 1992, 31, 267–274 Search PubMed
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211–5233 RSC
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1 CrossRef CAS
- M. Nakamura, J. Suzuki, F. Ota, T. Takada, K. Akagi and K. Yamana, Chem.–Eur. J., 2016, 22, 9121 CrossRef CAS PubMed
- A. Rybicka, G. Longhi, E. Castiglioni, S. Abbate, W. Dzwolak, V. Babenko and M. Pecul, ChemPhysChem, 2016, 17, 2931 CrossRef CAS PubMed
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 2, 925 CrossRef PubMed
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC
- J. Yuasa, T. Ohno, R. S. H. Tsumatori, H. Kamikubo, M. Kataoka, Y. Hasegawa and T. Kawai, Chem. Commun., 2013, 49, 4604 RSC
- G. Muller and J. P. Riehl, J. Fluoresc., 2005, 15, 553 CrossRef CAS PubMed
- G. Muller, Dalton Trans., 2009, 9692 RSC
- A. Moussa, C. Pham, S. Bommireddy and G. Muller, Chirality, 2009, 21, 497 CrossRef CAS PubMed
- T. Uchida, K. Nozaki and M. Iwamura, Chem.–Asian J., 2016, 11, 2415 CrossRef CAS PubMed
- B. T. Nguyen, A. J. Ingram and G. Muller, Chirality, 2016, 28, 325 CrossRef CAS PubMed
- H. P. J. M. Dekkers, in Circular dichroism, Principles and applications, ed. N. Berova, K. Nakanishi and R. W. Woody, Wiley, 2nd edn, 2000 Search PubMed
- E. Castiglioni, S. Abbate, F. Lebon and G. Longhi, Chirality, 2012, 74, 725 CrossRef PubMed
- E. Castiglioni, S. Abbate, F. Lebon and G. Longhi, Methods Appl. Fluoresc., 2014, 2, 024006 CrossRef
- P. R. Banks and D. M. Paquette, Bioconjugate Chem., 1995, 6, 447–458 CrossRef CAS PubMed
- R. C. Wolbers and G. Landrey, Preprints of the 15th Annual Meeting of American Institute for Conservation, Washington, 1987, pp. 168–202 Search PubMed
- S. Dallongeville, M. Richter, S. Schafer, M. Kuhlenthal, N. Garnier, C. Rolando and C. Tokarski, Analyst, 2013, 138, 5357–5364 RSC
- G. Crosby, W. Perkins and D. Klassen, J. Chem. Phys., 1965, 43, 1498–1503 CrossRef CAS
- G. D. Fasman, Circular dichroism and the conformational analysis of biomolecules, Springer Science & Business Media, 2013 Search PubMed
- F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed
- E. M. Sánchez Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef PubMed
- B. Le Guennic, W. Hieringer, A. Görling and J. Autschbach, J. Phys. Chem. A, 2005, 109, 4836–4846 CrossRef CAS PubMed
- H. G. Brittain and F. S. Richardson, J. Phys. Chem., 1976, 80, 2590–2592 CrossRef CAS
- C. Duce, E. Bramanti, L. Ghezzi, L. Bernazzani, I. Bonaduce, M. P. Colombini, A. Spepi, S. Biagi and M. R. Tine, Dalton Trans., 2013, 42, 5975–5984 RSC
- C. Duce, L. Ghezzi, M. Onor, I. Bonaduce, M. P. Colombini, M. R. Tine' and E. Bramanti, Anal. Bioanal. Chem., 2012, 402, 2183–2193 CrossRef CAS PubMed
- L. Ghezzi, C. Duce, L. Bernazzani, E. Bramanti, M. P. Colombini, M. R. Tiné and I. Bonaduce, J. Therm. Anal. Calorim., 2015, 122, 315–322 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Fluorescence analysis and experimental details. See DOI: 10.1039/c6ra14795j |
| This journal is © The Royal Society of Chemistry 2016 |