
Enhanced cycling stability and rate capability of Bi2O3-coated Li1.2Mn0.54Ni0.13Co0.13O2 cathode materials for lithium-ion batteries
Lin Zhou,
Huali Wu,
Mijie Tian,
Qiaoji Zheng,
Chenggang Xu and
Dunmin Lin*
College of Chemistry and Materials Science, Sichuan Normal University, Chengdu 610066, China. E-mail: ddmd222@sicnu.edu.cn; Fax: +86 28 84760802; Tel: +86 28 84760802
First published on 18th July 2016
Abstract
Li-rich layered oxide of Bi2O3-coated Li1.2Mn0.54Ni0.13Co0.13O2 was fabricated via a combined method of sol–gel and wet chemical coating processes. The crystal structure of Li1.2Mn0.54Ni0.13Co0.13O2 has no obvious change after the coating of 2 wt% Bi2O3. Bi2O3 is homogenously coated on the surface of Li1.2Mn0.54Ni0.13Co0.13O2 particles. The Bi2O3-coated Li1.2Mn0.54Ni0.13Co0.13O2 electrode presents an enhanced cycling stability, better rate performance and lower charge transfer resistance. After the coating of 2 wt% Bi2O3, the initial coulombic efficiency of the material has been improved to 76.9% compared to 72.6% for the bare Li1.2Mn0.54Ni0.13Co0.13O2. After 100 cycles at 0.1C, the Bi2O3-coated Li1.2Mn0.54Ni0.13Co0.13O2 electrode retains a discharge capacity of 182.9 mA h g−1 with capacity retention of 73.5%, which are much higher than those of Li1.2Mn0.54Ni0.13Co0.13O2 (146.9 mA h g−1 and 58.8%). In addition, the Bi2O3-coated Li1.2Mn0.54Ni0.13Co0.13O2 electrode exhibits better rate capability with discharge capacities of 155.8, 124.8 and 89.2 mA h g−1 at 1, 2 and 5C, respectively, superior to that of Li1.2Mn0.54Ni0.13Co0.13O2. The enhanced cycling stability and rate capability should be ascribed to the existence of Bi2O3 on the surface of the active material, which can efficiently restrain the side reactions between the electrode and electrolyte, stabilize the structure of Li1.2Mn0.54Ni0.13Co0.13O2 and improve the lithium ion diffusion.
1. Introduction
Rechargeable lithium-ion batteries (LIBs) are widely-used as a clean power source for various luggable electric devices including cellphones, laptops and electric tools due to their high energy density.1–3 Recently, they are also expanding rapidly in the commercial application of hybrid electric vehicles (HEVs) and electric vehicles (EVs).4 As is known, the electrochemical performances of LIBs rely heavily on cathode materials. However, the traditional commercial cathode materials (e.g., layered LiCoO2, olivine LiFeO4 and spinel LiMn2O4) deliver relatively low specific capacities (100–160 mA h g−1) and thus cannot fulfill the increasing demands for power density and energy density of HEVs and EVs.5 Therefore, there is an urgent demand to exploit a new cathode material with high specific capacity, great cycling stability and rate capability for new generation rechargeable LIBs.Recently, Li-rich layered cathode materials xLi2MnO3·(1 − x)LiMO2 (0 < x < 1, M = Mn, Ni, Fe, Mn0.33Ni0.33Co0.33…) have attracted considerable attention because of their higher discharge capacities (>200 mA h g−1), higher working potential (4.6–4.8 V vs. Li/Li+), lower cost, less toxicity and higher safety than the traditional cathode materials.6–9 However, these materials also suffer from several problems, for instance, their large irreversible capacity loss (ICL) in the 1st cycle, inferior rate performance and fast capacity decline, limiting the actual adhibition of the Li-rich cathodes.6,10,11 The large ICL is ascribed to the removal of Li2O from Li2MnO3 phase accompanies with the removing of oxygen-ion vacancies in the 1st charging process, which results in the decrease of intercalation active sites for Li+ in succeeding discharging processes;12,13 the inferior rate capability is related to the poor electronic conductivity and low lithium ion diffusion coefficient of Li2MnO3 component;6,14,15 and the structure evolution from layered to spinel during the further cycle causes the fast capacity decline of Li-rich cathodes.12,16,17 Many investigation have been carried out to solve these drawbacks of the Li-rich layered cathodes, including structure and morphology controlling,18,19 mild acidic treatment,20–22 cationic substitution23–26 and the formation of composite cathode with lithium-free insertion hosts materials.27,28 Among these investigations, surface modification has been demonstrated to be an available method to enhance the cycle stability and rate capability of cathodes. Numerous previous studies have shown that coating layer can protect the cathode materials against the attacking of hydrofluoric acid by separating them from electrolyte, suppress the oxygen loss by reducing the activity of oxygen ions and retain the oxygen-ion vacancies.29–31 Various compounds, such as metallic phosphates,32,33 oxides,34–36 fluorides and so on,11,37–39 have been used as coating layer materials.
Bi2O3 has been applied to electric ceramics, solid electrolytes and photoelectric materials owing to its special physical properties and crystal structure. A schematic of monoclinic Bi2O3 is shown in Fig. 1. The pure Bi2O3 is a monoclinic phase with 25% oxygen vacancies at room temperature, which suggests that Bi2O3 may act as good oxygen storage material. From Fig. 1, Bi2O3 possesses an ordered defect fluorite structure with Bi layers paralleling to (100) plane and arranged alternately with O layers.40 In general, this structure may provide fast diffusion paths for lithium ions. In addition, it has been noted that Bi2O3 has the highest ion conductivity among all oxide ion conductors (1 × 10−2 S cm−1 at 500 °C and 1 S cm−1 at 750 °C),41 which may improve the rate capability of cathode materials. Bi2O3 has been regarded as an effective surface coating material for the enhancement in the columbic efficiency, cyclability and rate capability of lithium spinel oxides (e.g., LiNi0.5Mn1.5−xTixO4, LiMn1.5Ni0.42Zn0.08O4 and LiMn1.42Ni0.42Co0.16O4).42–44 T. Noguchi et al. found that the cycle behavior and storage property of LiNi0.5Mn1.5−xTixO4 are improved by Bi oxide surface treatment (85% and 70% capacity retentions after 500 cycles at 20 °C and 40 °C, respectively).42 J. Liu et al. have employed Bi2O3, Al2O3 and ZnO as coating materials for LiMn1.5Ni0.42Zn0.08O4 and found that Bi2O3-modified cathode exhibits better rate capability than the uncoated sample and the rate capability degrades in the following order: Bi2O3-coated sample > Al2O3-coated sample > ZnO-coated sample > pristine sample.43 In addition, D. Bhuvaneswari et al. reported that Bi2O3 coated LiNi0.4Mn0.4Co0.2O2 electrode delivers much higher discharge capacity (175 mA h g−1 with 89% capacity retention) after 100 cycles compared with the bare sample (149 mA h g−1 and 66%) and the extent of enhancement in electrochemical performance decreases in the following order: Bi2O3-coated sample > Al2O3-coated sample > In2O3-coated sample > pristine sample.45 Obviously, Bi2O3 can greatly enhance the cycle performance and rate capability of cathodes for LIBs. It has be also noted that as a classic Li-rich layered cathodes, Li1.2Mn0.54Ni0.13Co0.13O2 (or LMNC, for short, equivalently 0.5Li2MnO3·0.5LiMn0.33Ni0.33Co0.33O2) has been extensively studied25,29 due to its large specific capacity.46 But the LMNC cathode exhibits a large ICL in 1st cycle, inferior rate capability and fast capacity decline. Based on the above mentioned, it can be reasonably anticipated that Bi2O3 can be used as a promising surface modifier for LMNC cathode material to improve its cyclability and rate capability. For surface modification of cathodes, it has been frequently shown that too little amount of coating cannot effectively improve the electrochemical performance,36 while excess coating will decrease the capacity of cathode material due to the increased lithium diffusion path and lowered electronic tunneling rate.47 Many researchers have reported that 2 wt% is the appropriate coating ratio for Li-rich cathode.38,39 Therefore, 2 wt% Bi2O3 is used to coat LMNC as a suitable coating amount.
 | ||
| Fig. 1 The schematic of monoclinic Bi2O3. | ||
In this work, Li-rich cathode material LMNC was prepared via a sol–gel method and then Bi2O3-modified LMNC was prepared using wet chemical coating process. The structure, morphology and electrochemical properties of LMNC and Bi2O3-coated LMNC samples were investigated in detail. Our results show that Bi2O3-coated LMNC cathode material exhibits higher capacity retention, better cyclic performance and rate capability than the uncoated material.
2. Experimental
2.1. Sample preparation
LMNC powders were fabricated via a sol–gel method. Stoichiometric amounts of Mn(CH3COO)2·4H2O (99%), Ni(CH3COO)2·4H2O (98%) and Co(CH3COO)2·4H2O (99.5%) were dissolved into distill water. Afterwards, the mixed solution of C6H8O7·H2O (99.5%) and Li(CH3COO)2·2H2O (99%, 5% excess) with a molar ratio about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were slowly dripped into the former solution under continually stirring. The pH value of the resulting mixtures was adjusted to ∼7.5 by adding NH3·H2O (25–28%). The mixture was heated at 80 °C to obtain purple gels. The prepared gels were heat treated at 120 °C for 12 h in a vacuum oven, sequentially preheated in muffle furnace at 450 °C for 5 h. The resulting powders were pressed into disk samples and sintered in air at 900 °C for 10 h to get the LMNC powders.
:1 were slowly dripped into the former solution under continually stirring. The pH value of the resulting mixtures was adjusted to ∼7.5 by adding NH3·H2O (25–28%). The mixture was heated at 80 °C to obtain purple gels. The prepared gels were heat treated at 120 °C for 12 h in a vacuum oven, sequentially preheated in muffle furnace at 450 °C for 5 h. The resulting powders were pressed into disk samples and sintered in air at 900 °C for 10 h to get the LMNC powders.
Bi2O3-coated LMNC (LMNC-BO) was synthesized via a wet chemical coating process. 2 wt% Bi2O3 was coated on the surface of LMNC. Stoichiometric amounts of Bi(NO3)3·5H2O (99%) was dissolved in distill water. The prepared LMNC powders were dispersed in the Bi(NO3)3·5H2O solution. Ammonium hydroxide was dripped into the mixture. Afterward, the mixture was stirred at 80 °C to evaporate water. The resulting product was sintered at 500 °C for 5 h in air to obtain the LMNC-BO powders.
2.2. Sample characterizations
The crystal structure of LMNC and LMNC-BO materials were measured via X-ray diffraction (XRD) analysis with Cu Kα radiation (Smart Lab, Rigaku) at a scan rate of 1° min−1 between 10° and 80°. The microstructures of LMNC and LMNC-BO were examined using a transmission electron microscopy (TEM, JEM2100). The morphology, element composition and distribution were observed by a field-emission scanning electron microscope (FE-SEM, JSM-7500) equipped with an energy dispersive spectrum X-ray detector (EDS). The element composition of samples was measured by an inductively coupled plasma emission spectrometry (ICP-AES, Optima 7000 DV, Pe).2.3. Electrochemical measurements
The cell cathodes were fabricated by blending active material, conductive agents (acetylene black) and PVDF binder (at a mass ratio of 8:1:1) into NMP solvent with thoroughly stirring to get a homogenous slurry. The black and ropy slurry were coated onto Al current collectors and heat treated at 120 °C for 12 h. CR-2025 coin-type half-cells were assembled and sealed in an Ar-filled glove box (MB-Labstar, Germany). The electrolyte consists of 1 mol L−1 LiPF6 dissolved in ethylene carbonate–dimethyl carbonate (at a volume ratio of 1:1). Metallic lithium plates were used as reference electrodes and Celgard 2400 were the separators. All the cells were placed for 12 h before electrochemical testing to make the electrolyte fully permeate active materials. The galvanostatic charge and discharge tests were measured using cell test system (LAND CT-2001A, Wuhan, China) between 2.0 and 4.8 V (vs. Li/Li+) at different rates at room temperature. The cyclic voltammograms (CV) for the cells were recorded on an electrochemical station (CHI660E, Shanghai, China) at a scanning rate of 0.1 mV s−1 in the voltage range of 2.0–4.8 V. Electrochemical impedance spectroscopy (EIS) measurements of the cells were carried out after 100 cycles at 0.1C and monitored at the electrochemical station in the frequency range of 0.01–100000 Hz at 5 mV voltage amplitude. Zview2 software was used for the simulation of the EIS results.
3. Result and discussion
Fig. 2 presents the XRD patterns of the LMNC and LMNC-BO materials. Both samples exhibit typical XRD diffraction patterns of Li-rich layered oxides, indicating that Bi2O3 coating layer has no obvious effect on the crystalline structure of LMNC. Similar to previous investigations,6,10,11,20,29 it can be seen that for LMNC and LMNC-BO materials, the strong diffraction peaks indicate a hexagonal layered α-NaFeO2 structure (space group symmetry: R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), and the extra diffraction peaks between 20° and 25° are corresponding to the LiMn6 cation arrangement in transition metal layer of monoclinic Li2MnO3 (space group symmetry: C2/m). Both the materials can prove to be a well-layered structure by the completely splitting of (006)/(012) and (118)/(110) peaks.23,39 In general, the I(003)/I(104) intensity ratio (R) is a crucial pointing of the cation mixing between Ni2+ and Li+ due to their similar ionic radius.36 The value of R for the bare LMNC is 1.17 (<1.2), indicating serious cation mixing. After coating with Bi2O3, the value of R increases to 1.68 (>1.2), implying a much lower degree of cation mixing which is favorable to the enhancement of electrochemical cycling stability.48 As shown in Fig. 2, no diffraction peak of Bi2O3 is observed, which may be due to the excessively low content of Bi2O3 on the surface of LMNC.
m), and the extra diffraction peaks between 20° and 25° are corresponding to the LiMn6 cation arrangement in transition metal layer of monoclinic Li2MnO3 (space group symmetry: C2/m). Both the materials can prove to be a well-layered structure by the completely splitting of (006)/(012) and (118)/(110) peaks.23,39 In general, the I(003)/I(104) intensity ratio (R) is a crucial pointing of the cation mixing between Ni2+ and Li+ due to their similar ionic radius.36 The value of R for the bare LMNC is 1.17 (<1.2), indicating serious cation mixing. After coating with Bi2O3, the value of R increases to 1.68 (>1.2), implying a much lower degree of cation mixing which is favorable to the enhancement of electrochemical cycling stability.48 As shown in Fig. 2, no diffraction peak of Bi2O3 is observed, which may be due to the excessively low content of Bi2O3 on the surface of LMNC.
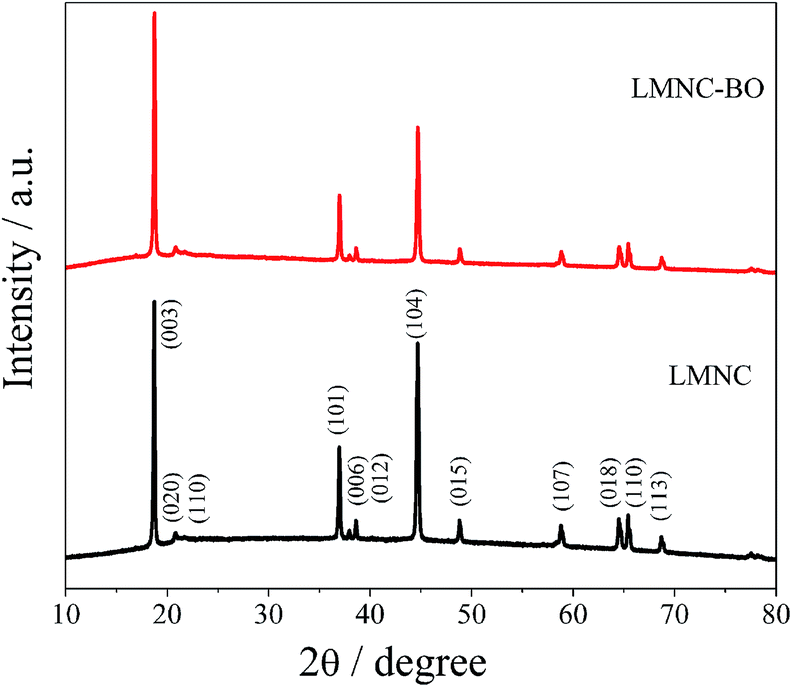 | ||
| Fig. 2 XRD patterns of LMNC and LMNC-BO. | ||
The element composition of the LMNC and LMNC-BO samples is computed by ICP-AES. The element content of LMNC and LMNC-BO samples is listed in Table 1. Both samples present a close element composition to the target stoichiometry of Li1.2Mn0.54Ni0.13Co0.13O2, except for Li element. The Li content is a little bit more than the target stoichiometry. This is caused by the excess 5% of Li source added for fear of the evaporation of Li at high temperature.
| Sample | Li | Mn | Ni | Co | Bi |
|---|---|---|---|---|---|
| LMNC | 1.321 | 0.540 | 0.130 | 0.129 | 0 |
| LMNC-BO | 1.251 | 0.540 | 0.130 | 0.129 | 0.007 |
The SEM images and EDS spectrums of the LMNC and LMNC-BO samples are shown in Fig. 3. As shown in Fig. 3a and b, both of LMNC and LMNC-BO are made of small particles with the size range of 100–400 nm. The pristine LMNC particles have a clear edge with obvious agglomeration. After Bi2O3 coating, there is a more serious agglomeration phenomenon in the LMNC-BO sample which may facilitates the charge transport property.49 From the EDS spectrums in Fig. 3c and d, the characteristic peaks of O, Mn, Ni and Co can be observed in both of the samples, while the peak of Bi can be only observed in Fig. 3d, indicating the presence of Bi in the LMNC-BO. Fig. 4 gives the EDS elemental mapping images of Ni, Mn, Co, O and Bi in the obtained LMNC-BO particles. Elements of Ni, Mn, Co and O distribute uniformly in LMNC-BO samples. As can be seen, the homogeneous distribution of Bi element can confirm the successful coating of Bi2O3 on the surface of LMNC-BO.
 | ||
| Fig. 3 SEM images of (a) LMNC and (b) LMNC-BO; EDS spectrums of (c) LMNC and (d) LMNC-BO. | ||
 | ||
| Fig. 4 EDS elemental mapping images of LMNC-BO. | ||
To further observe the morphology and microstructure of the particles, the TEM, HRTEM and fast Fourier transform pattern (FFT) images of the LMNC and LMNC-BO particles are shown in Fig. 5. From Fig. 5a and b, both of the samples consist of small quasi-sphere particles. It can be seen from Fig. 5c that the LMNC particles have a smooth edge with clear interference fringes extending to margin. A thin Bi2O3 layer is observed on the particle edge of LMNC-BO (Fig. 5d), which is consistent with the EDS spectrum and EDS elemental mapping results and further proves the existence of Bi2O3. The interference fringe spacings are about 0.48 nm and 0.43 nm for the LMNC and the LMNC-BO particles, respectively, which correspond to the interplanar distance of the (003) plane of the layered Rm phase and the (020) plane of the C/2m phase, respectively, which coincide with the results of insert FFTs.
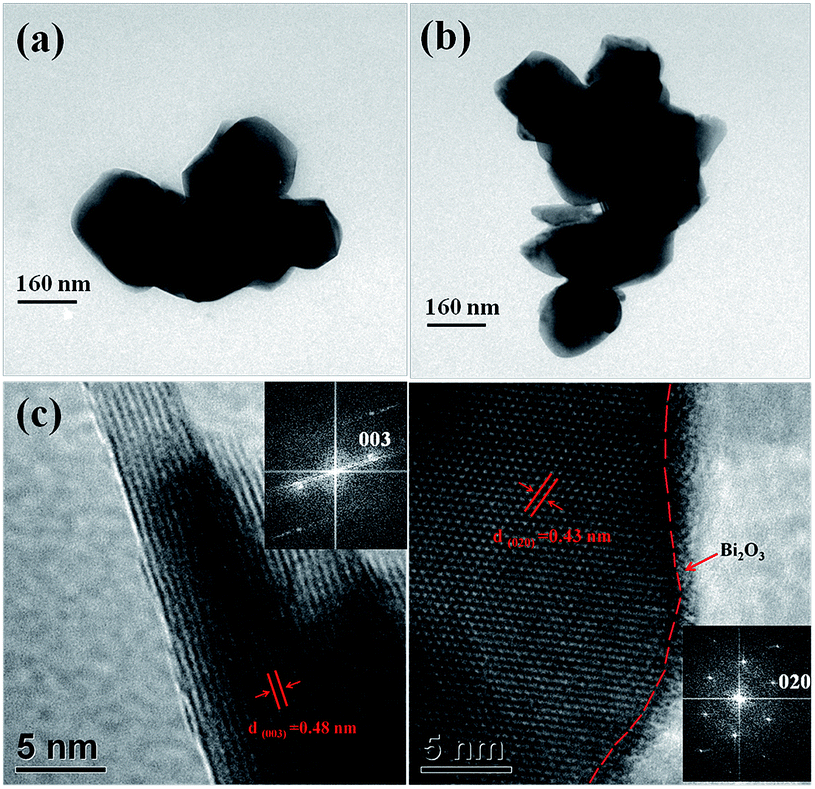 | ||
| Fig. 5 TEM images of (a) LMNC and (b) LMNC-BO; HRTEM images of (c) LMNC and (d) LMNC-BO (the insert is the corresponding fast Fourier transform pattern). | ||
The initial charge/discharge curves of LMNC and LMNC-BO electrodes at 0.1C (1C = 250 mA h g−1) are shown in Fig. 6. As can be seen, there is a long plateau at ∼4.5 V in the 1st charge profile for both of the samples, which is connected with the irreversible elimination of Li2O from the structure.8 However, after the coating of Bi2O3, the LMNC-BO electrode displays a shorter plateau than the pristine sample at 4.5 V, which suggests that Bi2O3 layer could restrain the migration of oxygen-ion vacancies and decrease the irreversible removal of oxygen, resulting in lower charge capacity during the initial charge process.30,39,50 The charge/discharge capacities are 344.5/250 and 323.6/249 mA h g−1 for the LMNC and LMNC-BO electrodes in the 1st cycle, respectively, and the corresponding initial coulombic efficiency are 72.6% and 76.9%, respectively. Although the initial discharge capacity has not been obviously improved, the LMNC-BO electrode exhibits a much lower ICL of 74.6 mA h g−1 than that for bare LMNC (94.5 mA h g−1).
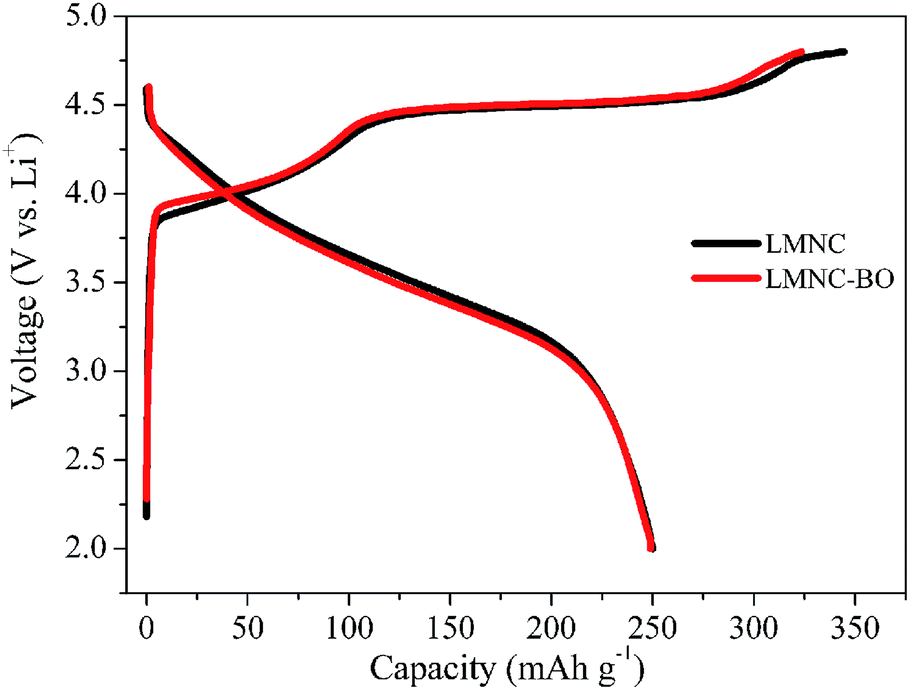 | ||
| Fig. 6 Initial charge/discharge curves of LMNC and LMNC-BO electrodes at 0.1C between 2.0 and 4.8 V. | ||
Fig. 7 exhibits the cycling performance of LMNC and LMNC-BO electrodes at 0.1C. The discharge capacity of LMNC electrode decreases rapidly from 250 to 167 mA h g−1 in the initial 20 cycles because of the large structural rearrangement.51 After 100 cycles, the discharge capacity of LMNC electrode fades to 146.9 mA h g−1, showing a quite low capacity retention (58.8%). The cyclic stability of LMNC has been improved after surface coating with Bi2O3 layer. Although the discharge capacity of LMNC-BO electrode also fades quickly from 249 to 215.5 mA h g−1 in the initial 10 cycles, it always shows higher values than that for LMNC in the subsequent cycles. The LMNC-BO electrode still presents the specific capacity of 182.9 mA h g−1 and retains 73.5% of the initial discharge capacity in the 100th cycle. For the present materials, the Bi2O3 layer can hinder active material from directly contacting electrolyte, and thus protects the active materials from the erosion in electrolyte and stabilize the structure of active material. This leads to the improvement in the cycling stability of the material.
 | ||
| Fig. 7 Cycling performance of LMNC and LMNC-BO electrodes at 0.1C between 2.0 and 4.8 V. | ||
The charge/discharge curves for LMNC and LMNC-BO cathodes in the 2nd, 10th, 50th and 100th cycles are presented in Fig. 8. The discharge capacities for LMNC and LMNC-BO electrodes fade quickly before 50 cycles, and the LMNC-BO electrode delivers much higher discharge capacity after 100 cycles compare with the LMNC electrode. For both of the electrodes, the discharge voltage plateaus shift to lower voltage plateau with the increase of cycle times, which implies the increasing of polarization upon cycling. As the indication of the black arrow in Fig. 8a, the midpoint voltage of the LMNC cathode decreases from 3.45 V for the 2nd cycle to 2.91 V for the 100th cycle, while that of the LMNC-BO electrode fades from 3.47 V to 2.94 V (Fig. 8b). Obviously, the discharge capacity fading and voltage decay for the LMNC electrode can be reduced by surface modification with Bi2O3.
 | ||
| Fig. 8 Charge/discharge curves for (a) LMNC and (b) LMNC-BO electrodes in the 2nd, 10th, 50th and 100th cycles at 0.1C. | ||
Rate performance of the LMNC and LMNC-BO electrodes at various rates are shown in Fig. 9. It can be seen that LMNC electrode has poor rate performance. Although the bare LMNC and LMNC-BO electrodes present close discharge capacities at 0.1C and deliver the maximum capacities of 256.8 and 257.1 mA h g−1, respectively, the difference in the capacities between the two electrodes increases quickly as the rate value increases. For the LMNC electrode, serious capacity fading happens at high rates, and it delivers the maximum discharge capacities of 207.2, 154.3, 103.4, 65.9 and 24 mA h g−1 at 0.2, 0.5, 1, 2 and 5C, respectively. Unlike the LMNC, the LMNC-BO electrode yields the maximum discharge capacities of 224.2, 189.1, 155.8, 124.8 and 89.2 mA h g−1 at 0.2, 0.5, 1, 2 and 5C, respectively. The discharge capacities at high rates for LMNC-BO electrode are much higher than that for LMNC electrode. As can be clearly seen from Fig. 9, the rate performance of LMNC-BO electrode is obviously enhanced after Bi2O3 coating.
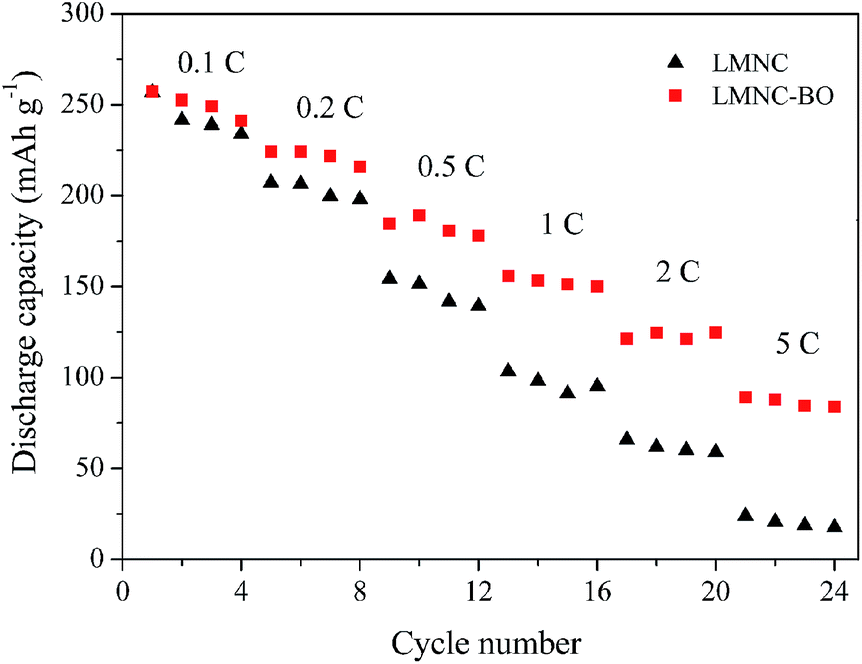 | ||
| Fig. 9 Rate performance of the LMNC and LMNC-BO electrodes at various rates between 2.0 and 4.8 V. | ||
The cycle voltammograms (CV) of the LMNC and LMNC-BO electrodes at 1st, 2nd and 5th cycles are shown in Fig. 10. As can be seen, the LMNC electrode exhibits characteristic cycle voltammograms of Li-rich oxides cathodes (Fig. 10a), but the LMNC-BO electrode presents sharper redox peaks (Fig. 10b). From Fig. 10a, the LMNC sample presents two anodic peaks in the 1st charging process, corresponding to the two potential plateaus in the initial charge profile. The first anodic peak located at 4.1 V is related to the oxidation of Ni2+/Co3+ to Ni4+/Co4+, while the second anodic peak at 4.6 V is connected with the elimination of Li2O from Li2MnO3. However, the first anodic peak for the LMNC-BO electrode moves to higher voltage region (4.2 V) and the second anodic peak moves to lower voltage region (4.5 V), resulting in the overlap of 4.2 V and 4.5 V anodic peaks and the indistinctness of 4.2 V anodic peak (in the black dotted box). This phenomenon suggests that the Bi2O3 coating can lower the polarization at high voltage region that is favorable to the extraction/insertion of lithium ions.52 Moreover, the oxygen loss and the corrosion of active material in the electrolyte are suppressed to some extent after Bi2O3 coating, leading to the enhancement of initial coulombic efficiency and variation of anodic peak in the 1st cycle. However, the 4.6 V anodic peak disappears in the followed cycles for both of the electrodes. The subsequent redox processes are similar for the two samples. In the initial cathodic process, two cathodic peaks at ∼3.8 V and ∼4.3 V can be observed, relating to the reduction of Ni4+ → Ni2+ and Co4+ → Co3+, respectively. Moreover, in the 2nd and 5th discharge process, a new cathodic peak appears after the activation of Li2MnO3 in the 1st cycle at ∼3.25 V related to the reduction of Mn4+/Mn3+.38 The initial anodic process for the samples is in accordance with the analysis of the initial charge process, implying that Bi2O3 layer can prevent active materials from the erosion of electrolyte and stabilize the structure of the active material.
 | ||
| Fig. 10 Cycle voltammograms of (a) LMNC and (b) LMNC-BO electrodes at 1st, 2nd and 5th cycles between 2.0 and 4.8 V at a scan rate of 0.1 mV s−1. | ||
EIS analysis is carried out to learn about the intrinsic influence of Bi2O3 layer on the electrochemical properties for LMNC. Fig. 11 exhibits the EIS patterns of the LMNC and LMNC-BO electrodes after 100 cycles and the used equivalent circuit models. As shown in Fig. 11, the Nyquist plot is consisted of two semicircles in high-mid-frequency region and a slope in low-frequency region. The first semicircle is associated with the lithium ion diffusion through the surface layer (including solid electrolyte interface (SEI) film and coating layer) and another semicircle is related to the charge transfer resistance in the interface of electrode/electrolyte, while the slope is assigned to the Warburg impedance which is correlated to the lithium ion diffusion process in electrode materials.53 The simulated electrochemical parameters from EIS spectra are shown in Table 2. Rs, Rsl and Rct represent the internal resistance of the cell, the surface layer resistance and the charge transfer resistance, respectively. As can be seen, the Rs and Rsl values are very small for both of the electrodes after 100 cycles, indicating that the two investigated materials have slight ohmic polarization.54,55 Obviously, the uncoated LMNC electrode shows a Rct value larger than 10000 Ω. As for the LMNC-BO electrode, the Rct value is much smaller than that of LMNC electrode, which clearly reveals that the Bi2O3 layer can reduce the charge transfer resistance. The lithium ion diffusion coefficient was calculated from EIS spectra using the method reported elsewhere.56 The lithium ion diffusion coefficient of LMNC-BO electrode (2.8719 × 10−13 cm2 s−1) is higher than that of LMNC electrode (2.0727 × 10−14 cm2 s−1), implying that the Bi2O3 coating can greatly enhance lithium ion diffusion. That's because that Bi2O3 has high ion conductivity and the monoclinic structure can provide fast lithium ion diffusion paths. Therefore, the enhancement of rate performance can be due to the huge reduction of charge transfer resistance and improved lithium ion diffusion.
 | ||
| Fig. 11 (a) Electrochemical impedance spectroscopy (EIS) of LMNC and LMNC-BO electrodes after 100 cycles (the insert is the enlarged view in the high frequency region), (b) the equivalent circuit for the impedance spectra. | ||
| Sample | Rs (Ω) | Rsl (Ω) | Rct (Ω) |
|---|---|---|---|
| LMNC | 5 | 50 | 15818 |
| LMNC-BO | 9 | 93 | 933 |
To further comprehend the influence of coating on the structure of LMNC material, the ex situ XRD analysis has been carried out. Fig. 12 presents the ex situ XRD patterns of LMNC and LMNC-BO electrodes at different discharging states. Compared with Fig. 2, the extra diffraction peaks between 20° and 25° for both samples disappear at discharging state, implying that the monoclinic structure is destroyed during charging and discharging processes.57 It can be clearly observed that the (003) and (101) peaks of LMNC electrode become gradually weak and broad with the process of discharging (Fig. 12a), while there is no obvious difference in the (003) and (101) peaks of LMNC-BO electrode (Fig. 12b). These suggest that the structural stability of LMNC material is improved by Bi2O3 coating; as a result, improved cycle performance is obtained in Bi2O3-doped material.
 | ||
| Fig. 12 Ex situ XRD patterns of (a) LMNC and (b) LMNC-BO electrodes at different discharging states. | ||
4. Conclusions
Bi2O3-coated LMNC cathode material is prepared via a combined method of sol–gel and wet chemical processes. Compared with LMNC, the Bi2O3-coated LMNC cathode exhibits lower initial irreversible capacity loss, better cycling stability and rate performance. After surface coating with Bi2O3, the initial coulombic efficiency increases from 72.6% to 76.9%. The capacity retention of the bare sample is only 58.8%, while for Bi2O3-coated LMNC electrode, the significantly improved value of 73.5% is exhibited. The LMNC sample only presents a discharge capacity of 24 mA h g−1 at 5C, but the Bi2O3-coated sample delivers a much higher value of 89.2 mA h g−1. The improved initial coulombic efficiency and cyclic stability for Bi2O3-coated LMNC are due to the retention of oxygen-ion vacancies, the suppression of side reactions between cathode and electrolyte and the improved structural stability of active material. The enhanced rate performance is attributed to the reduced charge transfer resistance and improved lithium ion diffusion. Therefore, surface modification with Bi2O3 may be a promising way to improve the electrochemical performances of Li-rich layered cathode materials.Acknowledgements
This work was supported by large precision instrument projects of Sichuan Normal University (DJ 2015-36 and DJ 2015-44).Notes and references
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- Y. Nishi, Chem. Rec., 2001, 1, 406–413 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- Q. Xie, C. Zhao, Z. Hu, Q. Huang, C. Chen and K. Liu, RSC Adv., 2015, 5, 77324–77331 RSC.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., R, 2012, 73, 51–65 CrossRef CAS.
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- A. Ito, D. Li, Y. Ohsawa and Y. Sato, J. Power Sources, 2008, 183, 344–346 CrossRef CAS.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- Y. D. Zhang, Y. Li, X. H. Xia, X. L. Wang, C. D. Gu and J. P. Tu, Sci. China: Technol. Sci., 2015, 58, 1809–1828 CrossRef CAS.
- D. Y. W. Yu, K. Yanagida and H. Nakamura, J. Electrochem. Soc., 2010, 157, A1177–A1182 CrossRef CAS.
- Y. K. Sun, M. J. Lee, C. S. Yoon, J. Hassoun, K. Amine and B. Scrosati, Adv. Mater., 2012, 24, 1192–1196 CrossRef CAS PubMed.
- Z. H. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef CAS.
- C. S. Johnson, J.-S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, J. Electrochem. Soc., 2004, 6, 1085–1091 CAS.
- H. Yu, Y. Wang, D. Asakura, E. Hosono, T. Zhang and H. Zhou, RSC Adv., 2012, 2, 8797–8807 RSC.
- H. Z. Zhang, Q. Q. Qiao, G. R. Li, S. H. Ye and X. P. Gao, J. Mater. Chem., 2012, 22, 13104–13109 RSC.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- S. H. Kang, P. Kempgens, S. Greenbaum, A. J. Kropf, K. Amine and M. M. Thackeray, J. Am. Chem. Soc., 2007, 17, 2069–2077 CAS.
- D. Wang, I. Belharouak, L. H. Ortega, X. Zhang, R. Xu, D. Zhou, G. Zhou and K. Amine, J. Power Sources, 2015, 274, 451–457 CrossRef CAS.
- S. J. Shi, Z. R. Lou, T. F. Xia, X. L. Wang, C. D. Gu and J. P. Tu, J. Power Sources, 2014, 257, 198–204 CrossRef CAS.
- G. Xu, J. Li, Q. Xue, X. Ren, G. Yan, X. Wang and F. Kang, J. Power Sources, 2014, 248, 894–899 CrossRef CAS.
- S. H. Kang, C. S. Johnson, J. T. Vaughey, K. Amine and M. M. Thackeray, J. Electrochem. Soc., 2006, 153, A1186–A1192 CrossRef CAS.
- S. H. Kang and M. M. Thackeray, J. Electrochem. Soc., 2008, 155, A269–A275 CrossRef CAS.
- W. He, D. Yuan, J. Qian, X. Ai, H. Yang and Y. Cao, J. Mater. Chem. A, 2013, 1, 11397–11403 CAS.
- Y. L. Wang, X. Huang, F. Li, J. S. Cao and S. H. Ye, RSC Adv., 2015, 5, 49651–49656 RSC.
- Q. Li, G. Li, C. Fu, D. Luo, J. Fan and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 10330–10341 CAS.
- X. Jin, Q. Xu, H. Liu, X. Yuan and Y. Xia, Electrochim. Acta, 2014, 136, 19–26 CrossRef CAS.
- F. Wu, Z. Wang, Y. Su, N. Yan, L. Bao and S. Chen, J. Power Sources, 2014, 247, 20–25 CrossRef CAS.
- K. L. Sun, C. Peng, Z. H. Li, Q. C. Xiao, G. T. Lei, Q. Z. Xiao, Y. H. Ding and Z. H. Hu, RSC Adv., 2016, 6, 28729–28736 RSC.
- L. N. Cong, X. G. Gao, S. C. Ma, X. Guo, Y. P. Zeng, L. H. Tai, R. S. Wang, H. M. Xie and L. Q. Sun, Electrochim. Acta, 2014, 115, 399–406 CrossRef CAS.
- G. Xu, J. Li, Q. Xue, Y. Dai, H. Zhou, X. Wang and F. Kang, Electrochim. Acta, 2014, 117, 41–47 CrossRef CAS.
- Y. Wu and A. Manthiram, Solid State Ionics, 2009, 180, 50–56 CrossRef CAS.
- B. Liu, Q. Zhang, S. He, Y. Sato, J. Zheng and D. Li, Electrochim. Acta, 2011, 56, 6748–6751 CrossRef CAS.
- Q. Q. Qiao, H. Z. Zhang, G. R. Li, S. H. Ye, C. W. Wang and X. P. Gao, J. Mater. Chem. A, 2013, 1, 5262–5268 CAS.
- S. J. Shi, J. P. Tu, Y. Y. Tang, X. Y. Liu, Y. Q. Zhang, X. L. Wang and C. D. Gu, Electrochim. Acta, 2013, 88, 671–679 CrossRef CAS.
- S. J. Shi, J. P. Tu, Y. J. Zhang, Y. D. Zhang, X. Y. Zhao, X. L. Wang and C. D. Gu, Electrochim. Acta, 2013, 108, 441–448 CrossRef CAS.
- E. Han, Y. Li, L. Zhu and L. Zhao, Solid State Ionics, 2014, 255, 113–119 CrossRef CAS.
- Q. L. Xie, Z. B. Hu, C. H. Zhao, S. R. Zhang and K. Y. Liu, RSC Adv., 2015, 5, 50859–50864 RSC.
- C. Lu, H. Wu, Y. Zhang, H. Liu, B. Chen, N. Wu and S. Wang, J. Power Sources, 2014, 267, 682–691 CrossRef CAS.
- X. Liu, J. Liu, T. Huang and A. Yu, Electrochim. Acta, 2013, 109, 52–58 CrossRef CAS.
- P. Shuk, H.-D. Wiemhöfer, U. Guth, W. Göpel and M. Greenblatt, Solid State Ionics, 1996, 89, 179–196 CrossRef CAS.
- N. X. Jiang, E. D. Wachsman and S. H. Jung, Solid State Ionics, 2002, 150, 347–353 CrossRef CAS.
- T. Noguchi, I. Yamazaki, T. Numata and M. Shirakata, J. Power Sources, 2007, 174, 359–365 CrossRef CAS.
- J. Liu and A. Manthiram, J. Electrochem. Soc., 2009, 156, A66–A72 CrossRef CAS.
- J. Liu and A. Manthiram, Chem. Mater., 2009, 21, 1695–1707 CrossRef CAS.
- D. Bhuvaneswari, G. Babu and N. Kalaiselvi, Electrochim. Acta, 2013, 109, 684–693 CrossRef CAS.
- C. Venkateswara Rao, J. Soler, R. Katiyar, J. Shojan, W. C. West and R. S. Katiyar, J. Phys. Chem. C, 2014, 118, 14133–14141 CAS.
- Y. Y. Sun, F. Li, Q. Q. Qiao, J. S. Cao, Y. L. Wang and S. H. Ye, Electrochim. Acta, 2015, 176, 1464–1475 CrossRef CAS.
- H. Z. Zhang, Q. Q. Qiao, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2014, 2, 7454–7460 CAS.
- J. Shojan, V. R. Chitturi, J. Soler, O. Resto, W. C. West and R. S. Katiyar, J. Power Sources, 2015, 274, 440–450 CrossRef CAS.
- J. Z. Kong, C. L. Wang, X. Qian, G. A. Tai, A. D. Li, D. Wu, H. Li, F. Zhou, C. Yu, Y. Sun, D. Jia and W. P. Tang, Electrochim. Acta, 2015, 174, 542–550 CrossRef CAS.
- M. Jiang, B. Key, Y. S. Meng and C. P. Grey, Chem. Mater., 2009, 21, 2733–2745 CrossRef CAS.
- W. Yuan, H. Z. Zhang, Q. Liu, G. R. Li and X. P. Gao, Electrochim. Acta, 2014, 135, 199–207 CrossRef CAS.
- J. M. Zheng, X. B. Wu and Y. Yang, Electrochim. Acta, 2011, 56, 3071–3078 CrossRef CAS.
- C. Yang, Q. Zhang, W. Ding, J. Zang, M. Lei, M. Zheng and Q. Dong, J. Mater. Chem. A, 2015, 3, 7554–7559 CAS.
- S. Guo, H. Yu, P. Liu, X. Liu, D. Li, M. Chen, M. Ishida and H. Zhou, J. Mater. Chem. A, 2014, 2, 4422–4428 CAS.
- S. Y. Yang, G. Huang, S. J. Hu, X. H. Hou, Y. Y. Huang, M. Yue and G. T. Lei, Mater. Lett., 2014, 118, 8–11 CrossRef CAS.
- Y. X. Wang, K. H. Shang, W. He, X. P. Ai, Y. L. Cao and H. X. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 13014–13021 CAS.
| This journal is © The Royal Society of Chemistry 2016 |