
Highly thermostable lanthanide metal–organic frameworks exhibiting unique selectivity for nitro explosives†
Shu-Na Zhaoab,
Xue-Zhi Songab,
Min Zhuab,
Xing Mengab,
Lan-Lan Wuab,
Shu-Yan Song*a,
Cheng Wang*a and
Hong-Jie Zhanga
aState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun, 130022, P. R. China. E-mail: songsy@ciac.ac.cn; cwang@ciac.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 21st November 2014
Abstract
Three isostructural 3D metal–organic frameworks [Ln(L)1.5(DEF)]n (Ln = Gd (1), Eu (2), Tb (3), H2L = 9,9-diethylfluorene-2,7-dicarboxylic acid) have been solvothermally synthesized and structurally characterized by single-crystal X-ray diffraction. The main framework of compounds 1–3 show high thermal stability to 400 °C but with a distortion or shrinking of the crystal lattice between 200 °C and 250 °C. The high red emission intensity and the microporous instinct of the solvent-free [Eu(L)1.5]n (2a) indicate that it can be potentially used as luminescent sensor. It was then applied in the detection of organic solvent molecules. Notably, it exhibits high sensitivity for 2,4,6-trinitrophenol (TNP) with Ksv constant 6.24 × 104 M−1 through luminescence quenching experiments.
Introduction
In the past two decades, the selective and rapid detection of high explosives and explosive-like substances has drawn great interest due to its importance in homeland security, civilian safety, and environmental protection.1 Although many methods have been developed for explosive detection, these detection techniques are limited by their equipment demands and cost drawbacks.2 However, fluorescence detection achieved by changes in optical signal response has proven to be an excellent candidate for the rapid detection of explosives. Compared to the traditional methods, fluorescence detection has obvious advantages and gains more attention owing to its high sensibility, simplicity, short response time, and its ability to be employed both in solution and solid phase.3 In fact, the materials usually used for fluorescence detection are still defective in the respects of stability, toxicity, sensitivity, and biodegradability, thus it is a challenging task to synthesize novel materials for fluorescence detection of explosives.Metal–organic frameworks (MOFs), all called porous coordination polymers (PCPs) are an emerging class of crystalline porous materials assemblied of inorganic metal ions and clusters and polydentate organic linker ligands. They have been extensively studied for their structural diversity and potential applications in gas storage and separation,4 heterogeneous catalysis,5 drug delivery,6 energy storage and conversion,7 and chemical sensing.8 The different recognition/binding events with guest substrates confined by the tunable pore sizes and functionalized pore surfaces, which can be transduced into externally optical signals, have enabled MOFs to be the excellent candidate for fluorescence detection materials. The pioneering work of Li et al.9 and subsequent works10 have demonstrated the great promise of MOFs for the sensing and detection of explosives. In general, many explosives such as nitroaromatics, nitramines are highly electron deficient organic compounds, and often act as good electron acceptors,2a thus MOFs with electron donating conjugated fluorophores are excellent candidates for the sensing and detection of such explosives, as the efficient excition migration intrinsic to the long-range conjugation results in high sensitivity for fluorescence quenching by electron acceptors.11 The fluorescence quenching chemosensing technology has been proven to be a simple, sensitive, and convenient way to detect the nitro explosives.12
Herein, we selected 9,9-diethylfluorene-2,7-dicarboxylic acid (H2L) and Eu3+ to synthesize porous MOFs for detection of explosives based on the following reasons: (i) H2L as the fluorene derivative is an excellent electron donor ligand, which demonstrated high sensing performance for nitro explosives due to the ‘molecular wire’ effect.13 (ii) H2L is a rigid ligand, advantageous for the formation of porous structures. (iii) Eu3+ ions can be used as the sensitive optical signal through its pure and sharp red-emission arising from characteristic 4f electronic transitions.14
In this work, we report three highly thermostable lanthanide MOFs, [Ln(L)1.5(DEF)]n (Ln = Gd (1), Eu (2), Tb (3)), in which the main framework can be stable up to 400 °C. The solvent-free phase [Eu(L)1.5]n (2a) could be obtained by direct heating of compound 2 at 250 °C. Then, the investigation of the ability of the solvent-free phase 2a for nitro explosive sensing has been accomplished through luminescence quenching experiments, indicating that 2a can detect nitroaromatic explosives fast and sensitively, especially for the detection of 2,4,6-trinitrophenol (TNP) with Ksv value 6.24 × 104 M−1.
Experimental section
The 9,9-diethylfluorene-2,7-dicarboxylic acid (H2L) was synthesized according to the literature.15 All of the other starting materials employed were purchased from commercial sources and used as received without further purification.Synthesis of [Gd(L)1.5(DEF)]n (1)
A solution containing H2L (0.0234 g, 0.075 mmol), Gd(NO3)3·6H2O (0.0223 g, 0.05 mmol) in 5.0 mL of DEF and 0.5 mL of H2O was sealed in a Teflon-lined autoclave and heated at 80 °C under autogenous pressure for three days and then allowed to cool to room temperature. The crystals were washed with ethanol and air-dried. Yield: 45% (based on Gd3+). Anal. calcd for C67H70N2O14Gd2 (Mr: 1441.75): C, 55.82%; H, 4.89%; N, 1.94%. Found: C, 56.16%; H, 4.32%; N, 1.61%.Synthesis of [Eu(L)1.5(DEF)]n (2)
Compound 2 was synthesized following the same synthetic procedure as that for compound 1 expect that Eu(NO3)3·6H2O was used instead of Gd(NO3)3·6H2O. Yield: 52% (based on Eu3+). Anal. calcd for C67H70N2O14Eu2 (Mr: 1431.22): C, 56.23%; H, 4.93%; N, 1.96%. Found: C, 56.01%; H, 4.57%; N, 1.81%.Synthesis of [Tb(L)1.5(DEF)]n (3)
Compound 3 was synthesized following the same synthetic procedure as that for compound 1 expect that Tb(NO3)3·6H2O was used instead of Gd(NO3)3·6H2O. Yield: 47% (based on Tb3+). Anal. calcd for C67H70N2O14Tb2 (Mr: 1445.16): C, 55.69%; H, 4.88%; N, 1.94%. Found: C, 55.31%; H, 4.97%; N, 2.06%.General method
Fourier transform infrared (FT-IR) spectroscopy was obtained with a Bruker TENSOR 27 Fourier transform infrared spectrometer with the KBr pellet technique and operating in the transmittance mode in the 4000–400 cm−1 region. Elemental analyses were determined with a VarioEL analyzer. Thermogravimetric analysis (TGA) was performed on a Netzsch STA 449F3 TG/DTA instrument under air atmosphere. The samples were heated from about 40 °C to 800 °C with a heating rate of 10 °C min−1. The experimental powder X-ray diffraction data (PXRD) were collected on a Bruker D8-FOCUS diffractometer equipped with Cu Kα1 (λ = 1.5406 Å; 1600 W, 40 kV, 40 mA) with the step of 0.02°. Temperature-dependent PXRD datas were recorded on a Bruker D8-ADVANCE X-ray powder diffractometer using Cu Kα radiation. The simulated PXRD patterns were calculated by using single-crystal X-ray diffraction data and processed by the free Mercury v1.4 program provided by the Cambridge Crystallographic Data Center. The fluorescence excitation and emission spectra were recorded at room temperature with a Hitachi F-4500 spectrophotometer equipped with a 150 W Xenon lamp as an excitation source. The photomultiplier tube (PMT) voltage was 700 V, the scan speed was 1200 nm min−1. UV-VIS spectra were recorded on a SHIMADZU UV-VIS-IR spectrophotometer. The luminescence decay curves were obtained by using a Lecroy Wave Runner 6100 Digital Oscilloscope (1 GHz) with a tunable laser (pulse width 4 ns, gate 50 ns) as the excitation source (Continuum Sunlite OPO).X-ray crystallography
The X-ray intensity data for the three compounds were collected on a Bruker SMART CCD diffractometer with graphite monochromatized Mo-Kα radiation (λ = 0.71073 Å, 45 kV, 35 mA). Data integration and reduction were processed with SAINT software.16 Multiscan absorption corrections were applied with the SADABS program.17 The crystal structure was solved by means of direct methods and refined employing full-matrix least-squares on F2 (SHELXTL-97).18 All the hydrogen atoms were generated geometrically and refined isotropically using the riding model. All non-hydrogen atoms were refined with anisotropic displacement parameters.Results and discussion
Structural characterization
Solvothermal reaction of Gd(NO3)3 and H2L in N,N-diethylformamide (DEF) produces block crystals of [Gd(L)1.5(DEF)]n. Single-crystal X-ray analysis reveals that compound 1–3 are isostructural, and hence only the results of 1 are given in the ensuing discussion. Compound 1 crystallizes in the monoclinic space group C2/c. The asymmetric unit contaions one crystallographically independent Gd3+ ion, one and a half completely deprotonated L2− anions, one terminally coordinated DEF molecule. The Gd3+ ion is surrounded by eight oxygen atoms from six different L2− ligands and one DEF molecule, showing a distorted square antiprismatic geometry (Fig. S1, ESI†) The Gd–O bond distances range from 2.285(3) to 2.563(3) Å, which are comparable to those reported for other gadolinium–oxygen donor compounds.19 The coordination modes of carboxyl groups belonging to L2− ligand are μ2-η1:η1-bridging and μ2-η2:η1-bridging modes (Fig. S2, ESI†). Furthermore, two crystallographically equivalent Gd atoms are connected each other by four L2− ligands to give rise to a binuclear gadolinium building block (the adjacent Gd⋯Gd separation is 3.838(10) Å). These binuclear units are bridged by carboxylate groups, forming infinite 1D chains along the c axis with Gd⋯Gd distances ranging from 3.838(10) to 5.369(12) Å (Fig. 1a). Moreover, the 1D chains are interconnected by L2− ligands to result in a 3D non-interpenetrating framework (Fig. 1b). The terminal DEF molecules are expected to be removed during thermal activation, leading to the formation of solvent-free MOFs [Ln(L)1.5]n (Ln = Ga, Eu, Tb) with open Ln3+ sites for their recognition of small molecules (Fig. 1c). PLATON analysis revealed that the 3D porous structure contains large voids of 1513.2 Å3, which represents 23.3% of the volume of the unit cell (Fig. S3, ESI†).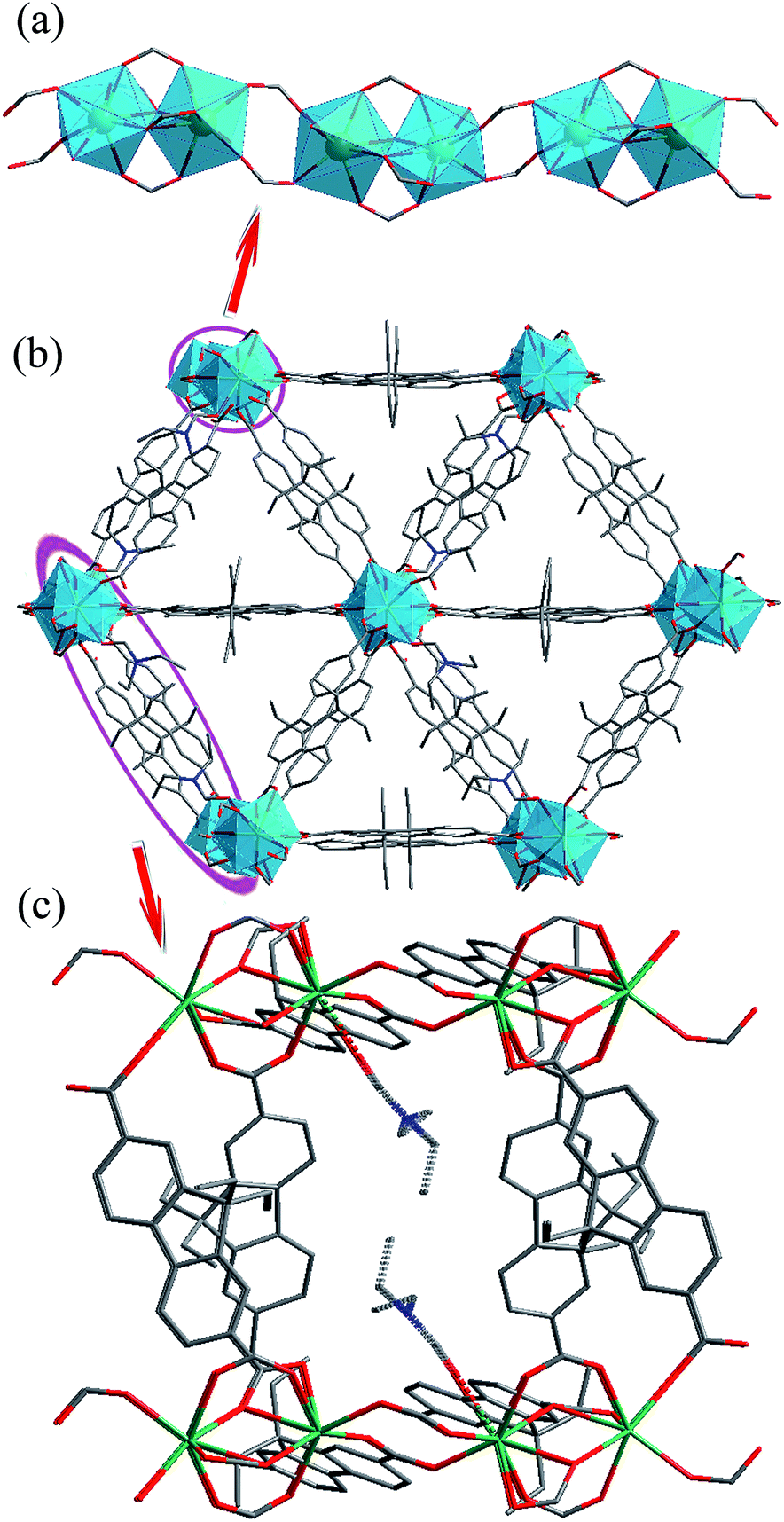 | ||
| Fig. 1 (a) Infinite 1D chains; (b) packing representation of 1 as viewed along the c axis; (c) 1D micropore of about 13.98(24) Å × 5.37(12) Å. The blue polyhedra in (a) and (b) represent coordination spheres of Gd sites, while grey, blue, red, and green spheres vertices represent C, N, O, and Gd centers, respectively. | ||
Thermal stabilities
Thermal gravimetric analyses (TGA) of compounds 1, 2, 3, and 2a were performed under air atmosphere from room temperature to 800 °C with a heating rate of 10 °C min−1. As seen in Fig. S3,† the TGA result reveals that compound 1 is stable up to 200 °C, and then shows a gradual weight loss of 14.85 wt% in the temperature range from 200 °C to 285 °C corresponding to the release of coordinated DEF molecules (calcd 14.01 wt%). The decomposition process is followed by a plateau of stability from 290 °C to 430 °C, then the framework rapidly collapses. Compounds 2 and 3 have the similar profiles to that of compound 1, thus it will not be described in detail. The solvent-free phase 2a could be obtained by direct heating of 2 at 250 °C for 24 h. The TGA value shows that 2a is stable up to 435 °C with no obvious weight loss (Fig. S4, ESI†), indicating that the coordinated DEF molecules are totally removed without the collapse of the framework. It is further confirmed by the IR spectra. In the IR spectra, the absence of the strong vibration 1659 cm−1 in 2a, which is corresponded to the coordinated DEF molecules, implies a complete removing of DEF molecules (Fig. S5, ESI†).20 The framework integrity was then examined by powder X-ray diffraction (PXRD). 2a remains highly crystallized and retains the main framework features but with lost or shift of several peaks and occurrence of a few new peaks. This is probably due to the distortion or shrinking of the crystal lattice to some degree in response to heating and removal of coordinated DEF molecules, which is commonly observed in a lot of MOF structures (Fig. S6 and S7, ESI†).21Guest-dependent luminescent properties
The excitation spectrum of 2a monitored under the characteristic emission (615 nm) of the Eu3+ ion exhibits a broad band with a maximum at around 340 nm and a shoulder peak at 310 nm. The emission spectrum of 2a in the solid state excited at 340 nm exhibits the luminescence peaks at 578, 591, 615, 650, 699 nm, which could be attributed the characteristic transitions of the Eu3+ ion: 5D0 → 7FJ (J = 0, 1, 2, 3, 4), respectively (Fig. S10, ESI†). The strong blue light emission in free H2L completely disappears, leading us to infer that H2L ligand is an excellent chromophore for sensitization of the Eu3+ ion.The bright-red luminescence of 2a primarily drove us to investigate its potential for sensing common organic solvent molecules. Finely ground samples of 2a of known amount were immersed in different organic solvents (DMF, ethanol, 2-propanol, ethylacetate, 1-propanol, methanol, acetonitrile, 1,4-dioxane, cyclohexane, chloroform, tetrahydrofuran, dichloromethane, acetone), treated with ultrasonication, and then aged to form stable emulsions prior to fluorescence measurements. As shown in Fig. 2 and S11, ESI,† the photoluminescence intensity is largely dependent on the solvent molecules, particularly in the case of acetone, which exhibit the most noticeable quenching effect. Such solvent-dependent luminescent properties are of significant interest in the sensing of acetone, which is very harmful to human beings. Therefore, the effect of acetone on the luminescent intensity of 2a has been examined in more detail.
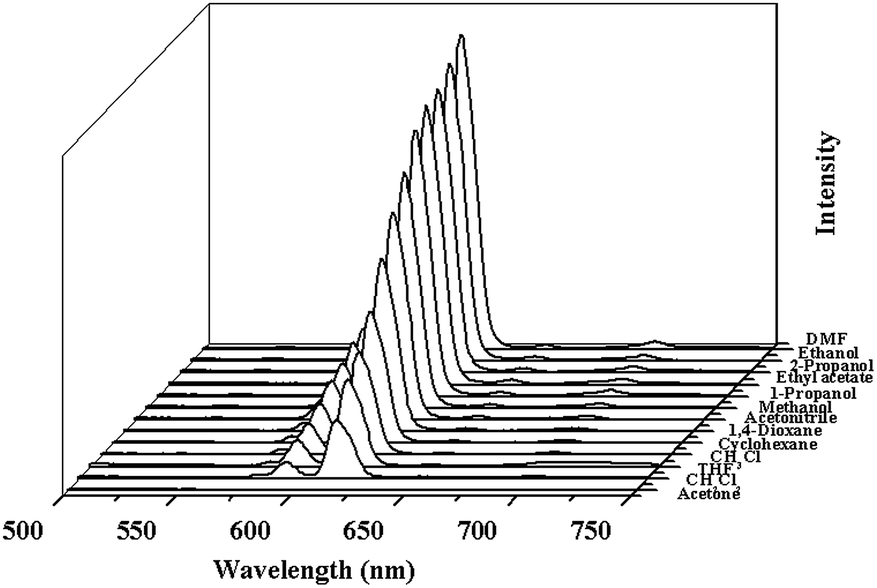 | ||
| Fig. 2 The photoluminescence spectra of the 2a samples that were introduced into various pure solvents. Excitation: 310 nm. | ||
The 2a was dispersed in DMF as the standard suspension, while the acetone content was gradually increased to monitor the emissive response (Fig. S12, ESI†), and it almost disappeared at an acetone content of 1.8 vol%. The decreasing trend of luminescent intensity of the 5D0 → 7F2 transition of Eu3+ at 615 nm versus the volume ratio of acetone could be well fitted with a first-order exponential decay (Fig. S12, ESI†), indicating that luminescence quenching of 2a by acetone is diffusion-controlled.22 The physical interaction of the solute and solvent plays a vital role in such fluorescence quenching effects of organic solvent molecules. Upon illumination, there is competition of absorption of the light source energy between the solvent molecules and organic ligands, resulting in a decrease (even quenching) in the PL intensity.23
Such solvent-dependent luminescence properties are very fascinating and important for selective sensing of solvent molecules, leading us to an exploration of more organic molecules, especially for aromatic compounds. So, the luminescence responses of 2a to various aromatic compounds were ascertained by dispersing identical volumes of different aromatic compounds into a standard emulsion of 2a in DMF. Strong fluorescence quenching effect was only observed for nitrobenzene (NB) (Fig. 3a). To examine sensing sensitivity towards NBs in more detail, a batch of emulsion of 2a with gradually increasing NB contents in DMF was prepared to monitor the emissive response (Fig. 3b). The luminescence intensity decreased to 50% at 0.022 vol% (1.83 mM), and complete quenching was received at 0.095 vol% (7.9 mM). As a result, the selective luminescence quenching behavior for NB may be probably due to the electron transfer from the electron-donating ligands to the electron-deficient NB molecules.
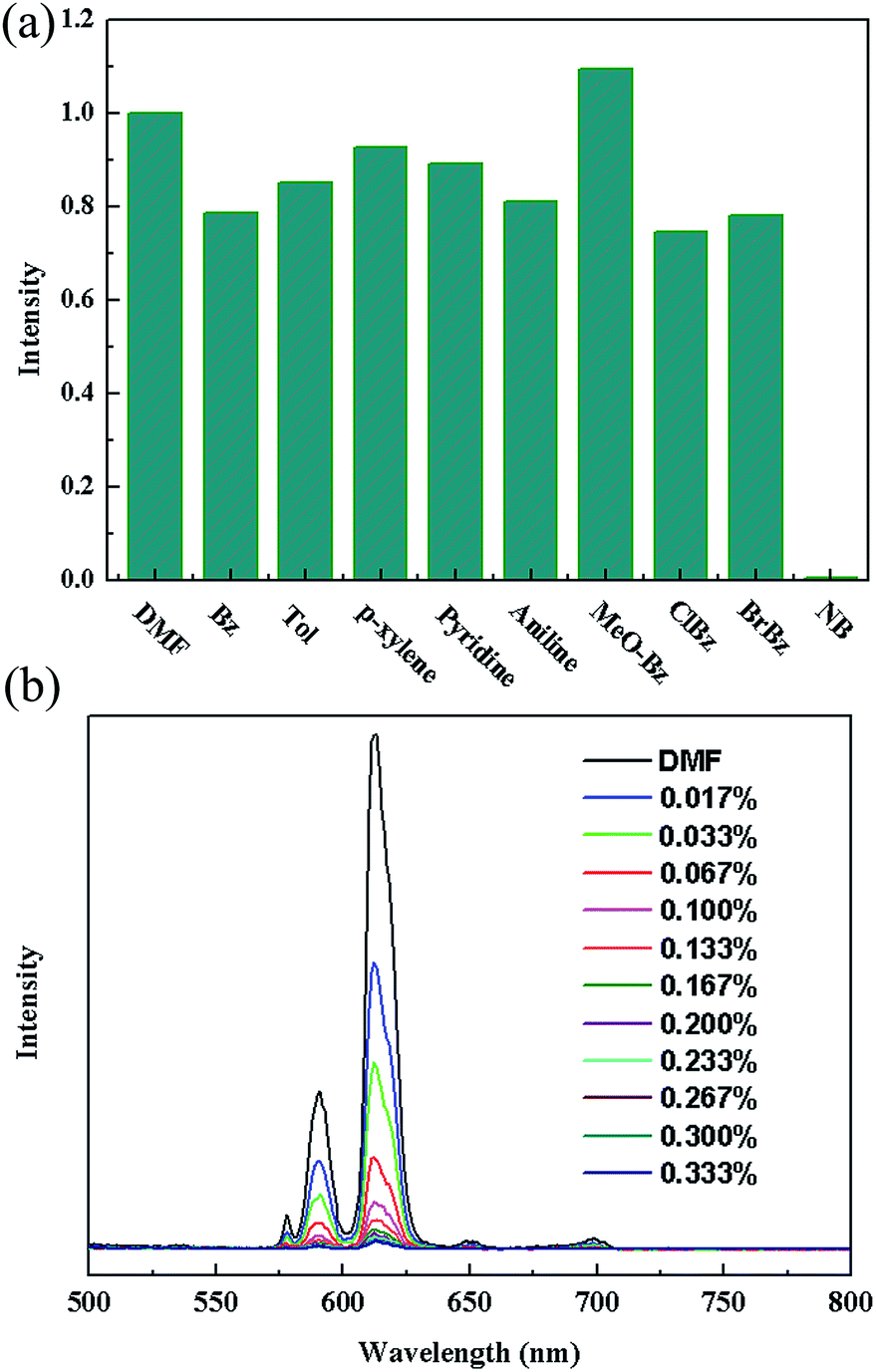 | ||
| Fig. 3 (a) The 5D0 → 7F2 transition intensities of in different solvents. Excitation: 340 nm. (b) The photoluminescence spectra of 2a DMF emulsion in the presence of various contents of NB. | ||
The luminescence quenching behavior is primary related to the electron-withdrawing nitro-group, inspiring us to investigate the potential of 2a towards sensing a series of nitroaromatic explosives, such as 2,4,6-trinitrophenol (TNP), 4-nitrophenol (4-NP), 3-nitrophenol (3-NP), o-nitrotoluene (o-NT), m-nitrotoluene (m-NT), nitrobenzene (NB) and nitromethane (NM). All seven nitro compounds can weaken the photoluminescent intensity of the 2a emulsion to a varying degree. The most effective quencher is TNP with the quenching percentage (QP) of 94.61%, whereas the quenched efficiencies of 4-NP, 3-NP, o-NT, m-NT, NB, NM is 90.99%, 63.04%, 40.54%, 29.21%, 10.86%, 5.94%, respectively (Fig. 4). The fluorescence-quenching titration was used to investigate the sensitivity and selectivity of 2a for sensing nitroaromatics in this work. The fluorescence quenching by TNP could be easily discerned at as low as 5 μM concentration (Fig. S16, ESI†). On the other hand, all other nitroaromatics showed petite effect on fluorescence intensity of 2a. These results demonstrate that 2a has high selectivity for TNP compared to other nitroaromatics. And it is comparable to the reported known MOFs as sensors for nitroaromatics.3a,13b,24 Further, the fluorescence quenching efficiency was analyzed using the Stern–Volmer (SV) equation, (I0/I) = Ksv[A] + 1, where I0 and I are the fluorescence intensities before and after addition of the analyte, respectively, Ksv is the quenching constant (M−1), [A] is the molar concentration of the analyte. The linear nature of the SV plot of TNP can be ascribed to either a static or dynamic quenching process. The quenching constant for TNP was found to be 6.24 × 104 M−1, which is comparable to the know organic polymers (Fig. 5).25
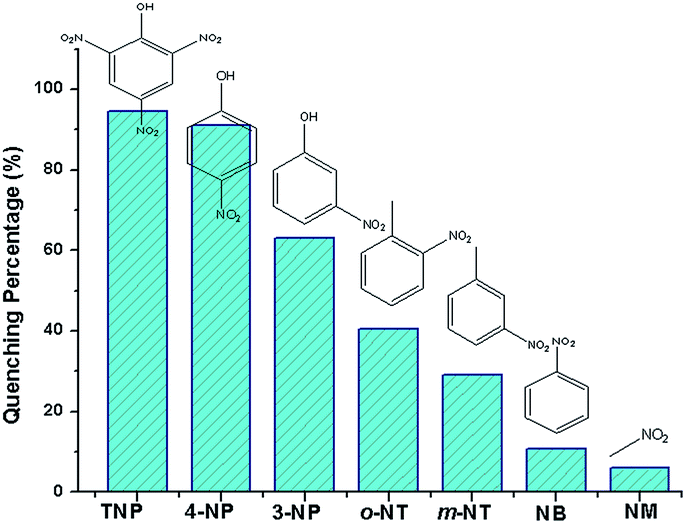 | ||
| Fig. 4 Luminescence quenching percentage when 2a was dispersed in seven different nitro explosives in the DMF (excited and monitored at 340 nm and 615 nm, respectively). | ||
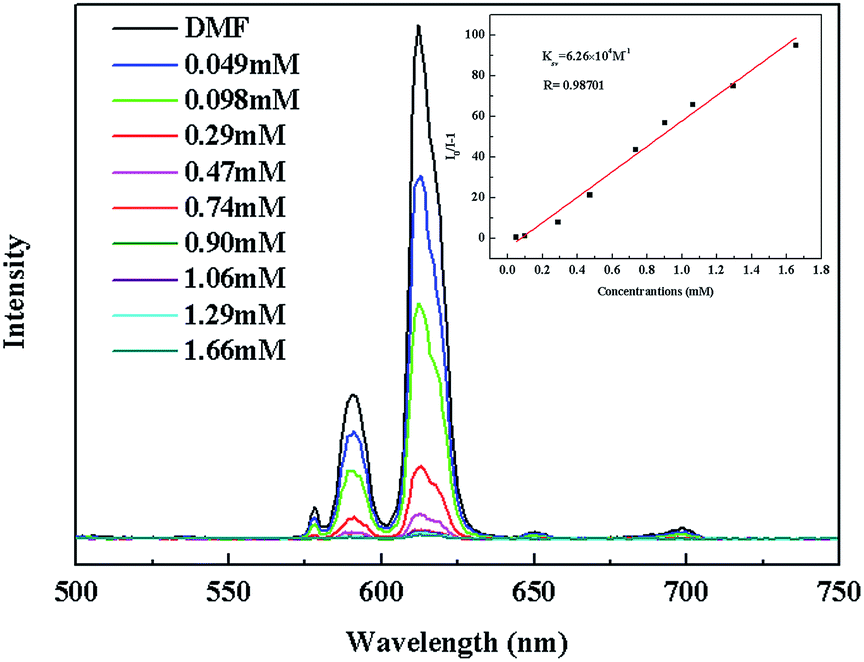 | ||
| Fig. 5 The photoluminescence spectra of 2a DMF emulsion in the presence of various contents of TNP solvent. The insert is the Stern–Volmer plot for the quenching of 2a by TNP. | ||
The remarkable sensing performance of 2a in suspension motivated us to investigate its sensing capabilities in the detection of TNP in vapor phase. The PL spectra of 2a deposited on a quartz slide was monitored, before and after exposing it to the equilibrated vapor of TNP at a specified exposure time (0.5, 1, 2.5, 5, 7.5, 10, 15, and 20 min; Fig. S21 and 22, ESI†). Rapid response to the TNP vapor was observed; within 0.5 min, a quenching percentage of nearly 23% was reached. At longer exposure time, the quenching percentage almost reached a constant of about 27%.
To understand the origin of the high selectivity of 2a for TNP, the mechanism of quenching was investigated. Usually, the conduction band (CB) of electron rich MOF lies higher than the lowest unoccupied molecular orbitals (LUMOs) energies of nitro analytes and upon excitation the excited electron from CB transfers to the LUMOs orbitals of nitro analytes, thus quenching the fluorescence intensity.12,26 Another possible reason for the quenching is stronger excited light absorption by the analytes, as was observed for acetone. As exhibited in the UV-vis absorption spectra (Fig. S23, ESI†), the absorption efficiencies at 340 nm follow the order TNP > 4-NP > 3-NP > o-NT > m-NT ≈ NB ≈ NM. In addition, luminescence lifetime of 2a is a reasonable value 0.759 ms, in contrast, 2a@nitro compounds (TNP, 4-NP, 3-NP, o-NT, m-NT, NB, NM) have decay time of 0.470, 0.506, 0.532, 0.539, 0.601, 0.670, 0.682 ms respectively (Fig. S24–S31, ESI†), also indicating that the energy migration from ligand to Eu ions have been suppressed and the energy have transferred for ligand to the electron-deficient nitro compounds.27 As a result, the combination of electron transfer and excited light absorption give rise to the fluorescence quenching, making 2a one of the best sensitive fluorescence-based sensing materials for TNP detection.
Conclusions
In conclusion, a highly thermostable luminescent MOF [Eu(L)1.5(DEF)]n and its solvent-free phase [Eu(L)1.5]n was successfully synthesized. The dispersed emulsion of 2a in DMF exhibits strong fluorescence emission, which could be quenched by nitro compounds, especially for TNP with Ksv constant 6.24 × 104 M−1. The high selectivity and high sensitivity of the fluorescence response of 2a to TNP indicate that 2a could be used as an efficient fluorescence sensor for TNP.Acknowledgements
This work was supported by the financial aid from the National Natural Science Foundation of China (Grant no. 91122030, 21210001, 21221061 and 51372242), and the National Key Basic Research Program of China (no. 2014CB643802).Notes and references
-
(a) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC
; (b) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed
-
(a) Y. Salinas, R. Martinez-Manez, M. D. Marcos, F. Sancenon, A. M. Costero, M. Parra and S. Gil, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC
-
(a) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881–2885 CrossRef CAS PubMed
- H. Wang, K. X. Yao, Z. Zhang, J. Jagiello, Q. Gong, Y. Han and J. Li, Chem. Sci., 2014, 5, 620–624 RSC
-
(a) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC
- Y. Wang, J. Yang, Y. Y. Liu and J. F. Ma, Chem.–Eur. J., 2013, 19, 14591–14599 CrossRef CAS PubMed
-
(a) S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed
-
(a) M. Zhang, G. Feng, Z. Song, Y. P. Zhou, H. Y. Chao, D. Yuan, T. T. Tan, Z. Guo, Z. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed
- A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef CAS PubMed
-
(a) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
- Y. N. Gong, L. Jiang and T. B. Lu, Chem.
Commun., 2013, 49, 11113–11115 RSC
- S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed
-
(a) K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed
-
(a) J. C. Rybak, M. Hailmann, P. R. Matthes, A. Zurawski, J. Nitsch, A. Steffen, J. G. Heck, C. Feldmann, S. Gotzendorfer, J. Meinhardt, G. Sextl, H. Kohlmann, S. J. Sedlmaier, W. Schnick and K. Muller-Buschbaum, J. Am. Chem. Soc., 2013, 135, 6896–6902 CrossRef CAS PubMed
-
(a) B. Tsuie, J. L. Reddinger, G. A. Sotzing, J. Soloducho, A. R. Katritzkyb and J. R. Reynolds, J. Mater. Chem., 1999, 9, 2189–2200 RSC
- SAINT, Program for Data Extraction and Reduction, Bruker AXS, Inc., Madison, WI, 2001 Search PubMed
- G. M. Sheldrick, SADABS, University of Göttingen, Göttingen, Germany 1996 Search PubMed
- G. M. Sheldrick, SHELXL-97, Program for Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed
-
(a) S. J. Wang, Y. W. Tian, L. X. You, F. Ding, K. W. Meert, D. Poelman, P. F. Smet, B. Y. Ren and Y. G. Sun, Dalton Trans., 2014, 43, 3462–3470 RSC
-
(a) S. Viswanathan and A. d. Bettencourt-Dias, Inorg. Chem., 2006, 45, 10138–10146 CrossRef CAS PubMed
-
(a) M. Guo and Z.-M. Sun, J. Mater. Chem., 2012, 22, 15939–15946 RSC
-
(a) B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS
-
(a) J. M. Zhou, W. Shi, N. Xu and P. Cheng, Inorg. Chem., 2013, 52, 8082–8090 CrossRef CAS PubMed
- Ref. 3
(a) L. V. Meyer, F. Schonfeld and K. Muller-Buschbaum, Chem. Commun., 2014, 50, 8093–8108 RSC
-
(a) A. Saxena, M. Fujiki, R. Rai and G. Kwak, Chem. Mater., 2005, 17, 2181–2185 CrossRef CAS
-
(a) H. Sohn, M. J. Sailor, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2003, 125, 3821–3830 CrossRef CAS PubMed
- X.-Y. Dong, R. Wang, J.-Z. Wang, S.-Q. Zang and T. C. W. Mak, J. Mater. Chem. A, 2014 10.1039/c4ta04421e
Footnote |
| † Electronic supplementary information (ESI) available: Information of materials and measurements, synthesis and characterization data, supplementary data and figures, TGA and powder X-ray diffraction analyses. CCDC 1019319. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra13773f |
| This journal is © The Royal Society of Chemistry 2015 |