
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGelation behavior of short protected peptides in organic medium†
Thomas M.
FitzSimons
 a,
Israt Jahan
Duti
a,
Nathaniel
Conrad
a,
Paridhi
Agrawal
b,
Alexandria
Niemoeller
b,
Emily
Guinn
b,
Jingyao
Wang
b,
Anastasiia
Vasiukhina-Martin
b and
Adrianne M.
Rosales
*a
a,
Israt Jahan
Duti
a,
Nathaniel
Conrad
a,
Paridhi
Agrawal
b,
Alexandria
Niemoeller
b,
Emily
Guinn
b,
Jingyao
Wang
b,
Anastasiia
Vasiukhina-Martin
b and
Adrianne M.
Rosales
*a
aMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, USA. E-mail: arosales@che.utexas.edu
bEli Lilly and Company, Indianapolis, USA
First published on 16th May 2025
Abstract
Peptide gelators have been widely explored in aqueous systems due to their prevalence in biological and medical applications. However, gelation behavior of peptides is less understood in organic medium, despite the extensive use of organic solvents in solid phase peptide synthesis, hybrid synthesis protocols, and the formation and application of organogels. Here, peptides with a short aqueous gelator sequence, lysine-tyrosine-phenylalanine or KYF, were investigated for the ability to form gels in dichloromethane (DCM) when their side groups are protected. Protected KYF-containing peptides formed gels in DCM at similar concentrations to deprotected KYF peptides in water. Structural characterization via Fourier transform infrared spectroscopy indicated the presence of antiparallel β-sheets in both the protected and the deprotected KYF gels. However, transmission electron microscopy and dynamic light scattering indicated the protected KYF gels in DCM consisted of short, anisotropic particles at the mesoscale, whereas the deprotected KYF gels in water showed entangled fibers. The protected KYF gels in DCM exhibited similar rheological properties to colloidal gels, namely an increasing resistance to flow at higher shear rates, a shear thinning profile, and a gel-to-fluid transition with increasing strain. Altogether, this study provides critical insights on the assembly behavior and structure of a tripeptide motif and its variants in organic medium, which can facilitate optimizing the processing conditions of similar peptides in organic solvents during synthesis or end-use applications.
Introduction
Synthetic peptides and peptide-drug conjugates play a major role in the pharmaceutical industry as therapeutic candidates to treat diseases ranging from diabetes to cancer.1–6 Unlike larger proteins and antibodies, these peptides are typically synthesized using solid phase peptide synthesis (SPPS), which enables efficient access to low molecular weights and facile incorporation of non-natural amino acids.7 After SPPS, the peptides are typically treated in organic solvent systems for cleavage from the resin or conjugation to other peptides or molecules. This step involves processing challenges, especially because the reaction conditions are heavily dependent on peptide sequence. In addition, sequence effects during workup can lead to higher order assembly, followed by aggregation or gelation, thereby decreasing peptide yield and purity or resulting in an inability to process and isolate the peptide sequences.8Numerous studies have investigated the aqueous conditions affecting peptide aggregation or gelation, including primary peptide sequence,9,10 length,11,12 pH,13 temperature,14 and salt concentration,15 but there is limited knowledge about the effects of organic solvent on peptide gel formation. Though peptide assembly in organic medium is reported in the literature,16–22 it is unclear how the mechanisms that describe gelation in water impact gelation in organic systems. In aqueous systems, certain amino acid groups have emerged as particularly prone to gelation, such as two consecutive phenylalanines protected by an N-fluorenylmethoxycarbonyl (Fmoc) group, or Fmoc-FF.23–28 In water, Fmoc-FF self-assembles into hydrogels via π-stacking of the fluorenyl groups and β-sheet formation, which are mechanisms that have been observed in other aqueous peptide gelators as well.29,30 Salt also mediates these types of interactions in water; for instance, strong hydrophobic interactions are strengthened in the presence of high salinity,15,31 while strong electrostatic interactions are weakened.32,33 In the absence of water, e.g., in organic solvents, however, the degree to which peptide associative interactions change is still largely unexplored.
One of the parameters that can influence peptide gelation in organic solvents is the presence of side chain protecting groups on the peptide. These groups typically mask hydrophilic functionalities on the peptide chain, such as carboxylic acids, thiols, and amines. They are necessary during SPPS or fragment condensation protocols to ensure linear peptide chains via coupling at the N-terminus. The majority of side chain protecting groups are hydrophobic,34 making them compatible with organic solvent systems for post-synthesis treatment. Even in the presence of these protecting groups, however, peptide gelation in organic solvent, i.e., organogel assembly, can still occur.18,35 This creates challenges in the post-synthesis procedures and affects the overall yield of peptide synthesis. Better understanding of protected peptide assembly leading to gelation in organic medium can facilitate the design of peptide synthesis and isolation processes to avoid gelation risk.
Beyond considerations during post-synthesis cleavage and workup, deeper knowledge on peptide gelation in organic solvent can also assist in their applications as well. One particularly active area of research is the use of organogels for water remediation. Organic-based pollutants, such as oils or dyes, are a constant threat to marine ecosystems, but peptides have emerged as useful tools to combat these pollutants.36–38 For example, non-natural β-amino acid based protected tripeptides are reported to entrap organic solvents and oil (e.g., diesel, kerosene) from organic-aqueous mixture, leading to the formation of organogels to aid easy removal of organics for oil spill recovery.36 In that study, the peptides assembled into antiparallel β-sheet structures in organic, which then formed nanofibers resulting in gel formation. Moreover, organogels can be used to stabilize different nanostructures, such as quantum dots or graphene sheets.39,40 In one example with quantum dots, organogels of FF dipeptides formed fibrillar structures in chloroform and aromatic solvents.39 Given both the considerations during synthesis and processing, as well as the aforementioned utility of organogels, further research investigating the structure formation of peptides and subsequent gelation pathways in organic solvents is warranted.
In this study, we investigated the ability of short, protected peptide sequences to form higher order structures and gels in dichloromethane (DCM), a common and extensively used solvent in peptide synthesis and cleavage protocols. The protected sequences were based on a short deprotected sequence, lysine–tyrosine–phenylalanine or KYF, which can form fibrillar gels in aqueous environments.9 We varied the peptide sequences by adding non-polar amino acids and polar amino acids with protected side chains and repeating the KYF sequence. The gelation behavior, secondary structure, morphology, and mechanics of these peptides in DCM were characterized and compared to the same properties of the deprotected KYF gels in water. All protected peptides formed small, anisotropic structures that packed to form a gel in DCM at sufficiently high concentrations, though the size and the mechanics of these gels varied with peptide sequences. Together, this study highlights how an aqueous peptide gelator translates its gelation behavior to organic medium, benefiting the post-synthesis treatment design and applications of peptides in organics.
Materials and methods
Peptide synthesis
Peptides (KYF, pKYF, pKFYK, pKYFE, pKYFIL, pKYFKYF) were synthesized via standard Fmoc solid phase synthesis (Fig. S1A, ESI†) on a Prelude X automated peptide synthesizer from Gyros Protein Technologies. All peptides were made on a 2-chlorotrityl chloride resin using Fmoc-protected amino acids. Peptide residues utilized Fmoc-protected amino acids (250 μM, 5× molar excess) coupled using HCTU activator (250 μM, 5× molar excess) and NMM (500 μM, 10× molar excess). Fmoc groups were removed using 20% piperidine in DMF. Cleavage of deprotected peptides used a cleavage cocktail of 95% TFA/2.5% TIPS/2.5% water for 4 hours (Fig. S1B, ESI†). Cleavage of protected peptides used a cleavage cocktail of 80% DCM/10% trifluoroethanol/10% acetic acid for 4 hours (Fig. S1C, ESI†). After cleavage, the molecular masses of the peptides were verified using ESI,†via an Agilent Technologies 6125B Single Quadrupole LC-MS with an Agilent ZORBAX Eclipse Plus C18 narrow bore column (Fig. S2, ESI†). All peptides were used as is to mimic processing conditions directly after cleavage from the resin.Peptide gelation
All peptide sequences show signs of gelation at a concentration of 30 mM, but in order to ensure stable gel formation, all peptide gels were formulated at a concentration of 40 mM. KYF samples were made in ultrafiltered water, and the pH was adjusted to pH 7 using 1 M NaOH and 1 M HCl solutions. All protected peptide samples were made using filtered DCM. Immediately after the addition of solvent, all samples were sonicated for 10 seconds. pKYF, pKYFKYF, and pKYFIL gels formed within seconds of dissolution into DCM. These samples were used immediately thereafter. pKYFE and pKYFK samples only showed noticeable gelation after one day, so all experiments using these samples were performed after one day to ensure all samples were tested at tgelation = 0.Transmission electron microscopy (TEM)
All TEM samples were prepared on a 200 mesh carbon type B grid purchased from Ted Pella. The staining protocol is as follows: gently touching the grid to the gelled sample surface, two washes with pure water each followed by blotting with filter paper, one wash with 2% methylamine vanadate stain followed by blotting, and finally floating the sample side of the grid on a droplet of 2% methylamine vanadate stain for approximately one minute followed by complete blotting of the sample. TEM samples were imaged using a FEI Tecnai transmission electron microscope at 80 kV with a magnification of 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
Scanning electron microscopy (SEM)
Scanning electron microscopy was carried out on a Zeiss Supra 40V scanning electron microscope. Samples were dissolved in the corresponding solvent and dried on a mica surface attached with conductive tape to the SEM pin mount. Graphite paint was added along the edges to assist with electron transfer. Samples were then sputtered with a Pt/Pd coating 4 nm thick using a Cressington 208 Benchtop Sputter Coater.Dynamic light scattering (DLS)
Dynamic light scattering data were acquired on a Zetasizer Nano ZS using 173° backscattering angle to minimize the effect of multiple scattering. All peptides were in DCM at 2 mM concentration. To study the effect of concentration, 5 mM, 10 mM, and 20 mM solutions, 40 mM gels, and diluted 2 mM solutions created from the 40 mM gels were also prepared for pKYF peptides. The DLS instrument outputs the second-order correlation function, g2(t), as raw data. The Seigert relation, g2(t) = 1 + β|g1(t)|2, was used to calculate the first-order correlation function, g1(t), where β ≡ instrument-dependent coherence factor = 0.9. To extract relaxation time constants, g1(τ) were analyzed by fitting either single (eqn (1)) or double (eqn (2)) exponential decay fits using MATLAB's non-linear fitting code ‘fitnlm’ with default optimization settings.| g1(τ) = Aexp(−τΓ) + c | (1) |
| g1(τ) = A1exp(−τΓ1) + A2exp(−τΓ2) | (2) |
CONTIN was also performed on g1(τ) data in order to confirm the existence of one or two dominant modes of relaxation, with the mean of the distributed mode consistent with fitted constants of eqn (1) and/or eqn (2).
Rheometry
Rheometry data were collected on a TA Instruments Discovery Hybrid Rheometer DHR20 rheometer. All experiments were performed at 25 °C using a 20 mm diameter 2° stainless steel cone geometry with a solvent trap on a Peltier plate. Frequency sweeps from 0.1–100 rad s−1 were performed at 1% strain, strain sweeps from 1–1000% strain were performed at a frequency of 1 rad s−1, and shear rate sweeps data were taken at equilibrium with a maximum sampling time of 30 seconds. For samples dissolved in DCM, the samples were taken from storage at −20 °C and pipetted onto the Peltier plate at 5 °C to minimize solvent evaporation while the solvent trap equilibrated the vapor environment around the sample.Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR)
ATR-FTIR results were obtained on a Thermo Fisher Nicolet iS10 with a diamond crystal plate. All samples were first dissolved at a concentration of 40 mM. The samples were then placed on the plate and allowed to evaporate to form a film. Each sample was subjected to 48 scans with a resolution of 0.482 cm−1. All spectra are baseline corrected with background removed. Second derivative calculations were done using Origin (2019) software.Results and discussion
In this study, we selected peptide sequences based on a short tripeptide, lysine (K)–tyrosine (Y)–phenylalanine (F) (KYF), to investigate their gelation behavior in dichloromethane (DCM). The KYF peptide sequence is reported to demonstrate self-assembly in aqueous environments, leading to hydrogel formation.9 The derivative of KYF, pentapeptide KYFIL, with two additional nonpolar amino acids, leucine and isoleucine, can also form gels in phosphate buffered saline at pH 7.4.10,13 The self-assembly of these peptides has been attributed to the formation of β-sheets and subsequent fibrillization.9,10,13 At a sufficient concentration, these fibers can entangle to form a gel. Though KYF is not soluble in DCM, its protected version, pKYF, is soluble in DCM (Fig. S3, ESI†). Therefore, we studied the gelation of protected peptides pKYF and its variants in this study. Here in pKYF, the lysine group is protected with a tert-butyloxycarbonyl (Boc) group and the tyrosine is protected with a tert-butyl (tBu) group (Fig. 1A). Because β-sheet assembly involves the formation of an extensive hydrogen bonding network between the backbone amines and carbonyls of adjacent peptide strands, side chain alterations may be accommodated as long as the balance of solubility and steric interactions is not disrupted.41 Thus, we hypothesized that the inclusion of relatively small protecting groups (Boc or tBu) may still permit β-sheet assembly for pKYF motifs in DCM.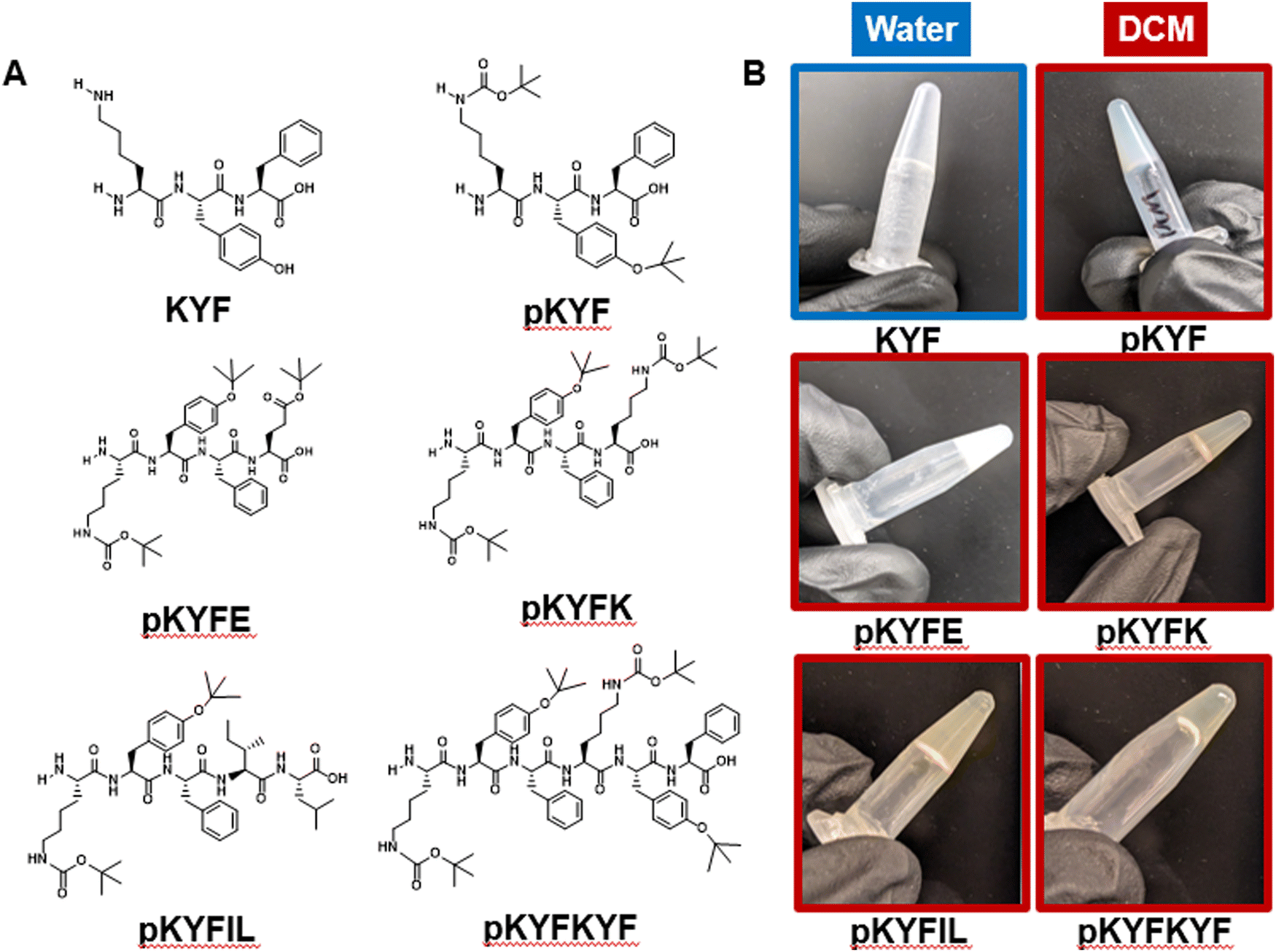 | ||
| Fig. 1 (A) Chemical structures of synthesized peptides. Lysine (K) residues contain a tert-butyloxycarbonyl (Boc) protecting group, and tyrosine (Y) and glutamic acid (E) residues contain tert-butyl (tBu) and tert-butyl ester (OtBu) protecting groups, respectively. (B) Vial inversion test of peptide samples at 40 mM. All samples are in DCM, except KYF, which is in water at pH 7. Images were taken at the initial onset of gelation for each sample, as observed by visual inspection. | ||
To investigate whether pKYF and its derivatives could still form gels in DCM, we synthesized and characterized five protected peptides: pKYF, pKYFE, pKYFK, pKYFIL, and pKYFKYF (Fig. 1A). These five sequences were chosen to study the effect of amino acid additions and repeats on gelation. For example, pKYFE adds a protected glutamic acid, which includes a two-carbon spacer and a tert-butyl ester protecting group on the side chain, whereas pKYFK adds a protected lysine, which includes a four-carbon spacer and a Boc group in the side chain. In addition, pKYFIL is the protected analog of the deprotected KYFIL peptide. Finally, pKYFKYF repeats the parent sequence allowing us to study the effect of the protected peptide length in gelation. All peptides contained a free N-terminus and a carboxylic acid at the C-terminus.
We investigated the gelation behavior of all synthesized peptides in DCM and compared their behavior to that of the unprotected KYF peptide in water at equivalent concentrations. None of the protected peptides were soluble in water. At a concentration of 40 mM, KYF formed gels in water at neutral pH (Fig. 1B), as has been reported previously.9 The gelation behavior was qualitatively assessed using a vial inversion test, which confirmed the ability of KYF to form a self-supporting solid that resists flow (Fig. 1B). At the same concentration of 40 mM in DCM, all of the five protected peptides also formed gels, as shown by the vial inversion test (Fig. 1B). All peptides formed gels that were translucent, single-phase solids. Both pKYFE and pKYFK formed gels after one day of dissolving the sample, while the rest of the peptides formed gels within seconds of dissolution.
To understand whether the protected peptides formed gels due to fibrillization, we performed transmission electron microscopy (TEM) on each gel. As a control, we also imaged a hydrogel formed from deprotected KYF in water, which showed a fibrillar structure (Fig. 2A and Fig. S4, ESI†), consistent with previous reports.9 However, each of the protected peptides showed significantly different morphologies than deprotected KYF (Fig. 2A and Fig. S4, ESI†). The protected peptides showed formation of anisotropic rod-like structures with lengths of 200–500 nm, with the exception of pKYFKYF peptide. The pKYFKYF gel showed a more disordered, but non-fibrillar, morphology that was smaller than that of the other protected peptides. To corroborate these measurements, scanning electron microscopy (SEM) was performed on dried gels (Fig. 2B). The KYF gels in water indicated a fibrillar nanostructure, whereas the protected peptide gels in DCM showed anisotropic morphologies, similar to the observation from TEM images. These data suggest that gelation in DCM is due to dense packing of the assembled anisotropic structures forming a colloidal-type gel, which is a markedly different gelation mechanism from the KYF motif in water. The underlying mechanism governing structure formation, however, remains to be elucidated and was next probed using Fourier transform infrared (FTIR) spectroscopy.
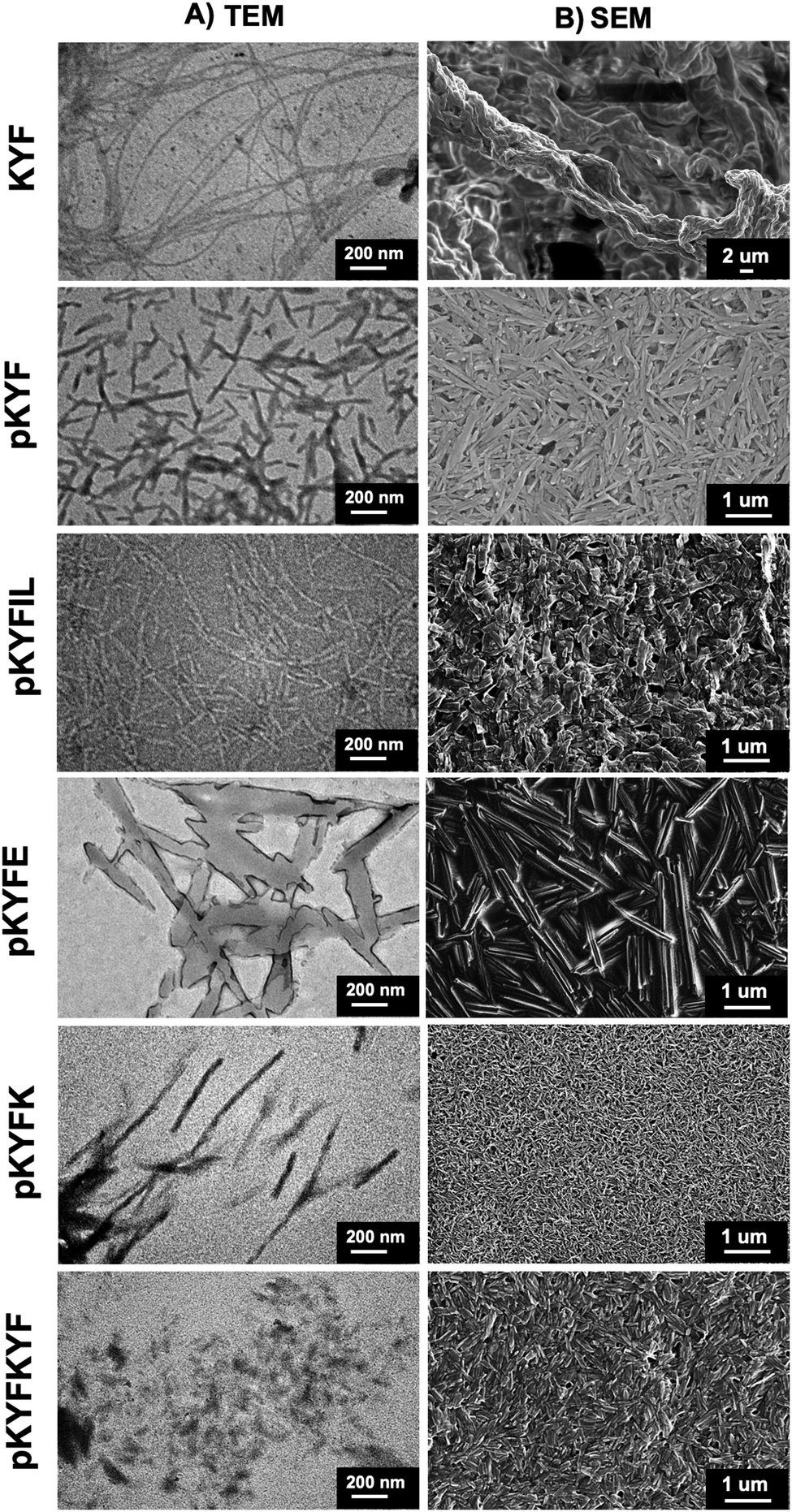 | ||
| Fig. 2 The images of gels dried from water for deprotected KYF or DCM for all other peptides obtained from (A) transmission electron microscopy and (B) scanning electron microscopy. The deprotected KYF gels showed entangled fibers, whereas the protected peptide gels show rod-like structures. | ||
Despite the difference in mesoscale morphology between the deprotected KYF and the protected peptides, we hypothesized β-sheet assembly may still drive formation of the anisotropic rod-like structures. Previous studies showed that aqueous KYF and KYFIL samples assemble into β-sheets by tracking the amide I absorption via FTIR spectroscopy.9,10,13,42 The parallel β-sheet peptides display absorbances at 1610–1640 cm−1, whereas antiparallel β-sheets show absorbances both at 1610–1640 cm−1 and 1670–1690 cm−1.43–45 For the peptides with random coil structures, the amide I absorbance band is observed between 1640–1650 cm−1.10,43
Each of the peptides investigated here displayed absorbance peaks corresponding to specific secondary structures. KYF peptides in water showed absorbances between 1610–1640 cm−1 and at 1680 cm−1 (Fig. 3A), as supported by the sharp peaks observed in the second derivative of the absorbances (Fig. 3B). This suggests that KYF forms antiparallel β-sheets in water. We note that the peak ∼1650 cm−1 in the KYF peptide FTIR spectra indicates the potential presence of some random coil structures. Similar to the KYF in water, all the protected peptides in DCM showed significant absorbances at 1610–1640 cm−1 and 1670–1690 cm−1 (Fig. 3), indicating their antiparallel β-sheet configuration, though these peaks displayed shifts within the absorbance bands for each peptide. The absorbance intensity also varied for each peptide, where the pKYFK showed lowest absorbance at 1610–1640 cm−1 but highest absorbance at 1670–1690 cm−1. Taken together, the FTIR spectra indicated the protected peptides still form β-sheets in organic solvent, though the β-sheet content and composition were not quantifiable from the FTIR data without further modeling.
 | ||
| Fig. 3 (A) FT-IR spectra of peptide films dried from water for KYF or DCM for all other peptides. The absorbances at 1610–1640 cm−1 and 1670–1690 cm−1 (highlighted) indicate the formation of antiparallel β-sheets for all peptides. (B) Second derivative traces of the FT-IR data. Negative peaks from the baseline correspond to sharpened peaks in Fig. 3A. | ||
To further investigate the relative size and dispersity of the assembled peptides in situ, we conducted dynamic light scattering (DLS) experiments on each peptide sequence. We first investigated the peptides at a dilute concentration of 2 mM, which is lower than their gelation concentrations of approximately 40 mM. For each peptide, we obtained the first-order correlation function g1(τ) in order to assess how quickly they diffuse through the solution (Fig. 4A). Three out of five protected sequences, pKYF, pKYFK, pKYFIL, result in solutions with a g1(τ) showing only one dominant mode of relaxation, while the other two sequences, pKYFE and pKYFKYF, show two dominant modes of relaxation. CONTIN analysis further confirms this observation (Fig. S5, ESI†). Accordingly, we estimated the diffusive relaxation times of each sample using either single or double exponential decay fits (Fig. 4B). These relaxation times are ultimately correlated with the hydrodynamic size of the assembled peptide.46 From this analysis, we can deduce that the sequences with one mode of relaxation must have a rather monodisperse population of peptide assemblies. We did not directly estimate a hydrodynamic size of the peptides, due to their varied morphologies and non-spherical shapes of aggregates observed by TEM for each peptide. Such varied non-spherical morphologies of peptide aggregates may lead to the emergence of translational and rotational diffusive modes. Accordingly, for the pKYFE and pKYFKYF solutions possessing two relaxation modes, we are unable to discern if there are two monodisperse populations of peptide aggregates or one monodisperse population of peptide aggregate with a rotational and translational diffusive mode.
 | ||
| Fig. 4 (A) Dynamic light scattering (DLS) of all protected peptides in DCM at a concentration of 2 mM. (B) Comparison of the first relaxation times from 4A. (C) Comparison of pKYF at three concentrations below the gelation concentration. All three samples in solution form exhibit similar DLS curves as their corresponding gels. (D) Comparison of pKYF samples, one as a gel at 40 mM and one as the same gel diluted to 2 mM. The diluted gel has a similar profile to the sample originally created at 2 mM. The error bars represent standard error bars of the fitted parameters. | ||
To understand whether increased concentration changes the peptide assembly size, pKYF was analyzed at concentrations of 5 mM, 10 mM, and 20 mM in DCM, which were below the gelation concentration (Fig. 4C). Each concentration demonstrated similar relaxation profiles, suggesting that the assemblies have a similar size and that increased concentration did not alter the assembly size. However, at 40 mM, the relaxation spectrum for pKYF displayed two relaxation modes (Fig. 4D), and the secondary relaxation time was very long at 4000 ms due to gelation. This long relaxation time does not accurately reflect the characteristics of the assembled peptides, but rather indicates kinetic arrest of the assembled peptides in the gel. Notably, the primary relaxation mode corresponded to a relaxation time of 0.096 ± 0.016 ms, which was comparable to the dilute samples (0.19 ± 0.004 ms), providing further evidence that the gels were formed via interaction of smaller peptide assemblies.
We next studied how the initial concentration corresponds to the peptide assembly size. The peptide pKYF was dissolved at the expected gelation concentration of 40 mM and diluted to the original tested concentration of 2 mM, using vigorous mixing with vortexing and sonication. Once diluted, the sample relaxation looked nearly identical to those of the samples that started at the dilute concentration (Fig. 4D). This result suggests that even at a high initial concentration, the peptide assembly size does not significantly change. Instead, the elevated concentration leads to stronger interactions between the assembled peptides, leading to gelation. We note that without sufficient mixing after gel dilution, there are some large aggregates of the assembled peptides that do not disperse (Fig. S6, ESI†); however, these large aggregates disappear with sufficient mixing.
Finally, we investigated the rheological properties of each protected peptide gel using a rotational rheometer to understand how these solids respond to deformation. For KYF, pKYF, pKYFIL, and pKYFKYF, gels were measured immediately after dissolution in water or DCM, due to their rapid assembly. For pKYFE and pKYFK, samples were dissolved in DCM and stored overnight in the refrigerator to prevent DCM evaporation, then measured the next day due to their slower rate of gelation. We first performed a frequency sweep for each peptide, which informed the dependence of the viscoelastic behavior of the materials on the deformation rate (Fig. 5A and Fig. S7–S8, ESI†). In general, each peptide gel demonstrated an increase in the storage modulus with increasing frequency. The storage modulus can be interpreted as the measure of the resistance of a sample to deformation. Therefore, as the frequency increases, there is an increased resistance to flow, in part due to the inability of the material to relax on the timescale of deformation.
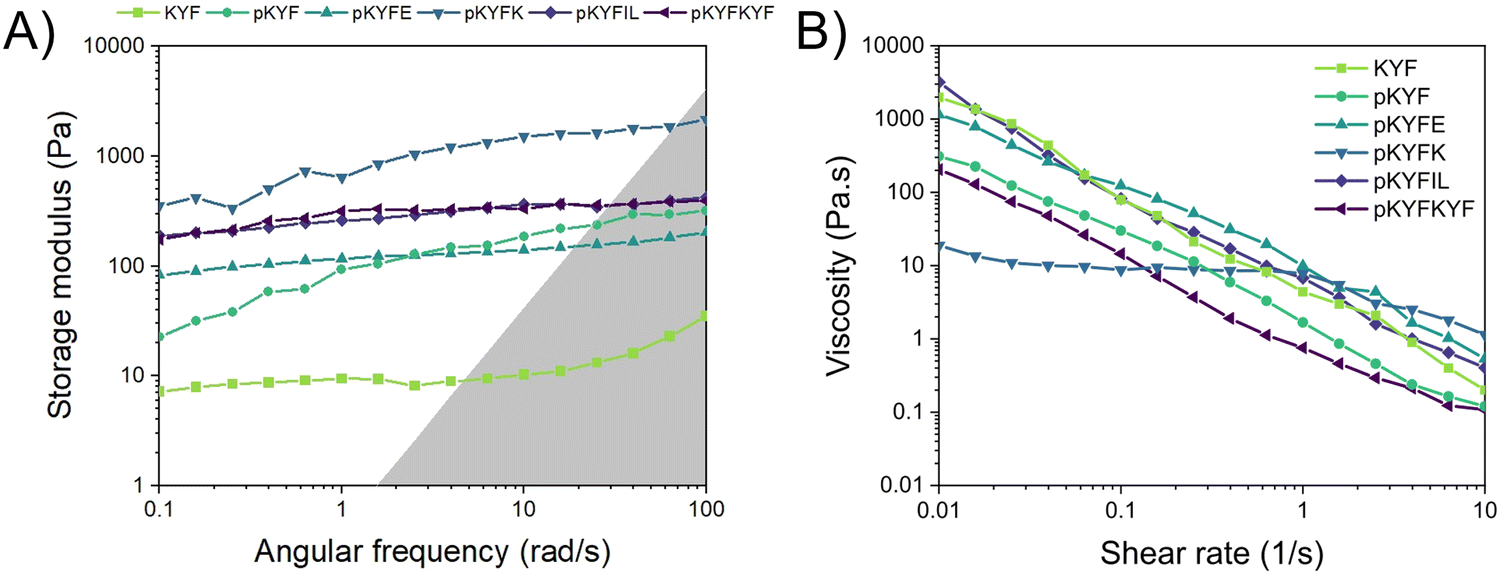 | ||
| Fig. 5 (A) Representative frequency sweeps of 40 mM peptide gels using a strain of 1%. The highlighted region represents the area where the machine inertia dominates the measurement. pKYFK gels show the highest storage moduli. (B) Representative shear rate sweeps of 40 mM peptide gels with equilibrium achieved for each shear tested. At low shear rates, pKYFK gels exhibit lower viscosity compared to other gels. At high shear rate, all gels demonstrated shear thinning behavior, with pKYKF peptide gels showing shear thinning behavior at higher shear rates relative to the other peptide gels. | ||
Despite the overall similarity in the trend of the storage moduli with frequency, the peptide gels displayed significant differences in the magnitude of their moduli. Notably, the KYF gel had the lowest modulus of all the tested peptide gels, indicating a lower resistance to deformation. Due to its fibrillar morphology, the fibers may have more conformational flexibility and therefore not resist shear as much as a gel composed of rigid rod-like particles.47 Within the protected peptide gels, the pKYFK gel indicated a much higher storage modulus than the rest of the tested samples. This result could be associated with the secondary structure of the peptides. The FTIR spectra of peptides show pKYFK has the lowest absorbance at 1610–1640 cm−1 and the highest absorbance at 1670–1690 cm−1 among all peptides. Though all the peptides are antiparallel β-sheets, pKYFK peptides showed distinct peak intensity relative to other peptides in FTIR spectroscopy. This suggests that there might be a difference in the β-sheets features of pKYFK compared to other sequences, which could impact the packing of pKYFK peptides, resulting in increased rigidity of the peptide assemblies. The increased rigidity would lead the gel to resist flow more than a gel composed of softer particles. Thus, the frequency sweep results corroborate the observations seen via FTIR spectroscopy for the pKYFK gel, relative to the other protected peptide sequences.
As colloidal gels can fluidize under external forces, the samples were further analyzed with a flow sweep test, which varies the shear rate in continuous rotation and reports the viscosity (Fig. 5B). All of the protected peptide gels exhibited similar behavior, with the exception of pKYFK. Every sample demonstrated clear shear-thinning behavior, or a decrease in viscosity with increasing shear rate. In this continuous flow experiment, higher shear rates likely cause the individual assembled structures to align,48,49 which lowers the overall viscosity of the material in the direction of flow. In the case of pKYFK, more rigid assemblies may be more difficult to align, which would increase the shear rate required to observe shear thinning behavior. At sufficiently high shear rates, however, the material behaves in a similar fashion to the other protected peptide gels (Fig. 5B).
The final mechanical characterization performed was a strain sweep, which is an oscillatory test at a fixed frequency (1 rad s−1) but with increasing strain (Fig. 6A–F). Similar to the frequency sweep and shear rate sweep results, all of the protected peptide gels apart from pKYFK exhibited similar behavior. At low strains, all peptide gels behaved like an elastic solid, as evidenced by the high ratio of the shear storage modulus to the shear loss modulus at lower strain. As the strain increased, however, the shear storage moduli decreased below the shear loss moduli for each peptide gel, indicating a solid-to-fluid transition. The high strains lead to yield and flow of the constituent particles in the gel and prevent the assembled particles from forming a system-spanning network. Again, pKYFK displayed some deviation from the other peptides – specifically, the crossover from a more elastic regime to a more viscous regime occurred below the tested strain of 1%. Because pKYFK may form more rigid assemblies with respect to the other protected peptides as indicated from FTIR spectroscopy, the rigid structure could prevent the storage of elastic energy. Upon application of strain, these rigid peptide assemblies will instead flow past one another, resulting in the dissipation of the applied strain energy like a viscous fluid. These observations correlate with the fact that pKYFK also displayed the highest modulus.
 | ||
| Fig. 6 Strain sweeps (1–1000%) of each peptide sequence at a frequency of 1 rad s−1. (A) KYF (B) pKYF (C) pKYFE (D) pKYFK (E) pKYFIL (F) pKYFKYF. At low stain, all peptides gels except pKYFK show an elastic solid behavior and at high strain the shear storage moduli decreased below the shear loss moduli for each peptide gel, indicating a solid-to-fluid transition. For pKYFK, This transition below the tested strain of 1%. | ||
Conclusion
We investigated the gelation behavior of protected KYF peptide and its derivatives in DCM and compared it to the gelation behavior of aqueous KYF peptides. In water, deprotected KYF peptides form gels through fibrillization of β-sheets, whereas this study shows that protected KYF peptides in DCM form a system-spanning network of smaller assembled peptide particles that led to gelation at elevated concentration (approximately 40 mM). The peptide assemblies indicated antiparallel β-sheet formation, however, it was not clear how solvent, peptide interactions, and the presence of protecting groups led to the formation of smaller assembled particles instead of long fibrils. The protected peptide gels exhibited similar mechanical profiles to colloidal suspensions, namely an increasing resistance to flow at higher shear rates, a shear thinning profile, and a transition from more elastic-like behavior to more viscous-like behavior with increasing oscillatory strains. Thus, this research highlights how aqueous gelator peptides may preserve gelation behavior in organic medium despite the presence of protecting groups, although there are significant differences in the mechanism of assembly, structure, morphology, and gel properties. Future work involves modeling and simulation to understand the mechanism of gelation in organic medium, and exploring the effects of different parameters such as peptide structure, solvent, and time on gelation. Altogether, the knowledge gained from this research can help improve the post-synthesis processing of peptides in organic solvent, as well as provide guidelines for designing organogels for wide range of applications.Author contributions
T. M. F., P. A., A. N., E. G., J. W., A. V., and A. M. R. conceived the idea. T. M. F. and I. D. designed and conducted experiments and performed associated analysis. N. C. analyzed dynamic light scattering data. T. M. F, I. D., N. C., and A. M. R. wrote the manuscript with input from all authors. P. A., A. N., E. G., J. W., A. V., and A. M. R. supervised the work.Data availability
The data supporting this article, including LC-MS data for synthesized peptides, solubility and gelation results in different organic medium, additional TEM data for the formulated gels, dynamic light scattering data for a diluted gel, and rheology data for the formulated gels, have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was primarily funded by Eli Lilly & Company, and additional support was provided by the National Institute of General Medical Sciences of the National Institutes of Health (R35GM138193, A. M. R.).References
- R. Galyean, H. Tariga, S. Alagarsamy, G. Croston, J. Heitzmann, A. Kohan, H. Wi, P. J. Rivi and C. D. Schteingart, New, Potent, Selective, and Short-Acting Peptidic V 1a Receptor Agonists, J. Med. Chem., 2011, 54, 4388–4398 Search PubMed.
- K. Fosgerau and T. Hoffmann, Peptide Therapeutics: Current Status and Future Directions, Drug Discovery Today, 2015, 20(1), 122–128, DOI:10.1016/j.drudis.2014.10.003.
- E. Fisher, K. Pavlenko, A. Vlasov and G. Ramenskaya, Peptide-Based Therapeutics for Oncology, Pharm. Med., 2019, 33(1), 9–20, DOI:10.1007/s40290-018-0261-7.
- J. Lindberg, J. Nilvebrant, P. Å. Nygren and F. Lehmann, Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy, Molecules, 2021, 26(19), 1–16, DOI:10.3390/molecules26196042.
- Y. Wang, A. G. Cheetham, G. Angacian, H. Su, L. Xie and H. Cui, Peptide–Drug Conjugates as Effective Prodrug Strategies for Targeted Delivery, Adv. Drug Delivery Rev., 2017, 110–111, 112–126, DOI:10.1016/j.addr.2016.06.015.
- B. M. Cooper, J. Iegre, D. H. O’Donovan, M. Ölwegård Halvarsson and D. R. Spring, Peptides as a Platform for Targeted Therapeutics for Cancer: Peptide-Drug Conjugates (PDCs), Chem. Soc. Rev., 2021, 50(3), 1480–1494, 10.1039/d0cs00556h.
- J. Wang, P. Agrawal, M. R. Berglund, J. M. Groh, M. E. Kopach, K. D. Seibert and S. K. Viswanath, Quality by Design of Solid-phase Peptide/Protein Coupling Reaction via Mechanistic Reaction Kinetics Modeling Approach, AIChE J., 2024, 70(8), e18453, DOI:10.1002/aic.18453.
- J. Wang, A. Niemoeller and S. Viswanath, Kinetic Model-Aided Process Optimization via Integrated in-Situ FTIR and Real-Time Oscillatory Shear Rheometry: A Case Study on Impurity Control and Gelation Mitigation for a Solid Phase Peptide Cleavage Process, Chem. Eng. J., 2023, 476, 146928, DOI:10.1016/j.cej.2023.146928.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Exploring the Sequence Space for (Tri-)Peptide Self-Assembly to Design and Discover New Hydrogels, Nat. Chem., 2015, 7(1), 30–37, DOI:10.1038/nchem.2122.
- I. J. Duti, J. R. Florian, A. R. Kittel, C. D. Amelung, V. P. Gray, K. J. Lampe and R. A. Letteri, Peptide Stereocomplexation Orchestrates Supramolecular Assembly of Hydrogel Biomaterials, J. Am. Chem. Soc., 2023, 145(33), 18468–18476, DOI:10.1021/jacs.3c04872.
- H. Reiersen, A. R. Clarke and A. R. Rees, Short Elastin-like Peptides Exhibit the Same Temperature-Induced Structural Transitions as Elastin Polymers: Implications for Protein Engineering, J. Mol. Biol., 1998, 283(1), 255–264, DOI:10.1006/jmbi.1998.2067.
- H. Nuhn and H.-A. Klok, Secondary Structure Formation and LCST Behavior of Short Elastin-Like Peptides, Biomacromolecules, 2008, 9(10), 2755–2763, DOI:10.1021/bm800784y.
- J. D. Tang, C. Mura and K. J. Lampe, Stimuli-Responsive, Pentapeptide, Nanofiber Hydrogel for Tissue Engineering, J. Am. Chem. Soc., 2019, 141(12), 4886–4899, DOI:10.1021/jacs.8b13363.
- M. Gkikas, R. K. Avery and B. D. Olsen, Thermoresponsive and Mechanical Properties of Poly(L-proline) Gels, Biomacromolecules, 2016, 17, 399–406, DOI:10.1021/acs.biomac.5b01168.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Salt-Triggered Peptide Folding and Consequent Self-Assembly into Hydrogels with Tunable Modulus, Macromolecules, 2004, 37(19), 7331–7337, DOI:10.1021/ma0491762.
- M. J. Krysmann, V. Castelletto and I. W. Hamley, Fibrillisation of Hydrophobically Modified Amyloid Peptide Fragments in an Organic Solvent, Soft Matter, 2007, 3(11), 1401, 10.1039/b709889h.
- M. J. Krysmann, V. Castelletto, J. E. McKendrick, L. A. Clifton, P. J. F. Harris and S. M. King, Self-Assembly of Peptide Nanotubes in an Organic Solvent, Langmuir, 2008, 24(15), 8158–8162, DOI:10.1021/la800942n.
- G. Aykent, C. Zeytun, A. Marion and S. Özçubukçu, Simple Tyrosine Derivatives Act as Low Molecular Weight Organogelators, Sci. Rep., 2019, 9(1), 4893, DOI:10.1038/s41598-019-41142-z.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Zamith Cardoso, P. C. Griffiths and D. J. Adams, The Effect of Solvent Choice on the Gelation and Final Hydrogel Properties of Fmoc–Diphenylalanine, Soft Matter, 2015, 11(5), 927–935, 10.1039/C4SM02256D.
- A. Del Giudice, A. Rüter, N. V. Pavel, L. Galantini and U. Olsson, Self-Assembly of Model Amphiphilic Peptides in Nonaqueous Solvents: Changing the Driving Force for Aggregation Does Not Change the Fibril Structure, Langmuir, 2020, 36(29), 8451–8460, DOI:10.1021/acs.langmuir.0c00876.
- C. Lagadec and D. K. Smith, Synthetically Accessible, Tunable, Low-Molecular-Weight Oligopeptide Organogelators, Chem. Commun., 2011, 47(1), 340–342, 10.1039/C0CC01449D.
- C. K. Rouse, A. D. Martin, C. J. Easton and P. Thordarson, A Peptide Amphiphile Organogelator of Polar Organic Solvents, Sci. Rep., 2017, 7(1), 43668, DOI:10.1038/srep43668.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Fmoc-Diphenylalanine Self Assembles to a Hydrogel via a Novel Architecture Based on π–π Interlocked β-Sheets, Adv. Mater., 2008, 20(1), 37–41, DOI:10.1002/adma.200701221.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Nanostructured Hydrogels for Three-Dimensional Cell Culture through Self-Assembly of Fluorenylmethoxycarbonyl-Dipeptides, Adv. Mater., 2006, 18(5), 611–614, DOI:10.1002/adma.200501522.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Fmoc-Diphenylalanine Self-ASsembly Mechanism Induces Apparent pK a Shifts, Langmuir, 2009, 25(16), 9447–9453, DOI:10.1021/la900653q.
- Y. Wang, Q. Geng, Y. Zhang, L. Adler-Abramovich, X. Fan, D. Mei, E. Gazit and K. Tao, Fmoc-Diphenylalanine Gelating Nanoarchitectonics: A Simplistic Peptide Self-Assembly to Meet Complex Applications, J. Colloid Interface Sci., 2023, 636, 113–133, DOI:10.1016/j.jcis.2022.12.166.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Rigid, Self-Assembled Hydrogel Composed of a Modified Aromatic Dipeptide, Adv. Mater., 2006, 18(11), 1365–1370, DOI:10.1002/adma.200501765.
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Fmoc-Modified Amino Acids and Short Peptides: Simple Bio-Inspired Building Blocks for the Fabrication of Functional Materials, Chem. Soc. Rev., 2016, 45(14), 3935–3953, 10.1039/C5CS00889A.
- V. P. Gray, C. D. Amelung, I. Jahan, E. G. Laudermilch, R. A. Letteri and K. J. Lampe, Biomaterials via Peptide Assembly: Design, Characterization, and Application in Tissue Engineering, Acta Biomater., 2022, 140, 43–75, DOI:10.1016/j.actbio.2021.10.030.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, Self-Assembly of Peptide-Amphiphile Nanofibers: The Roles of Hydrogen Bonding and Amphiphilic Packing, J. Am. Chem. Soc., 2006, 128(22), 7291–7298, DOI:10.1021/ja060573x.
- J. Léonil, D. Mollé, S. Bouhallab and G. Henry, Precipitation of Hydrophobic Peptides from Tryptic Casein Hydrolysate by Salt and pH, Enzyme Microb. Technol., 1994, 16(7), 591–595, DOI:10.1016/0141-0229(94)90124-4.
- S. Ghosh, G. Pandit, S. Debnath, S. Chatterjee and P. Satpati, Effect of Monovalent Salt Concentration and Peptide Secondary Structure in Peptide-Micelle Binding, RSC Adv., 2021, 11(58), 36836–36849, 10.1039/d1ra06772a.
- R. Perez-Jimenez, R. Godoy-Ruiz, B. Ibarra-Molero and J. M. Sanchez-Ruiz, The Efficiency of Different Salts to Screen Charge Interactions in Proteins: A Hofmeister Effect?, Biophys. J., 2004, 86(4), 2414–2429, DOI:10.1016/S0006-3495(04)74298-8.
- V. Gavva, O. Al Musaimi, C. Bent and D. R. Williams, Determining the Hydrophobicity Index of Protected Amino Acids and Common Protecting Groups, Separations, 2023, 10(8), 456, DOI:10.3390/separations10080456.
- G. Palui, J. Nanda, S. Ray and A. Banerjee, Fabrication of Luminescent CdS Nanoparticles on Short-Peptide-Based Hydrogel Nanofibers: Tuning of Optoelectronic Properties, Chem. – Eur. J., 2009, 15(28), 6902–6909, DOI:10.1002/chem.200900149.
- M. Konda, I. Maity, D. B. Rasale and A. K. Das, A New Class of Phase-Selective Synthetic β-Amino Acid Based Peptide Gelator: From Mechanistic Aspects to Oil Spill Recovery, ChemPlusChem, 2014, 79(10), 1482–1488, DOI:10.1002/cplu.201402120.
- M. Chetia, S. Debnath, S. Chowdhury and S. Chatterjee, Self-Assembly and Multifunctionality of Peptide Organogels: Oil Spill Recovery, Dye Absorption and Synthesis of Conducting Biomaterials, RSC Adv., 2020, 10(9), 5220–5233, 10.1039/C9RA10395C.
- S. Debnath, A. Shome, S. Dutta and P. K. Das, Dipeptide-Based Low-Molecular-Weight Efficient Organogelators and Their Application in Water Purification, Chem. – Eur. J., 2008, 14(23), 6870–6881, DOI:10.1002/chem.200800731.
- X. Yan, Y. Cui, Q. He, K. Wang and J. Li, Organogels Based on Self-Assembly of Diphenylalanine Peptide and Their Application to Immobilize Quantum Dots, Chem. Mater., 2008, 20(4), 1522–1526, DOI:10.1021/cm702931b.
- B. Adhikari, J. Nanda and A. Banerjee, Pyrene-Containing Peptide-Based Fluorescent Organogels: Inclusion of Graphene into the Organogel, Chem. – Eur. J., 2011, 17(41), 11488–11496, DOI:10.1002/chem.201101360.
- K. H. Chan, B. Xue, R. C. Robinson and C. A. E. Hauser, Systematic Moiety Variations of Ultrashort Peptides Produce Profound Effects on Self-Assembly, Nanostructure Formation, Hydrogelation, and Phase Transition, Sci. Rep., 2017, 7(1), 12897, DOI:10.1038/s41598-017-12694-9.
- S. Fleming, P. W. J. M. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Assessing the Utility of Infrared Spectroscopy as a Structural Diagnostic Tool for β-Sheets in Self-Assembling Aromatic Peptide Amphiphiles, Langmuir, 2013, 29(30), 9510–9515, DOI:10.1021/la400994v.
- S. Krimm and J. Bandekar, in Vibrational Spectroscopy and Conformation of Peptides, Polypeptides, and Proteins, Advances in Protein Chemistry, Elsevier, 1986, vol. 38, pp. 181–364 DOI:10.1016/S0065-3233(08)60528-8.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schöllkopf, M. T. Bowers, G. Von Helden and K. Pagel, An Infrared Spectroscopy Approach to Follow β-Sheet Formation in Peptide Amyloid Assemblies, Nat. Chem., 2017, 9(1), 39–44, DOI:10.1038/nchem.2615.
- Y. Zou, Y. Li, W. Hao, X. Hu and G. Ma, Parallel β-Sheet Fibril and Antiparallel β-Sheet Oligomer: New Insights into Amyloid Formation of Hen Egg White Lysozyme under Heat and Acidic Condition from FTIR Spectroscopy, J. Phys. Chem. B, 2013, 117(15), 4003–4013, DOI:10.1021/jp4003559.
- N. C. Santos and M. A. Castanho, Teaching Light Scattering Spectroscopy: The Dimension and Shape of Tobacco Mosaic Virus, Biophys. J., 1996, 71(3), 1641–1650, DOI:10.1016/S0006-3495(96)79369-4.
- V. K. Lai, C. R. Frey, A. M. Kerandi, S. P. Lake, R. T. Tranquillo and V. H. Barocas, Microstructural and Mechanical Differences Between Digested Collagen-Fibrin Co-Gels and Pure Collagen and Fibrin Gels Victor, Acta Biomater., 2012, 8(11), 4031–4042, DOI:10.1016/j.actbio.2012.07.010.Microstructural.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Injectable Solid Hydrogel: Mechanism of Shear-Thinning and Immediate Recovery of Injectable β-Hairpin Peptide Hydrogels, Soft Matter, 2010, 6(20), 5143, 10.1039/c0sm00642d.
- I. W. Hamley, S. Burholt, J. Hutchinson, V. Castelletto, E. R. Da Silva, W. Alves, P. Gutfreund, L. Porcar, R. Dattani, D. Hermida-Merino, G. Newby, M. Reza, J. Ruokolainen and J. Stasiak, Shear Alignment of Bola-Amphiphilic Arginine-Coated Peptide Nanotubes, Biomacromolecules, 2017, 18(1), 141–149, DOI:10.1021/acs.biomac.6b01425.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00275c |
| This journal is © The Royal Society of Chemistry 2025 |