
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEDA complex photochemistry as a strategy for C–S bond formation
Helena F.
Piedra†
 ,
Illán
Tagarro†
and
Manuel
Plaza
*
,
Illán
Tagarro†
and
Manuel
Plaza
*
Departamento de Química Orgánica e Inorgánica and Instituto Universitario de Química Organometálica “Enrique Moles” and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: plazamanuel@uniovi.es
First published on 28th March 2025
Abstract
This review highlights the use of photochemically excited electron donor–acceptor (EDA) complexes as a sustainable, modern approach to carbon–sulfur bond formation. C–S bonds are essential in various fields, including pharmaceuticals, materials science, and agrochemicals, yet traditional synthetic methods often face challenges such as harsh conditions and high costs. EDA complexes, formed through non-covalent interactions between electron-rich donors and electron-deficient acceptors, undergo visible-light-induced single-electron transfer (SET) to generate reactive radical intermediates. These intermediates enable efficient, selective, and environmentally friendly C–S bond formation under mild conditions. The article explores recent examples of practical applications of these reactions, including their mechanism, providing a comprehensive understanding of these cutting-edge methods and their potential to advance sustainable synthetic chemistry.
 Helena F. Piedra | Helena Fernandez Piedra earned her BSc in Chemistry from the University of Oviedo in 2020. She then moved to Madrid to pursue a Master's in Organic Chemistry at the Autonomous University of Madrid, which she completed in 2021. Following her master's, she returned to Oviedo, where she is currently pursuing a PhD under the supervision of Prof. Carlos Valdés and Dr Manuel Plaza. Her research focuses on the development of novel C–S bond-forming alkenylation and dienylation reactions, utilizing photochemical activation of halogen bonding complexes. |
 Illán Tagarro | Illán Tagarro Peña completed in 2024 a BS in Chemistry at the University of Oviedo. He is currently completing his master studies at the same university under the supervision of Prof. Carlos Valdés and Dr Manuel Plaza. His research is devoted to the development of multicomponent transformations oriented to C–S bond formation that rely on photochemical activation of EDA complexes. |
 Manuel Plaza | Manuel Plaza finished his BS in Chemistry and master's studies at the University of Oviedo in 2014. In 2018, he completed his PhD in Synthesis and Chemical Reactivity at the same university under the supervision of Prof. Carlos Valdés, and subsequently undertook postdoctoral training in photochemistry for three years with Prof. Thorsten Bach at the Technical University of Munich. In 2021, he moved back to the University of Oviedo, where he started his independent career as a Margarita Salas group leader. Currently, he is a Ramón y Cajal assistant professor at the same university. His research focuses on the development of photochemical cross-coupling reactions and photobiocatalytic processes. |
1. General introduction
Carbon–sulfur bonds hold immense significance in organic chemistry due to their widespread occurrence in biologically active molecules, functional materials, and natural products.1 Organic compounds featuring C–S bonds (representative classes are shown in Fig. 1) are essential components in pharmaceuticals, agrochemicals, and industrial applications, making their efficient and selective synthesis a cornerstone of modern research.2 While traditional methodologies for C–S bond formation, such as nucleophilic substitution,3 metal-catalyzed cross-coupling reactions,4 and radical-mediated processes,5 have undergone substantial advancements, many of these approaches are constrained by harsh reaction conditions, reliance on expensive catalysts, or the use of stoichiometric reagents. These limitations often hinder sustainability and restrict compatibility with sensitive functional groups. | ||
| Fig. 1 Representative families of sulfur-containing organic molecules. | ||
In recent years, photochemistry has emerged as a transformative tool in organic synthesis, offering novel pathways to access diverse radical-based chemical transformations,6 including C–S bond formation.7 A particularly promising innovation in this domain is the utilization of electron donor–acceptor (EDA) complexes to drive photochemical reactions.8 EDA complexes are non-covalent assemblies formed through supramolecular interactions between an electron-rich donor and an electron-deficient acceptor. Upon absorption of visible light, these complexes are promoted to an electronically excited state, characterized by charge-transfer interactions. This excitation triggers single electron transfer (SET) from the donor to the acceptor, generating a radical ion pair (Scheme 1b). These radical ions are highly reactive intermediates, capable of engaging in a variety of bond-forming processes, including C–C, C–heteroatom, and, notably, C–S bond construction.9 The SET process is facilitated by the alignment of frontier molecular orbitals (HOMO and LUMO) of the donor and acceptor, ensuring efficient electron transfer. The photochemical strategies based on EDA complexes often circumvent the need for metals, employ readily available and inexpensive starting materials, and operate under mild, ambient conditions. Such features align with the principles of green chemistry, emphasizing sustainability and reduced environmental impact.10 Moreover, the ability to utilize visible light as a renewable and abundant energy source further highlights the appeal of EDA complex-based photochemical reactions in contemporary synthesis.11
 | ||
| Scheme 1 Activation modes through the photochemistry of EDA complexes for C–S bond formation. | ||
Recent studies have illustrated that photochemically activated EDA complexes enable C–S bond formation with remarkable efficiency, selectivity, and functional group compatibility. This review article aims to provide an overview of the burgeoning field of C–S bond-forming reactions enabled by photochemical excitation of EDA complexes, delving into the reaction mechanisms proposed by the authors for such transformations, with a particular focus on the synthesis of small molecules.12 By advancing the understanding of these innovative strategies, this field has the potential to influence a wide array of disciplines and drive sustainable advancements in synthetic chemistry.
2. Creation of C–S bonds through photochemical excitation of EDA complexes
2.1. General mechanisms
As outlined in the introduction, light-driven excitation of an EDA complex typically facilitates a photoinduced electron transfer (PET) from the electron donor to the electron acceptor within the complex. This process results in the formation of a radical ion pair, which can evolve through various pathways to generate new chemical bonds (Scheme 1a).In the context of photochemical activation of EDA complexes to form C–S bonds, two general mechanisms can be distinguished. In the first, an electron acceptor 1 forms an EDA complex with a nucleophilic sulfur source 2 as the electron donor (Scheme 1b). Upon photochemical excitation, the EDA complex 3 generates a radical anion 4 and a sulfur-centered radical 5. The radical anion 4, being unstable, undergoes irreversible fragmentation to produce a new radical species 6 and eliminating a leaving group (LG). Subsequent radical recombination between this newly formed radical 6 and the sulfur-centered radical 5 leads to the formation of a new C–S bond, yielding the final product 7. Alternatively, the electron acceptor 1 can be activated using a sacrificial electron donor 8, which does not contain a sulfur motif (Scheme 1c). In this case, the resulting EDA complex 9 generates the radical anion 4 and a radical cation 10 upon excitation. As before, the radical anion 4 fragments to form the radical intermediate 6, while the salt 11 is produced as a byproduct. It should be noted at this point that catalytic amounts of the sacrificial donor can be used, and this will be contemplated later through specific examples. The radical intermediate 6 is then intercepted by a sulfur-containing radical trapping agent, forming a new C–S bond and yielding the final product 13. These complementary approaches have been explored extensively by various research groups to synthesize diverse organosulfur compounds, leveraging the unique advantages of EDA complex photochemistry. In the subsequent sections of this review, these reactions will be categorized based on the type of C–S bond formed (Csp2–S or Csp3–S). While the mechanistic discussion will be addressed in detail in most of the cases, the analysis of the experimental work that led the authors to propose each mechanism is beyond the scope of this review. Readers seeking a deeper understanding of the specific studies are encouraged to consult the original publications or a review by Prof. Stephenson on mechanistic investigations of EDA complexes.13
2.2. C(sp2)–S bond forming reactions
The most extensively developed and successful chemistry for the photochemical activation of EDA complexes aimed at carbon–sulfur bond formation has undoubtedly focused on C(sp2)–S bonds.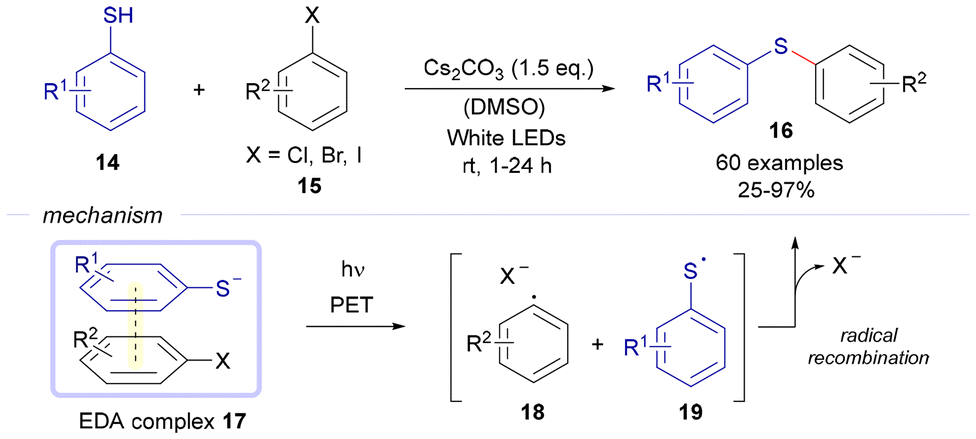 | ||
| Scheme 2 Photochemical catalyst-free preparation of aryl thioethers 16 from aryl halides 15 and thiophenols 14. | ||
Following this work, numerous transformations utilizing organic halides as electron acceptors and nucleophilic sulfur species as electron donors were reported. Notably, in 2019, Yang and co-workers developed a synthetic protocol for the preparation of S-aryl dithiocarbamates 21 from aryl halides 15, amines 20, and carbon disulfide (Scheme 3).15 Mechanistically, the reaction begins with the generation of an anionic species via deprotonation of the amines 20 under basic conditions. This anionic intermediate then reacts with carbon disulfide to form a sulfur-centered anionic species 22, which acts as the electron donor in the EDA complex 23, formed with the organic halide 15 as the electron acceptor. Upon photochemical activation, the EDA complex undergoes fragmentation, generating radical intermediates 24 and 18. These radicals subsequently recombine to yield the desired S-aryl dithiocarbamates 21.
 | ||
| Scheme 3 Light-driven synthesis of aryl S-aryl dithiocarbonates 21 from aryl halides 15, amines 20 and carbon disulfide. | ||
Following the first work by Yang and colleagues, additional EDA-complex-mediated transformations were developed, employing intermediates 22 as electron donors while varying the electron-accepting precursors. In 2022, the same group explored the use of Katriztky salts as electron acceptors (via EDA complex 25; Scheme 4a).16 More recently, different research groups employed thianthrenium salts as alternative reagents in this type of transformation (via EDA complex 26; Scheme 4b).17 Due to the mechanistic similarities with Yang's earlier work, a detailed discussion of these reactions will be omitted.
 | ||
| Scheme 4 Dithiocarbamates as electron donors in the reaction between Katriztky salts or thianthrenium salts. | ||
In 2021, Sekar and co-workers introduced a novel method for the direct synthesis of diverse heteroaryl thioethers using a halogen-bonding-initiated photochemical reaction.18 This reaction is initiated by the photochemical excitation of a halogen-bonding (HaB) complex, which is a specific type of EDA complex. Electron-acceptor molecules that possess a covalently bonded halogen atom typically exhibit a positive electrostatic potential region along the axis of the covalent bond, referred to as a σ-hole.19 This feature enables the possibility of a non-covalent attractive interaction between an electron-acceptor molecule and an electron donor or nucleophile. This interaction involves a partial n → σ* charge transfer, where electrons from a non-bonding orbital of the electron donor (HaB acceptor, electron donor) are transferred to an antibonding orbital (σ*) of the electron acceptor (HaB donor).20 The nature of the halogen bond influences significantly the electron transfer efficiency within halogen-bonding-assisted EDA complexes. Due to the greater size of the σ-hole, stronger halogen bonds are formed when more polarizable halogen atoms are involved as the halogen-bond donor (e.g., iodine over bromine or chlorine). Additionally, the electronic environment of the halogen-bond donor (such as electron-withdrawing substituents) modulates the strength of the interaction, thereby impacting the overlap between frontier molecular orbitals with the halogen-bond acceptor, therefore facilitating electron transfer. These factors collectively dictate the efficiency of the halogen-bonding complex formation and subsequent reactivity. In particular, the transformation reported by Sekar, outlined in Scheme 5, proceeds in two distinct steps. Initially, the heterocyclic compound undergoes iodination. Subsequently, a photochemically driven cross-coupling reaction between the resulting heteroaryl iodide (from 27, 29 or 31) and a thiol leads, respectively, to the formation of azaindoles 28, quinolines 30 and isoquinolines 32. For example, in the transformation aiming at isoquinoline formation, the reaction mechanism suggests that a halogen-bonding complex 33, formed between the heteroaryl iodide and the thiolate anion, serves as the precursor for cross-coupling. Photoinduced fragmentation of this complex generates radicals 34 and 19, which recombine to produce in this case the thioether 32.
 | ||
| Scheme 5 One-pot strategy for the direct synthesis of heteroaryl thioethers via iodination followed by C–S bond formation. TBHP: tert-butylhydroperoxide. | ||
Following the previous work, a different C–S cross-coupling strategy for the synthesis of different thiochromanes via photoinduced electron transfer was also reported by the same research group (Scheme 6).21 In this approach, 2-iodochalcones 35 were combined with potassium ethyl xanthate 36 to form the HaB complex 42. Upon photoexcitation with visible light, the HaB complex underwent electron transfer and fragmentation, generating radical species 43 and 44. The rapid recombination of these radicals resulted in the formation of carbonodithiolate 45. This intermediate then decomposed, facilitated by an additional molecule of potassium ethyl xanthate, producing a new thiolate 46, which subsequently transformed into thiochromane 37 through an intramolecular Michael addition. Using a similar pathway, a three-component reaction involving o-iodobenzaldehydes 38, sodium ethyl xanthate, and α,β-unsaturated carbonyls 39 led to the formation of thiochromenes 40 and thiochromanols 41. In these processes, the thiolate intermediate, generated via the HaB-photochemically activated reaction, underwent a sequence of intermolecular Michael addition and intramolecular aldol reaction, yielding the condensed heterocyclic products 40 or 41.
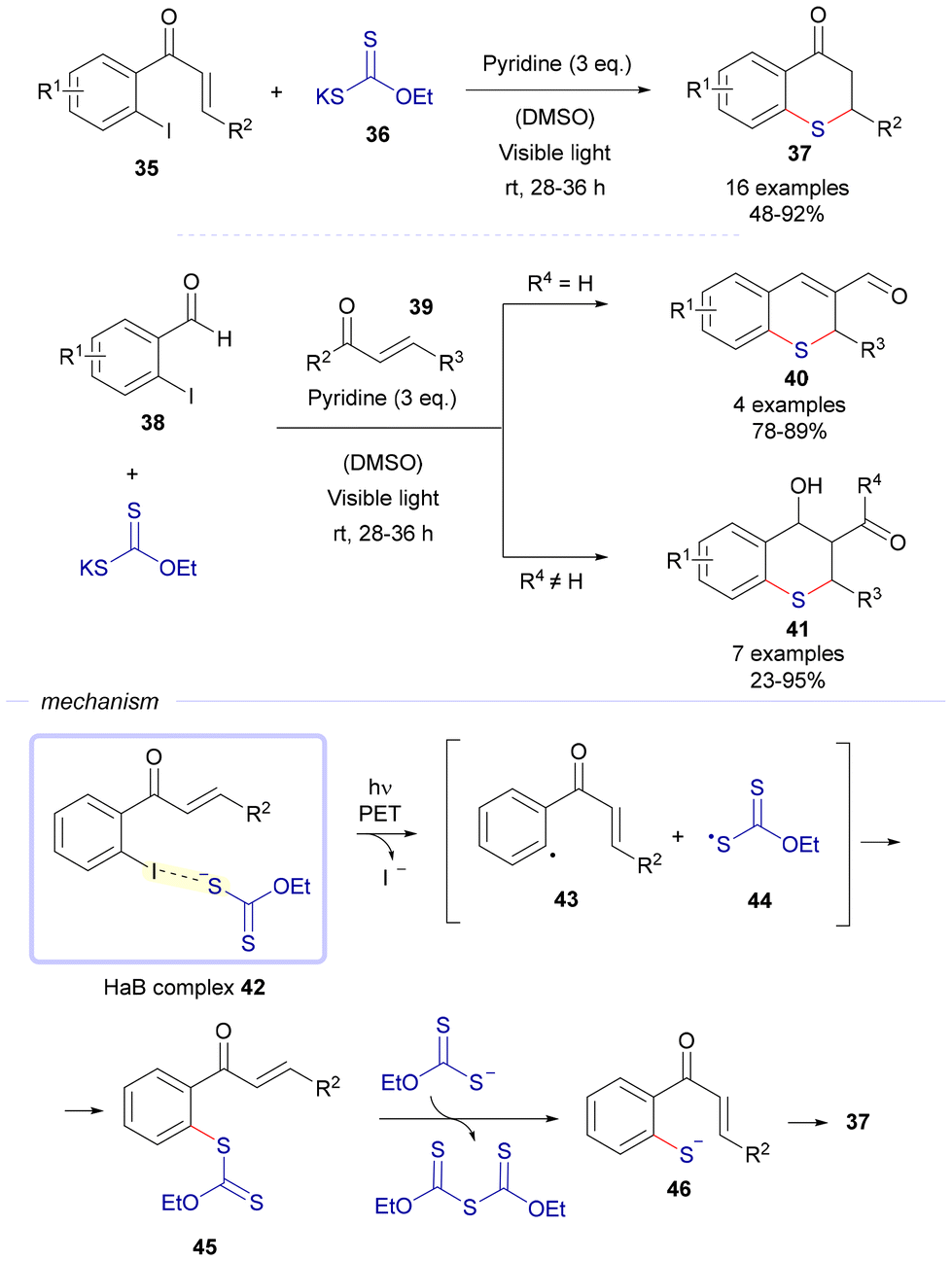 | ||
| Scheme 6 Photochemical synthesis of compounds 37, 40 and 41via activation of halogen-bonding complexes. | ||
Again in 2021, Xia and Wang reported a C–S bond forming reaction via photochemical excitation of EDA complexes formed between aryl diazonium salts 47 and sulfinate salts 48 (Scheme 7).22 In this case, the light-driven excitation of the EDA complex 50 affords the formation of the aryl radical 18 and the ambidentate sulfonyl radical 51, which rapidly recombine to create the aryl sulfones 49. It is important to highlight the novel role of diazonium salts as electron acceptors in this kind of photochemical transformations oriented to C–S bond formation.
 | ||
| Scheme 7 Sulfonylation transformations of aryl diazonium salts 47via light-driven excitation of EDA complexes 50. | ||
Alternatively, in 2022, Molander and co-workers demonstrated the use of aryl thianthrenium salts as electron acceptors for C–S bond formation via photochemical excitation of EDA complexes (Scheme 8). That same year, the same group reported a cross-coupling strategy involving thiophenols 14 or sulfinate salts 48, which act as nucleophilic electron donors, with aryl thianthrenium salts 52 to synthesize aryl thioethers 16,23 and sulfones 49,24 respectively. Mechanistically, the reaction is initiated by light-driven excitation of the EDA complex 53. Photoinduced electron transfer followed by fragmentation generates aryl-centered radical 18 and the corresponding sulfur-centered radical, with thianthrene (TT) forming as a byproduct in stoichiometric amounts. The recombination of these radical intermediates yields the final products, either aryl thioethers 16 or sulfones 49, depending on the sulfur donor employed. Given the significance of thianthrenium salts as electron acceptors in photochemical processes,25 these transformations mark an important milestone in C–S bond formation via this strategy. Noticeable, starting from the same reaction precursors, the same year the group of Zhang reported an application of this transformation to the preparation of bioactive and DNA-encoded molecules.26
 | ||
| Scheme 8 Thioetherification and sulfonylation reactions of thianthrenium salts 52via EDA complex photochemistry. | ||
In 2022, Liu and co-workers developed a [3 + 2] cyclization reaction between chalcones 54 and 2-mercaptobenzoimidazoles 55 under basic conditions for the preparation of imidazo[2,1-b]thiazoles 56 (Scheme 9).27 In the presence of Cs2CO3, 2-mercaptobenzimidazole generates the corresponding thiol anion intermediate 58. Chalcone 54 subsequently reacts with this anionic intermediate, forming an EDA complex 57. Upon exposure to visible light, the EDA complex undergoes an intermolecular single electron transfer, resulting in radical intermediate 60 and radical anion species 59. The radical anion 59 then reacts with compound 55, yielding radical 61 and regenerating intermediate 60. Radical 61 is quenched by O2, forming 54. At the same time, the peroxyradical released in this step abstracts a hydrogen from 55, generating H2O2 and radical 60. Afterwards, radical intermediate 60 attacks compound 54 to produce the benzylic radical 62. This radical 62 is subsequently transformed into the cationic intermediate 63 through the action of H2O2. Finally, cationic intermediate 63 undergoes nucleophilic cyclization to form intermediate 64, which is aromatized to yield the desired products 56.
 | ||
| Scheme 9 Synthesis of imidazothiazoles 56via EDA complex photochemistry. | ||
The same year, Bugaenko and Karchava reported a simple procedure for the thioesterification of aryl halides 15 with potassium thiocarboxylates 65 under photochemical conditions (Scheme 10).28 Mechanistically, the transformation is initiated by the formation of an EDA complex 67 between both reaction starting materials 15 and 65. This aggregate undergoes light-driven fragmentation into the radical species 18 and 68, which subsequently recombine to afford the thioesters 66. It should be noted that in 2024, the group of Sharma reported a similar transformation, in which instead of aryl halides, aryl thianthrenium salts were employed as electron acceptors.29
 | ||
| Scheme 10 Light-driven thioesterification reactions of aryl halides 15 with potassium thiocarboxylate salts 65. | ||
It should be noted that, very recently, the group of Liu reported a similar reaction for thioesterification based on EDA complex formation with thioester salts, but this time employing thianthrenium salts as electron acceptors.30 Since the reaction proceeds in a similar way to the thioesterification reported by Bugaenko and Karchava, a detail discussion will be omitted.
Again in 2022, Wang and Wang reported a visible-light-driven protocol for the preparation of aryl xanthates 71 and aryl dithiocarbamates 73 through the reaction of dibenzothiophenium salts 69 with either sodium xanthogenates 70 or dithiocarbamates 72, respectively (Scheme 11).31 These salts are synthesized through the reaction of either alcohols or amines with CS2 in basic media. The reaction is started in both cases by the formation of an EDA complex 74 between the electron acceptor part 69 and the corresponding nucleophilic sulfur source. Photoexcitation of this aggregate leads to the intermolecular SET event responsible for the fragmentation of the complex, creating the aryl radical 18 and the sulfur-centered radical 75, along with stoichiometric amounts of dibenzothiophene (DBT). The radical intermediates recombine to afford, depending on the specific reaction, either 71 or 73 as the final products.
 | ||
| Scheme 11 Synthesis of aryl xanthates 71 and dithiocarbamates 73 driven by visible light through the EDA complex 74. | ||
In 2022, Guo and Chen reported more progress in this field with the development of a novel method for synthesizing aryl disulfides. Their approach involved the use of aryl halides 15 and potassium sulfide, employing an EDA complex-mediated strategy (Scheme 12).32 Notably, while their method is also applicable to the preparation of aryl thioethers, this aspect is beyond the scope of this review as it requires the use of a photocatalyst. The transformation begins with the formation of an EDA complex 77 between the aryl halide and the nucleophilic sulfur anion S2−. Visible-light-driven fragmentation of the complex then occurs to create the aryl radical 18 and the sulfur radical anion 78. A radical recombination of these two species leads to the formation of a sulfur thiophenolate 79, which forms a different EDA complex 80 with another molecule of the aryl halide 15. Again, at the specific irradiation wavelength of the reaction, a photochemical fragmentation into the corresponding aryl radical 18 and sulfur-centered radical 19 takes place. The first radical recombines with the intermediate 78 to ensure the formation of another equivalent of the aryl thiphenolate 79, while radical recombination of two different units of 19 create the final products of the reaction, the aryl disulfides 76.
 | ||
| Scheme 12 Construction of C–S bonds using inorganic sulfur salts and aryl halides via photoinduced activation of EDA complexes. | ||
In 2023, Xia and Wang reported a β-trifluoromethylthiolation reaction of tertiary amines 81 and enamines 90 using N-trifluoromethylthiophthalimide 82.33 Since these reactions follow distinct pathways, each transformation is presented separately in Scheme 13. For the trifluoromethylthiolation of amines 81, the process begins with the photochemical activation of the EDA complex 84, leading to its fragmentation into a phthalimide anion 85, a trifluoromethylthio radical 86, and an amine radical cation 87. The latter undergoes hydrogen atom abstraction by radical 86, generating the iminium species 88. Subsequent deprotonation of 88 by either 85 or sodium carbonate produces an enamine intermediate 89, which then reacts with the electrophilic trifluoromethylthiophthalimide 82 to yield the final products 83. In the case of the trifluoromethylthiolation of enamines 90, the reaction proceeds through the formation of an EDA complex 92. Upon light-driven excitation, this complex dissociates into the phthalimide anion 85, the trifluoromethylthio radical 86, and a radical cation intermediate 93. Radical recombination between 93 and 86 generates the iminium cation 94, which undergoes base-mediated deprotonation by either 85 or sodium carbonate to afford the final products 91.
 | ||
| Scheme 13 Photochemical trifluoromethylthiolation reactions of tertiary amines 81 and enamines 90. HAT: hydrogen atom transfer. | ||
In 2024, Hu and Xu reported a cyclization reaction of alkynes to furnish 3-sulfonylindoles 98 and vinyl sulfone oxindoles 99, depending on which starting material, 95 or 96, respectively, was used in combination with a sodium sulfinate 97 (Scheme 14).34 In order to illustrate the mechanism of this transformation, the case of the synthesis of compounds 98 will be contemplated. In this case, the corresponding sodium sulfinate reacts with the starting material 95 in the presence of K2CO3 to form an EDA complex 100. Under light exposure, the EDA complex undergoes an intermolecular single electron transfer process from the donor 97 to the acceptor 95, leading to the generation of a sulfonyl radical 102 and an N-sulfonamide radical anion 101. The fragmentation of radical anion 101 yields a sulfonyl anion and an N-centered radical intermediate 103. A 5-exo-dig cyclization of intermediate 103 results in the formation of an indole radical intermediate 104. Subsequently, intermediate 104 and sulfonyl radical 102 undergo selective radical–radical coupling to produce the target compound 98.
 | ||
| Scheme 14 EDA complex-facilitated cascade cyclization of alkynes for the synthesis of 3-sulfonylindoles 98 and vinyl sulfone oxindoles 99. | ||
Also in 2024, the group of Ramesh reported an EDA-complex-mediated photochemical thioetherification reaction of halogenated pyrazole-5-amines 105 with thiophenols 14 under basic conditions (Scheme 15).35
 | ||
| Scheme 15 Photochemical synthesis of thiolated pyrazole-5-amines 106via formation of EDA complexes. | ||
Mechanistically, the reaction is believed to proceed through the formation of an EDA complex 107 between the deprotonated thiophenol and the electron acceptor, the corresponding compound 105. The attractive force that is responsible for the creation of such aggregate could be due to halogen-bonding and/or π–π interactions. Subsequent light-driven fragmentation of the complex leads to the formation of two different radical intermediates: a carbon-centered radical 108 and a sulfur-centered radical 19. An eventual radical recombination step grants the creation of the final sulfurated heterocycles 106.
In 2024, Yang and co-workers reported a one-pot multicomponent reaction which makes use of isothiocyanates 107, isocyanides 108, and thianthrenium salt-functionalized arenes 52 to access trisubstituted imidazoles 109 (Scheme 16).36 The transformation is started by the base-mediated deprotonation of isocyanides 108, which initially undergo a [3 + 2] cycloaddition with aryl isothiocyanates 107, resulting in the formation of intermediate 110. Through tautomerism, sulfur anions 111 are generated, which can behave as electron donors. These anions then form an EDA complex 112 in solution with thianthrenium salts 52. Upon exposure to visible light, the ground-state EDA complex transitions to its excited state, triggering an intermolecular single electron transfer from sulfur anions 111 to thianthrenium salts 52. This leads to the release of thianthrene and the formation of an aryl radical 18 along with a thiyl radical 113. Finally, a radical coupling between 18 and 113 produces the target products 109.
 | ||
| Scheme 16 One-pot multicomponent reaction of isothiocyanates 107, isocyanides 108, and thianthrenium salts 52 to prepare trisubstituted imidazoles 109. | ||
In the realm of C(sp2)–S bond-forming reactions, our research group has pioneered synthetic methodologies leveraging the generation of highly reactive alkenyl radicals. Our approach employs alkenyl halides as starting materials, which serve as the electron acceptor component in EDA complexes, specifically those stabilized by halogen-bonding interactions. Various nucleophilic sulfur-based species (including thiols 115, phosphorothioate diester salts 117, sulfinates 83, and thiocyanate salts) are utilized to enable streamlined thioetherifications,37 carbothiophosphorylations,38 sulfonylations,39 and thiocyanations,40 respectively. In parallel to the development by our group of the sulfonylation reaction, the group of Wang and Luo developed a transformation starting from the same reaction precursors, alkenyl bromides 114 and sulfinate salts 83.41 However, different reaction conditions (irradiation from 390 nm LEDs, 16 h) to ours (427 nm LEDs, 0.5 h) were reported. All in all, these transformations are distinguished by their remarkable functional group tolerance, high stereoselectivity, operational simplicity, and scalability. Detailed mechanistic investigations were conducted to confirm both the radical nature of the process and the role of the halogen-bonding EDA complex as the initiator. Under visible light, the fragmentation of the corresponding HaB complex 121 generates an alkenyl radical 122 and a sulfur-centered radical 123. Subsequent radical recombination of these intermediates culminates in the formation of the specific C(sp2)–S bond in each of the described transformations in Scheme 17.
 | ||
| Scheme 17 Thioetherifications, carbothiophosphorylation, sulfonylation and thiocyanation reactions of alkenyl bromides 114 based on photochemical activation of HaB complexes. | ||
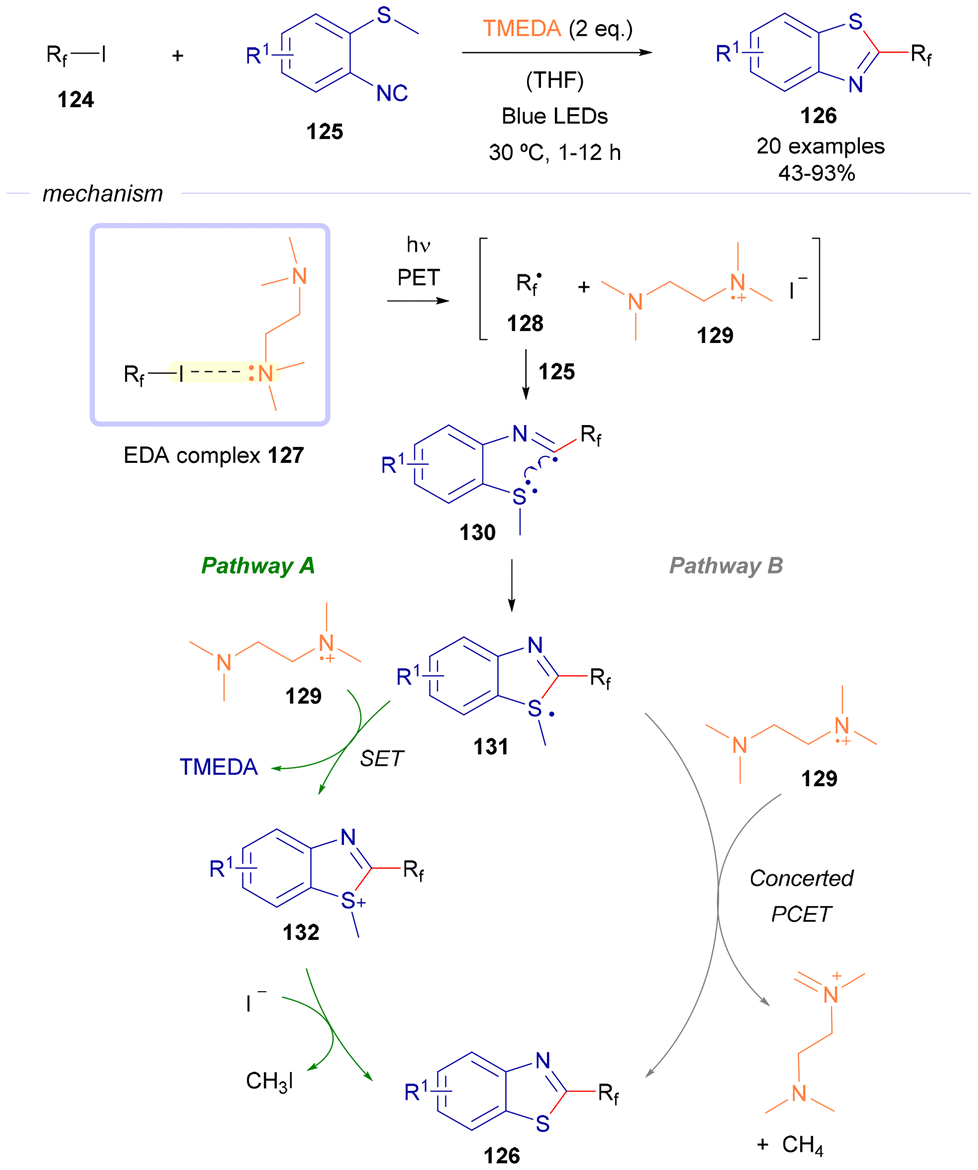 | ||
| Scheme 18 Synthesis of 2-perfluoroalkylbenzothiazoles 126 based on the employment of TMEDA as the sacrificial donor. | ||
Also the same year, in 2019, Quian and Xuan reported a decarboxylative sulfonylation of cinnamic acids based on light-driven activation of EDA complexes.43 A conceivable reaction pathway is illustrated in Scheme 19. Initially, EDA complex 136 was generated through the interaction between an aryl sulfonate phenol ester 134 and dimethylacetamide (DMA) in the presence of Cs2CO3. Exposure to visible light activated the complex (136*), which then underwent single-electron transfer, yielding radical anion species 138 alongside a carbon-centered radical intermediate, 137. The subsequent fragmentation of 136 released a tosyl radical, which reacted with the double bond of the cinnamic acid to form a benzyl radical, 140. Oxidation of benzyl radical 140 by the excited complex 136* led to the formation of intermediate 141 while regenerating radical anion 138. Finally, 141 underwent decarboxylation, resulting in the formation of the desired vinyl sulfone product 135.
 | ||
| Scheme 19 Preparation of alkenyl sulfones 135 through the initial formation of an EDA complex 136 between 134 and DMA. | ||
In 2022, three years later, Huang and Zhang reported a comparable transformation to the one previously described, substituting cinnamic acids with alkynes to achieve an oxosulfonylation reaction.44 Given the similarity in the reaction mechanism to the one discussed earlier, a detailed mechanistic explanation will not be included here.
Interestingly, the group of Tang reported in 2021 a photochemical reaction via EDA complexes where catalytic amounts of the sacrificial donor were used.45 This new activation mode within the EDA complex photochemistry was used for the cross-coupling between indoles 142 and thiophenols 14 to furnish the final products 143, where B(C6F5)3 served as a catalyst to facilitate such process (Scheme 20). Mechanistically, the process begins with the formation of an EDA complex 144 between indole 142 and B(C6F5)3. Upon exposure to visible light, a single-electron transfer takes place within the EDA complex, leading to the creation of a radical anion 145 and a radical cation 146. Intermediate 145 reacts with oxygen to form the oxygenated intermediate 147, which abstracts a hydrogen atom from 14 to produce intermediate 149 and a sulfur-centered radical. Protonation of complex 149 results in the formation of hydrogen peroxide, and B(C6F5)3 is regenerated, completing the catalytic cycle. Hydrogen peroxide oxidizes 14 and can also generate sulfur radical 19 along with water. Intermediate 146 interacts with 19 to yield intermediate 148, which is subsequently deprotonated to produce the final product 143. All in all, this approach introduces a novel reaction framework that employs B(C6F5)3 as a single-electron oxidant.
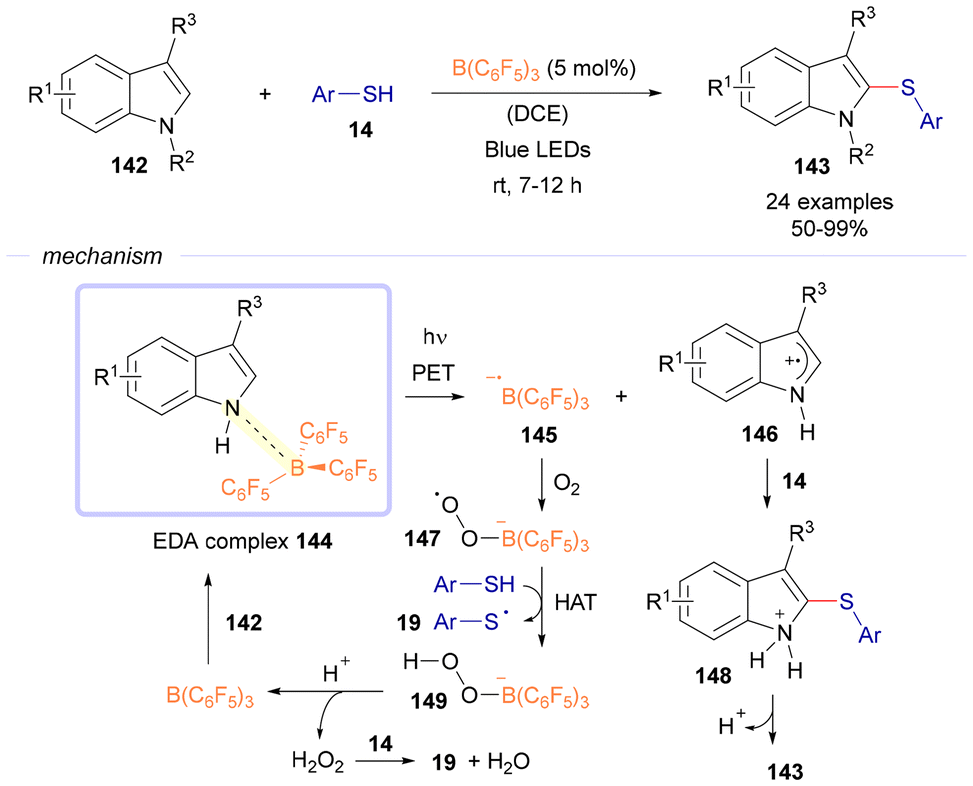 | ||
| Scheme 20 Photochemical C–S bond forming reactions between indoles 142 and thiols 14 catalyzed by B(C6F5)3. | ||
In 2021, Li and co-workers developed a novel strategy for C–S bond formation utilizing the photochemistry of EDA complexes in the presence of surfactants (Scheme 21).46 The method enabled the preparation of alkenyl or alkyl sulfones 152 by combining sulfonyl chlorides 151, NaI, catalytic amounts of cetyltrimethylammonium bromide (CTAB) as a surfactant, and alkenes or alkynes 150 as radical trapping agents. Mechanistically, the reaction proceeds via the formation of an EDA complex 153 in water, where the aromatic sulfonyl chloride acts as the electron acceptor and iodide serves as a sacrificial donor for photochemical activation. The surfactant assists in the assembly of the EDA complex 153via encapsulation of the different starting materials. Upon irradiation with blue light, the EDA complex undergoes fragmentation, generating two distinct radical species, 51 and I−. These radicals are subsequently intercepted by the alkenes or alkynes, resulting in a 1,2-difunctionalization of the unsaturation and yielding the final sulfone products 152.
 | ||
| Scheme 21 Assistance of surfactants in EDA complex formation for the preparation of sulfones 152 from sulfonyl chlorides 151, NaI and alkenes/alkynes. | ||
The assistance of sodium iodide as a sacrifical electron donor in EDA complex photochemistry oriented to C–S bond formation was also exploited by the group of Jiang and Li to carry out sulfonylation reactions of alkenes 155 or alkynes 157, rendering respectively alkenyl sulfones 156 or alkynyl sulfones 158 (Scheme 22).47 In these reactions, thiosulfonates 154 were employed as the electrophilic species responsible for the formation of the EDA complex 159 with sodium iodide. Photochemical excitation of the aggregate creates the sulfonyl radical 51 and iodide radical intermediates. Then, depending on the radical-trapping agent, two different transformations can occur based on an atom transfer radical addition process. In the radical intermediates are intercepted with alkenes 155, a new intermediate 160 is formed, which upon elimination of HI forges the alkenyl sulfones 156. On the other hand, if alkynes are used, the intermediate 161 is created, which also can undergo elimination of HI to furnish the corresponding alkynyl sulfones 158. It should be noted that in this last case, due to the elimination step, overall, a C(sp)–S bond is formed in the transformation to forge compounds 158.
 | ||
| Scheme 22 Synthesis of alkenyl or alkynyl sulfones by visible light through an EDA complex 159 with assistance of NaI. | ||
In 2023, the group of Mancheño reported a visible-light driven thiolation reaction of 2-chlorothiophenes 162 employing disulfides 163 as the sulfur source (Scheme 23).48 The mechanism of this transformation is believed to proceed through the generation of the EDA complex 165, formed between the 2-cholothiophene (electron acceptor) and the diisopropylethylamine (DIPEA, sacrificial electron donor). Photochemical excitation of this complex forms the corresponding radical cation amine 167 and the carbon-centered radical 166, which arises from the reduction of the C–X bond. At this point, the first radical intermediate is deprotonated with the assistance of potassium carbonate, and reacts with the starting disulfide 163 to form a new carbon-centered radical 168 and a sulfur-centered radical 169. Intermediate 168 is believed to start a radical chain propagation through its reaction with another equivalent of 162, generating in this way an additional equivalent of the radical intermediate 166 and 167. At this point, 167 undergoes a single electron transfer event with 169, generating the sulfur-centered cationic species 170 and an equivalent of 168. Intermediate 170 undergoes fragmentation, eliminating a thiophenolate anion and generating the final products of the reaction 164. The existence of this radical chain propagation mechanism is supported by the quantum yield measurement (ϕ = 4).
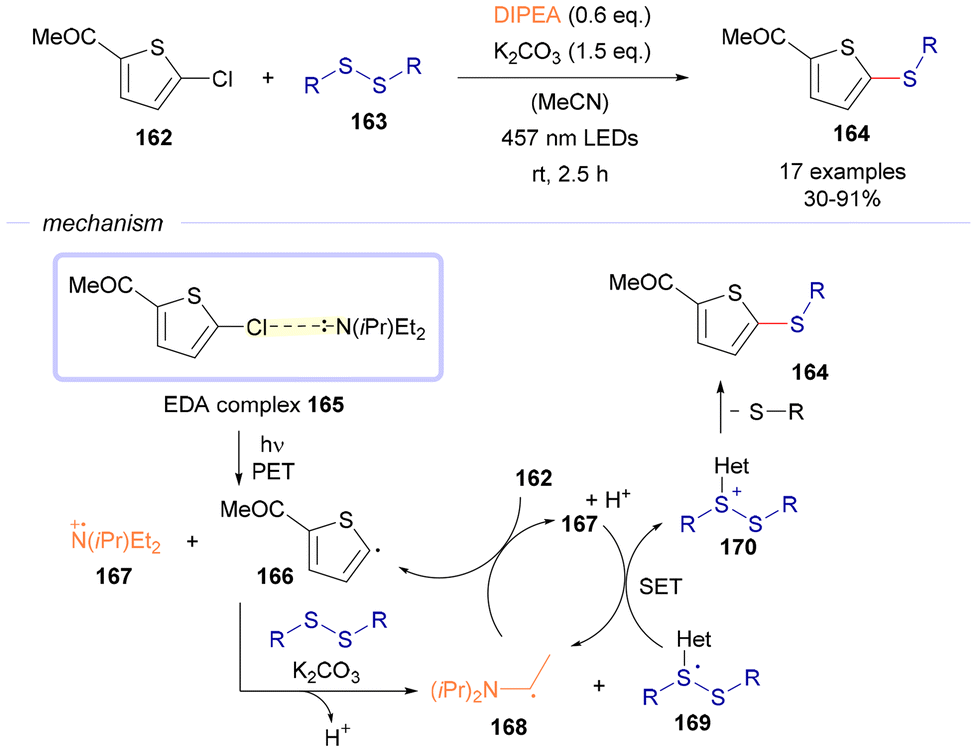 | ||
| Scheme 23 Photoactivation of alkylamine-based EDA complexes for the thiolation of 2-chlorothiophenes 162. | ||
In summary, this section highlights how photochemical activation of EDA complexes serves as a powerful strategy for C(sp2)–S bond construction. To date, the developed methodologies incorporate a variety of electron acceptor molecules (primarily organic halides and sulfonium salts) paired with nucleophilic electron donors, typically sulfur-centered anionic species such as thiolates or sulfinates. Alternatively, the use of sacrificial agents (e.g., amines, enolates, or iodide anions) in combination with sulfur sources has also been demonstrated as an effective approach for C(sp2)–S bond formation within EDA complex photochemistry.
2.3. C(sp3)–S bond forming reactions
In addition to the significant progress made in the photochemical construction of C(sp2)–S bonds, substantial advancements have also been achieved in analogous methodologies for forming C(sp3)–S bonds. As in the previous section, the following subsections will be organized based on the type of photochemical transformation. | ||
| Scheme 24 Photochemical cascade reactions for the construction of 3,4-dihydro-2H-1,4-thiazine-1,1-dioxides 173. TMS: trimethylsilyl. | ||
In 2019, the group of Liao reported a thioetherification reaction aiming at the formation of C(sp3)–S bonds via EDA complex photochemistry (Scheme 25).50 In this case, Katriztky salts 180 were used as electron acceptors in combination with different thiols 104, seeking the formation of an EDA complex 182 between both reactants to carry on a thioetherification transformation. Fragmentation of this complex upon exposure to blue light drives the formation of two different radical species, 183 and 184, along with the elimination of a pyridinium salt as a byproduct of the reaction. At this point two different paths are proposed to explain the formation of the final products of the reaction 181. Path A consists of a recombination of the carbon-centered radical 183 with an equivalent of the corresponding thiolate anion, resulting in the creation of a radical anion 185. This species reduces a different molecule of the starting material 180 through a single electron transfer, generating an additional equivalent of the radical intermediate 183 along with the thioether 181. Path B shows the direct recombination of the radical species 183 and 184 to forge the thioethers 181. To the best of our knowledge, this is one of the first examples in which Katritzky salts were used as electron acceptors in C–S bond forming reactions driven by the formation of EDA complexes.51
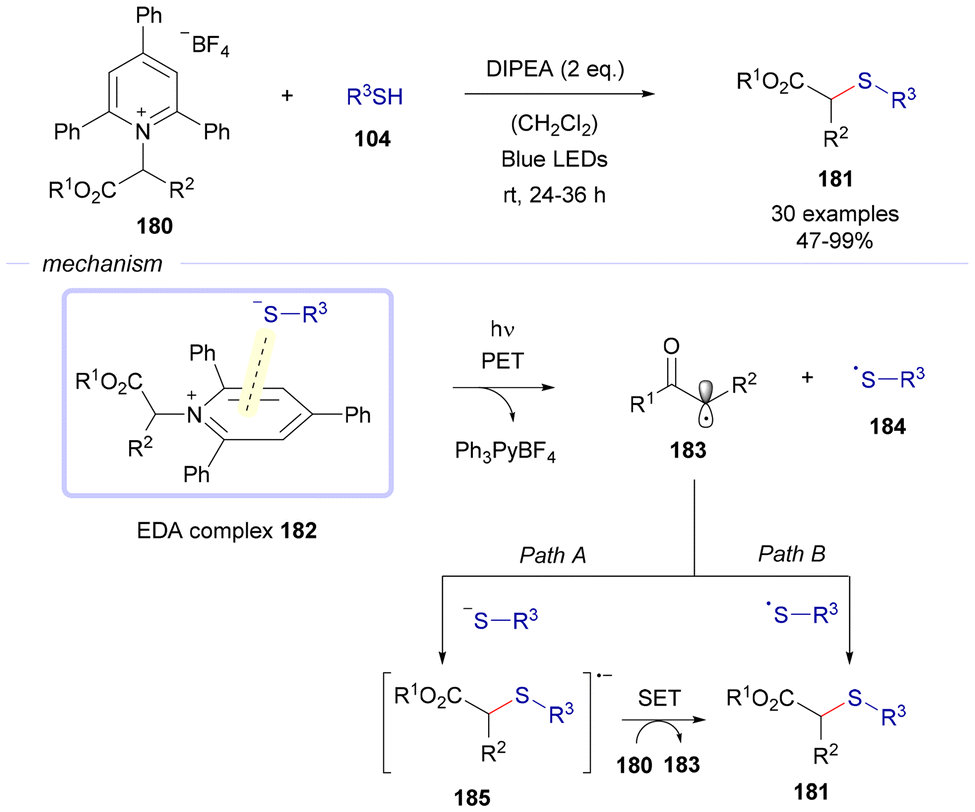 | ||
| Scheme 25 Thioetherification reactions of Katriztky salts 180 through the formation of EDA complexes 182. | ||
In 2021, both Akayima's and Xia's research groups independently developed comparable photochemical methods for activating C(sp3)–H bonds in the synthesis of sulfides. Xia and co-workers specifically explored the thiolation of etheric, benzylic, and allylic substrates 186 using thiophenols 14 (Scheme 26).52 Their approach involved the photochemical excitation of the halogen-bonding complex 189. In this study, 2,6-dimethyl iodobenzene 187 serves as the electron acceptor. Upon exposure to light, the typical sequence of photoexcitation, PET, and fragmentation produces radicals 19 and 190. Due to the steric hindrance of the 2,6-disubstituted iodide, direct radical recombination is prevented. Instead, the phenyl radical 190 abstracts a hydrogen atom from the Csp3–H bond of substrate 186, which is present in excess, leading to the formation of the Csp3-centered radical 192. This radical then recombines with the thiyl radical 19, yielding thioethers 188 in moderate to good yields. In contrast, Akayima's research proposes that the transformation is initiated by an EDA complex 194 between 4-bromoacetophenone 193 and an aromatic thiolate 14 (Scheme 27).53 Unlike Xia's study, no HaB complex was considered in this case. Instead, the authors noted that the reaction does not occur with alkanethiols, highlighting the crucial role of π–π stacking interactions in the EDA complex 194. In this case, photochemical excitation of the aggregate 194 leads to the formation of the radical species 19 and 195. The carbon-centered radical intermediate 195 abstracts a hydrogen from 186, forging 192 and forming 196 as a byproduct. Eventually, a radical recombination step between 192 and 19 grants the formation of the final products 188.
 | ||
| Scheme 26 Xia's approach to the C(sp3)–H bond functionalization reactions for the preparation of thioethers 188. | ||
 | ||
| Scheme 27 Akiyama's work on C(sp3)–S bond forming transformations through the activation of C(sp3)–H bonds. | ||
Again in 2021, the group of Molander reported a protocol for decarbonylative trifluoromethylthiolation of via activation of EDA complexes (Scheme 28).54 In this study, dihidropyridines (DHPs) 197 reacted with N-(trifluoromethylthio)phtalimide 82 to afford he creation of trifluoromethylthiolated molecules 198. While extensive mechanistic studies and multiple pathways are contemplated by the authors, a simplified description will be made. Interested readers are encouraged to consult the original publication for details. The reactions proceed via the formation of the EDA complex 199, which light-driven fragmentation grants the creation of two different radical ion intermediates, the radical cation 200 and the radical anion 201. Intermediate 200 decomposes into a pyridinium cation 202 and a carbon-centered radical 203. Simultaneously, the radical anion 201 fragments into a phtalimide anion 85 and sulfur-centered radical 86 through reduction of the N–S bond. Radical recombination of intermediates 203 and 86 grant the formation of the final products 198.
 | ||
| Scheme 28 Trifluoromethylthiolation reactions through photochemical activation of EDA complexes 199. | ||
Also in 2021, the research group of Song reported a photochemical transformation which makes use of redox active esters 204 as electron acceptors for thiolation reactions with thiophenols 14 (Scheme 29).55 When a thiolate anion is generated under basic media, the formation of an EDA complex 206 takes place, which fragmentation produces the sulfur-centered radical 19 and the radical anion 207. Redox active esters are known to be radical precursors upon one electron reduction.56 The corresponding radical anion 207 that is initially formed fragments into phthalimide anion 85 and the carbon-centered radical 208. Eventually, radical recombination between 208 and 19 forges the final thioethers 205.
 | ||
| Scheme 29 Visible-light-driven thioetherification reactions with redox active esters 204 and thiophenols 14. | ||
In 2022, the group of Hu developed a photochemical protocol for the S-trifluoromethylation of thiophenols 14 with trifluoromethyl phenyl sulfone 209 (Scheme 30).57 The transformation is initiated by the formation of an EDA complex 211 between sulfone 209 and the corresponding thiphenolate anion from 14, allowing for the expected formation of the sulfonyl radical 212 and sulfur-centered radical 19. Intermediate 212 is unstable, decomposing into a trifluoromethyl radical 213 and a sulfinate anion. Radical recombination between intermediates 213 and 19 renders the desired S-trifluoromethylated thiophenols 210.
 | ||
| Scheme 30 Visible-light-driven trifluoromethylation of thiphenols 14 with trifluoromethyl phenyl sulfone 209. | ||
In 2023, Hitomi and co-workers discovered an EDA-complex driven transformation between benzylidenemalononitriles 214 and thioethers 215 for the construction of aryl alkyl thioethers 216 (Scheme 31).58 Photochemical fragmentation of the EDA complex 217, formed between both reactants, grants the formation of the radical anion 218 and the radical cation 219. By losing a proton, intermediate 219 evolves to the carbon-centered radical 220, which recombines with 218. After protonation, the final products 216 are formed.
 | ||
| Scheme 31 Formation of aryl alkyl thioethers 216 by photochemical excitation of EDA complexes 217. | ||
Also the same year, the group of Shen reported multicomponent transformation for the 1,2-difunctionalization of styrenes 144 with thiophenols 14 and 4-cyanopyridines 221 (Scheme 32).59 The reaction is initiated by the deprotonation under basic conditions of the corresponding thiophenol 14, rendering a thiophenolate anion which aggregates with compound 221 to form an EDA complex 223. Visible-light-driven excitation of this complex causes its fragmentation into the radical anion 224 and the sulfur-centered radical 19. The latter intermediate adds to the alkene 144, forming an alkyl-centered radical 225. Radical recombination between 225 and 224 creates a new intermediate 226, in which aromatization of the pyridine core through elimination of a cyanide group forges the final products 222. In 2024, the Shi group reported a distinct 1,2-difunctionalization of alkenes based on similar reactivity. However, their method employed an iron salt and used trifluoromethylarenes instead of 4-cyanopyridines.60 To avoid redundancy, a detailed mechanistic discussion will not be provided.
 | ||
| Scheme 32 Difunctionalization reaction of styrenes 144 to create compounds 213. | ||
In 2023, Renzi's group reported a thiol–ene reaction between alkenes 227 and thiophenols 14, driven by the formation of EDA complexes (Scheme 33).61 In this process, a molecule of 14 pairs with its deprotonated form to assemble an EDA complex 229. Upon photochemical excitation, this complex generates a radical anion 230 and a sulfur-centered radical 19, which subsequently adds to olefin 227, forming an alkyl-centered radical 231. This intermediate can then evolve via two pathways: hydrogen abstraction from radical anion 230, yielding the final product 228 and a thiophenolate anion; or hydrogen abstraction from 14, also forming 228 while regenerating radical 19. It should be noted that photochemical thiol–ene and thiol–yne reactions represent a powerful method for the construction of organosulfur compounds, where also photocatalytic versions have been recently developed by the groups of König and Ananikov.62
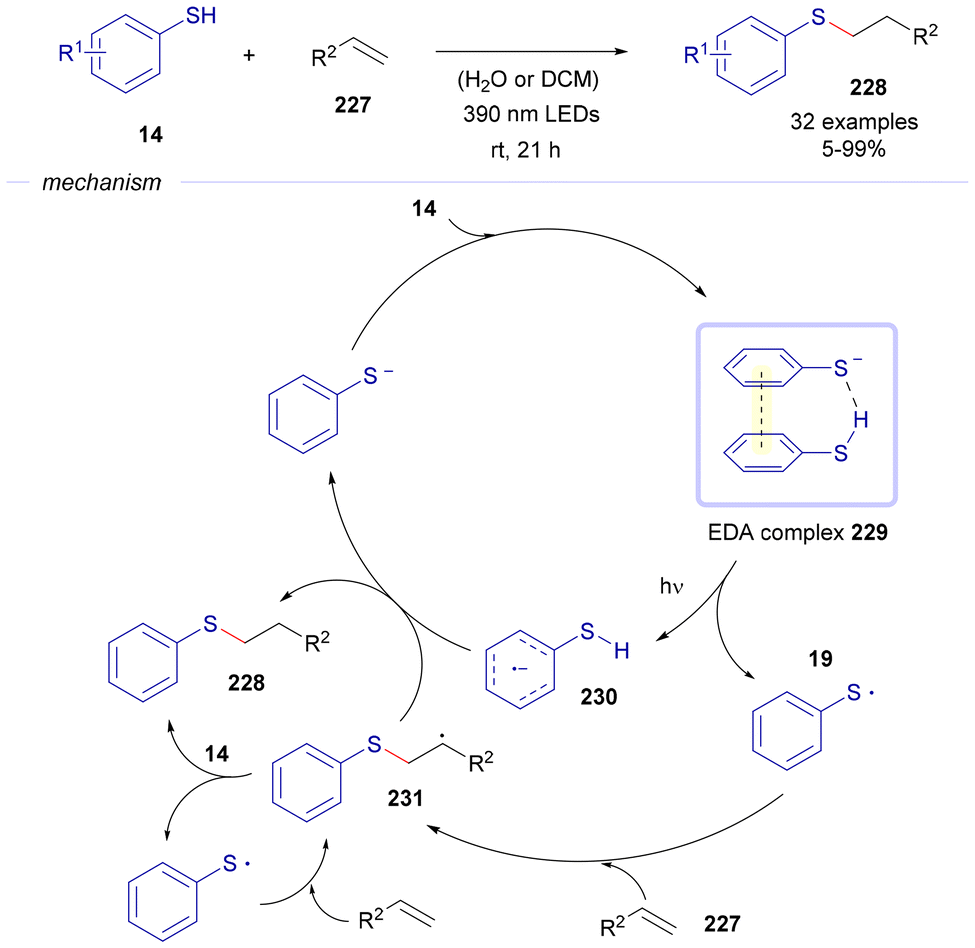 | ||
| Scheme 33 Thiol–ene transformations of alkenes 227 into sulfides 228. | ||
In 2024, Yamaguchi and Itoh developed a photochemical halogen-bonding assisted ATRA/cyclization reaction between thioethers 232 and homoallyl bromides 233 for the construction of tricyclic heterocycles 234 (Scheme 34).63 Initially, the halogen-bonding complex 235 forms through the interaction between 233 and either DIPEA or methanol, which act as electron donors (abbreviated as Y in Scheme 33). Upon exposure to light, this complex enters an excited state, leading to the breaking of the C–Br bond, resulting in the formation of the alkyl radical 236 along with either an ammonium radical cation or a methoxy radical. The radical 236 then undergoes an addition reaction with 232, forming a new C–C bond and generating radical intermediate 237. This intermediate subsequently undergoes an intramolecular radical 5-exo-trig cyclization, leading to the formation of radical intermediate 238. Next, radical species 238 reacts with 236, producing the brominated compound 239 while simultaneously regenerating the amine. Additionally, there is a possibility of a radical chain mechanism where the carbon-centered radical 238 interacts with 235, leading to the formation of brominated compound 239 and the regeneration of 238. Finally, the intermediate 239 formed via these pathways undergoes a second cyclization step, ultimately yielding the desired tricyclic compound 234.
 | ||
| Scheme 34 Photochemical halogen-bonding-assisted reaction between thioethers 232 and homoallyl bromides 233 for the preparation of tricyclic heterocycles 234. | ||
Also in 2024, Tong and Zhong developed a visible-light-driven reaction between different oxime esters 240 or 242 and thiophenols 14 to forge either compounds 241 or 243 (Scheme 35).64
 | ||
| Scheme 35 Photochemical EDA-complex-mediated reactions with oxime esters to form compounds 241 and 243. | ||
The reactions take place through the formation of an EDA complex 244 between both starting materials. Upon photoinduced electron transfer from the thiophenolate anion to the corresponding oxime ester, the latter is fragmentated through reduction of the N–O bond. If oximes 240 are used, a new carbon-centered radical 245 is formed, which recombines with the sulfur-centered radical 19 to forge compounds 243. On the other hand, when oxime precursors 242 are employed, a different radical species 246 is created, which arises from a previous 5-exo-trig cyclization. This intermediate recombines with 19 to create the final products 243.
Also in 2024, Zhou and Li reported a 1,2-difunctionalization of alkenes 144 for the synthesis of β-hydroxysulfides 247 (Scheme 36).65 The reaction begins with the formation of an EDA complex 248 between the formate anion and thiosulfone 143. Upon photochemical fragmentation, this complex generates an oxygen-centered radical 249 and a sulfur-centered radical 19. Intermediate 249 abstracts a hydrogen atom from another equivalent of the formate anion, yielding formic acid and radical anion species 251. Simultaneously, radical 19 undergoes a series of hydrogen atom transfer events, ultimately reducing to the corresponding thiophenolate anion. Alternatively, radical 19 can add to olefin 144, forming a carbon-centered radical 252. Quenching of this intermediate by O2 produces peroxy radical 253, which is then converted into intermediate 254via HAT from formic acid. Intermediate 254 is subsequently reduced by 253, generating oxygen-centered radical 257. A final hydrogen atom abstraction from the formate anion leads to the formation of the final product 247, along with the regeneration of radical anion species 251.
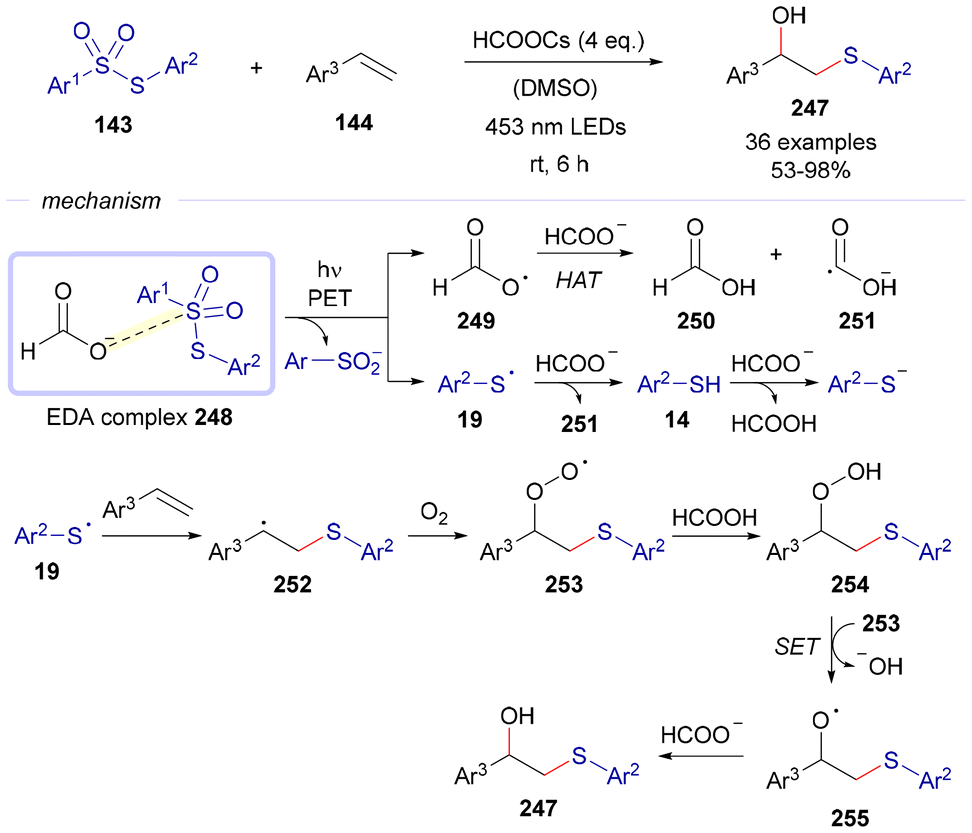 | ||
| Scheme 36 1,2-Difunctionalization reactions of alkenes 144 for the preparation of β-hydroxysulfides 247. | ||
One of the most recent works in the field of photochemical C(sp3)–S bond formation with hydrogen-bonding EDA complexes has been developed by Li and Wen (Scheme 37).66 In this study, a regioselective (Markovnikov or anti-Markovnikov) 1,2-difunctionalization of alkenes derived from lactams 256 produces in a selective manner thioethers 257, 258 or 259. Interestingly, the choice of the solvent for the reaction is critical to dictate the regiochemistry of this transformation, where charge transfer (CT) can be favoured over energy charge transfer (En-CT) depending on the solvent. Initially, exposure to the EDA complex 260 to visible light triggers a rapid electron transfer within the THF/DBE solvent system, resulting in the generation of radical anion 261 and radical cation 262. The Markovnikov hydrothiolation product 257 is efficiently obtained through radical cross-coupling of these intermediates in the THF/DBE mixture. In contrast, the formation of the anti-Markovnikov hydrothiolation product 258 appears to be linked to a slower En-CT process that occurs when the H-bonding EDA complex 260 is exposed to light in an ether-based solvent. Additionally, sulfur radical 264 and radical anion 261 are produced under visible light irradiation when the reaction is conducted in THF with K2HPO4 acting as a base, being the formation of EDA complex 263 critical. The carbon-centered radical 268 emerges from the oxidation of carbanion intermediate 267 by sulfur radical 264, while intermediate 265 itself is generated through the anti-Markovnikov cross-coupling of 261 and 264. Subsequently, the carbon-centered radical 266 reacts with O2, forming the peroxy radical intermediate 267. The desired anti-Markovnikov hydroxysulfenylation product 259 is then produced via its interaction with compound 104, releasing sulfoxide radical 268.
 | ||
| Scheme 37 Light-driven excitation of hydrogen-bonding EDA-complexes enables the preparation of compounds 257, 258 and 259. | ||
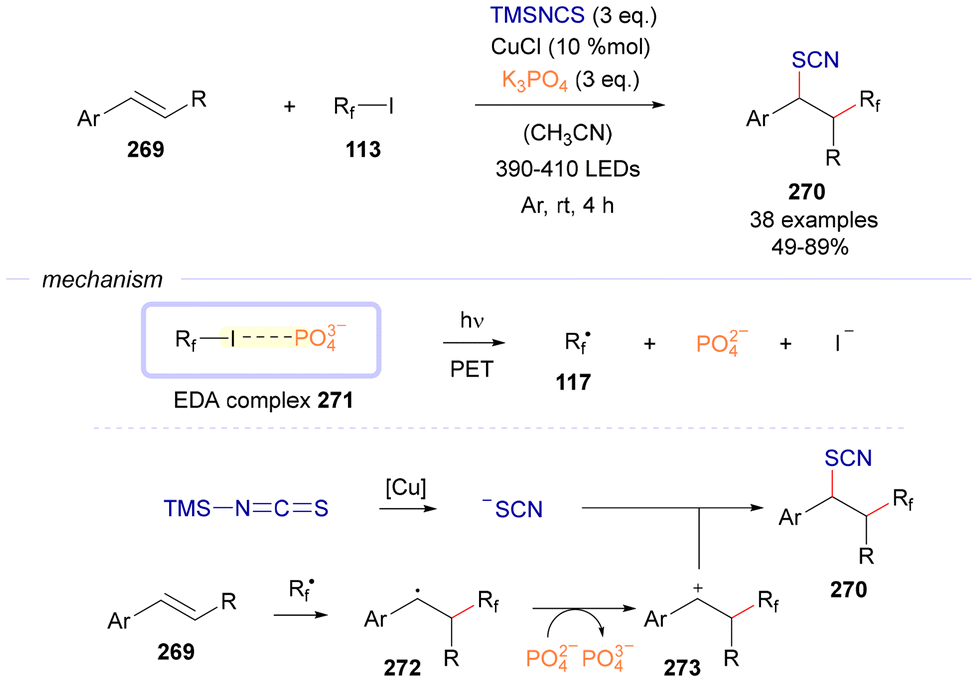 | ||
| Scheme 38 Fluoroalkylthiocyanation of alkenes 269 based on the employment of K3PO4 as the sacrificial donor. | ||
In 2024, Liu's group developed a multicomponent, visible-light-driven perfluoroalkylation–thiolation reaction of alkenes, utilizing sacrificial donors to form electron donor–acceptor complexes (Scheme 39).68 In this approach, thiosulfones 143 serve as the thiolating agent, while DIPEA acts as a sacrificial donor within the EDA complex 275, in combination with perfluoroalkyl iodides 113. Photochemical fragmentation of the aggregate generates the perfluoroalkyl radical 117 and the DIPEA radical cation. Radical addition of 117 to olefin 218 produces an alkyl-centered radical intermediate 276, which can undergo two main pathways to yield the final products 274. Pathway A contemplates a reduction of intermediate 276 by the DIPEA radical cation, affording the carbanionic species 277. At this point a nucleophilic substitution on the thiosulfone 143 takes place, forming compound 274 and a sulfinate anion. On the other hand, pathway B is based on a radical substitution reaction of 276 on 143, generating this way the thioether 274, along with a sulfonyl radical 51. The latter radical intermediate is transformed into the corresponding sulfinate anion through reduction from the DIPEA radical cation.
 | ||
| Scheme 39 1,2-Perfluoroalkylation/thiolation reactions of alkenes 218 for the preparation of thioethers 274. | ||
A different transformation involving EDA complexes for C(sp3)–S bond formation was reported by Wu and Wang (Scheme 40).69 In this work, thiosulfones 278 again serve as the thiolating agent, this time in the thiolation of benzyl chlorides 279.
 | ||
| Scheme 40 Thioetherification reactions of benzyl chlorides 279 to form the benzyl thioethers 280. | ||
The cross-coupling reaction proceeds via the formation of an EDA complex 281, where DIPEA acts as the electron donor, and thiosulfone 278 serves as the electron acceptor. Upon light-driven fragmentation, the complex breaks into a sulfur-centered radical 282, a DIPEA radical cation, and a sulfinate anion. The sulfinate anion then deprotonates the DIPEA radical cation, producing a carbon-centered radical 283. This radical undergoes a halogen atom transfer (XAT) reaction with benzyl chloride 279, abstracting a chlorine atom and forming the benzyl radical 285, along with stoichiometric amounts of compound 284. Finally, radical recombination between 282 and 285 yields the benzyl thioethers 280.
The final study covered in this section was recently reported by the group of Patel (Scheme 41).70 In this study, functionalization reactions of alkynes 157 were explored with thiosulfonates 286.
 | ||
| Scheme 41 Photochemical regioselective vicinal and oxidative geminal functionalization of alkynes 157. | ||
Depending on the reaction conditions, an oxidative functionalization to form compounds 288 or vicinal difunctionalization to create products 287 can be achieved. For the mechanistic discussion, only the C(sp3)–S bond forming reaction will be contemplated. In this case, the thiosulfonate 286 forms an EDA complex 289 with Et3N. Upon exposure to visible light, this complex transitions into an excited state (289*), leading to a single electron transfer from Et3N to 286. This process generates a radical anion 290, which subsequently undergoes S–S bond cleavage, producing a sulfonyl radical 291 and a thiolate anion 292. The thiolate anion 292 is then oxidized by the triethylamine radical cation, yielding a thiyl radical 293, which dimerizes to form diphenyl disulfide 294. Simultaneously, the sulfonyl radical 291 reacts with the acetylene 157, forming a vinyl sulfonyl radical 295. This radical is intercepted by molecular oxygen to produce a peroxo intermediate 296, which subsequently interacts with moisture (H2O) in the reaction medium, forming a hydroxoperoxyl intermediate 297 and releasing hydroxyl radicals. The intermediate 297 then reacts with the hydroxyl radical, generating intermediate 298 and releasing H2O2. Following this, a hydrogen atom transfer from H2O2 to 298 results in the formation of an enol intermediate 299, which undergoes tautomerization to yield the β-keto sulfone 300. Finally, in the presence of Et3N, the β-keto sulfone reacts either with thiosulfonate 290 or with the in situ generated diphenyl disulfide 294, leading to the formation of the trifunctionalized product 287 (path a). Alternatively, product 287 can also be formed via radical–radical cross-coupling between intermediate 298 and the thiyl radical 293 (path b).
In summary, recent years have seen significant advancements in C(sp3)–S bond-forming reactions via photochemical excitation of EDA complexes. Several research groups have focused on developing methodologies that leverage various nucleophilic donors (primarily thiols, thioethers, and thiosulfonates) in combination with electron acceptors such as Katriztky salts, organic halides, redox-active esters, and dihydropyridines. Additionally, strategies employing sacrificial donors, particularly amines and inorganic bases, have emerged as effective approaches for facilitating these transformations.
3. Conclusions
The photochemical activation of electron donor–acceptor (EDA) complexes has emerged as a highly efficient and sustainable strategy for C–S bond formation. This review has demonstrated how EDA complexes facilitate single-electron transfer (SET) processes under mild conditions, enabling the selective construction of C–S bonds without the need for harsh reagents, expensive catalysts, or metal-based systems. The discussed methodologies highlight key advantages of EDA complex photochemistry, including improved functional group tolerance, operational simplicity, and alignment with green chemistry principles. These features make it an attractive alternative to traditional approaches, such as nucleophilic substitution and metal-catalyzed cross-coupling. Continued research in this area will likely uncover new methodologies and applications, further solidifying the role of EDA complex photochemistry as a transformative tool for sustainable C–S bond formation.Author contributions
H. F. P and I. T. contributed equally to the literature review, analysis, and drafting of the figures and schemes of the manuscript. M. P. conceptualized and supervised the overall study and wrote the manuscript. All authors discussed the content, contributed to the writing, and reviewed the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A “Severo Ochoa” predoctoral fellowship (BP21/050) to H. F. P. by FICYT (Principality of Asturias) and a “Ramón y Cajal” postdoctoral grant to M. P. (MCINN-24-RYC2022-035485-I) by Ministerio de Ciencia e Innovación of Spain (Agencia Estatal de Investigación) are gratefully acknowledged. Financial support by Ministerio de Ciencia e Innovación of Spain (Agencia Estatal de Investigación: MCINN-23-PID2022-140635NB-I00/MCIN/AEI/10.13039/501100011033/FEDER, UE) is also acknowledged.References
- (a) D. A. Boyd, Sulfur and Its Role In Modern Materials Science, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CAS; (b) H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Sulfur Chemistry in Polymer and Materials Science, Macromol. Rapid Commun., 2019, 40, 1800650 Search PubMed; (c) N. Wang, P. Saidhareddy and X. Jiang, Construction of sulfur-containing moieties in the total synthesis of natural products, Nat. Prod. Rep., 2020, 37, 246–275 Search PubMed; (d) A. Francioso, A. Baseggio Conrado, L. Mosca and M. Fontana, Chemistry and Biochemistry of Sulfur Natural Compounds: Key Intermediates of Metabolism and Redox Biology, Oxid. Med. Cell. Longevity, 2020, 2020, 1–27 CrossRef PubMed.
- (a) E. A. Ilardi, E. Vitaku and J. T. Njardarson, Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals To Reveal Opportunities for Drug Design and Discovery, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (b) J. Yu and X. Jiang, Synthesis and perspective of organosulfur chemicals in agrochemicals, Adv. Agrochem, 2023, 2, 3–14 CrossRef; (c) P. Sang, Q. Chen, D.-Y. Wang, W. Guo and Y. Fu, Organosulfur Materials for Rechargeable Batteries: Structure, Mechanism, and Application, Chem. Rev., 2023, 123, 1262–1326 CrossRef CAS PubMed; (d) Y. Zheng, W. Lu, T. Ma and S. Huang, Recent advances in photochemical and electrochemical strategies for the synthesis of sulfonyl fluorides, Org. Chem. Front., 2024, 11, 217–235 RSC.
- L. Senatore, E. Ciuffarin, A. Fava and G. Levita, Nucleophilic substitution at sulfur. Effect of nucleophile and leaving group basicity as probe of bond formation and breaking, J. Am. Chem. Soc., 1973, 95, 2918–2922 CrossRef CAS.
- Selected reviews: (a) P. Annamalai, K. Liu, S. Singh Badsara and C. Lee, Carbon-Sulfur Bond Constructions: From Transition-Metal Catalysis to Sustainable Catalysis, Chem. Rec., 2021, 21, 3674–3688 CrossRef CAS PubMed; (b) I. P. Beletskaya and V. P. Ananikov, Transition-Metal-Catalyzed C–S, C–Se, and C–Te Bond Formations via Cross-Coupling and Atom-Economic Addition Reactions. Achievements and Challenges, Chem. Rev., 2022, 122, 16110–16293 Search PubMed.
- (a) R. S. Glass, Sulfur Radicals and Their Application, Top Curr. Chem. Z, 2018, 376, 22 Search PubMed; (b) N. Amri and T. Wirth, Recent Advances in the Electrochemical Synthesis of Organosulfur Compounds, Chem. Rec., 2021, 21, 2526–2537 CAS; (c) Z. Wu and D. A. Pratt, Radical approaches to C–S bonds, Nat. Rev. Chem., 2023, 7, 573–589 Search PubMed; (d) B. Dong, J. Shen and L.-G. Xie, Recent developments on 1,2-difunctionalization and hydrofunctionalization of unactivated alkenes and alkynes involving C–S bond formation, Org. Chem. Front., 2023, 10, 1322–1345 RSC.
- Selected recent reviews: (a) S. Crespi and M. Fagnoni, Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy, Chem. Rev., 2020, 120, 9790–9833 Search PubMed; (b) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CAS; (c) A. Das and K. R. Justin Thomas, Generation and Application of Aryl Radicals Under Photoinduced Conditions, Chem. – Eur. J., 2024, 30, e202400193 CAS; (d) X. Tian, Y. Liu, S. Yakubov, J. Schütte, S. Chiba and J. P. Barham, Photo- and electro-chemical strategies for the activations of strong chemical bonds, Chem. Soc. Rev., 2024, 53, 263–316 CAS; (e) R. Bhanja, S. K. Bera and P. Mal, Photocatalyst- and Transition-Metal-Free Light-Induced Formation of Carbon-Chalcogen Bonds, Adv. Synth. Catal., 2024, 366, 168–182 CrossRef CAS; (f) H. F. Piedra and M. Plaza, Advancements in visible-light-induced reactions via alkenyl radical intermediates, Photochem. Photobiol. Sci., 2024, 23, 1217–1228 CrossRef CAS PubMed.
- Selected reviews: (a) A. Wimmer and B. König, Photocatalytic formation of carbon–sulfur bonds, Beilstein J. Org. Chem., 2018, 14, 54–83 CrossRef CAS PubMed; (b) W. Guo, K. Tao, W. Tan, M. Zhao, L. Zheng and X. Fan, Recent advances in photocatalytic C–S/P–S bond formation via the generation of sulfur centered radicals and functionalization, Org. Chem. Front., 2019, 6, 2048–2066 RSC; (c) C. Cavedon, P. H. Seeberger and B. Pieber, Photochemical Strategies for Carbon–Heteroatom Bond Formation, Eur. J. Org. Chem., 2020, 1379–1392 CrossRef CAS; (d) H. Li, Y. Liu and S. Chiba, Leveraging of Sulfur Anions in Photoinduced Molecular Transformations, JACS Au, 2021, 1, 2121–2129 CrossRef CAS PubMed; (e) D. Yang, Q. Yan, E. Zhu, J. Lv and W.-M. He, Carbon–sulfur bond formation via photochemical strategies: An efficient method for the synthesis of sulfur-containing compounds, Chin. Chem. Lett., 2022, 33, 1798–1816 CrossRef CAS; (f) J. Feng, Y. Zhang, X. Wang, J. Liu, V. Benazzi, K. Lu, X. Zhao and S. Protti, Recent Advances in Visible-Light-Driven C–S Bond Formation, Adv. Synth. Catal., 2023, 365, 3413–3431 CrossRef CAS; (g) A. K. Sahoo, D. Barik, B. Dam and B. K. Patel, Metal-Free Visible-light Mediated C–S Bond Formation, Asian J. Org. Chem., 2023, 12, e202300252 CAS.
- Selected reviews: (a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed; (b) Y. Yuan, S. Majumder, M. Yang and S. Guo, Recent advances in catalyst-free photochemical reactions via electron-donor-acceptor (EDA) complex process, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS; (c) L. Zheng, L. Cai, K. Tao, Z. Xie, Y. Lai and W. Guo, Progress in Photoinduced Radical Reactions using Electron Donor-Acceptor Complexes, Asian J. Org. Chem., 2021, 10, 711–748 CAS; (d) Y. Sempere, M. Morgenstern, T. Bach and M. Plaza, Reactivity and selectivity modulation within a molecular assembly: recent examples from photochemistry, Photochem. Photobiol. Sci., 2022, 21, 719–737 CAS.
- Reviews on examples of photochemical radical-based reactions enabled by EDA complex preformation: (a) H. F. Piedra, C. Valdés and M. Plaza, Shining light on halogen-bonding complexes: a catalyst-free activation mode of carbon–halogen bonds for the generation of carbon-centered radicals, Chem. Sci., 2023, 14, 5545–5568 CAS; (b) L. Van Dalsen, R. E. Brown, J. A. Rossi-Ashton and D. J. Procter, Sulfonium Salts as Acceptors in Electron Donor-Acceptor Complexes, Angew. Chem., 2023, 135, e202303104 Search PubMed.
- P. Anastas and N. Eghbali, Green Chemistry: Principles and Practice, Chem. Soc. Rev., 2010, 39, 301–312 CAS.
- H. E. Bonfield, T. Knauber, F. Lévesque, E. G. Moschetta, F. Susanne and L. J. Edwards, Photons as a 21st century reagent, Nat. Commun., 2020, 11, 804 CAS.
- During the submission process of this review, a different review of the photochemistry of EDA complexes to create C–S bonds was reported: S. Wang, L. Wang, J. Cui, L. Zhang, Q. Zhang, C. Ke and S. Huang, Recent progress in C–S bond formation via electron donor–acceptor photoactivation, Org. Biomol. Chem., 2025, 23, 1794–1808 CAS.
- A. K. Wortman and C. R. J. Stephenson, EDA photochemistry: Mechanistic investigations and future opportunities, Chem, 2023, 9, 2390–2415 CAS.
- B. Liu, C.-H. Lim and G. M. Miyake, Visible-Light-Promoted C–S Cross-Coupling via Intermolecular Charge Transfer, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS PubMed.
- G. Li, Q. Yan, Z. Gan, Q. Li, X. Dou and D. Yang, Photocatalyst-Free Visible-Light-Promoted C(sp2)–S Coupling: A Strategy for the Preparation of S-Aryl Dithiocarbamates, Org. Lett., 2019, 21, 7938–7942 CrossRef CAS PubMed.
- X. Li, W. Cui, Q. Deng, X. Song, J. Lv and D. Yang, Three-component reaction access to S-alkyl dithiocarbamates under visible-light irradiation conditions in water, Green Chem., 2022, 24, 1302–1307 RSC.
- (a) M. Liu, Y. Qian, Y. Wu and F. Zhang, Multicomponent synthesis of di-aryl dithiocarbamates via electron donor–acceptor photoactivation with thianthrenium salts, Green Chem., 2023, 25, 3852–3856 RSC; (b) H. Xu, X. Li, J. Ma, J. Zuo, X. Song, J. Lv and D. Yang, An electron donor–acceptor photoactivation strategy for the synthesis of S-aryl dithiocarbamates using thianthrenium salts under mild aqueous micellar conditions, Chin. Chem. Lett., 2023, 34, 108403 CrossRef CAS; (c) Y. Tang, Y. Cai, Z. Xie, Z. Gao, X. Chen and J. Yi, Multicomponent reactions to access S-aryl dithiocarbamates via an electron donor–acceptor complex under open-to-air conditions, Org. Biomol. Chem., 2024, 22, 1378–1385 RSC.
- A. Nandy, I. Kazi, S. Guha and G. Sekar, Visible-Light-Driven Halogen-Bond-Assisted Direct Synthesis of Heteroaryl Thioethers Using Transition-Metal-Free One-Pot C–I Bond Formation/C–S Cross-Coupling Reaction, J. Org. Chem., 2021, 86, 2570–2581 CrossRef CAS PubMed.
- Reviews and selected publications on the importance of halogen-bonding interactions: (a) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Halogen bonding in solution: thermodynamics and applications, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Halogen Bonding in Supramolecular Chemistry, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (d) R. L. Sutar and S. M. Huber, Catalysis of Organic Reactions through Halogen Bonding, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS; (e) M. Breugst and J. J. Koenig, σ-Hole Interactions in Catalysis, Eur. J. Org. Chem., 2020, 5473–5487 CrossRef CAS; (f) A. C. Keuper, K. Fengler, F. Ostler, T. Danelzik, D. G. Piekarski and O. García Mancheño, Fine-Tuning Substrate–Catalyst Halogen–Halogen Interactions for Boosting Enantioselectivity in Halogen-Bonding Catalysis, Angew. Chem., Int. Ed., 2023, 62, e202304781 CrossRef CAS PubMed.
- A. Karpfen, Charge-Transfer Complexes between NH3 and the Halogens F2, ClF, and Cl2: An ab Initio Study on the Intermolecular Interaction, J. Phys. Chem. A, 2000, 104, 6871–6879 CrossRef CAS.
- N. Sundaravelu, A. Nandy and G. Sekar, Visible Light Mediated Photocatalyst Free C–S Cross Coupling: Domino Synthesis of Thiochromane Derivatives via Photoinduced Electron Transfer, Org. Lett., 2021, 23, 3115–3119 CrossRef CAS PubMed.
- X. Liang, Y. Li, Q. Xia, L. Cheng, J. Guo, P. Zhang, W. Zhang and Q. Wang, Visible-light-driven electron donor–acceptor complex induced sulfonylation of diazonium salts with sulfinates, Green Chem., 2021, 23, 8865–8870 RSC.
- M. J. Cabrera-Afonso, A. Granados and G. A. Molander, Sustainable Thioetherification via Electron Donor–Acceptor Photoactivation Using Thianthrenium Salts, Angew. Chem., Int. Ed., 2022, 61, e202202706 CrossRef CAS PubMed.
- A. Granados, M. J. Cabrera-Afonso, M. Escolano, S. O. Badir and G. A. Molander, Thianthrenium-enabled sulfonylation via electron donor-acceptor complex photoactivation, Chem. Catal., 2022, 2, 898–907 CrossRef CAS PubMed.
- H. Meng, M.-S. Liu and W. Shu, Organothianthrenium salts: synthesis and utilization, Chem. Sci., 2022, 13, 13690–13707 RSC.
- Y. Zhang, S. Xia, W. Shi, B. Lin, X. Su, W. Lu, X. Wu, X. Wang, X. Lu, M. Yan and X. Zhang, Radical C–H Sulfonation of Arenes: Its Applications on Bioactive and DNA-Encoded Molecules, Org. Lett., 2022, 24, 7961–7966 CrossRef CAS PubMed.
- Z. Chen, F. Xue, Y. Zhang, W. Jin, B. Wang, Y. Xia, M. Xie, A. Abdukader and C. Liu, Visible-Light-Promoted [3 + 2] Cyclization of Chalcones with 2-Mercaptobenzimidazoles: A Protocol for the Synthesis of Imidazo[2,1-b]thiazoles, Org. Lett., 2022, 24, 3149–3154 CrossRef CAS PubMed.
- A. Volkov, D. I. Bugaenko, A. V. Bogdanov and A. V. Karchava, Visible-Light-Driven Thioesterification of Aryl Halides with Potassium Thiocarboxylates: Transition-Metal Catalyst-Free Incorporation of Sulfur Functionalities into an Aromatic Ring, J. Org. Chem., 2022, 87, 8170–8182 Search PubMed.
- R. I. Patel, B. Saxena and A. Sharma, General electron–donor–acceptor complex mediated thioesterification reaction via site-selective C–H functionalization using aryl sulfonium Salts, Green Chem., 2024, 26, 10265–10274 RSC.
- B. Zhao, Y.-X. Liu, P.-P. Liang, G.-Q. Hu and J.-H. Liu, S-Arylation of Thioic S-Acid Using Thianthrenium Salts via Photoactivation of Electron Donor–Acceptor Complex, J. Org. Chem., 2024, 89, 12508–12513 CrossRef CAS PubMed.
- M. Zhang, B. Wang, Y. Cao, Y. Liu, Z. Wang and Q. Wang, Visible-Light-Driven Synthesis of Aryl Xanthates and Aryl Dithiocarbamates via an Electron Donor–Acceptor Complex, Org. Lett., 2022, 24, 8895–8900 CrossRef CAS PubMed.
- F. Yang, G.-C. He, S.-H. Sun, T.-T. Song, X.-T. Min, D.-W. Ji, S.-Y. Guo and Q.-A. Chen, Selective C–S Bond Constructions Using Inorganic Sulfurs via Photoinduced Electron Donor–Acceptor Activation, J. Org. Chem., 2022, 87, 14241–14249 CrossRef CAS PubMed.
- Y. Song, Z. Jiang, Y. Zhu, T.-Y. Sun, X.-F. Xia and D. Wang, Visible-light-induced dehydrogenative β-trifluoromethylthiolation of tertiary amines and direct β-trifluoromethylthiolation of enamides, Org. Chem. Front., 2023, 10, 5284–5290 RSC.
- M. Li, Q. Tan, X. Lyu, X. Guo, H. Wang, Z. Hu and X. Xu, EDA Complex-Promoted Cascade Cyclization of Alkynes Enabling the Rapid Assembly of 3-Sulfonylindoles and Vinyl Sulfone Oxindoles, Org. Lett., 2024, 26, 5799–5804 CrossRef CAS PubMed.
- M. Shanthi, K. Perumal, S. Sivalingam, A. Puhazhendhi, P. Kumar Mandali and S. Ramesh, Transition-Metal-Free and Photocatalyst-Free Sulfenylation of Halopyrazolamines under Visible-Light Irradiation via Electron Donor–Acceptor Complexes, Synlett, 2024, 313–318 CAS.
- J. Zuo, X. Li, Y. Shi, J. Lv and D. Yang, Synthesis of Sulfur-Containing Trisubstituted Imidazoles by One-Pot, Multicomponent Reaction via Electron Donor–Acceptor Complex Photoactivation, Org. Lett., 2024, 26, 3541–3546 Search PubMed.
- (a) H. F. Piedra and M. Plaza, Photochemical halogen-bonding assisted generation of vinyl and sulfur-centered radicals: stereoselective catalyst-free C(sp2)–S bond forming reactions, Chem. Sci., 2023, 14, 650–657 CAS; (b) H. F. Piedra, C. Valdés and M. Plaza, A Continuous-Flow Protocol for the Synthesis of Alkenyl Thioethers Based on the Photochemical Activation of Halogen-Bonding Complexes, Synlett, 2025, 36, 325–328 Search PubMed.
- H. F. Piedra, V. Gebler, C. Valdés and M. Plaza, Photochemical halogen-bonding assisted carbothiophosphorylation reactions of alkenyl and 1,3-dienyl bromides, Chem. Sci., 2023, 14, 12767–12773 CAS.
- H. F. Piedra, C. Valdés and M. Plaza, Synthesis of Vinyl and 1,3-Dienyl Sulfones Enabled by Photochemical Excitation of Halogen-Bonding Complexes, Adv. Synth. Catal., 2024, 366, 1422–1429 Search PubMed.
- H. F. Piedra and M. Plaza, Visible-light-driven synthesis of alkenyl thiocyanates: novel building blocks for assembly of diverse sulfur-containing molecules, Chem. Sci., 2024, 15, 19077–19083 CAS.
- Z. Jiang, K. You, H. Wu, M. Xu, T. Wang and J. Luo, Photochemical Halogen-Bonding Promoted Synthesis of Vinyl Sulfones via Vinyl and Sulfonyl Radicals, Org. Lett., 2024, 26, 636–641 Search PubMed.
- Y. Liu, X.-L. Chen, K. Sun, X.-Y. Li, F.-L. Zeng, X.-C. Liu, L.-B. Qu, Y.-F. Zhao and B. Yu, Visible-Light Induced Radical Perfluoroalkylation/Cyclization Strategy To Access 2-Perfluoroalkylbenzothiazoles/Benzoselenazoles by EDA Complex, Org. Lett., 2019, 21, 4019–4024 CAS.
- Q.-Q. Ge, J.-S. Qian and J. Xuan, Electron Donor–Acceptor Complex Enabled Decarboxylative Sulfonylation of Cinnamic Acids under Visible-Light Irradiation, J. Org. Chem., 2019, 84, 8691–8701 CAS.
- W. Xie, P. Ma, Y. Zhang, L. Xi, S. Qiu, X. Huang, B. Yang, Y. Gao and J. Zhang, Visible Light-Induced Highly Regioselective and Stereoselective Oxysulfonylation of Alkynes for the Synthesis of (E)-β-Phenoxy Vinylsulfones, Org. Lett., 2022, 24, 6099–6104 Search PubMed.
- W. Yuan, J. Huang, X. Xu, L. Wang and X.-Y. Tang, B(C6F5)3-Catalyzed Electron Donor–Acceptor Complex-Mediated Aerobic Sulfenylation of Indoles under Visible-Light Conditions, Org. Lett., 2021, 23, 7139–7143 CAS.
- L. Lin, Z. Yang, J. Liu, J. Wang, J. Zheng, J.-L. Li, X. Zhang, X.-W. Liu, H. Jiang and J. Li, Visible-light-induced surfactant-promoted sulfonylation of alkenes and alkynes with sulfonyl chloride by the formation of an EDA-complex with NaI in water at room temperature, Green Chem., 2021, 23, 5467–5473 CAS.
- K. Cao, N. Zhang, L. Lin, Q. Shen, H. Jiang and J. Li, The Synthesis of (E)-Vinyl and Alkynyl Sulfones by the Formation of an Electron Donor-Acceptor Complex Using Thiosulfonates and Sodium Iodide Under Visible Light, Adv. Synth. Catal., 2023, 366, 207–213 Search PubMed.
- J. C. Herrera-Luna, M. C. Pérez-Aguilar, L. Gerken, O. García Mancheño, M. Consuelo Jiménez and R. Pérez-Ruiz, Effective Formation of New C(sp2)–S Bonds via Photoactivation of Alkylamine-based Electron Donor-Acceptor Complexes, Chem. – Eur. J., 2023, 29, e202203353 CrossRef CAS PubMed.
- R. Mao, Z. Yuan, Y. Li and J. Wu, N-Radical-Initiated Cyclization through Insertion of Sulfur Dioxide under Photoinduced Catalyst-Free Conditions, Chem. – Eur. J., 2017, 23, 8176–8179 CrossRef CAS PubMed.
- M. Yang, T. Cao, T. Xu and S. Liao, Visible-Light-Induced Deaminative Thioesterification of Amino Acid Derived Katritzky Salts via Electron Donor–Acceptor Complex Formation, Org. Lett., 2019, 21, 8673–8678 CrossRef CAS PubMed.
- J. T. M. Correia, V. A. Fernandes, B. T. Matsuo, J. A. C. Delgado, W. C. De Souza and M. W. Paixão, Photoinduced deaminative strategies: Katritzky salts as alkyl radical precursors, Chem. Commun., 2020, 56, 503–514 RSC.
- T. Li, K. Liang, J. Tang, Y. Ding, X. Tong and C. Xia, A photoexcited halogen-bonded EDA complex of the thiophenolate anion with iodobenzene for C(sp3)–H activation and thiolation, Chem. Sci., 2021, 12, 15655–15661 RSC.
- T. Uchikura, Y. Hara, K. Tsubono and T. Akiyama, Visible-Light-Driven C–S Bond Formation Based on Electron Donor–Acceptor Excitation and Hydrogen Atom Transfer Combined System, ACS Org. Inorg. Au, 2021, 1, 23–28 CAS.
- A. Lipp, S. O. Badir, R. Dykstra, O. Gutierrez and G. A. Molander, Catalyst-Free Decarbonylative Trifluoromethylthiolation Enabled by Electron Donor-Acceptor Complex Photoactivation, Adv. Synth. Catal., 2021, 363, 3507–3520 CAS.
- Y.-P. Cai, F.-Y. Nie and Q.-H. Song, Visible-Light-Mediated Alkylation of Thiophenols via Electron Donor–Acceptor Complexes Formed between Two Reactants, J. Org. Chem., 2021, 86, 12419–12426 CAS.
- P. Niu, J. Li, Y. Zhang and C. Huo, One-Electron Reduction of Redox-Active Esters to Generate Carbon-Centered Radicals, Eur. J. Org. Chem., 2020, 5801–5814 CAS.
- Z. Wei, Z. Lou, C. Ni, W. Zhang and J. Hu, Visible-light-promoted S-trifluoromethylation of thiophenols with trifluoromethyl phenyl sulfone, Chem. Commun., 2022, 58, 10024–10027 CAS.
- P. K. Roy and Y. Hitomi, A Simple Method for the Formation of α-Thioalkyl Radicals and the Promotion of Carbon-Carbon Bond Formation by Photoirradiation of Electron Donor-Acceptor Complexes, Asian J. Org. Chem., 2023, 12, e202300378 CAS.
- J. Shen, J. Li, M. Chen and Y. Chen, Photocatalyst-free, metal-free, visible light-induced thiolation/pyridylation of styrenes using an electron donor–acceptor complex as a bifunctional reagent, Org. Chem. Front., 2023, 10, 1166–1172 CAS.
- L. Yuan, Z. Wang, W. Zhuang, X. Li, C. Shi, X. Li and D. Shi, Photocatalyst-free, metal-free, visible light-induced thiolation/pyridylation of styrenes using an electron donor–acceptor complex as a bifunctional reagent, Org. Lett., 2024, 26, 7066–7071 CAS.
- P. Renzi, M. Rusconi, G. Ghigo and A. Deagostino, Purple-Light Promoted Thiol-ene Reaction of Alkenes, Adv. Synth. Catal., 2023, 365, 4623–4633 CrossRef CAS.
- N. S. Shlapakov, A. D. Kobelev, J. V. Burykina, A. Yu. Kostyukovich, B. König and V. P. Ananikov, Reversible Radical Addition Guides Selective Photocatalytic Intermolecular Thiol-Yne-Ene Molecular Assembly, Angew. Chem., Int. Ed., 2024, 63, e202314208 CrossRef CAS PubMed.
- E. Yamaguchi, M. Murai and A. Itoh, Halogen-Bonding-Enabled Photoinduced Atom Transfer Radical Addition/Cyclization Reaction Leading to Tricyclic Heterocycles, J. Org. Chem., 2024, 89, 6555–6563 CrossRef CAS PubMed.
- M. Lu, L. Jiang, Y. Xu, S. Li, Q. Tong and J. Zhong, A General Electron Donor-Acceptor Photoactivation Using Oxime Esters Enabled Divergent Thioetherifications, Chin. J. Chem., 2024, 42, 2751–2756 CrossRef CAS.
- Q. Shen, X. Peng, J. Sui, M. Peng, X. Liu, H. Jiang, R. Tan, M. Zhou and J. Li, Formate-Mediated Synthesis of β-Hydroxysulfides from Olefins and Thiosulfonates via EDA Complex Strategy under Visible Light Irradiation in Air, Chin. J. Chem., 2024, 42, 3129–3134 CrossRef CAS.
- T. Yan, J. Yang, K. Yan, Z. Wang, B. Li and J. Wen, A General Photoactive H-Bonding EDA Complex Model Drives the Selective Hydrothiolation and Hydroxysulfenylation of Carbonyl Activated Alkenes, Angew. Chem., Int. Ed., 2024, e202405186 CAS.
- Z. Li, S. Wang, Y. Huo, B. Wang, J. Yan and Q. Guo, Visible light-driven fluoroalkylthiocyanation of alkenes via electron donor–acceptor complexes, Org. Chem. Front., 2021, 8, 3076–3081 CAS.
- L. Liu, Q. Wang, Y. Li, M. Liu, B. Liu, Q. Li and K. Feng, Photodriven Radical Perfluoroalkylation–Thiolation of Unactivated Alkenes Enabled by Electron Donor–Acceptor Complex, Org. Lett., 2024, 26, 2271–2275 CAS.
- C. Gong, J. Huang, L. Cai, Y. Yuan, T. Pu, M. Huang, S.-H. Wu and L. Wang, Visible-Light-Promoted Thiolation of Benzyl Chlorides with Thiosulfonates via a Photoactive Electron Donor–Acceptor Complex, J. Org. Chem., 2024, 89, 9450–9461 CAS.
- D. Barik, N. Chakraborty, A. K. Sahoo, H. N. Dhara and B. K. Patel, Electron-donor–acceptor (EDA) complex-driven regioselective vicinal and oxidative geminal functionalization of alkynes, Chem. Commun., 2024, 60, 12577–12580 CAS.
Footnote |
| † Both authors contributed equally. |
| This journal is © the Partner Organisations 2025 |