Molecular engineering of antiaromatic orangarin-based sensitizers for high-performance dye-sensitized solar-cell applications
Received
3rd September 2025
, Accepted 15th December 2025
First published on 23rd December 2025
Abstract
To meet increasing energy demand, several molecules have been explored as dyes for dye-sensitized solar-cell (DSSC) applications. However, such designed cells are yet to attain a solar-power conversion efficiency (η) of more than 13%. To achieve this, new molecules should be investigated. In this work, for the first time, an antiaromatic metal-free molecule is explored for DSSC applications. We consider the antiaromatic orangarin core and load it with different donor and acceptor moieties at rationally chosen places that can enhance the absorption of sunlight. In this way, we design ten potential candidates for use as dyes in DSSCs. The photovoltaic properties, including the light-harvesting efficiency (LHE), open-circuit voltage, maximum short-circuit current density, fill factor, and η of all the designed molecules are calculated and analyzed using the state-of-the-art density functional theory and time-dependent density functional theory methods. Our study reveals that the antiaromatic nature of the core indeed increases the absorption strengths of the dyes and hence enhances the LHE and η up to an incredible value of 26%. The present study clearly demonstrates that an antiaromatic core should be explored further for DSSC application.
1. Introduction
Today, as the world rapidly advances in energy innovation, renewable energy (RE) is an essential source for generating sustainable energy.1–3 Renewable energy is crucial for a sustainable future because it offers a cleaner, more environment-friendly alternative to traditional fossil fuels. Renewable sources of energy include solar, wind, hydropower, biomass, geothermal, and ocean energy. Among these, solar energy is considered the best renewable source due to its abundance, accessibility, sustainability, and low cost.4,5 The Earth receives approximately 3.8 million EJ (1 EJ = 1018 Joule) of solar energy annually and just one hour of this energy would be enough to meet the world's total energy consumption for an entire year, highlighting solar power's vast and sustainable potential.6 Solar energy can be harvested in several ways, including using photovoltaic (PV) cells7,8 that convert sunlight directly into electricity, solar thermal power systems,9 and solar water heating.10 PV cells are generally considered the most versatile and widely used technology due to their scalability, ease of installation, and direct electricity generation, making them the most effective source for distributed and decentralized energy production.11,12 The evolution of PV technologies is classified under three generations.13 The first-generation (wafer-based technology) PV cells are made from conventional silicon (Si)-based materials, and include traditional Si solar panels, mono-Si, and multi-Si.14,15 These cells offer high efficiency, but come with higher costs. The second-generation (thin-film technology) PV cells use materials like cadmium telluride or copper indium gallium selenide, offering flexibility and lower production costs, but moderate efficiency. Examples of second-generation PV cells also include gallium arsenide and cadmium sulfide.16,17 The third-generation (organic technology) solar cells, such as dye-sensitized solar cells (DSSCs), organic photovoltaics (OPVs), quantum-dot solar cells (QDSCs), and perovskite solar cells, are a more advanced and emerging technology that promises enhanced flexibility and high efficiency.18,19
The concept of using a dye-sensitized semiconductor to capture sunlight and generate electricity, known as the DSSC or Grätzel cell, was first co-developed by Brian O'Regan and Michael Grätzel in 1988. They advanced the technology at the École Polytechnique Fédérale de Lausanne, culminating in the first high-efficiency DSSC in 1991.20 The key components of a DSSC,21 as shown in Fig. 1, are: a transparent conducting electrode that allows light to pass and conducts electricity, TiO2 nanoparticles with an adsorbed organic dye that absorbs light and generates electrons, an electrolyte (I−/I−3) to facilitate electron flow and regeneration of the dye, and a counter electrode (platinum or carbon) to collect electrons and complete the circuit.
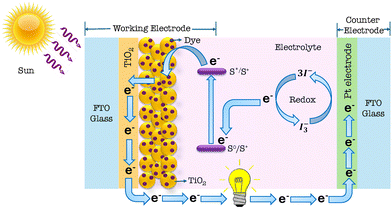 |
| | Fig. 1 Schematic diagram illustrating the working principle of a DSSC. | |
As sunlight strikes the dye on the TiO2 film, electrons from the dye are injected into the conduction band of the semiconductor (TiO2). These electrons diffuse through the mesoporous TiO2 layer towards the anode. The electrons are then transferred to the electrolyte at the counter electrode, which regenerates the dye. Finally, the electrolyte passes electrons back to the counter electrode, completing the circuit and allowing the process to repeat.
In a DSSC, the sensitizer plays a crucial role in capturing light and converting it into electrical energy. Researchers have mainly developed two types of sensitizers, organic22–25 and inorganic.26–29 The former include both natural and synthetic variants, while the latter ones are derived from metal complexes such as ruthenium (Ru), osmium, inorganic quantum dots and porphyrin. Ru-based dyes, such as N3,30 N719,31 and black dye,32 are among the best performing inorganic dyes. However, they suffer from severe drawbacks, such as lower absorption and the low abundance of Ru. To overcome these limitations, researchers explored and developed organic materials as sensitizers for DSSC. In recent years, porphyrin-based dyes, because of their easy and cost-effective synthesis and improved light-harvesting efficiency, have gained significant attention as promising sensitizers for DSSCs. The 18-π-electron conjugated system of porphyrin facilitates better absorption across a broader spectrum ranging from visible to near-infrared (NIR) wavelengths. One of the first reports highlighting the application of porphyrin in DSSCs is the work by Kay et al. in 1993, where the resultant devices were found to have a maximum power conversion efficiency (η) of 2.6%.33 In later reports, Wang et al. studied the photophysical and electrochemical properties of a series of novel green Zn-porphyrin-based dyes that achieve an η of 5.6%.34 Further, in 2007, Campbell et al. synthesized a novel porphyrin derivative containing an electron-accepting unit, which showed an η of 7.1%.35 In 2010, Bessho et al. tailored YD2 dyes by co-sensitization with an organic dye, which achieved an η of 11%.36 In 2014, Mathew et al. reported a porphyrin-dye-based sensitizer, SM315, with a cobalt(II/III) redox shuttle; the DSSC achieved a high open-circuit voltage (VOC) of 0.91 V, short-circuit current density (JSC) of 18.1 mA cm−2, fill factor of 0.78, and η of 13%.37 In 2015, Xie et al. engineered a porphyrin-based co-sensitizer with an improved JSC from 18.83 to 20.33 mA cm−2, resulting in an efficiency of 11.5%.38 In 2018, Kato et al. reported a new porphyrin-based dye (DIPDAB2) with an improved near-IR absorption, better current generation and optimized yield of about 10% solar conversion efficiency.39 In 2019, Imahori et al. synthesized monomeric porphyrin-based sensitizers, namely DfZnP and DfZnP-iPr; the optimized dye DfZnP-iPr achieved 10.1% η, and its co-sensitization with LEG4 further raised the efficiency to 10.7% for fused porphyrin DSSCs.40 Recently, in 2023, Chen et al. synthesized novel double-fence porphyrin dyes by incorporating an indacenodithiophene group as a donor. One of these dyes, namely YS7, is reported to have an η of 11.5%.41 Very recently, in 2025, Faraghally et al. introduced various acceptors, such as benzothiadiazole, benzotriazole, and cyanoacrylic acid, into porphyrin dyes to enhance their photovoltaic properties. Their strategy gives a maximum overall efficiency of 9.9%.42 All these studies indicate that porphyrin-based molecules are potential candidates for designing photosensitizers for DSSC applications. However, these sensitizers face certain limitations, such as limited NIR absorption, relatively small light harvesting efficiency (LHE), dye aggregation, and small electron injection efficiency.43 Researchers have adopted several strategies, such as incorporating electron-rich donor groups,44–46 electron-deficient acceptor groups,47–49 and dimerization50–52 to overcome said limitations.
In recent years, expanded porphyrins have drawn attention in the field of DSSCs.53–57 For example, in 2019, Borges-Martínez et al. carried out a theoretical study on BODIPY-oxasmaragdyrin dyads for DSSC applications. In this work, acetylene-bridged dyads showed enhanced charge separation, panchromatic absorption, and favorable energy alignment for electron injection and dye regeneration.54 In 2019, Pino-Rios et al.,55 in a computational study of meso- and beta-halogenated expanded porphyrins (sapphyrin and orangarin), revealed that in meso-substituted systems, halogenation reduces symmetry, stabilizes the frontier molecular orbitals, and decreases the energy gap leading to an increase in Q-band oscillator strength, and improving the LHE. The same group in 2021 conducted a theoretical investigation on sapphyrin derivatives modified by replacing imine units with oxazolone groups. These modifications resulted in enhanced NIR absorption, improved electron injection, increased aromaticity, and better charge transport properties, highlighting their potential as efficient chromophores for DSSCs.56 Very recently, in 2023, Menéndez et al. studied fused core-modified porphyrins (pentaphyrin, hexaphyrin, heptaphyrin, and octaphyrin) as potential dyes for DSSCs, showing strong absorption in the visible to NIR range and efficient electron injection. Pentaphyrin and hexaphyrin showed better dye regeneration, hexaphyrin and heptaphyrin had strong aromaticity from planarity, and pentaphyrin and octaphyrin exhibited superior charge transport, making them promising for solar cells.57 To the best of our knowledge, none of these studies address the computation or measurement of the photovoltaic properties of expanded porphyrin systems. Furthermore, there are several other expanded and contracted porpyrin systems that can be explored for DSSC applications.
Being inspired by the work of Pino et al.(2019), in the current work we focus on designing the first antiaromatic orangarin-based sensitizers that enhance the performance of DSSCs. To achieve this, several modifications have been made, including altering the donor groups and varying their positions simultaneously. The donor moieties were chosen based on findings from previous experimental studies,58–64 where their performance was systematically evaluated in combination with porphyrin.
2. Theory
To evaluate the PV performance of a DSSC, it is essential to consider the following factors.65–67
2.1. Power conversion efficiency (η)
The η of a DSSC is a measure of how effectively the cell converts sunlight into electrical energy. It is calculated using the following formula:68,69| | 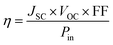 | (1) |
where JSC is the short-circuit current density (mA cm −2), VOC is the open-circuit voltage (V), FF is the fill factor (a measure of the solar cell's quality), and Pin is the power of the incident light (mW cm−2).
2.2. Short-circuit current density (JSC)
J
SC is the photocurrent density generated when a solar cell is short-circuited. For a DSSC, JSC indicates how efficiently the cell converts light into charge carriers. A higher JSC signifies better charge collection and improved light-to-electricity conversion, and is given as:70| | 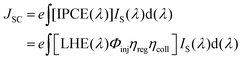 | (2) |
where e is the elementary charge, and Φinj,71ηreg, and ηcoll72 are, respectively, the electron injection, dye regeneration, and electron collection efficiencies. IS(λ) under AM1.5G conditions represents the standard solar photon flux, measured in units of mA cm−2 nm−1. When JSC is maximum, i.e., JmaxSC, the values of Φinj, ηreg, ηcoll are set to unity.| | | IPCE(λ) = LHE(λ) × Φinj× ηreg × ηcoll | (3) |
where Φinj, ηreg, ηcoll are unity for the ideal case, when JSC is maximum, i.e., JmaxSC. LHE is expressed as:65where ε(λ) is the molar absorption coefficient at a certain wavelength λ, and Γ is the surface loading of the dye, i.e., the product of the concentration of the dye and thickness of the TiO2 layer.
2.3. Open-circuit voltage (VOC)
V
OC is the maximum voltage a solar cell generates when exposed to light under open circuit conditions, and is calculated as:73where ELUMO is the energy of the LUMO of the considered dye, and ECBM is the conduction band minimum of TiO2. It is important to note that the theoretical value of VOC is reported in eV as eVOC.
2.4. Exciton binding energy (EBE)
EBE is the energy needed to separate an exciton into a free electron and hole. It is evaluated as:67| | | EBE = ELUMO − EHOMO − E0−0 | (6) |
where ELUMO, EHOMO respectively are the energies of the LUMO and HOMO of the dye, and E0−0 denotes the vertical excitation energy associated with the maximum absorption wavelength of the dye.
2.5. Fill factor (FF)
FF is defined as the ratio of the maximum power output of a device to its theoretical maximum power, and is expressed as:74| | 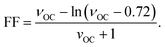 | (7) |
νOC in the above equation depends upon VOC and temperature. The relation is given as| | 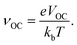 | (8) |
2.6. Rate of intermolecular electron transfer
The rate of electron transfer within a system as determined from Marcus theory is given as:66,75,76| |  | (9) |
where, ΔG0inj/rec is the change in Gibbs free energy for the process (electron injection/recombination) and is given as77
A is a unit-less constant with a value of 2.5 × 10−2, β is known as the attenuation factor and its value is estimated from empirical values (0.5 Å−1).66r is the electron transfer distance, where rinj is the distance between the electron accepting group (the acid anchoring group of the dye) and the TiO2 surface, and rrec is the distance between the electron donating group (the donor group of the dye) and the TiO2 surface,65 with their values provided in the SI file (Table S5). Λ is the reorganization energy corresponding to the combined structural changes in the donor and acceptor molecules during charge transfer. It is given as
where
Λe and
Λh are the reorganization energies for the electron and hole, respectively.
| | 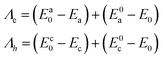 | (13) |
here
E0 is energy of the optimized neutral molecule, and
Ea/c0 is the energy of the cation or anion evaluated at the geometry of the optimized neutral molecule.
Ea/c refers to the total energy of the fully optimized cation or anion, while
E0a/c represents the energy of the neutral molecule calculated at the optimized geometry of the corresponding charged species. Further, the change in Gibbs free energy for dye regeneration (Δ
Greg) is given as
78| | | ΔGreg = Eredox − Edye | (14) |
where
Eredox is the redox potential of the electrolyte (for I
−/I
−3, the value is −4.8 eV)
79 and
Edye is the oxidation potential of the dye in the ground state and is given as the HOMO of the dye.
80
3. Computational details
The ground state geometries of all the dyes were fully optimized in acetonitrile solvent using B3LYP/6-311+G(d,p) with Grimme's D3 empirical dispersion correction with Becke–Johnson damping,81,82 and the conductor-like polarizable continuum model (C-PCM).83 To ensure the obtained structures correspond to a true minimum, a vibrational frequency analysis was carried out at the same level. The absence of any imaginary frequency confirmed that the optimization resulted in a stable configuration. After geometry optimization and frequency calculations, the reorganization energy was determined by optimizing the neutral and charged ground-state geometries of the cation and anion at the same level of theory. Single-point energy calculations were then performed for these optimized structures. In the next step, the optical absorption spectra of all the molecules were simulated in acetonitrile solvent using the TD-CAM-B3LYP/6-311+G(d,p) method. The CAM-B3LYP functional is a combination of the hybrid functional B3LYP and a long-range correction term, designed to better describe charge transfer excitations, which are important for accurately predicting optical properties.84 The charge density difference (CDD) plot and distance of the charge transfer (dCT)85 are calculated using the MN1586,87/6-311+G(d,p) level of theory. MN15 is a long-range exchange correlation functional well-known88 for charge transfer studies. Compared to the CC2 model, this functional gives relatively accurate ground- and excited-state electronic densities.88 All these calculations were performed using the Gaussian 16 program package.89
To study the adsorption of the dyes on the TiO2 surface, which is essential for understanding the nature of interactions between the two, we considered a (TiO2)38 cluster and optimized the geometries of the cluster, the dye, and dye-cluster systems using the DMol390,91 program in Materials Studio Version 7.0. These calculations employed the generalized gradient approximation (GGA)92,93 method with the Perdew–Burke–Ernzerhof (PBE)94,95 exchange–correlation functional and the double numerical polarized (DNP)96 basis set, in acetonitrile solvent using the Conductor-like Screening Model (COSMO).97,98 To enhance the computational efficiency, a Fermi smearing of 0.005 Hartree and a global cutoff of 5.2 Å were applied. The convergence criteria were set to 1 × 10−5 Hartree for energy, 2 × 10−3 Hartree per Å for force, and 5 × 10−3 Å for displacement. Subsequently, all dyes were adsorbed onto the (TiO2)38 surface in a bidentate bridging configuration, where both oxygen atoms of the –COOH anchoring group coordinate with two titanium atoms. This mode of binding is considered the most stable for a carboxyl group as a anchoring group.99,100 From the energy of the optimized structures, the adsorption energy and the parameters required for photovoltaic properties were calculated for the dye@(TiO2)38 surface.
4. Result and discussion
4.1. Naming convention used in this work
The structures of the 10 donor–acceptor-substituted orangarin molecules considered for this study are shown in Fig. 2. Five different donors, viz. carbazole (CBZ), bis(4-hexyl)phenyl amine (DPA), phenothiazine (PTZ), phenothiazine glycol (GLY) and triarylamine (TAA), and a fixed acceptor, namely benzoic acid, are considered to design the 10 molecules. The donor moieties are chosen based on previous studies,58–64 where it was shown that porphyrin-based DSSC systems having these groups display better efficiency. Orangarin is considered as the core for attaching the donor and acceptor groups. The acceptor is attached at one of the meso positions and the donor (D) is varied between positions 4 (D′) and 7 (D″), as shown in Fig. 2. The molecules are named as ODnm, where O represents orangarin core, D represents one of the donors (CBZ, DPA, PTZ, GLY, or TAA), and n and m represent, respectively, the positions of the acceptor and donor groups, as shown in Fig. 2. The rationale behind choosing positions 4 and 7 for the donors is supported by our initial assessment of one of the donors placed successively at all possible positions in the orangarin core (except 1, 2, 10, and 11).
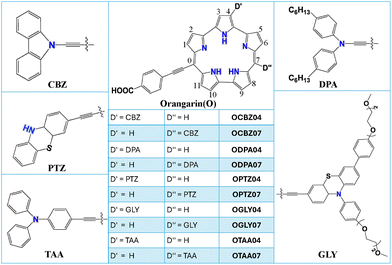 |
| | Fig. 2 Representation of the 10 different orangarin-based dyes considered for this study. | |
4.2. Frontier molecular orbital energy alignment
In a DSSC, when light is absorbed by the dye molecules, the dye is excited as an electron moves from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). The excited electron is then injected into the conduction band of the TiO2 semiconductor. For efficient electron injection, the LUMO energy level of the dye must be higher than the conduction band edge of TiO2, which is typically around −4.0 eV. After injection, the dye molecule is left in an oxidized state and must be regenerated by accepting an electron from the redox electrolyte; for this to happen effectively, the HOMO level of the dye should be lower than the redox potential of the I−/I−3 electrolyte, which is approximately −4.8 eV.101 The energies of the HOMO and LUMO, and the corresponding energy gaps for all the ten dyes are illustrated in Fig. 3. It is evident that the LUMOs of all the dyes are positioned above the conduction band of TiO2, facilitating efficient electron injection, and the HOMO levels are situated below the redox potential of the electrolyte, indicating favorable conditions for rapid regeneration of the dye molecules.
 |
| | Fig. 3 Energy levels and HOMO–LUMO gaps of all the 10 dyes, calculated at the TD-CAM-B3LYP/6-311+G(d,p)/C-PCM level of theory in acetonitrile solvent. | |
4.3. UV-visible absorption and charge transfer analysis
Absorption studies play a vital role in DSSCs by assessing the dye's ability to capture solar energy efficiently. For optimal performance, the dye should strongly absorb light in the visible region (≈400–700 nm), where sunlight intensity peaks, to maximize photon absorption and enhance overall solar energy conversion (Table 1).
Table 1 Calculated data for electronic transitions in terms of excitation energies (in eV), wavelengths (in nm), oscillator strengths, and orbital contributions for the dyes at the TD-CAM-B3LYP/6-311+G(d,p)/C-PCM level of theory in acetonitrile solvent
| Dye |
Excited state |
Excitation energy (eV) |
Wavelength (λ) (nm) |
Oscillator strength |
Contribution |
| SM315 |
S3 |
2.60 |
477 |
1.06 |
H → L+1 (46%) |
| OCBZ04 |
S2 |
2.56 |
483 |
1.60 |
H → L+1 (62%) |
| OCBZ07 |
S2 |
2.48 |
499 |
1.50 |
H−1 → L (54%) |
| ODPA04 |
S3 |
2.49 |
497 |
1.42 |
H → L+1 (65%) |
| ODPA07 |
S2 |
2.39 |
517 |
1.82 |
H−1 → L (63%) |
| OPTZ04 |
S2 |
2.50 |
496 |
1.29 |
H → L+1 (49%) |
| OPTZ07 |
S2 |
2.25 |
550 |
1.87 |
H−1 → L (58%) |
| OGLY04 |
S3 |
2.52 |
492 |
1.29 |
H → L+1 (54%) |
| OGLY07 |
S4 |
2.10 |
590 |
1.09 |
H−2 → L (61%) |
| OTAA04 |
S3 |
2.50 |
496 |
1.91 |
H → L+1 (64%) |
| OTAA07 |
S2 |
2.17 |
571 |
2.10 |
H−1 → L (59%) |
The simulated absorption profiles of all the ten dyes and SM315, displayed in Fig. 4, indicate that all of these dyes absorb strongly in the UV-vis range. The strongest absorption peak always corresponds to either the S0 → S2, S0 → S3 or S0 → S4 transition and is red-shifted in the order of CBZ < DPA < PTZ < TAA < GLY, which correlates well with the increasing conjugation. We further noticed that OD07 dyes consistently show a larger red-shift compared to their OD04 counterparts. Notably, OGLY07 shows the largest red-shift among the considered systems. This trend can be explained by examining the excitation energies. The excitation energy of the OD04 dyes does not change significantly, whereas the same for the OD07 systems gradually decreases on moving from OCBZ07 to OGLY07, resulting in the largest oscillator strength for OCBZ07.
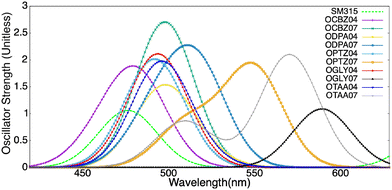 |
| | Fig. 4 UV-visible spectra of the dyes calculated at the TD-CAM-B3LYP/6-311+G(d,p)/C-PCM level of theory in acetonitrile solvent. | |
A study by Woller et al.102 on orangarin reveals that its antiaromatic character arises from a fully conjugated 20 π-electron macrocyclic system, formed by three fused pyrrole rings and the meso-carbon bridges that connect them. This extended conjugation pathway includes both the OD04 positions (β-positions of the pyrrole units) and the OD07 positions (meso-carbons), enabling continuous π-electron delocalization throughout the ring system. Notably, both OD04 and OD07 are integral components of this conjugated circuit and play a direct role in the molecule's antiaromatic behavior. These positions exhibit higher one photon absorption strengths compared to other sites, reflecting their enhanced electronic involvement. The researchers also analyzed the anisotropy of the induced current density (AICD) plots, which showed a paratropic (anticlockwise) ring current, which is consistent with the presence of antiaromatic systems.102 Orangarin displays a stronger current density within the inner circuit, particularly at higher AICD isovalues. The OD07 position lies within this inner circuit, meaning that electron donors at this site directly contribute to the antiaromatic ring current. This antiaromaticity introduces electronic instability by raising the energy of the delocalized π-system, which in turn significantly affects the molecule's optical absorption. Specifically, it leads to a red shift in the absorption spectrum due to a narrowing of the gap between the involved orbital pairs. An orbital analysis of the strongest peaks revealed that in all the OD04 systems, the HOMO → LUMO+1 is the most contributing orbital transition involving a shift of electron density from the orangarin core to the acceptor moiety. However, the strongest peak in all the OD07 systems is dominated by HOMO−1 → LUMO and HOMO−2 → LUMO transitions, where the electron density is moved from the donor moiety to the acceptor moiety via the orangarin core, indicating a long-range charge transfer in these systems. The orbital pictures are presented in Fig. 5. To get further insight into the charge transfer mechanism, we calculated the CDD and dCT for all the dyes and SM315, as shown in Fig. 6, where the CDD is evaluated as the difference between the total electron densities of the excited and ground states, and dCT is the distance between the barycenter of the positive and negative charge densities. Here, the CDD plots represent the distribution of electron and hole densities, where the green color represents the electron-deficient regions and yellow color represents the electron-rich regions. A large spatial distribution of the electron and hole densities signifies a stronger intramolecular charge transfer (ICT) character, while their overlap indicates a localized excitation process. Among the investigated dyes, it was observed that a more distinct delocalization of electron and hole densities was observed for OPTZ04, OPTZ07, and OTAA07 compared to SM315, confirming that the excited-state transition is primarily ICT in nature. This is also corroborated by the higher dCT values for the respective systems. This pronounced charge separation highlights the role of the donor and acceptor groups in enhancing the ICT process, which is crucial for improving charge transport and overall photovoltaic performance.
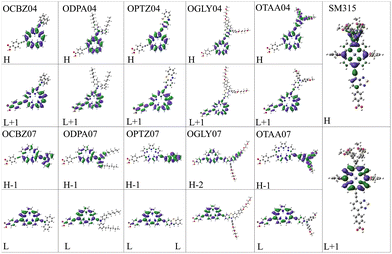 |
| | Fig. 5 Most contributing pairs of orbitals involved in the strongest one photon absorption peak in all the designed dyes, calculated at the TD-CAM-B3LYP/6-311+G(d,p)/C-PCM level of theory in acetonitrile solvent. | |
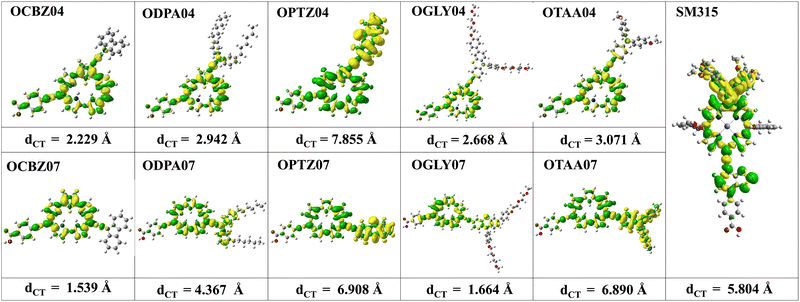 |
| | Fig. 6 Plot showing the charge density difference (CDD) and charge transfer distance (dCT), calculated at the MN15/6-311+G(d,p) level of theory. Here, yellow zones represents electron-rich regions and green zones represent electron-deficient regions. An iso-contour value of 0.0004 a.u. was used for the dCT plot. | |
4.4. Adsorption of dyes on a (TiO2)38 cluster
In an actual solar cell, the dye is adsorbed on a semiconductor, say TiO2. This adsorption may affect the relevant photophysical properties of the dyes. To mimic the actual scenario, we calculated the adsorption energy and corresponding change in orbital energies of all the ten dyes adsorbed on a TiO2 cluster. Based on a literature survey,103–105 we considered the (TiO2)38 cluster for adsorbing our dyes. The dyes are attached to the (TiO2)38 surface through a bidentate binding between the two oxygen atoms of the –COOH anchoring group of the dye and two neighbouring titanium atoms of the (TiO2)38 surface. The adsorption energy (Eads) was calculated using:| | | Eads = Edye+TiO2 − (Edye + ETiO2) | (15) |
where Edye is total energy of the isolated dye, ETiO2 is the total energy of the bare (TiO2)38 cluster and Edye+TiO2 is the total energy of the dye-(TiO2)38 system. The calculated HOMO, LUMO, HOMO–LUMO gap, and Eads after adsorption are listed in Table 2.
Table 2 Calculated HOMOs, LUMOs, band gaps, Ti–O bond lengths (in bidendate bridging), and Eads (kcal mol−1) of the dye@(TiO2)38 clusters, calculated using the TD-CAM-B3LYP/6-311+G(d,p)/LANL2DZ/C-PCM level of theory for dyes optimised using GGA/PBE/DNP in acetonitrile solvent using the DMol3 package
| Dye@TiO2 |
HOMO (eV) |
LUMO (eV) |
Band gap (eV) |
Ti–O (Å) |
Ti–O (Å) |
E
ads (kcal mol−1) |
| SM315@TiO2 |
−6.10 |
−2.45 |
3.65 |
2.16 |
2.12 |
−12.50 |
| OCBZ04@TiO2 |
−5.94 |
−2.90 |
3.04 |
1.29 |
1.59 |
−13.44 |
| OCBZ07@TiO2 |
−5.86 |
−2.91 |
2.94 |
1.31 |
1.60 |
−12.03 |
| ODPA04@TiO2 |
−5.75 |
−2.88 |
2.87 |
1.38 |
1.41 |
−11.93 |
| ODPA07@TiO2 |
−5.80 |
−2.84 |
2.96 |
2.14 |
2.17 |
−9.21 |
| OPTZ04@TiO2 |
−5.87 |
−2.90 |
2.97 |
2.10 |
2.14 |
−5.81 |
| OPTZ07@TiO2 |
−5.85 |
−2.98 |
2.87 |
1.62 |
1.84 |
−8.29 |
| OGLY04@TiO2 |
−5.88 |
−2.90 |
2.98 |
2.09 |
2.14 |
−10.62 |
| OGLY07@TiO2 |
−5.85 |
−2.99 |
2.87 |
2.13 |
2.14 |
−10.13 |
| OTAA04@TiO2 |
−5.85 |
−2.91 |
2.94 |
2.15 |
2.16 |
−13.09 |
| OTAA07@TiO2 |
−5.83 |
−2.96 |
2.87 |
1.75 |
1.40 |
−9.44 |
The results indicate that all the dye-(TiO2)38 systems possess negative values for Eads, indicating a more stable interaction between the two. Among all the dyes, OCBZ04 exhibits a slightly higher Eads value along with a shorter Ti–O bond length, indicating a strong interaction with the TiO2 surface. The interaction strength follows the trend: OCBZ04 (−13.44 kcal mol−1) > OTAA04 (−13.09 kcal mol−1) > OCBZ07 (−12.03 kcal mol−1) > ODPA04 (−11.93 kcal mol−1) > OGLY04 (−10.62 kcal mol−1) > OGLY07 (−10.13 kcal mol−1) > OTAA07 (−9.44 kcal mol−1) > ODPA07 (−9.21 kcal mol−1) > OPTZ07 (−8.29 kcal mol−1) > OPTZ04 (−5.81 kcal mol−1). A further analysis of the HOMO, LUMO, and corresponding energy gap as shown in Table 2 indicates that the HOMO–LUMO gap increases with the adsorption of the dyes on the (TiO2)38 surface.
4.5. Photovoltaic properties of dyes on a (TiO2)38 cluster
The computed values of the photovoltaic properties mentioned in the theory section, viz. EBE, eVOC, JmaxSC, vOC, FF and η, of the dyes adsorbed on the (TiO2)38 surface are presented in Table 3. The ideal value of LHE is 1, representing 100% absorption of photons. The variation of LHE with the wavelength of the absorbed light is displayed in Fig. 7.
Table 3 Table showing the calculated photovoltaic properties of all studied dyes adsorbed on the (TiO2)38 cluster
| Dye@TiO2 |
EBE (eV) |
eV
OC (eV) |
J
maxSC (mA cm−2) |
v
OC
|
FF |
η (%) |
| SM315@TiO2 |
1.05 |
1.55 |
27.15 |
59.90 |
0.92 |
38.57 |
| OCBZ04@TiO2 |
0.48 |
1.10 |
18.60 |
42.54 |
0.89 |
18.25 |
| OCBZ07@TiO2 |
0.49 |
1.09 |
21.54 |
41.95 |
0.89 |
20.81 |
| ODPA04@TiO2 |
0.40 |
1.12 |
22.00 |
43.29 |
0.89 |
22.00 |
| ODPA07@TiO2 |
0.59 |
1.16 |
25.13 |
44.80 |
0.90 |
26.08 |
| OPTZ04@TiO2 |
0.47 |
1.10 |
23.78 |
42.50 |
0.89 |
23.21 |
| OPTZ07@TiO2 |
0.71 |
1.02 |
27.51 |
39.27 |
0.88 |
24.72 |
| OGLY04@TiO2 |
0.47 |
1.10 |
23.28 |
42.40 |
0.89 |
22.76 |
| OGLY07@TiO2 |
0.72 |
1.02 |
27.19 |
39.23 |
0.88 |
24.41 |
| OTAA04@TiO2 |
0.45 |
1.09 |
23.84 |
42.26 |
0.89 |
23.22 |
| OTAA07@TiO2 |
0.75 |
1.04 |
25.87 |
40.06 |
0.89 |
23.77 |
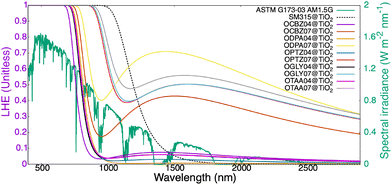 |
| | Fig. 7 LHE plot for all the considered dye@(TiO2)38 systems, against the AM 1.5G solar spectrum shown in green. | |
The plot reveals that for the dye@(TiO2)38 systems, the maximum value of the LHE is obtained in the wavelength range of 680 to 950 nm. In general, the LHE plots for the OD07 dyes are broader and red-shifted as compared to those for the OD04 dyes, suggesting better absorption in the solar spectrum and hence making them better candidates for DSSC applications. Among the OD07 dyes, OTAA07 shows greatest red-shift in its LHE plot.
To generate an electric current efficiently from the absorbed photons, the excitons thus created should be separated into electrons and holes. This can be done if their respective binding energy, i.e., EBE, is very small.106 In Table 3, we report that the EBE values for all the designed dyes range between 0.40 to 0.75 eV, lower than the value calculated for SM315 (1.0 eV), indicating a balance between exciton stability and charge carrier generation and correlating well with the previously reported values in the literature.67,80,107 The short-circuit current density is calculated as the maximum theoretical value (JmaxSC) when the incident photon-to-current efficiency (IPCE) across the absorption spectrum is 100%, i.e., when Φinj, ηreg and ηcoll are all taken to be unity. The experimental value47 of JSC for the best performing porphyrin-based DSSC dye (SM315) is 18.1 mA cm−2 as compared to the theoretical value of JmaxSC of 27.15 mA cm−2, as shown in Table 3. The values of JmaxSC for all the dye@(TiO2)38 systems considered in this work lie in the range of 18.60 mA cm−2 for OCBZ04 to 27.51 mA cm−2 for OPTZ07, suggesting that the JmaxSC values of the studied dyes are comparable to that of SM315. We observed that the OD07 dyes have larger values of JmaxSC than the respective OD04 siblings, which is directly related to the larger oscillator strength of the former than the latter. Amongst them, the OPTZ07 and OGLY07 dyes exhibit a comparable photoresponse to SM315, indicating similar light-harvesting characteristics.
To understand the η of the dyes, it is also important to analyze the eVOC, which depends upon the ECB and the number of electrons in the conduction band. All of our designed dyes possess high eVOC values, ranging from 1.02 eV to 1.16 eV (Table 3), which are comparable to that of the SM315 dye, as shown in Table 3. The largest value of eVOC (1.16 eV) is observed for ODPA07. An elevated eVOC reflects effective charge carrier separation and reduced recombination losses, which are critical for achieving high solar-cell efficiency. Our further computation reveals that the fill-factor and overall η are also the largest (0.90 and 26.08% respectively) for ODPA07. The values of η for the studied dyes range from 18.25% to 26.08%. The OD07 dyes possess larger η than their OD04 counterparts.
4.6. Rate of electron transfer between the dye and (TiO2)38
k
inj/rec can be understood using Marcus theory, as mentioned in the Theory section (eqn (9)). The calculated reorganisation energies, and the changes in Gibbs free energy for the two processes (electron injection and recombination) along with their respective calculated rates, are presented in Table 4. The changes in Gibbs free energy for the electron injection (ΔG0inj) and recombination (ΔG0rec) and dye regeneration (ΔG0reg) processes are crucial for ensuring efficient charge transfer and dye regeneration. These quantities are calculated from the energies of the HOMO (EHOMO) and LUMO (ELUMO) of the dye and the conduction band minimum (ECB) of TiO2.
Table 4 Gibbs free-energy change for electron injection (ΔG0inj) and electron recombination (ΔG0rec), reorganization energy (Λ), rate of electron injection (kinj), rate of electron recombination (krec), and Gibbs free-energy change for dye regeneration (ΔGreg) of the considered dyes with the (TiO2)38 cluster
| Dye@TiO2 |
ΔG0inj (eV) |
ΔG0rec (eV) |
Λ (eV) |
k
inj (1015 s−1) |
k
rec (1012 s−1) |
ΔGreg (eV) |
| SM315@TiO2 |
0.33 |
1.05 |
0.33 |
0.51 |
0.0003 |
0.25 |
| OCBZ04@TiO2 |
1.10 |
1.94 |
0.14 |
4.53 |
4.93 |
1.14 |
| OCBZ07@TiO2 |
1.09 |
1.86 |
0.13 |
5.39 |
1.01 |
1.06 |
| ODPA04@TiO2 |
1.12 |
1.75 |
0.09 |
9.91 |
11.9 |
0.95 |
| ODPA07@TiO2 |
1.16 |
1.80 |
0.14 |
5.22 |
0.68 |
1.00 |
| OPTZ04@TiO2 |
1.10 |
1.87 |
0.07 |
13.7 |
13.4 |
1.07 |
| OPTZ07@TiO2 |
1.02 |
1.85 |
0.11 |
6.33 |
1.38 |
1.05 |
| OGLY04@TiO2 |
1.10 |
1.88 |
0.07 |
14.9 |
29.5 |
1.08 |
| OGLY07@TiO2 |
1.02 |
1.85 |
0.13 |
4.53 |
0.95 |
1.05 |
| OTAA04@TiO2 |
1.09 |
1.85 |
0.06 |
17.3 |
24.0 |
1.05 |
| OTAA07@TiO2 |
1.04 |
1.83 |
0.10 |
6.73 |
1.31 |
1.03 |
According to a study by Fu et. al,67 ΔG0inj > 0.2 eV leads to Φinj ≈1, indicating an efficient electron injection from the dye to TiO2. Table 4 shows that our designed dyes have ΔG0inj ranging from 1.02 eV to 1.16 eV and ΔG0rec ranging from 1.75 eV to 1.94 eV. The respective values for SM315 are 0.33 eV and 1.05 eV.
The reorganization energy for all the investigated dyes lies within the range of 0.06 eV to 0.14 eV, lower than that of the SM315 dye, thus indicating a relatively low energy barrier for electron transfer. Such low Λ values further suggest that minimal structural rearrangement is required during the electron transfer process, thereby promoting efficient and rapid charge transfer from the donor to the acceptor, as shown in Fig. 8. This also helps in minimizing charge accumulation on the dye molecules, which is beneficial for improving overall device performance. Further, the kinj is about 103 times higher than the krec, with both being higher than those of the SM315 dye. This combination is favorable for efficient charge separation, as it ensures rapid electron transfer from the dye to the (TiO2)38 cluster while minimizing charge loss due to recombination. As a result, the efficient intermolecular electron transfer validates the improved performance of the engineered dyes and highlights their effectiveness in enhancing overall device efficiency. Apart from efficient electron injection into the conduction band of (TiO2)38, it is also very important that the oxidized dye be reduced by the electrolyte and regenerated. The results of ΔGreg for all the dye@(TiO2)38 systems are presented in Table 4. From the table, it is observed that all the designed dyes have ΔGreg > 0.95 eV, which exceeds the minimum threshold criterion of 0.60 eV108 as reported in various experimental studies,109 thus indicating better recombination for the studied dyes than SM315. Thus, all our tailored dyes exhibit efficient charge regeneration and a reduced possibility of charge recombination.
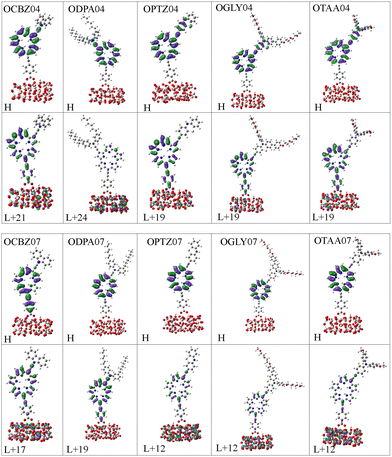 |
| | Fig. 8 HOMO and LUMO orbitals showing charge transfer for dyes adsorbed on the (TiO2)38 surface, calculated at the TD-CAM-B3LYP/6-311+G(d,p)/C-PCM level of theory. | |
5. Conclusion
In conclusion, using the state-of-the-art DFT and TDDFT methods, we designed ten orangarin-based molecules that can be used as photosensitizers (dyes) in DSSCs. To the best of our knowledge, this is the first study where an antiaromatic core devoid of any metal is used for designing a DSSC-dye. For this purpose, we attached five donor moieties at the two most promising positions (4 and 7) in the orangarin core and studied all the DSSC-relevant properties of the ten designed molecules. Our study reveals that all the ten dyes absorb strongly in the visible region where the solar spectrum AM1.5G displays maximum irradiance. Due to the antiaromatic nature of the orangarin core, the substitution of a donor at the 7th position places it in the inner circuit and contributes to the ring-current. This leads to a red-shift in the absorption profile and larger oscillator strength for the OD07 molecules. All the ten dyes display negative adsorption energy, reflecting good binding with the (TiO2)38 surface. The adsorption also leads to an increase in the HOMO–LUMO gap and hence their absorption falls within the visible region. The study of the photovoltaic properties suggests that all the OD07 dyes have larger open-circuit voltage, maximum short-circuit current density and overall η. Almost all of the dyes have η larger than 20%, with a maximum value of 26.08% for ODPA07. Our study suggests that an antiaromatic molecule can also be used for designing a highly efficient dye for DSSC applications.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: 1. HOMO, LUMO, oscillator strength, wavelength, major orbital contribution, and dihedral angles of all triarylamine-based dyes considered in this work, calculated at the B3LYP/6-311+G(d,p) level of theory using the Gaussian 16 program. 2. HOMO and LUMO orbital diagram for orangarin-triarylamine derivatives, calculated at the TD-CAM-B3LYP/6-311+G(d,p) level of theory using the Gaussian 16 program. 3. Energy levels of the HOMO and LUMO, and energy gap of dye@TiO2 systems calculated at the TD-CAM-B3LYP/6-311+G(d,p)/LANL2DZ/C-PCM level of theory in acetonitrile, using the Gaussian 16 program. 4. The optimized Cartesian coordinates of all dyes and dye@TiO2 systems considered in this work, which are optimized using the GGA/PBE/DNP level of theory, in acetonitrile solvent using Materials Studio software. 5. Table showing the values of electron transfer distance for recombination (rrec) and injection (rinj). 6. HOMO and LUMO orbitals showing charge transfer for SM315 adsorbed on the (TiO2)38 surface, calculated at the TD-CAM-B3LYP/6-311+G(d,p)/LANL2DZ/C-PCM level of theory. 7. The transition dipole moment integral values, oscillator strengths and excitation energies (in eV) computed for the studied dyes. See DOI: https://doi.org/10.1039/d5cp03388h.
Acknowledgements
The authors acknowledge the Wrocław Center for Networking and Supercomputing for granting computational resources. BT acknowledges the Indian Institute of Technology Bhilai for a fellowship. NC acknowledges DST India for her INSPIRE fellowship (grant no. IF220460). SR acknowledges Dr Alam for offering an internship at IIT Bhilai.
References
- P. A. Østergaard, N. Duic, Y. Noorollahi and S. Kalogirou, Renewable Energy, 2022, 199, 1145–1152 CrossRef.
- Q. Hassan, P. Viktor, T. J. Al-Musawi, B. Mahmood Ali, S. Algburi, H. M. Alzoubi, A. Khudhair Al-Jiboory, A. Zuhair Sameen, H. M. Salman and M. Jaszczur, Renewable Energy Focus, 2024, 48, 100545 CrossRef.
- E. T. Sayed, A. G. Olabi, A. H. Alami, A. Radwan, A. Mdallal, A. Rezk and M. A. Abdelkareem, Energies, 2023, 16, 1415 CrossRef CAS.
- N. Panwar, S. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 Search PubMed.
- I. Dincer and C. Acar, Int. J. Energy Res., 2015, 39, 585–606 CrossRef.
- M. Grätzel, Nature, 2001, 414, 338–344 Search PubMed.
- A. S. Al-Ezzi and M. N. M. Ansari, Appl. Syst. Innov., 2022, 5, 67 Search PubMed.
- A. R. Jordehi, Renewable Sustainable Energy Rev., 2016, 61, 354–371 Search PubMed.
- K. M. Powell, K. Rashid, K. Ellingwood, J. Tuttle and B. D. Iverson, Renewable Sustainable Energy Rev., 2017, 80, 215–237 Search PubMed.
- M. Raisul Islam, K. Sumathy and S. Ullah Khan, Renewable Sustainable Energy Rev., 2013, 17, 1–25 Search PubMed.
- E. K. Solak and E. Irmak, RSC Adv., 2023, 13, 12244–12269 Search PubMed.
- H. Meddeb, M. Götz-Köhler, N. Neugebohrn, U. Banik, N. Osterthun, O. Sergeev, D. Berends, C. Lattyak, K. Gehrke and M. Vehse, Adv. Energy Mater., 2022, 12, 2200713 CrossRef CAS.
- R. A. Marques Lameirinhas, J. P. N. Torres and J. P. de Melo Cunha, Energies, 2022, 15, 1823 CrossRef CAS.
- S. R. Wenham and M. A. Green, Prog. Photovolt.: Res. Appl., 1996, 4, 3–33 CrossRef CAS.
- L. C. Andreani, A. Bozzola, P. Kowalczewski, M. Liscidini and L. Redorici, Adv. Phys. X, 2019, 4, 1548305 CAS.
- M. M. A. Moon, M. F. Rahman, J. Hossain and A. B. M. Ismail, Adv. Mater. Res., 2019, 1154, 102–111 Search PubMed.
- O. Vigil-Galán, M. Courel, J. Andrade-Arvizu, Y. Sánchez, M. Espndola-Rodrguez, E. Saucedo, D. Seuret-Jiménez and M. Titsworth, J. Mater. Sci.: Mater. Electron., 2015, 26, 5562–5573 CrossRef.
- N. Shah, A. Shah, P. Leung, S. Khan, K. Sun, X. Zhu and Q. Liao, Processes, 2023, 11, 1852 CrossRef CAS.
- M. Neukom, S. Züfle, S. Jenatsch and B. Ruhstaller, Sci. Technol. Adv. Mater., 2018, 19, 291–316 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- J. Wang, X. Xie, G. Weng, Y. Yuan, J. Zhang and P. Wang, ChemSusChem, 2018, 11, 1460–1466 CrossRef CAS PubMed.
- Y. Ren, D. Sun, Y. Cao, H. N. Tsao, Y. Yuan, S. M. Zakeeruddin, P. Wang and M. Gratzel, J. Am. Chem. Soc., 2018, 140, 2405–2408 CrossRef CAS PubMed.
- J. Wang, H. Wu, L. Jin, J. Zhang, Y. Yuan and P. Wang, ChemSusChem, 2017, 10, 2962–2967 CrossRef CAS PubMed.
- Y. Ren, Y. Li, S. Chen, J. Liu, J. Zhang and P. Wang, Energy Environ. Sci., 2016, 9, 1390–1399 RSC.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS PubMed.
- Q. Yu, Y. Wang, Z. Yi, N. Zu, J. Zhang, M. Zhang and P. Wang, ACS Nano, 2010, 4, 6032–6038 Search PubMed.
- L. Han, A. Islam, H. Chen, C. Malapaka, B. Chiranjeevi, S. Zhang, X. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057–6060 Search PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS PubMed.
- Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Han, Jpn. J. Appl. Phys., 2006, 45, L638 CrossRef CAS.
- A. Kay and M. Graetzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- Q. Wang, W. M. Campbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2005, 109, 15397–15409 CrossRef CAS PubMed.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang and M. Grätzel,
et al.
, J. Phys. Chem. C, 2007, 111, 11760–11762 CrossRef CAS.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- Y. Xie, Y. Tang, W. Wu, Y. Wang, J. Liu, X. Li, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2015, 137, 14055–14058 CrossRef CAS PubMed.
- N. Kato, S. Moribe, M. Shiozawa, R. Suzuki, K. Higuchi, A. Suzuki, M. Sreenivasu, K. Tsuchimoto, K. Tatematsu, K. Mizumoto, S. Doi and T. Toyoda, J. Mater. Chem. A, 2018, 6, 22508–22512 RSC.
- Y. Kurumisawa, T. Higashino, S. Nimura, Y. Tsuji, H. Iiyama and H. Imahori, J. Am. Chem. Soc., 2019, 141, 9910–9919 CrossRef CAS PubMed.
- C.-C. Chen, Y.-H. Chen, V. S. Nguyen, S.-Y. Chen, M.-C. Tsai, J.-S. Chen, S.-Y. Lin, T.-C. Wei and C.-Y. Yeh, Adv. Energy Mater., 2023, 13, 2300353 CrossRef CAS.
- F. A. Faraghally, Y.-H. Chen, T.-Z. Lee, Y.-D. Chen, T.-C. Wei and C.-Y. Yeh, Sustainable Energy Fuels., 2025, 9, 2369–2379 RSC.
- T. D. Santos, A. Morandeira, S. Koops, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, G. Wallace, J. C. Earles, K. C. Gordon, D. Officer and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 3276–3279 CrossRef.
- Y. Wang, B. Chen, W. Wu, X. Li, W. Zhu, H. Tian and Y. Xie, Angew. Chem., 2014, 126, 10955–10959 CrossRef.
- K. Zeng, W. Tang, C. Li, Y. Chen, S. Zhao, Q. Liu and Y. Xie, J. Mater. Chem. A, 2019, 7, 20854–20860 RSC.
- C. Li, L. Luo, D. Wu, R. Jiang, J. Lan, R. Wang, L. Huang, S. Yang and J. You, J. Mater. Chem. A, 2016, 4, 11829–11834 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- L. Cabau, C. V. Kumar, A. Moncho, J. N. Clifford, N. López and E. Palomares, Energy Environ. Sci., 2015, 8, 1368–1375 RSC.
- H. Zhou, J.-M. Ji, S. H. Kang, M. S. Kim, H. S. Lee, C. H. Kim and H. K. Kim, J. Mater. Chem. C, 2019, 7, 2843–2852 RSC.
- A. J. Mozer, M. J. Griffith, G. Tsekouras, P. Wagner, G. G. Wallace, S. Mori, K. Sunahara, M. Miyashita, J. C. Earles and K. C. Gordon,
et al.
, J. Am. Chem. Soc., 2009, 131, 15621–15623 CrossRef CAS PubMed.
- J.-W. Shiu, Y.-C. Chang, C.-Y. Chan, H.-P. Wu, H.-Y. Hsu, C.-L. Wang, C.-Y. Lin and E. W.-G. Diau, J. Mater. Chem. A, 2015, 3, 1417–1420 RSC.
- N. Kato, S. Moribe, M. Shiozawa, R. Suzuki, K. Higuchi, A. Suzuki, M. Sreenivasu, K. Tsuchimoto, K. Tatematsu and K. Mizumoto,
et al.
, J. Mater. Chem. A, 2018, 6, 22508–22512 RSC.
- B. Vaz and M. Pérez-Lorenzo, Nanomaterials, 2023, 13, 1097 CrossRef CAS PubMed.
- M. Borges-Martínez, D. Alvarez, N. Montenegro-Pohlhammer, M. I. Menéndez, R. López and G. Cárdenas-Jirón, J. Phys. Chem. C, 2019, 123, 19362–19375 CrossRef.
- R. Pino-Rios and G. Cárdenas-Jirón, J. Photochem. Photobiol., A, 2019, 385, 112052 CrossRef CAS.
- R. Pino-Rios, N. Montenegro-Pohlhammer and G. Cardenas-Jiron, J. Phys. Chem. A, 2021, 125, 2267–2275 CrossRef CAS PubMed.
- M. I. Menéndez, N. Montenegro-Pohlhammer, R. Pino-Rios, R. Urzúa-Leiva, S. Morales-Lovera, M. Borges-Martnez, K. Granados-Tavera, R. López and G. Cárdenas-Jirón, J. Chem. Phys., 2023, 158, 164305 CrossRef PubMed.
- C.-W. Lee, H.-P. Lu, C.-M. Lan, Y.-L. Huang, Y.-R. Liang, W.-N. Yen, Y.-C. Liu, Y.-S. Lin, E. W.-G. Diau and C.-Y. Yeh, Chem. – Eur. J., 2009, 15, 1403–1412 CrossRef CAS PubMed.
- S. H. Kang, S. Y. Jung, Y. W. Kim, Y. K. Eom and H. K. Kim, Dyes Pigm., 2018, 149, 341–347 CrossRef CAS.
- J.-M. Ji, S. H. Kim, H. Zhou, C. H. Kim and H. K. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 24067–24077 CrossRef CAS PubMed.
- Y. Wang, B. Chen, W. Wu, X. Li, W. Zhu, H. Tian and Y. Xie, Angew. Chem., 2014, 126, 10955–10959 CrossRef.
- Y. Liu, H. Lin, J. T. Dy, K. Tamaki, J. Nakazaki, D. Nakayama, S. Uchida, T. Kubo and H. Segawa, Chem. Commun., 2011, 47, 4010–4012 Search PubMed.
- H.-L. Jia, M.-D. Zhang, W. Yan, X.-H. Ju and H.-G. Zheng, J. Mater. Chem. A, 2016, 4, 11782–11788 Search PubMed.
- K. Zeng, W. Tang, C. Li, Y. Chen, S. Zhao, Q. Liu and Y. Xie, J. Mater. Chem. A, 2019, 7, 20854–20860 RSC.
- G. Consiglio, A. Gorcy ski, S. Petralia and G. Forte, Dalton Trans., 2023, 52, 15995–16004 RSC.
- W. Zhang, L. Wang, L. Mao, J. Jiang, H. Ren, P. Heng, H. Ågren and J. Zhang, J. Phys. Chem. C, 2020, 124, 3980–3987 CrossRef CAS.
- Y. Fu, T. Lu, Y. Xu, M. Li, Z. Wei, H. Liu and W. Lu, Dyes Pigm., 2018, 155, 292–299 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- O. S. Al-Qurashi and N. Wazzan, ACS Omega, 2021, 6, 8967–8975 CrossRef CAS PubMed.
- T. W. Hamann and J. W. Ondersma, Energy Environ. Sci., 2011, 4, 370–381 RSC.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, J.-E. Moser, R. Humphry-Baker, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291–1302 CrossRef CAS.
- X. Zarate, S. Schott-Verdugo, A. Rodriguez-Serrano and E. Schott, J. Phys. Chem. A, 2016, 120, 1613–1624 CrossRef CAS PubMed.
- M. A. Green, Solid-State Electron., 1981, 24, 788–789 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 Search PubMed.
- T. J. Meade, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 1989, 111, 4353–4356 CrossRef CAS.
- E. Maggio, N. Martsinovich and A. Troisi, J. Phys. Chem. C, 2012, 116, 7638–7649 CrossRef CAS.
- M. Xie, F.-Q. Bai, H.-X. Zhang and Y.-Q. Zheng, J. Mater. Chem. C, 2016, 4, 10130–10145 RSC.
- J. Preat, A. Hagfeldt and E. A. Perpete, Energy Environ. Sci., 2011, 4, 4537–4549 RSC.
- H. Kaur and N. Goel, Phys. Chem. Chem. Phys., 2024, 26, 29311–29327 RSC.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- Y. Takano and K. N. Houk, J. Chem. Theory Comput., 2005, 1, 70–77 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 Search PubMed.
- H. S. Yu, X. He and D. G. Truhlar, J. Chem. Theory Comput., 2016, 12, 1280–1293 Search PubMed.
- A. M. Grabarz and B. Osmialowski, Molecules, 2021, 26, 7434 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- Z. Wu and R. E. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235116 CrossRef.
- X. Hua, X. Chen and W. Goddard, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 16103 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 Search PubMed.
- N. Benedek, I. Snook, K. Latham and I. Yarovsky, Chem. Phys., 2005, 122, 144102 CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- B. Delley, Mol. Simul., 2006, 32, 117–123 CrossRef CAS.
- H. Park, E. Bae, J.-J. Lee, J. Park and W. Choi, J. Phys. Chem. B, 2006, 110, 8740–8749 CrossRef CAS PubMed.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CrossRef CAS PubMed.
- D. Cahen, G. Hodes, M. Grätzel, J. F. Guillemoles and I. Riess, J. Phys. Chem. B, 2000, 104, 2053–2059 CrossRef CAS.
- T. Woller, P. Geerlings, F. De Proft, B. Champagne and M. Alonso, Molecules, 2018, 23, 1333 CrossRef PubMed.
- R. B. Alnoman, E. Nabil, S. Parveen, M. Hagar, M. Zakaria and A. A. Hasanein, Molecules, 2021, 26, 7336 CrossRef CAS PubMed.
- A. Irfan, Comp. Theor. Chem., 2019, 1159, 1–6 CrossRef CAS.
- S. Namuangruk, S. Jungsuttiwong, N. Kungwan, V. Promarak, T. Sudyoadsuk, B. Jansang and M. Ehara, Theor. Chem. Acc., 2016, 135, 14 Search PubMed.
- B.-G. Kim, C.-G. Zhen, E. J. Jeong, J. Kieffer and J. Kim, Adv. Funct. Mater., 2012, 22, 1606–1612 CrossRef CAS.
- F. Arkan and M. Izadyar, Mater. Chem. Phys., 2017, 196, 142–152 CrossRef CAS.
- H. Kaur and N. Goel, J. Phys. Chem. C, 2024, 128, 13410–13421 CrossRef CAS.
- T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137–15140 CrossRef CAS.
Footnote |
| † These authors have contributed equally to this work. |
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Samarth
Razdan†
,
Samarth
Razdan†













