
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complex active site structures influence absorption spectrum of Chrimson wild type and mutants†
Katharina
Spies
 *ab,
Beatrix M.
Bold
a and
Marcus
Elstner
ac
*ab,
Beatrix M.
Bold
a and
Marcus
Elstner
ac
aInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Karlsruhe, Germany. E-mail: marcus.elstner@kit.edu; Fax: +49 721 608-44290; Tel: +49 721 608-0
bCNRS, ENS de Lyon, LCH, UMR 5182, 69342, Lyon cedex 07, France
cInstitute of Nano Technology (INT), Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 17th June 2025
Abstract
The red light-activated channelrhodopsin Chrimson is widely used in optogenetic applications, including vision and hearing restoration. Despite structural insights from X-ray crystallography and their identification of another red-shifted mutant (S169A), the molecular determinants of its large spectral shift remain incompletely understood. In this study, we present a computational analysis of wild type Chrimson and several mutants within a QM/MM approach. A key finding is the pronounced flexibility of the active site, where multiple conformations interconvert on the nano-second scale. We also highlight the role of residue S169, whose hydrogen bonding influences the torsional flexibility of the nearby counterion E165, thus affecting the prevalence of distinct structural motifs at the active site. The comparison of experimental and QM/MM MD-sampled absorption spectra supports the validity of our computational models. A direct hydrogen bond between counterions is identified as one of the key factors contributing to the red-shifted absorption spectrum, with increased occurrence observed in the red-shifted S169A mutant.
1 Introduction
Rhodopsins are light-sensitive proteins that enable organisms to detect and respond to light. Among them, microbial rhodopsins include channelrhodopsins (ChRs)—light-gated ion channels that conduct ions across cell membranes upon activation. ChRs are central to optogenetics, where they are used to modulate neuronal activity with high temporal and spatial precision by controlling membrane potential using light.1–3Channelrhodopsin 1 and 2 (ChR1, ChR2) from Chlamydomonas reinhardtii are activated by blue light.4,5 For optogenetic applications, however, red-light-activated ChRs are preferred due to deeper tissue penetration.6 Chrimson, a red-shifted ChR with an absorption maximum at 590 nm, was identified in the algae Chlamydomonas noctigama.
Although far red absorbing rhodopsins such as NeoRhodopsin (NeoR)7 and BestRhodopsin (BestR)8 have recently been discovered, Chrimson still remains the most red-shifted cationic channelrhodopsin in nature. Therapeutic applications of Chrimson include the restoration of sight9–11 and hearing,12–15 or the treatment of neural disorders.16,17 Important features of Chrimson are a high proton selectivity and rapid pH-dependent photocurrent kinetics.18
Like other ChRs,19 Chrimson features seven transmembrane (TM) α-helices with the N-terminus facing outward and the C-terminus facing inward. Its overall structure resembles C1C2, a chimera of ChR1 (helices 1–5) and ChR2 (helices 6–7). While Chrimson shares a similar ion gate architecture with other ChRs, its retinal binding pocket contains amino acids resembling those in bacteriorhodospin (bR), absent in blue-absorbing ChRs. Substituting these residues with those from C1C2 induces a blue shift in absorption.6 Similar effects are observed in bR and sensory rhodopsin II (SRII),20,21 where specific residues modulate the polarity distribution around the β-ionone ring, influencing the color tuning.
In Chrimson, several glutamic acid residues form key components of the ion pore (see Fig. 1): E124 and E125 at the inner gate, E132 and E300 at the central gate, and E139 and E143 at the outer gate.
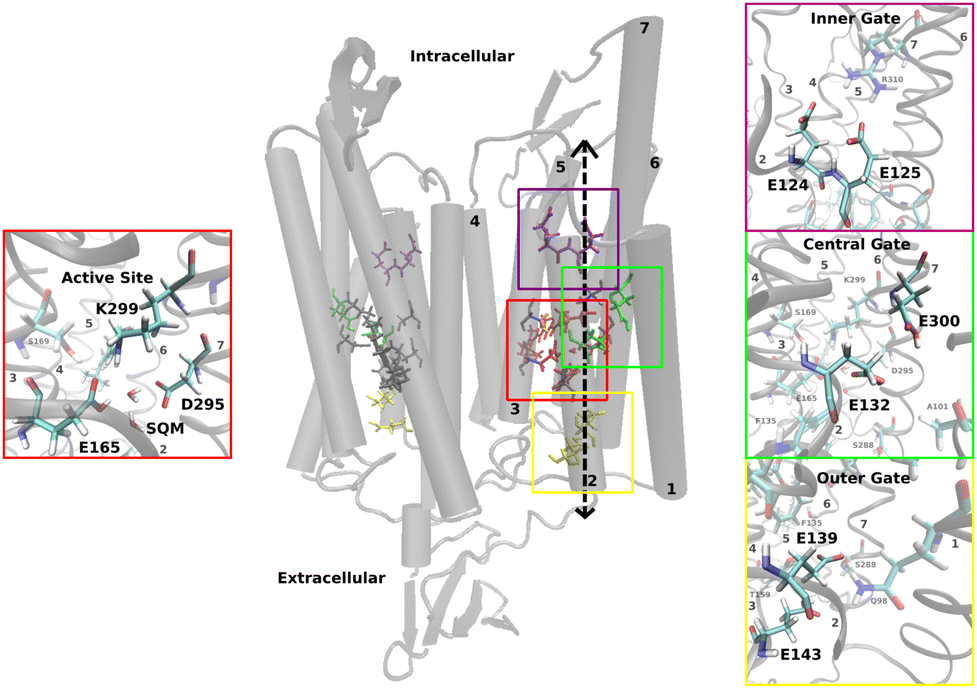 | ||
| Fig. 1 Structural model of the Chrimson dimer consisting of two homomonomers with seven transmembrane α-helices each and the chromophore retinal in the active site; zoom in on the molecules in the active site (red box) and the inner (purple box), middle (green box) and outer gates (yellow box) with the position of the protonatable glutamates. | ||
The active site is located at the center of each monomer and includes retinal, covalently bound to TM7 via a lysine residue (K299), along with two counterions, E165 and D295. The protonated retinal Schiff base (RSBH+) and its immediate environment, particularly the counterions and nearby water molecules, are critical factors for the opsin shift.6 This has been well established in various rhodopsins19,22 and recently reaffirmed in the near-infrared absorbing rhodopsins NeoR7,23 and BestR,8 both of which feature a counterion triad. In NeoR, a single negative charge near the RSBH+ – due to one deprotonated counterion – appears to cause the narrow, red-shifted absorption.7 Similarily, BestR's red-shifted absorption maximum at 661 ns is attributed to two protonated counterions, leaving one effective negative charge near the chromophore.8 A key contributor to Chrimson's red-shifted absorption is the unique protonation state of the counterions – unlike other ChRs, one remains protonated. Experimental evidence points to E165 as the protonated counterion,6,18,20 with its close proximity to D295 enabling strong hydrogen bonding. Chrimson's absorption spectrum is pH dependent, as in ChR1;24 titration experiments show that deprotonation of both counterions induces a hypsochromic (blue) shift.6
Combined quantum mechanical/molecular mechanical (QM/MM) methods have been widely used to gain detailed insights into the structure and function of photoreactive proteins, effectively complementing experimental observations. The status quo of these methods as well as current challenges have recently been reviewed, providing a comprehensive picture of the capabilities of these approaches.25 In this study, we apply QM/MM methods to investigate key structural features of wild type (wt) Chrimson – specifically the active site structure and the gates – as well as several mutants in these regions. For a considerable number of photoreactive proteins, the standard procedure involves initial equilibration of the overall system using molecular mechanics (MM) force fields, followed by a QM/MM geometry optimization with focus on the active site structure. This optimized structure can then serve as the basis for computing spectroscopic properties or initiating simulations of chromophore excited-state dynamics. While the active sites of these proteins are not static, they often exhibit stable ground state conformations, where thermal fluctuations average out to structures that closely resemble the QM/MM-optimized geometry. This behavior is observed, for example, in bR and proteorhodopsin (ppR), where the distances between active site residues result from such a unique global energy minimum.
The active site structure of ChRs, as previously discussed for ChR2,26 appears to challenge such a procedure. In the case of Chrimson, we encounter several modeling challenges that are addressed in this study. The key structural features involve extended, strongly hydrogen-bonded networks that exhibit considerable flexibility and support multiple hydrogen bonding patterns. As a result: (i) the protonation states are difficult to determine, (ii) an equilibration of the overall system with MM force fields may lead to unfavorable starting structures of the QM/MM methods,26 (iii) extensive QM/MM simulations on the ns-timescale are required to capture relevant dynamics and structural heterogeneity. However, such simulations are computationally prohibitive for DFT or ab initio methods, particularly when very large QM regions are involved. Additionally, the retinal chromophore itself presents a significant challenge for excited states methods.27,28
In a first step, to address the uncertainty in protonation states and hydrogen bonding patterns, we constructed several structural models and investigated their stability. These models were further evaluated by computing absorption spectra, also including important mutants. Our results reveal a highly flexible active site that exhibits multiple hydrogen bonding patterns, with their relative occurrence varying depending on the protonation states and specific mutations. This structural flexibility, which is characteristic for ChRs,21,26,29 is identified as a central factor contributing to the unique red shift absorption in Chrimson.
2 Methods
2.1 Chrimson models and system optimization
The Chrimson models used in this study are based on the X-ray structure of wt-Chrimson (PDB ID:5ZIH). Missing residues not visible in the crystal structure were added using the Swiss model.30 The resulting monomer was used to construct the initial structure of a Chrimson dimer. The dimer was embedded in a POPC (1-palmitoyl-2-oleoylphosphatidylcholine) lipid bilayer and solvated by water molecules in addition to those in the crystal structure, as shown in Fig. 2. | ||
| Fig. 2 Model of Chrimson: a protein dimer (purple) is inserted into a POPC lipid bilayer and surrounded by water molecules on both the cytoplasmic and extracellular sides. The retinal chromophores (yellow) are covalently attached to K299 side chains. (a) Front view; (b) top view, rotated by 90°. | ||
Experimental studies6,18,20 suggest that the RSBH+ counterion E165 is protonated, which is one of the main factors responsible for the red shift. However, the protonation state of the RSBH+ counterions (E165 and D295) was modeled as follows: (i) residue E165 protonated and D295 deprotonated and (ii) residue E165 deprotonated and D295 protonated; in order to obtain a comprehensive theoretical study. Further protonation states of titrable amino acids were not determined by experimental studies, so additional PropKa31–33 and MCCE34–40 analyses were performed (details in Section A.1 in the ESI†). Protonation states of histidines were determined by comparison with ChR2. We decided on eight models, see Table 1, with different protonation states of the glutamates located in the putative ion transport pathway, based on the calculated pKa values and experimental suggestions made by comparing the absorption spectra of several Chrimson mutants.6,20 In model I, all glutamates are deprotonated, creating a highly negatively charged putative ion pathway. Protonation is then gradually increased. Models IV and V are based on protonation states suggested by MCCE and PropKa, respectively. Models VI–VIII build upon models III–V by additionally protonating E139, resulting in all glutamates in the central and outer gate being protonated in model VIII.
The mutants were constructed using the same procedure as described for the WT. The respective amino acids were substituted with the program PYMOL 2.5.41 and the rotamer with the highest probability and least structural disruption was selected.
2.2 System equilibration, optimization and production run
The simulation protocols are similar to those used in our previous publications on retinal proteins,26,28,29,42–45 using classical MD simulations for the system preparation and DFTB3/3ob for the QM region.The QM region is the same as that used for the QM/MM optimization. Different simulation protocols were also used depending on the model (details in Table A.4 in ESI† Section A.3) and distance restraints that were introduced in the classical equilibration were gradually removed to obtain smooth and well equilibrated QM/MM MD production runs. All models were simulated for a minimum of 1 ns. A more detailed analysis was performed for wt-Chrimson models IV and V, for which simulations with E165 protonated in the initial structure were run three times with the same starting structure and different velocities for a total simulation time of 6 ns. Residue S169 was additionally included in the QM region because it strongly influences the polarity and hydrogen bonding patterns in the active site. All Chrimson mutants were simulated with the protonation states of model V, and otherwise the simulations were performed as described above.
For comparison purposes, QM/MM simulations were performed for ChR229 and bR44 with the initial structures and force field parameters taken from our previous studies. Simulation parameters are identical to the Chrimson models. All QM/MM optimizations and simulations were performed using the DFTB3/3ob parameter set51,52 for the QM region, as implemented in the GROMACS package (version 2020).53–57 The remainder of the system was modeled using the CHARMM3648 force field. DFTB3/3ob offers a favorable balance between accuracy and computational efficiency for modeling rhodopsins, particularly in capturing complex hydrogen-bonding networks.26,43,46 The accuracy of DFTB/3ob is comparable to that of full DFT calculations with medium-size basis sets.51 This is supported by: (i) its reliable performance in describing hydrogen-bonding interactions and large water clusters as reviewed in ref. 58 (Chap. 7.2) (ii) its accurate estimation of proton affinities for a wide range of organic molecules, relevant for the description of relative energies of proton donor–acceptor complexes (iii) and its ability to reproduce proton transfer barriers,51,52 which are essential for evaluating reaction kinetics. DFTB3/3ob has been shown to achieve accuracy close to that of B3LYP with medium-sized basis sets across these relevant properties, indicating its ability to resolve the relative occurrence of hydrogen bonded motifs as reported in this manuscript, similar to those described in our earlier work.26,43,46
Distance restraints on the atoms in the active site were applied with the PLUMED library (version 2.5.1).59,60
All data was processed and visualized with Python63 using the Matplotlib,64 NumPy,65 pandas,66 and seaborn67 libraries.
2.3 Excitation energies
The ground state dynamics of the Chrimson active site were described by the DFTB3/3ob method within a QM/MM framework. Excitation energies were calculated in a subsequent step for the respective structures. Only the retinal (residue K299) was part of the QM region, whereas the rest of the protein was modeled by classical MM point charges. This restriction of the QM region to the chromophore and its covalently linked lysine side chain reduces computational cost and facilitates consistent comparison of the relative excitation energies across different proteins, models and mutants. While this simplification may limit the accuracy of absolute excitation energies – due to the exclusion of nearby residues that can participate in charge transfer, leading to blue-shifted energies – these effects are largely systematic within a given protein. Moreover, although a more extensive QM region and explicit treatment of polarization and dispersion would improve the absolute value,68–70 such corrections introduce uniform shifts that cancel out when comparing relative excitation energies. As a result, using only the chromophore in the QM region yields realistic excitation energy trends, as shown in previous studies on rhodopsins.26 Excitation energies were calculated with the time-dependent generalization of DFTB using a long-range corrected functional (LC-TD-DFTB71–74 with ob2 parameters75) for all QM/MM MD sampled structures (6000 snapshots for each model and mutant, respectively). In the following, LC-TD-DFTB/ob2 will be abbreviated by LC-DFTB. The electronic parameters differ to those used in ref. 28,72 and match the ones used in ref. 76,77. The SCC convergence criterium was set to 1 × 10−8 electrons, the range separation parameter to 0.3/a0 and the threshold of the screening method is set to 1 × 10−16. LC-DFTB is computationally very efficient and therefore suitable for the calculation of absorption spectra, where a large number of calculations is required.28 For comparison purposes excitation energies of two other rhodopsin proteins were computed: bR and ChR2. The structures used for QM/MM MD simulations were based on previous publications,28,29 and the snapshots (1000 snapshots in the case of bR and 6000 snaphopts for ChR2) used for the excited state calculations. In addition, 250 trajectory snapshots were chosen randomly from the QM/MM MD simulations of each system to calculate excitation energies on the TD-DFT level of theory using the ωB97X functional78 and def2-TZVP basis set.79 These calculations were performed using the ORCA program package version 5.0.3.80,813 Computational model of Chrimson
This section presents the computational model of Chrimson used to analyze the active site and the effects of mutations in the following sections. We initially examined all proposed models and discard the majority of them due to structural inconsistencies. As described in the Methods 2.2.1 and Section A.3 in the ESI,† special care is required when preparing initial configurations. Since classical force-fields are insufficient to preserve the complex hydrogen-bonding network of the active site, as discussed above, we applied an equilibration protocol that includes distance restraints, which were gradually released to enable fully unconstrained QM/MM MD simulations. The retinal chromophore, E165, D295, F135 and nearby water molecules, denoted as SQM (see Section 2.2.2) are included in the QM region.3.1 Protonation states of the counterions
Experimental studies at different pH values (6, 7.4 and 9) indicate a pH dependent protonation state for the counterions of the protonated Schiff base (RSBH+) and a hypsochromic shift in the absorption spectra due to the deprotonation of the counterions. The pKa of the counterion E165 is about 8, resulting in a protonated residue at physiological pH. This is identified as one of the key factors for the red shift of Chrimson,18,20 because a reduction of the negative charge near the RSBH+ destabilizes the ground state structure leading to absorption at higher wavelengths.19 K93 in ChR282,83 is a conserved residue in channelrhodopsins, as it stabilizes the deprotonated form of the counterion E123 through a salt bridge. In Chrimson, K93 is replaced by F135, whose hydrophobic side chain shields the active site from extracellular bulk solvent and increases the pKa of the glutamate, thus stabilizing its protonated form.20,84In our simulations, we considered two initial protonation states: (i) residue E165 protonated and D295 deprotonated, and (ii) residue E165 deprotonated and D295 protonated. The QM/MM MD simulations resulted in the proton to be found in close proximity to residue E165 regardless of the starting structure as shown in Fig. 3. In model (ii), proton transfer occurs immediately at the start of the QM/MM MD simulations, followed by a few back-and-forth events until approximately ≈ 250 ps (see Fig. C.2b and C.2d in ESI† Section C.2). After this point, E165 remains stably protonated with no further transfer. In contrast, when E165 is protonated in the initial structure (model i), no proton transfer to D295 is observed (see Fig. C.2a and C.2c in ESI† Section C.2). These results support experimental findings that E165 is the protonated counterion.6,18,20
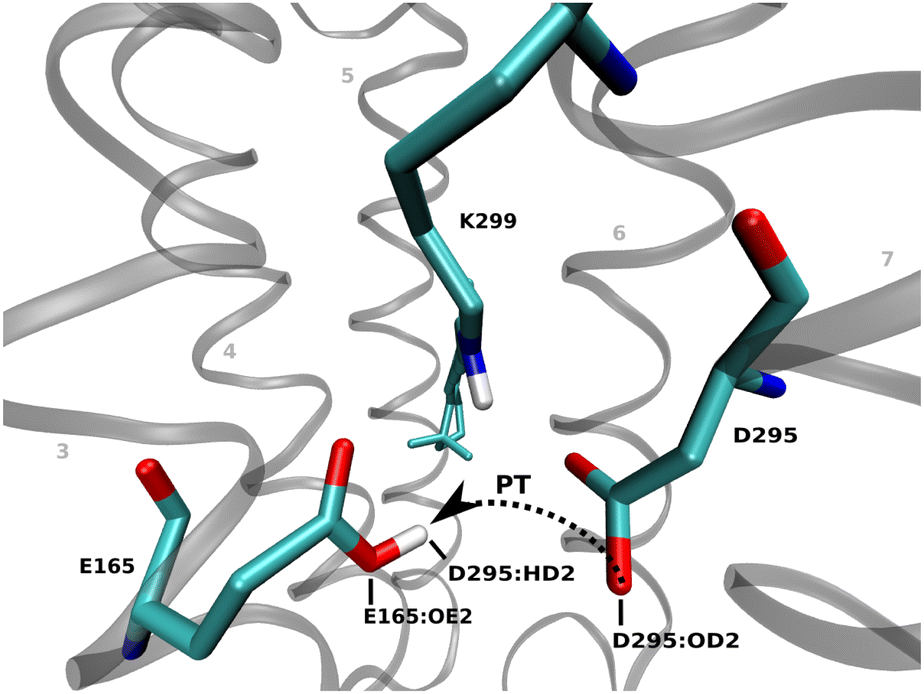 | ||
| Fig. 3 The residue D295 is the predominant counterion in Chrimson, while E165 is protonated in the ground state. | ||
3.2 Protonation states of the glutamates in the putative ion pathway
As stated in the Introduction and in Section 2.1, a challenge in simulating rhodopsin proteins is the uncertainty in protonation states of titrable amino acids. The ion-conducting pore in Chrimson is closed in the ground state by three gates, that are lined by glutamates, the inner gate (E124, E125), the central gate (E132, 300) and the outer gate (E139, E143).6 In order to investigate the protonation states of the amino acids in the putative ion pore, we consider several computational models as summarized in the Table 1 in Section 2.1. Since the glutamates E124 and E125 in the inner gate of Chrimson are homologous to E82 and E83 in ChR2 and both of them are deprotonated in ChR2,26 we assume these protonation states also for Chrimson. Our simulations show that deprotonation of E132, as in models I and II, leads to a destabilization of the active site, whereas deprotonation of E300 impedes hydrogen bonding to A101 and therefore a connection of helices 7 and 1 (see Fig. C.4b in ESI† Section C.3 for details). These observations lead to the conclusion that both, E132 and E300, must be protonated, excluding model I to III, to obtain a stable active site and to establish the experimentally observed linkage of helices 1 and 7.6 Regarding the outer gate, we observe that the protonation of E139 prevents the formation of hydrogen bonds with the amino group of Q98, which excludes models VI–VIII (see Fig. C.5b in ESI† Section C.3 for details).The determination of the protonation state of E143 is less straightforward, as models IV and V share similar structural motifs despite the difference in the protonation state of E143. Therefore, in the following sections we present a more detailed analysis of models IV and V.
4 Active site of Chrimson
The active site of Chrimson shows a particularly high flexibility, already reported for other channelrhodopsins, such as ChR2,26 which we will present in detail in Section 4.1. In addition, the unique red light absorption of Chrimson can be related to specific motifs in the active site, as shown in Section 4.2.4.1 Structural motifs
In contrast to bR, where the active site features a relatively stable water-bridged hydrogen bonding network between the RSBH+ and the two counterions D85 and D212,21,85 Chrimson exhibits a highly flexible active site, fluctuating between distinct structural motives similar to those observed in ChR2.26Fig. 4(a) shows the Chrimson active site as determined by X-ray diffraction.6 The reported interatomic distance between the oxygens of the counterions is 3.3 Å, while the distances from the retinal nitrogen to the oxygens of E165 and D295 are 3.0 Å and 3.6 Å, respectively. These values appear relatively long, given that E165 interacts with a positively charged retinal and a negatively charged D295, where hydrogen bond lengths are typically shorter than 3 Å.86 In other retinal proteins, the distance between the RSBH+ nitrogen and the oxygen of a nearby water molecule or counterion commonly falls below <2.9 Å. For example, in bR, the hydrogen bond between retinal and a nearby water molecule is about 2.8 Å; in phoborhodopsin (ppR), this distance is even shorter at 2.6 Å.21 In ChR226 the distance is approximately 2.8 Å, involving either a water molecule or the counterion E123. In the sodium pump ErNaR from Erythrobacter sp. HL-111, the RSBH+-counterion distance is ≈2.8 Å, while the distance between the two counterions is exceptionally short at just 2.2 Å87 We note that the interatomic distances observed in the crystal structure appear to be unusual for retinal proteins. Our QM/MM MD simulations, discussed below, suggest that the crystal structure may represent an average over multiple structural motifs. Notably, a water molecule positioned near the counterions in the crystal structure can enter the active site and dynamically rearrange the counterion-retinal complex. | ||
| Fig. 4 Active site structure of Chrimson. (a) The interatomic distance between the oxygens of the counterions in the active site of the Chrimson crystal structure (PDB-ID: 5ZIH)6 is 3.3 Å, while the distances of the RSBH+ to E165 and D295 are 3.0 Å and 3.6 Å, respectively. A water molecule (red) is in close proximity to the counterions. Two structural motifs in the active site structure are observed along the QM/MM MD simulations: the counterions are (b) in close proximity and permanently hydrogen-bonded (C1), or (c) a water molecule (SQM) is located between the two counterions with hydrogen bonds to both of them (C2). | ||
As a result, along the the QM/MM MD trajectories reveal two dominant structural motifs in the active site: either the counterion oxygens of E165 and D295 are within a distance of ≤3.0 Å of each other (Fig. 4(b), conformation C1), forming a strong hydrogen bond, or they are separated by ≥4.0 Å (Fig. 4(c), conformation C2), bridged by a hydrogen-bonded water molecule.
The average distance between the counterion oxygens is 2.8 Å in conformation C1 and 4.2 Å in conformation C2. A detailed analysis of these distances is provided in ESI† Section C.3. The 3.3 Å bond length observed in the crystal structure may reflect an average of these two conformations, particularly if they are similar populated. In model IV, conformation C1 is slightly more populated, while in model IV both conformations occur at approximately equal frequency (see Table 2). In C1, the proton is not shared between the two counterions: the hydrogen consistenly remains closer to E165, and no proton transfer is observed along the trajectory (see Fig. C.2a and C.2c in ESI† Section C.2). Nevertheless, strong hydrogen bonding is present (see Table 3).
| wtIV | wtV | S169A | S169D | |
|---|---|---|---|---|
| C1 | 58.4 | 49.1 | 61.4 | 6.1 |
| C2 | 41.6 | 50.9 | 38.6 | 93.9 |
| wtIV | wtV | |
|---|---|---|
| RSBH+⋯E165 | 23.6 | 23.0 |
| RSBH+⋯D295 | 39.1 | 30.0 |
| RSBH+⋯SQM | 0.4 | 14.8 |
| Total | 63.0 | 66.3 |
 | ||
| Fig. 5 The orientation of the E165 side chain changes according to the structural motif (pink: conformation C1; cyan: conformation C2). The black arrow indicates the torsional motion of the E165 side chain from one conformation to the other. Hydrogen bonds are indicated by dotted lines. | ||
The orientation of E165 alternates between a lateral conformation (C1), with a dihedral angle of −120° to −180°, where it forms a strong hydrogen bond with D295 and a vertical conformation (C2), with a dihedral angle of −80° to −110°, where a hydrogen-bonded water molecule (SQM) bridges the counterions. In both cases, the backbones of E165 and S169 remain hydrogen-bonded. However, an additional hydrogen bond between their side chains stabilizes the vertical orientation of E165, favoring conformation C2 (see Fig. C.3b and 3e in ESI† Section C.3).
4.2 Calculated absorption spectra
The excited-state spectra of rhodopsins are highly sensitive to the chromophore's environment: (i) steric interactions can induce a twist in the retinal structure, altering excitation energies, and (ii) hydrogen bonding and Coulomb interactions with charged and polar groups can affect the absorption spectrum drastically. Consequently, computed retinal excitation energies can offer valuable insight into the validity of a given protein structural model. While agreement with experimental absorption energies does not confirm a structure, significant discrepancies may suggest that the model is inaccurate. Given that the structures of bR and ChR2 are well established – and Chrimson shares many structural motifs with ChR2, while exhibiting even more red-shifted absorption with respect to (wrt.) bR – comparing these three systems provides meaningful insights to access the structural models developed in this work.6,21,26,29 When applying quantum chemical (QC) methods within QM/MM approaches, three primary sources of error must be considered: (i) QC methods often lack accuracy in absolute excitation energies. This is evident in the vacuum excitation energy, which typically exhibits a systematic error. However, this can be corrected by calibrating against accurate gas-phase reference values for the retinal chromophore. (ii) Accurately modeling the response to the electrostatic potential from the protein environment – the so-called opsin shift – is particularly challenging. The retinal chromophore is highly polarizable, and this poses a significant difficulty even for well-established quantum chemistry methods (see, e.g., ref. 28, 27). (iii) Finally, the effect of protein polarization and dispersion can contribute significantly to the excitation energy, as discussed in ref. 69, 70. Despite these known limitations, we apply LC-DFTB within an electrostatic embedding scheme to enable extensive conformational sampling. Due to the high flexibility of the active site, it is not feasible to identify few representative structures suitable for high-level quantum chemical calculations. As a result, our simulations are not intended to yield highly accurate absolute excitation energies. Instead, we focus on comparing relative trends across the three proteins. While LC-DFTB systematically underestimates excitation energy shifts, it reliably captures qualitative trends.Chrimson absorbs at 590 nm (2.10 eV) under neutral pH conditions, bR at 568 nm (2.18 eV) and ChR2 at 473 nm (2.62 eV).6 A previous benchmark of LC-DFTB has shown that opsin shifts are typically underestimated by about 50%. For example, the computed shift between bR and ChR2 is expected to be around 0.2 eV, compared to the experimental value of 0.4 eV. It is worth noting that most DFT methods, such as CAM-B3LYP or B3LYP, exhibit similar errors. Among tested functionals, ωB97 performed best, but still underestimates the shift by approximately 25%.28
Based on previous benchmarking, LC-DFTB is expected to yield a blue shift of 0.2 eV for ChR2 and a slight red shift of about 0.04 eV for Chrimson. The results summarized in Table 4 are consistent with these expectations. The red shift to ChR2 is underestimated, amounting to roughly half of the experimental value. In comparison, calculations using the ωB97X functional reproduce about 75% of the experimental bR-ChR2 shift, while the ChR2-Chrimson shift reaches 60–70% of the measured value, depending on the model. For the bR-Chrimson shift, both methods – LC-DFTB and ω97X – underestimate the red-shift, with model V even yielding a slight blue shift.
| bR | ChR2 | Chrimson | |||
|---|---|---|---|---|---|
| wtIV | wtV | ||||
| Exp6 | 2.18 | 2.62 | 2.1 | ||
| LC-DFTB | QM/MM | 2.52 | 2.75 | 2.51 | 2.54 |
| Δ Chrimson-bR | −0.01 | 0.02 | |||
| Δ Chrimson-ChR2 | −0.24 | −0.21 | |||
| ωB97X | QM/MM | 2.87 | 3.21 | 2.85 | 2.90 |
| Δ Chrimson-bR | −0.02 | 0.03 | |||
| Δ Chrimson-ChR2 | −0.36 | −0.31 | |||
In Section D in the ESI,† we also report opsin shifts and results from OM2/MRCI calculations, which – due to technical limitations – were performed without including the membrane. OM2/MRCI yields a qualitatively similar picture, though it overestimates the opsin shift, as expected. Nevertheless, since the overall trends are consistent with those from the other methods, these additional calculations support the validity of our modeling strategy.
The excitation energies are similar for both models IV and V, and the comparison with bR and ChR2 gives qualitatively correct spectral shifts. The comparison with bR reveals very similar results for the Chrimson models. Running our simulations and subsequent excitation energy calculations in triplicate, we observed a standard deviation of approximately 0.02 eV. When this variability is considered alongside the known underestimation of spectral shifts by the LC-DFTB method, the computed red shift of 0.01 eV appears slightly smaller than expected based on the combined systematic and statistical uncertainties. Nonetheless, the discrepancy is minor, and we consider both models IV and V suitable candidates for further investigation in mutation studies.
The excitation energies were correlated with the corresponding structural motifs in the active site to identify which conformations are responsible for the red-shifted absorption in Chrimson. Histograms of the excitation energies (see Fig. 6) are categorized by the structural motif present in the active site, as defined by the distance between the counterion oxygens. Conformation C1, characterized by a counterion oxygen–oxygen distance of less than 3 Å, clearly correlates with lower excitation energies and thus contributes to the red shift. In model IV, conformation C1 occurs more frequently, resulting in a slightly more pronounced red shift compared to model V.
 | ||
| Fig. 6 Histograms of the excitation energies in eV according to the structural motif in the active site. Condition for the configuration is the distance between the counterions E165 and D295. The mean values are indicated on the top right and the colors indicate the configuration of the active site. | ||
5 Chrimson mutants
Based on the wt-Chrimson structure discussed above, we investigated mutants of residue S169 and the two counterions. These positions are particularly interesting because their mutation perturbs the complex counterion network, altering the conformational distribution of hydrogen bonding patterns. Moreover, these changes lead to even more red-shifted variants, enhancing their potential for optogenetic applications.Models IV and V predict similar trends: while model IV shows a slightly better red-shift relative to bR, both models perform comparably with respect to ChR2. However, the analysis of small structural differences favors model V (see Section C.3 in the ESI† for details). Consequently, Chrimson mutants were constructed using the protonation states of the glutamates defined in model V.
A detailed structural analysis of the Chrimson mutants is provided in ESI† Section C.4. The primary goal of the mutant study is to validate the computational model from the previous section by comparing excitation energies of the mutants to those of Chrimson-wt.
5.1 Excitation energies of the Chrimson mutants
Experimental studies show that S169A is red-shifted compared to the wt and absorbs at 608 nm (2.04 eV), while mutation of either one of the counterions, E165Q and D295N, leads to a blue-shifted spectrum (2.25 eV and 2.30 eV, respectively).6Table 5 shows the results of the excited state calculations of the mutants compared to wt-Chrimson.| wtV | S169A | S169D | E165Q | D295N | |
|---|---|---|---|---|---|
| Exp6 | 2.10 | 2.04 | — | 2.25 | 2.30 |
| QM/MM | 2.54 | 2.48 | 2.53 | 2.57 | 2.61 |
| Δ wt-mutant | −0.06 | −0.01 | 0.03 | 0.07 |
In agreement with experiment, our calculations show a red-shifted absorption of S169A, while for S169D only a very small shift in the range of the standard deviation is found, as discussed in Section 4.2. As expected, the experimental shift of E165Q and D295N is qualitatively reproduced but underestimated, as discussed above. The deviation is partly due to the limitations of the applied excited states method, but the structural models of the mutations may also contribute.
5.2 Influence of structural motifs on the absorption of S169A
Fig. 6(c) shows histograms of excitation energies for different active site conformations in the S169A mutant, distinguished by the distance between the counterions. As in wt-Chrimson, conformation C1 is red-shifted relative to C2 and occurs more frequently (see Table 2 in Section 4). This shift can be attributed to the loss of one hydrogen bond: unlike serine in wt-Chrimson, alanine at position 169 lacks an additional proton donor, allowing only a single hydrogen bond between the backbones of E165 and A169. Nevertheless, both conformations in the mutant are more red-shifted than in wt-Chrimson, due to reduced polarity near the RSBH+.In summary, the study of the Chrimson mutants provides an interesting insight into altered hydrogen bonding patterns reflected in the experimental absorption shifts. Our analysis of the mutants validates the computational model proposed in Section 4.1 and confirms the findings regarding the red shift in Chrimson from Section 4.2: the presence of the direct hydrogen bond between the counterions shifts the absorption spectra towards lower excitation energies.
6 Conclusions
We applied a multiscale approach based on DFTB/MD simulations to investigate the active site of the red-light-sensitive channelrhodopsin Chrimson, which is of particular significance for optogenetic applications. Our study provides new insights into the molecular dynamics at the nanosecond timescale.Several studies have investigated the molecular origin of the red-shift in Chrimson,6,18,20 identifying three key factors: (i) the protonation of one of the counterion residues destabilizing the RSBH+ in the ground state; (ii) a biased distribution of polar residues near the β-ionone ring, similar to bR; and (iii) a rigid retinal chromophore stabilized by bulky side chains. The mutant S169A, reported in ref. 6, exhibits an even greater red-shift, attributed to a further reduction in active site polarity.
Our calculations successfully reproduce the characteristic red-shift of Chrimson relative to other ChRs. Moreover, we validate the reliability of our computational model by simulating several Chrimson mutants, recovering the experimentally observed spectral shifts. Consistent with experimental data, we confirm that E165 serves as the protonated counterion. As discussed above, the structure of the retinal binding pocket – particularly the residues in the active site – is a key determinant of color tuning in Chrimson. To investigate this, we examined the structural dynamics, which cannot be resolved by static techniques such as X-ray diffraction. As a result, we identified a multitude of active site conformations that deviate from the the deposited crystal structure. The active site of Chrimson exhibits structural heterogeneity comparable to that observed in ChR2,26 including water movement within the active site and pronounced fluctuations in the distances between the counterions and the retinal Schiff base.
Interestingly, contrary to the crystallographic structure,6 both counterions can approach the RSBH+ within ≤3 Å, and D295 exhibits a higher frequency of hydrogen bonding with the RSBH+ than E165. Two dominant structural motifs emerge along the trajectory: either the counterions are in close proximity (E165OE–D295OD distance ≤3 Å), or they are farther apart, bridged by a hydrogen-bonded water molecule. Notably, at the counterion-counterion distance of approximately 2.8 Å, strong hydrogen bonding is observed, although no proton sharing occurs. Our analysis reveals a correlation between structural motifs and optical properties: the conformation characterized by a reduced distance between the counterions correlates with longer-wavelength absorption and is more prevalent in the red-shifted mutant S169A. This enhanced occurrence is linked to the S169A substitution: the replacement of serin by alanine results in the loss of an additional hydrogen bond to E165 that restricts the torsional flexibility of E165, promoting the red-shifted conformation. These findings represent a significant step towards understanding structure–function relationships in color tuning, providing a mechanistic basis for rational design of optogenetic tools with tailored spectral properties.
Overall, our QM/MM simulations have provided a rationalization for the crystal structure as an average of the conformations found in our simulations. These conformations interconvert on the nanosecond timescale, likely contributing to the intermediate active site distances in the crystal structure, which appear slightly too long for strong hydrogen bonds.86
Our work discusses the various challenges associated with multi-scale modeling of ChRs, some of which are discussed in a recent review on photoreceptor modeling.25 One key issue is the treatment of multiple protonation states. In principle, future simulations may be based on constant pH molecular dynamics (CpHMD), as suggested in the review. Although approaches such as CpHMD followed by QM/MM simulations currently involve significantly higher computational costs, ongoing advances in hardware performance may make them increasingly feasible and help to reduce the uncertainty associated with using several models with different protonation states.
A major limitation, however, lies in the inability of current force fields to reliably model extended networks of strong hydrogen bonds, as demonstrated in this and previous studies.26,29 Similar challenges have also been reported in other systems.88 While the use of polarized charges,88 offers some improvements, their application remains problematic in highly flexible systems such as channel proteins. Consequently, we employed constrained MD simulations during initial equilibration and used large QM regions to accurately refine the active site structure.
Machine learning-based models may eventually help overcome this limitation; however, accurate equivariant neural network potentials currently exhibit computational demands comparable to the DFTB3 method used in this work. For such highly flexible active sites, there appears to be no viable substitute for fast QM or ML-based models combined with extensive sampling. The commonly used approach of relying on QM/MM-optimized structures – sufficient for many photoreceptors – is clearly inadequate in this case. An additional challenge arises from the complex electronic structure of retinal, which poses significant difficulties for accurate modeling.27,28
All TD-DFT methods, including those applying range-separated functionals, have been shown to have large errors considering shifts in excitation energies. Even the generally very well-performing ωB97 functional underestimates color shifts by approximately 25%, while other commonly used functionals such as B3LPY or CAM-B3LYP can exhibit errors of up to 50%.28
Future work in our group aims to extend LC-DFTB by implementing more accurate functionals, such as ωB97, into the DFTB framework. However, due to inherent limitations of the method, the potential for improvement may be limited. As an alternative, the development and application of machine learning models trained on post-Hartree–Fock methods seems to be a promising approach.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support by the German Research Foundation (DFG) through the Research Training Group 2450 “Tailored Scale-Bridging Approaches to Computational Nanoscience”. Additional support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster) is appreciated. We are grateful to Prof. Peter Hegemann and Dr Johannes Vierock for their insightful feedback and fruitful discussions that significantly shaped the direction of this study, especially regarding the Chrimson mutations.References
- F. Schneider, C. Grimm and P. Hegemann, Annu. Rev. Biophys., 2015, 44, 167–186 CrossRef CAS PubMed.
- K. Deisseroth and P. Hegemann, Science, 2017, 357, eaan5544 CrossRef PubMed.
- V. Emiliani, E. Entcheva, R. Hedrich, P. Hegemann, K. Konrad, C. Lüscher, M. Mahn, Z.-H. Pan, R. Sims, J. Vierock and O. Yizhar, Nat. Rev. Methods Primers, 2022, 2, 55 CrossRef CAS PubMed.
- F. Zhang, J. Vierock, O. Yizhar, L. E. Fenno, S. Tsunoda, A. Kianianmomeni, M. Prigge, A. Berndt, J. Cushman, J. Polle, J. Magnuson, P. Hegemann and K. Deisseroth, Cell, 2011, 147, 1446–1457 CrossRef CAS PubMed.
- H. Harz and P. Hegemann, Nature, 1991, 351, 489–491 CrossRef CAS.
- K. Oda, J. Vierock, S. Oishi, S. Rodriguez-Rozada, R. Taniguchi, K. Yamashita, J. S. Wiegert, T. Nishizawa, P. Hegemann and O. Nureki, Nat. Commun., 2018, 9, 3949 CrossRef PubMed.
- M. Broser, A. Spreen, P. E. Konold, E. Peter, S. Adam, V. Borin, I. Schapiro, R. Seifert, J. T. M. Kennis, Y. A. Bernal Sierra and P. Hegemann, Nat. Commun., 2020, 11, 5682 CrossRef CAS PubMed.
- A. Rozenberg, I. Kaczmarczyk, D. Matzov, J. Vierock, T. Nagata, M. Sugiura, K. Katayama, Y. Kawasaki, M. Konno, Y. Nagasaka, M. Aoyama, I. Das, E. Pahima, J. Church, S. Adam, V. A. Borin, A. Chazan, S. Augustin, J. Wietek, J. Dine, Y. Peleg, A. Kawanabe, Y. Fujiwara, O. Yizhar, M. Sheves, I. Schapiro, Y. Furutani, H. Kandori, K. Inoue, P. Hegemann, O. Béjà and M. Shalev-Benami, Nat. Struct. Mol. Biol., 2022, 29, 592–603 CrossRef CAS PubMed.
- J.-A. Sahel, E. Boulanger-Scemama, C. Pagot, A. Arleo, F. Galluppi, J. N. Martel, S. D. Esposti, A. Delaux, J.-B. de Saint Aubert, C. de Montleau, E. Gutman, I. Audo, J. Duebel, S. Picaud, D. Dalkara, L. Blouin, M. Taiel and B. Roska, Nat. Med., 2021, 27, 1223–1229 CrossRef CAS PubMed.
- A. Bi, J. Cui, Y.-P. Ma, E. Olshevskaya, M. Pu, A. M. Dizhoor and Z.-H. Pan, Neuron, 2006, 50, 23–33 CrossRef CAS PubMed.
- J. E. McGregor, T. Godat, K. R. Dhakal, K. Parkins, J. M. Strazzeri, B. A. Bateman, W. S. Fischer, D. R. Williams and W. H. Merigan, Nat. Commun., 2020, 11, 1703 CrossRef CAS PubMed.
- T. Shimano, B. Fyk-Kolodziej, N. Mirza, M. Asako, K. Tomoda, S. Bledsoe, Z. H. Pan, S. Molitor and A. G. Holt, Brain Res., 2013, 1511, 138–152 CrossRef CAS PubMed.
- M. Zerche, T. Moser and T. Mager, Laryngo-Rhino-Otologie, 2022 Search PubMed.
- B. Bali, E. Gruber-Dujardin, K. Kusch, V. Rankovic and T. Moser, Life Sci. Alliance, 2022, 5, e202101338 CrossRef CAS PubMed.
- M. Zerche, C. Wrobel, K. Kusch, T. Moser and T. Mager, Mol. Ther., Methods Clin. Dev., 2023, 29, 202–212 CrossRef CAS PubMed.
- V. Gradinaru, M. Mogri, K. R. Thompson, J. M. Henderson and K. Deisseroth, Science, 2009, 324, 354–359 CrossRef CAS PubMed.
- E. Kip, L. Bentall, C. Underwood, S. Hughes and L. Parr-Brownlie, Neuroscience, 2022, 507, 64–78 CrossRef CAS PubMed.
- J. Vierock, C. Grimm, N. Nitzan and P. Hegemann, Sci. Rep., 2017, 7, 9928 CrossRef PubMed.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed.
- D. Urmann, C. Lorenz, S. M. Linker, M. Braun, J. Wachtveitl and C. Bamann, Photochem. Photobiol., 2017, 93, 782–795 CrossRef CAS PubMed.
- M. Hoffmann, M. Wanko, P. Strodel, P. H. König, T. Frauenheim, K. Schulten, W. Thiel, E. Tajkhorshid and M. Elstner, J. Am. Chem. Soc., 2006, 128, 10808–10818 CrossRef CAS PubMed.
- R. Rajamani, Y. Lin and J. Gao, J. Comput. Chem., 2011, 32, 854–865 CrossRef CAS PubMed.
- R. Palombo, L. Barneschi, L. Pedraza-González, D. Padula, I. Schapiro and M. Olivucci, Nat. Commun., 2022, 13, 6652 CrossRef CAS PubMed.
- O. P. Ernst, P. A. S. Murcia, P. Daldrop, S. P. Tsunoda, S. Kateriya and P. Hegemann, J. Biol. Chem., 2008, 283, 1637–1643 CrossRef CAS PubMed.
- M. Mroginski, S. Adam, G. S. Amoyal, A. Barnoy, A. Bondar, V. A. Borin, J. R. Church, T. Domratcheva, B. Ensing, F. Fanelli, N. Ferré, O. Filiba, L. Pedraza-González, R. González, C. E. González-Espinoza, R. K. Kar, L. Kemmler, S. S. Kim, J. Kongsted, A. I. Krylov, Y. Lahav, M. Lazaratos, Q. NasserEddin, I. Navizet, A. Nemukhin, M. Olivucci, J. M. H. Olsen, A. Pérez De Alba Ortíz, E. Pieri, A. G. Rao, Y. M. Rhee, N. Ricardi, S. Sen, I. A. Solov’yov, L. De Vico, T. A. Wesolowski, C. Wiebeler, X. Yang and I. Schapiro, Photochem. Photobiol., 2021, 97, 243–269 CrossRef CAS PubMed.
- Y. Guo, F. E. Beyle, B. M. Bold, H. C. Watanabe, A. Koslowski, W. Thiel, P. Hegemann, M. Marazzi and M. Elstner, Chem. Sci., 2016, 7, 3879–3891 RSC.
- M. Wanko, M. Hoffmann, P. Strodel, A. Koslowski, W. Thiel, F. Neese, T. Frauenheim and M. Elstner, J. Phys. Chem. B, 2005, 109, 3606–3615 CrossRef CAS PubMed.
- B. M. Bold, M. Sokolov, S. Maity, M. Wanko, P. M. Dohmen, J. J. Kranz, U. Kleinekathöfer, S. Höfener and M. Elstner, Phys. Chem. Chem. Phys., 2020, 22, 10500–10518 RSC.
- Y. Guo, F. E. Wolff, I. Schapiro, M. Elstner and M. Marazzi, Phys. Chem. Chem. Phys., 2018, 20, 27501–27509 RSC.
- F. Kiefer, K. Arnold, M. Kunzli, L. Bordoli and T. Schwede, Nucleic Acids Res., 2009, 37, D387–D392 CrossRef CAS PubMed.
- H. Li, A. D. Robertson and J. H. Jensen, Proteins: Struct., Funct., Bioinf., 2005, 61, 704–721 CrossRef CAS PubMed.
- D. C. Bas, D. M. Rogers and J. H. Jensen, Proteins, 2008, 73, 765–783 CrossRef CAS PubMed.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- E. G. Alexov and M. R. Gunner, Biophys. J., 1997, 72, 2075–2093 CrossRef CAS PubMed.
- R. E. Georgescu, E. G. Alexov and M. R. Gunner, Biophys. J., 2002, 83, 1731–1748 CrossRef CAS PubMed.
- M. R. Gunner and E. Alexov, Biochem. Biophys. Acta, 2000, 1458, 63–87 CAS.
- J. Mao, K. Hauser and M. R. Gunner, Biochemistry, 2003, 42, 9829–9840 CrossRef CAS PubMed.
- Y. Song, J. Mao and M. R. Gunner, Biochemistry, 2003, 42, 9875–9888 CrossRef CAS PubMed.
- K. Hauser, J. Mao and M. R. Gunner, Biopolymers, 2004, 74, 51–54 CrossRef CAS PubMed.
- Z. Zhu and M. R. Gunner, Biochemistry, 2005, 44, 82–96 CrossRef CAS PubMed.
- Schrödinger, LLC.
- H. C. Watanabe, K. Welke, F. Schneider, S. Tsunoda, F. Zhang, K. Deisseroth, P. Hegemann and M. Elstner, J. Biol. Chem., 2012, 287, 7456–7466 CrossRef CAS PubMed.
- H. C. Watanabe, K. Welke, D. J. Sindhikara, P. Hegemann and M. Elstner, J. Mol. Biol., 2013, 425, 1795–1814 CrossRef CAS PubMed.
- K. Welke, J. S. Frähmcke, H. C. Watanabe, P. Hegemann and M. Elstner, J. Phys. Chem. B, 2011, 115, 15119–15128 CrossRef CAS PubMed.
- K. Welke, H. C. Watanabe, T. Wolter, M. Gaus and M. Elstner, Phys. Chem. Chem. Phys., 2013, 15, 6651 RSC.
- T. Wolter, M. Elstner, S. Fischer, J. C. Smith and A.-N. Bondar, J. Phys. Chem. B, 2015, 119, 2229–2240 CrossRef CAS PubMed.
- M. Eichinger, P. Tavan, J. Hutter and M. Parrinello, J. Chem. Phys., 1999, 110, 10452–10467 CrossRef CAS.
- J. Huang and A. D. MacKerell, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. OConnor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. J. MacKerell and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. Gaus, Q. Cui and M. Elstner, J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS PubMed.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef CAS PubMed.
- P. Bauer, B. Hess and E. Lindahl, 2022.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- A. S. Christensen, T. Kubař, Q. Cui and M. Elstner, Chem. Rev., 2016, 116, 5301–5337 CrossRef CAS PubMed.
- G. Bussi and G. A. Tribello, in Biomolecular Simulations, ed. M. Bonomi and C. Camilloni, Springer, New York, 2019, vol. 2022, pp. 529–578 Search PubMed.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. Stone, MSc thesis, Computer Science Department, University of Missouri-Rolla, 1998.
- G. Van Rossum and F. L. Drake, Python 3 Reference Manual, CreateSpace, Scotts Valley, CA, 2009 Search PubMed.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
- C. R. Harris, K. J. Millman, S. J. van der Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N. J. Smith, R. Kern, M. Picus, S. Hoyer, M. H. van Kerkwijk, M. Brett, A. Haldane, J. Fernández del Río, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke and T. E. Oliphant, Nature, 2020, 585, 357–362 CrossRef CAS PubMed.
- W. McKinney and others, Proceedings of the 9th Python in Science Conference, 2010, pp. 51-56.
- M. L. Waskom, J. Open Source Softw., 2021, 6, 3021 CrossRef.
- M. Wanko, PhD thesis, University of Braunschweig, 2009.
- M. Wanko, M. Hoffmann, T. Frauenheim and M. Elstner, J. Phys. Chem. B, 2008, 112, 11462–11467 CrossRef CAS PubMed.
- M. Wanko, M. Hoffmann, J. Frähmcke, T. Frauenheim and M. Elstner, J. Phys. Chem. B, 2008, 112, 11468–11478 CrossRef CAS PubMed.
- T. A. Niehaus and F. Della Sala, Phys. Status Solidi B, 2012, 249, 237–244 CrossRef CAS.
- J. J. Kranz and M. Elstner, J. Chem. Theory Comput., 2016, 12, 4209–4221 CrossRef CAS PubMed.
- J. J. Kranz, M. Elstner, B. Aradi, T. Frauenheim, V. Lutsker, A. D. Garcia and T. A. Niehaus, J. Chem. Theory Comput., 2017, 13, 1737–1747 CrossRef CAS PubMed.
- V. Lutsker, B. Aradi and T. A. Niehaus, J. Chem. Phys., 2015, 143, 184107 CrossRef CAS PubMed.
- V. Q. Vuong, J. Akkarapattiakal Kuriappan, M. Kubillus, J. J. Kranz, T. Mast, T. A. Niehaus, S. Irle and M. Elstner, J. Chem. Theory Comput., 2018, 14, 115–125 CrossRef CAS PubMed.
- N. Schieschke, B. M. Bold, P. M. Dohmen, D. Wehl, M. Hoffmann, A. Dreuw, M. Elstner and S. Höfener, J. Comput. Chem., 2021, 42, 1402–1418 CrossRef CAS PubMed.
- M. Sokolov, B. M. Bold, J. J. Kranz, S. Höfener, T. A. Niehaus and M. Elstner, J. Chem. Theory Comput., 2021, 17, 2266–2282 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- H. E. Kato, F. Zhang, O. Yizhar, C. Ramakrishnan, T. Nishizawa, K. Hirata, J. Ito, Y. Aita, T. Tsukazaki, S. Hayashi, P. Hegemann, A. D. Maturana, R. Ishitani, K. Deisseroth and O. Nureki, Nature, 2012, 482, 369–374 CrossRef CAS PubMed.
- O. Volkov, K. Kovalev, V. Polovinkin, V. Borshchevskiy, C. Bamann, R. Astashkin, E. Marin, A. Popov, T. Balandin, D. Willbold, G. Büldt, E. Bamberg and V. Gordeliy, Science, 2017, 358, eaan8862 CrossRef PubMed.
- H. Li, E. Govorunova, O. Sineshchekov and J. Spudich, Biophys. J., 2014, 106, 1607–1617 CrossRef CAS PubMed.
- K. Gerwert, E. Freier and S. Wolf, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 606–613 CrossRef CAS PubMed.
- D. Herschlag and M. M. Pinney, Biochemistry, 2018, 57, 3338–3352 CrossRef CAS PubMed.
- E. Podoliak, G. H. U. Lamm, E. Marin, A. V. Schellbach, D. A. Fedotov, A. Stetsenko, M. Asido, N. Maliar, G. Bourenkov, T. Balandin, C. Baeken, R. Astashkin, T. R. Schneider, A. Bateman, J. Wachtveitl, I. Schapiro, V. Busskamp, A. Guskov, V. Gordeliy, A. Alekseev and K. Kovalev, Nat. Commun., 2024, 15, 3119 CrossRef CAS PubMed.
- Z. Pang, M. Sokolov, T. Kubař and M. Elstner, Phys. Chem. Chem. Phys., 2022, 24, 2441–2453 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00762c |
| This journal is © the Owner Societies 2025 |