
Suppressing cation segregation on lanthanum-based perovskite oxides to enhance the stability of solid oxide fuel cell cathodes†
Hyunguk Kwona,
Wonyoung Lee*b and
Jeong Woo Han*a
aDepartment of Chemical Engineering, University of Seoul, Seoul 130-743, Republic of Korea. E-mail: jwhan@uos.ac.kr
bSchool of Mechanical Engineering, Sungkyunkwan University, Suwon 440-746, Republic of Korea. E-mail: leewy@skku.edu
First published on 15th July 2016
Abstract
The slow rate of oxygen reduction reaction (ORR) at the cathode side has been considered as a scientific challenge to improve the overall performance of solid oxide fuel cell (SOFC). Unfortunately, dopant cation enrichment on the surface of perovskite-type cathode materials often leads directly to the reduction in activity and stability of ORR. For this reason, we quantitatively assessed the main driving force of dopant segregation on LaBO3(001) surfaces (B = Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni) using density functional theory (DFT) calculations. Based on our findings, the minimization of elastic energy, which is closely related to the size of both A-site and B-site cations, plays an important role on the A-site dopant segregation. The degree of dopant segregation can be controlled by the proper choice of cations contained in LaBO3-type perovskite oxides. We therefore suggest a valuable principle that can be applied to design high performance cathode materials by suppressing the dopant cation enrichment at the surface.
1. Introduction
Solid oxide fuel cells (SOFCs) have attracted wide interest for efficient and clean electric power generation. In spite of many advantages (i.e. high efficiency, low emission, fuel flexibility, and so on), there are still several obstacles that SOFCs should overcome.1,2 In particular, the development of cathode materials with fast and stable oxygen reduction reaction (ORR) kinetics has been a significant challenge to improve the overall SOFC performance.3,4 In order to design highly active cathodes, it is necessary to understand the surface chemistry closely related to the rate of ORR.5–8 However, cation segregation phenomena have significantly influenced the surface chemistry via the changes in both cation concentrations and chemical phases. For example, the degree of Sr enrichment on La0.7Sr0.3MnO3 (LSM) surface can be controlled by temperature and oxygen pressure.9 The segregated Sr often induces the formation of secondary phases (e.g., SrO, SrCO3) on La1−xSrx-based perovskite surfaces,10 which can deteriorate the reactivity and stability of the cathode surfaces. Cai et al. reported that the electrochemical performance of La0.6Sr0.4CoO3−δ (LSC) films is reduced due to the formation of a chemically heterogeneous phase on the surface, but recovered after chemical etching.11 Chen et al. also demonstrated that a Sr excess layer, known to have a large band gap,12,13 suppresses the oxygen exchange reaction by blocking the tunneling of electrons from the bulk phase on the SrTi1−xFexO3−δ (STF) surface.14 Based on these examples, it turns out that a comprehensive understanding of the segregation behavior plays an important role in designing new cathode materials.Many researchers previously have analyzed the segregation behaviors of A-site cation dopants in SOFC cathode candidate materials such as LaSrMnO3,5–9 LaCaMnO3,15 LaSrCoO3,8,11,16 LaSrCoFeO3,17 and LaSrCrMnO3 (ref. 18) perovskites. Nevertheless, each of them has focused on investigating the phenomena at the particular materials or conditions, and thus has not identified a universal driving force that can be generally applied to the various perovskite oxides. Therefore, the development of robust cathode materials with optimal surface cation chemistry has been delayed without the fundamental understating of the segregation phenomena.
In our earlier study,19 both elastic and electrostatic contributions were quantitatively assessed as the main driving forces of cation rearrangements on the surfaces of manganite perovskites using density functional theory (DFT) calculations, and were also proved by sequential surface-sensitive experiments. We successfully demonstrated that the elastic energy of dopants depends on the size difference between the host and dopant cations in the A-site, and therefore (Rdopant − Rhost) can be a descriptor implying a main thermodynamic driving force under a specific electrostatic condition. However, it is not still a perfect descriptor for the elastic energy generated upon doping the A-site. The elastic energy may also be contributed by the properties of B-site cations because when a cation is doped in the A-site, it occupies the 12 coordinate cavities that are created by a BO3 network.20 Jung and Tuller reported that the surface segregation of lattice Sr cation is dependent upon the Fe concentration on the surface of SrTi1−xFexO3−δ (STF) because Fe atoms in the B-site may exert to the high compressive strain onto the Sr atoms in the A-site.21 Therefore, a new type of descriptor which can more reasonably capture the elastic energy originated from both A- and B-site cations must be devised to control the dopant segregation of (La,A)BO3 perovskite surface.
In this paper, we aim to extend aforementioned results19 by quantitatively assessing how B-site cation as well as the A-site cation contributes to the surface segregation of A-site dopant cation based on DFT calculations. It is noteworthy that we only consider the case that A-site dopant cation segregates toward the surface as experimentally reported. To examine the effect of elastic interactions, a wide range of A-site cation (Ca, Sr, and Ba) doped LaBO3 perovskites (B = Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni) that have been commonly used as cathode candidates3,18,22–25 are chosen. This paper is organized as follows. The segregation energy is firstly calculated as a quantitative indicator of dopant segregation. Then, we introduce a descriptor, which is change of free volume, to describe the effects of the A- or B-site cations on the elastic interactions of dopant cation. The descriptor can be used to elucidate the underlying principles of dopant segregation and predict the degree of surface segregation. Lastly, based on these results, we suggest possible ways to suppress the dopant segregation.
2. Computational methods
The Vienna Ab initio Simulation Package (VASP)26–28 was used to perform density functional theory (DFT) calculations. All calculations employed the Perdew–Burke–Ernzerhof (PBE) functional based on the generalized gradient approximation (GGA).29 The total energy calculations used a plane wave expansion with a cutoff of 400 eV and included spin-polarization to describe the magnetic properties. In our model system, Ca, Sr, and Ba (alkaline earth metals) with 2+ charge states were chosen as A-site dopant cations and Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni (first row 3d-transition metals) with 3+ charge states were chosen as the B-site cations. This selection was aimed to systemically introduce the elastic energy caused by depending on the size of A- and B-site cations. Table 1 shows the Shannon's ionic radii30 of cations where the charge state, coordination number, and spin state (only B-site cations) were considered.| A-Site | La | Ca | Sr | Ba |
|---|---|---|---|---|
| 1.360 | 1.340 | 1.440 | 1.610 |
| B-Site | Cr0.50Mn0.50 | Mn | Fe | Co0.25Fe0.75 | Co | Ni |
|---|---|---|---|---|---|---|
| 0.630 | 0.645 | 0.645 | 0.620 | 0.545 | 0.560 |
To choose the proper ionic radii from the database of Shannon's ionic radii, it is required to identify the spin state of B-site cation. For that, we compared our calculated magnetic moments per respective B-site cations with the reported values.31–37 For the mixed B-sites such as Cr0.50Mn0.50 and Co25Fe0.75, the weighted average of ionic radii were used.38,39 To avoid the self-interaction errors in the standard DFT-GGA for strongly correlated electronic materials such as mid-to-late first low transition metal oxides,40 the DFT+U approach41 was used with Ueff = 3.5 eV (Cr), 4.0 eV (Mn), 4.0 eV (Fe), 3.3 eV (Co), 6.4 eV (Ni).42
Periodic asymmetric (stoichiometric) slab models were employed to assess the energetics of dopant segregation with a vacuum of 15 Å in the direction of the surface normal. Dipole corrections43 were considered in z-direction to avoid the error that is derived in non-zero dipole models such as the asymmetric slabs. Since the (001) surface in perovskite-type oxides is the most stable at high operating temperatures of SOFC,44–46 the slab model was constructed by truncating the bulk perovskite structure along the (001) plane. Under the SOFC operating conditions, AO-terminated surfaces have been mainly observed in various perovskite oxides.47–49 We thus focused only on the AO-terminated surfaces to describe a segregated dopant cation on the top surface layer among the two types of surface terminations in the (001) surfaces of perovskites (AO and BO2). A (2 × 2) surface unit cell with six atomic layers was selected while one bottom layer is fixed as in their bulk positions. Compared to the previous reports,6,7,19,50–53 it is thought that our choices of AO-terminated surfaces and slab layer thickness are appropriate to analyze the trends in dopant segregation thermodynamics. All calculations for slab model used a 4 × 4 × 1 Monkhorst–Pack54 grid k-point mesh.
To calculate the change of free volume (ΔVf) upon the doping, a 2 × 2 × 2 supercell containing 40 total atoms were used with a Monkhorst–Pack grid54 of 4 × 4 × 4 k-points. In these calculations, we used (pseudo)cubic structures that are exhibited at high SOFC operating temperature,55,56 which have also been applied in previous DFT studies.57–59 Two La atoms were substituted with two dopant atoms to calculate the lattice constant (or free volume) of the doped perovskite that was clearly altered compared to that of the corresponding perfect perovskite (Fig. S1 of ESI†).
3. Results and discussion
3.1. Surface segregation energy of A-site dopant cation on LaBO3(001)
To quantify the segregation tendency of A-site dopants, we calculated surface segregation energy, that is the energy required for a dopant cation to segregate toward the surface as below;| Esegr. = Esurf − Ebulk, | (1) |
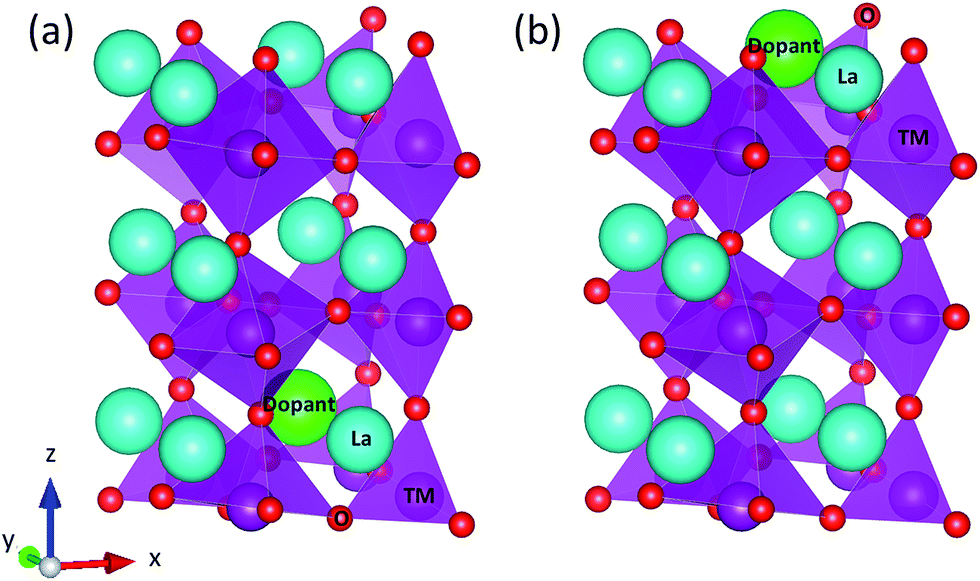 | ||
| Fig. 1 Schematic illustration of the doped LaBO3(001) slab models to calculate the segregation energy of a A-site cation dopant. (a) and (b) show slab models before and after the segregation of a A-site cation dopant, respectively. A-site host cations (La) are light blue spheres, A-site dopant cations (Ca, Sr, and Ba) are light green spheres, B-site transition metal (TM) cations are (Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni) purple spheres, and oxygen anions (O) are red spheres, respectively. | ||
Our DFT results for Esegr. on LaBO3(001) are summarized in Fig. 2. Our DFT values seem to be somewhat overestimated compared to the reported experimental data of Sr segregation enthalpy in the La1−xSrxMnO3 thin film.60 This inconsistency, which has also been observed in preceding DFT papers,6,7,19,50,52,53 may be attributed to the deviations in the reaction conditions (temperature, gas pressure), surface terminations, and dopant cation concentrations from the theoretical models. Nevertheless, the segregation energies of three dopant cations (Ca, Sr, and Ba) on LaMnO3(001) still show a similar tendency as shown in previous DFT studies19,51–53 even though there are several differences in the computational details such as the employed slab models and the calculating methods of the segregation energy. Here, we therefore focus on the tendency rather than the absolute values of the segregation energies.
 | ||
| Fig. 2 A trend in Esegr. of Ca, Sr, and Ba dopant cations on LaBO3(001) surfaces (B = Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni). The x-axis is configured in increasing the order of atomic number of the first row 3d-transition metals. The solid lines serve as a guide for the eye. | ||
Fig. 2 is plotted with regard to the increasing the atomic number of 3d-transition metals in the same period, where we found two interesting observations. First, the larger dopant cation segregates more strongly toward the surface. Second, a dopant cation tends to segregate more as larger transition metals occupy the B-site (Table 1). These trends indicate that the surface segregation of dopant cations significantly depends on the sizes of the A-site dopant and B-site transition metal cations.
3.2. Elastic energy contribution to the dopant segregation: change of free volume (ΔVf)
As mentioned above, we previously demonstrated that two key factors governing the dopant segregation on the surface of manganite perovskites are the elastic and electrostatic interactions.19 Elastic energy was induced by a size mismatch between the dopant and host cation, whereas electrostatic energy was generated by the charge interactions between a dopant and the other ions. Since A-site host cation (La), A-site dopant cation (Ca, Sr, and Ba), B-site cation (Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni), and atomic oxygen anion (O) have identical charge state, respectively, in LaBO3 perovskite system, the electrostatic contributions to dopant segregation can be considered constant. Indeed, our previous DFT results19 showed that the electrostatic energies in Ca, Sr, and Ba-doped LaMnO3 and SmMnO3 can be analytically calculated from same formal charges at each lattice site. Considering that electrostatic energy has nearly constant value in our (La,A)BO3 system, it is reasonable to focus only on the effect of elastic interactions to elucidate the underlying mechanism of the dopant segregation in this study.To explain the contribution of the elastic energy introduced by the A- and B-site cations in various lanthanum-based perovskites, we introduced the change of free volume (ΔVf). The free volume implies the unit cell volume that excludes the total volume of the constituent ions.61,62 Upon doping the A-site dopant cations (Ca, Sr, and Ba), the free volumes are changed depending on the sizes of A- or B-site cations. Therefore, ΔVf can be calculated by
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) doped) is the free volume of the perfect (or doped) bulk perovskite, aperfect (ordoped) is the lattice constant of the perfect (or doped) perovskite, and rhost (ordopant) is the Shannon's radius of the host (or dopant) cation. The lattice constants we used are summarized in Table S1 of ESI.† A more negative change in the free volume indicates that a dopant cation in the bulk phase is under the less stable condition due to the surrounding atoms, thereby generating a higher elastic energy of a dopant cation. Fig. 3 shows a linear relation between Esegr. and ΔVf in LaBO3 perovskites (B = Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni). For all cases, the dopant cations segregate toward the surface to minimize the elastic energy. Esegr. decreases with decreasing ΔVf due to the larger elastic energy driven by the substitution of dopant cations. It is noted that the segregation phenomena are dependent not only on the dopant cations doped on the A-site, but also B-site cations. Consequently, it is important to select the appropriate dopant and B-site cations in perovskite oxides for suppressing the dopant segregation. The full explanation of how to choose the cations is described in Section 3.5.
doped) is the free volume of the perfect (or doped) bulk perovskite, aperfect (ordoped) is the lattice constant of the perfect (or doped) perovskite, and rhost (ordopant) is the Shannon's radius of the host (or dopant) cation. The lattice constants we used are summarized in Table S1 of ESI.† A more negative change in the free volume indicates that a dopant cation in the bulk phase is under the less stable condition due to the surrounding atoms, thereby generating a higher elastic energy of a dopant cation. Fig. 3 shows a linear relation between Esegr. and ΔVf in LaBO3 perovskites (B = Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni). For all cases, the dopant cations segregate toward the surface to minimize the elastic energy. Esegr. decreases with decreasing ΔVf due to the larger elastic energy driven by the substitution of dopant cations. It is noted that the segregation phenomena are dependent not only on the dopant cations doped on the A-site, but also B-site cations. Consequently, it is important to select the appropriate dopant and B-site cations in perovskite oxides for suppressing the dopant segregation. The full explanation of how to choose the cations is described in Section 3.5.
 | ||
| Fig. 3 Surface segregation energy calculated by eqn (1) as a function of the change of free volume calculated by eqn (2). | ||
3.3. Contribution of additional factors to the dopant segregation
As shown in Fig. 3, although the changes of free volume are approximately zero (almost no elastic energies) in several materials, segregation energies are still negative. This implies that there exist additional contributions to the segregation energies except the elastic contribution. One extra contribution comes from the electrostatic interaction as mentioned above. Based on our previous report,19 a dopant cation has a charge of −1 upon replacing a host cation of 3+, thereby interacting with the other ions in the perovskite oxides even if they have no additional charged defects. In addition, the decrease in bond strength upon doping the cation can contribute to the surface segregation. From the previous reports,63–67 the difference in bond strength has been considered as one of driving forces for surface segregation in metal alloy system, demonstrating that an atom that weakly binds with the nearby atoms strongly segregates toward the surface. Battaile et al.68 also reported that the difference in bond strengths between cation and anion is an origin for surface segregation of solute cations in oxide solutions. In LaSrBO3 perovskite oxides, Deml et al.59 performed DFT calculations to estimate the bond strengths of La–O and Sr–O from the enthalpy of oxide formation which implies the average metal–oxygen bond strength. They demonstrated that bond strength of Sr2+–O is weaker than that of La3+–O from the reduction in enthalpy of oxide formation upon doping Sr in the A-site. It is therefore thought that in our system, the dopant2+–O bonds are weaker than La3+–O bonds, which facilitates the dopant cation to segregate toward the surface. Fig. 4 shows the segregation energy as a function of dopant–O bond length relative to La–O bond length in the bulk perovskites. The measurement details of the bond lengths are described in S2 section of ESI.† We previously showed that Sr–O bond length is closely related to the Sr segregation on (La,Sr)MnO3 surface.7 Likewise, in a particular (La,A)BO3 system, a larger A-site dopant cation that has larger bond distance with lattice oxygen tends to strongly segregate toward the surface due to the weaker bond strength (Fig. 4). This implies that the order of segregation tendency (Ba > Sr > Ca) caused by elastic contribution is fortified by the effects of bond strength. | ||
| Fig. 4 Calculated dopant segregation energy as a function of the dopant–O bond length relative to La–O. | ||
Nevertheless, elastic energy is a dominant governing factor to the dopant segregation than the bond strength. Only by dopant–O bond strength, we cannot explain the dependence of the dopant segregation on B-site cations (Fig. 4). For instance, the relative dopant–O bond length in Sr-doped La(Co or Ni)O3 is similar to that in Ba-doped La(Cr0.50Mn0.50, Mn, Fe, or Co0.25Fe0.75)O3 even though the segregation energy is significantly different. As a result, we conclude that the dopant segregation behavior is mainly determined by elastic contribution, and enforced by the bond strength of dopant–O.
3.4. Identification of useful descriptors to predict the dopant segregation behaviors
As mentioned above in Section 3.2, it is important to obtain the change of free volume that enables the simple estimation of the segregation energy. In eqn (2), the change of free volume can be decoupled into two terms: (1) size (volume) mismatch between the dopant and host cations on the A-site and (2) change in the unit cell volume [adoped3 − aperfect3]. The size mismatch between the A-site dopant and host cations (1) can be simply obtained from a database of Shannon's ionic radii, whereas the change in unit cell volume (2) can be calculated from the change in the lattice constant of perovskite before and after doping the A-site cation, which requires additional experimental measurements or computational calculations. For simple estimation of the change of free volume, and thus the segregation energy, therefore, the term of change in the unit cell needs to be replaced with the corresponding term that is easily accessible.
and (2) change in the unit cell volume [adoped3 − aperfect3]. The size mismatch between the A-site dopant and host cations (1) can be simply obtained from a database of Shannon's ionic radii, whereas the change in unit cell volume (2) can be calculated from the change in the lattice constant of perovskite before and after doping the A-site cation, which requires additional experimental measurements or computational calculations. For simple estimation of the change of free volume, and thus the segregation energy, therefore, the term of change in the unit cell needs to be replaced with the corresponding term that is easily accessible.
The change in unit cell volume term may be expressed in terms of the ionic radii of A- and B-site cations that are closely related to the lattice constant of perovskite oxides.69 Here, since the host cation (La) is constant in all perovskite oxides we examine, the change in unit cell volume may rely on the ionic radii of the dopant and B-site cations. The unit cell volume of LaBO3 perovskite is determined depending on the size of B-site cation, and it generally decreases with decreasing the size of B-site cation. If an A-site dopant cation, which is always larger than B-site cation, is substituted in the bulk LaBO3, thus, the unit cell with smaller B-site cations may increase more to expand the narrow internal space. For the same reason, expansion of the unit cell with a particular B-site cation may strongly induced upon a larger cation doping at the A-site. As a result, the ratio of size mismatch between the A-site dopant and B-site cation [(rdopant − rB)/rB] is linearly proportional to the change in unit cell volume, as shown in Fig. 5. Therefore, the change of free volume can be rewritten as
 | (3) |
 | ||
| Fig. 5 DFT-calculated change in the unit cell volume versus the ratio of size mismatch between the dopant and B-site cations from Shannon's ionic radii.30 | ||
Furthermore, based on the linear correlation between Esegr. and ΔVf in Fig. 3, the empirical equation of segregation energy is also obtained by linear fitting with two terms of eqn (3), and given as
 | (4) |
From eqn (4), the dopant enrichment at the surface can be quickly and easily predicted only with the information for the ionic radii of cations.
3.5. Suggested strategies to suppress the dopant segregation
Here, we discuss each contribution from the two terms of eqn (4) to the segregation energy. We firstly examine the correlation between the segregation energy and size mismatch on the A-site. As shown in Fig. 6a, the larger dopant cation (in order of Ba, Sr, and Ca) tends to more strongly segregate to the surface within the same B-site transition metal. According to previous reports which investigated the origin of the dopant segregation on the surfaces of manganite perovskites,19,51,52 this trend arises from the minimization of elastic energy induced by size mismatch between the dopant and host cations on the A-site. In this study, we more extensively support the previous arguments on a wide range of LaBO3 perovskite surfaces; a relatively small difference between the dopant and host cations can suppress the dopant segregation (see Table S2 of ESI†). Indeed, this result has been applied to improve the long-term stability of SOFC cathode materials. Yoo et al.70 expected the suppression of surface segregation on the surface of NdBaCo2O5+δ (NBCO) double-perovskite by only partially replacing Ba with Ca at the A-site. Then, they presented NdBa1−xCaxCo2O5+δ (NBCaCO) as a superior SOFC cathode material with high chemical stability. This example shows that our strategy to suppress the dopant segregation is successful. | ||
| Fig. 6 Contribution of the two terms in the predictive equation (eqn (4)) to the dopant segregation. Surface segregation energy as a function of (a) size mismatch between the A-site dopant and host cations and (b) size mismatch between the A-site dopant and B-site cations. The solid lines of (a) are used as a guide. | ||
More importantly, it is notable that the size of B-site cation also affects the segregation energy (Fig. 6b). When the host A-site cation is substituted with a particular dopant cation, the larger B-site cation leads to the higher compressive strain of dopant cations (a more negative change of free volume). Therefore, the dopant cation more strongly segregate with increasing the size of the B-site cation, as can be seen in Fig. 6b. We can conclude that using the small B-site transition metals alleviates the surface cation segregation. Unlike our computational model, the oxidation state of B-site cation often becomes closer to 2+ with the formation of oxygen vacancies at SOFC operating conditions.8 Since B2+ is larger than B3+ as listed in Table S3 of ESI,† in this case, dopant cation would more strongly segregate toward the surface due to the fact that the larger B-site cation tends to generate the stronger elastic energy, as we demonstrated in this study. Nevertheless, we expect that the changes of elastic energy depending on B-site cations in LaB2+O3 would not be significantly different from that in LaB3+O3 because the changes in the size of B2+ are similar to that of B3+ (see Table S3 of ESI†). Since the formation of B2+ accompanies the creation of oxygen vacancy, both elastic and electrostatic interactions would affect the dopant segregation at the same time. Although it is difficult to perfectly decouple the two contributions, we believe that creating B-site cation of 2+ state, instead of 3+, allows the dopant cation to more strongly segregate toward the surface by two reasons; stronger elastic interaction generated by larger B-site cation and stronger electrostatic interaction induced by surface oxygen vacancy created from the change in the oxidation state of B-site cation.
Based on our results, we suggest that among the elements we examined, Ca is the most suitable for the A-site dopant cation due to the smallest size mismatch with La host cation while Co or Ni may be a good choice for the B-site transition metal due to the smaller size. Combining these, the level of dopant segregation would be the lowest at the surface of Ca-doped La(Co or Ni)O3 perovskite among the materials we considered here. By just doping with the proper sizes of A- or B-site cations in perovskite oxides, SOFC cathode with better stability can be achieved. Since our proposals did not consider the kinetics or other factors of segregation that will be relevant in specific operating conditions, but only the thermodynamic aspect of segregation phenomena, we should be appropriately cautious. Nevertheless, our results still provide a useful basis for selecting the dopant elements where these other aspects of the dopant segregation could fruitfully be pursued. In fact, we previously demonstrated that at high temperatures, the tendency of dopant segregation was fairly predicted by just thermodynamics on the surface of manganite perovskites.19 However, the other contributions should be additionally considered to fully understand the dopant segregation phenomena, which will be a topic of our next study.
4. Conclusions
Cation segregation on perovskite oxide surfaces can often have an adverse effect on the SOFC cathode, especially the ORR. Thus, we quantitatively assessed the underlying mechanism of surface segregation to control the dopant enrichment at the Ca, Sr, and Ba doped-LaBO3(001) surfaces using DFT calculations, where B = Cr0.50Mn0.50, Mn, Fe, Co0.25Fe0.75, Co, and Ni. To demonstrate the thermodynamic driving force, we introduced the change of free volume (ΔVf). The minimization of elastic energy caused by the size of A- and B-site cations was determined to be a dominant factor governing the segregation energetics. This result, which expands our recent research,19 involves a significant finding in that the B-site cation also drives the surface segregation of dopants. The role of the change of free volume was strengthened by converting the lattice constant term into the ionic radius term, such as is done in eqn (3). By this alteration, the change of free volume (in other words, segregation energy) could easily be predicted from the ionic size of the cations. The useful principles were established for the development of new LaBO3-type SOFC cathodes hindering dopant segregation as follows: select (1) a dopant A-site cation that shows smaller size mismatch with the host A-site cation and (2) a smaller size of B-site cation. These principles are helpful for reducing the elastic energy of the dopant cations. We believe that our findings will provide good insight in devising advanced SOFC cathodes.Nevertheless, to develop new cathode materials with high stability, it is obvious that other factors should be also considered. Very recently, Tsvetkov et al. reported that reducing the concentration of surface oxygen vacancies in La0.8Sr0.2CoO3 by surface additive cations can suppress Sr segregation by weakening the electrostatic attractions between Sr and surface oxygen vacancies.71 This result clearly implies that the electrostatic interaction also plays an important role to dopant segregation. However, it is still a big challenge to find the cathode materials to simultaneously reduce both elastic and electrostatic interactions. In the aspect of elastic interaction, in this study, selecting a dopant A-site cation that exhibits smaller size mismatch with the host A-site cation (Ca) and a smaller size of B-site cation (Co or Ni) was proposed to hinder the dopant segregation. However, (surface) oxygen vacancies which may accelerate the dopant segregation are easily formed in La(Co or Ni)O3 among the candidate materials chosen in this study.72 It is therefore thought that there exists the opposite trend of contributions between elastic and electrostatic interactions to the dopant segregation, which brings us the fact that there will be an optimal point. Elucidating this more in detail will be our ultimate goal to design new cathode materials with high chemical and electrochemical stability.
Acknowledgements
The authors acknowledge support from the Global Frontier R&D Program on Center for Multiscale Energy System through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2014M3A6A7074784 and NRF-2014M3A6A7074785) and the KIST Institutional Program (2E26081-16-053), and the supercomputing resources, including technical support from Supercomputing Center/Korea Institute of Science and Technology Information (KSC-2015-C3-045).References
- E. D. Wachsman and S. C. Singhal, Am. Ceram. Soc. Bull., 2010, 89, 22–32 CAS
.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS PubMed
- Z. Shao and S. M. Haile, Nature, 2004, 431, 170–173 CrossRef CAS PubMed
- K. Katsiev, B. Yildiz, K. R. Balasubramaniam and P. A. Salvador, Appl. Phys. Lett., 2009, 95, 092106 CrossRef
- H. Jalili, J. W. Han, Y. Kuru, Z. Cai and B. Yildiz, ECS Trans., 2011, 35, 2097–2104 Search PubMed
- H. Jalili, J. W. Han, Y. Kuru, Z. Cai and B. Yildiz, J. Phys. Chem. Lett., 2011, 2, 801–807 CrossRef CAS
- Z. Cai, Y. Kuru, J. W. Han, Y. Chen and B. Yildiz, J. Am. Chem. Soc., 2011, 133, 17696–17704 CrossRef CAS PubMed
- T. T. Fister, D. D. Fong, J. A. Eastman, P. M. Baldo, M. J. Highland, P. H. Fuoss, K. R. Balasubramaniam, J. C. Meador and P. A. Salvador, Appl. Phys. Lett., 2008, 93, 151904 CrossRef
- P. A. W. van der Heide, Surf. Interface Anal., 2002, 33, 414–425 CrossRef CAS
- Z. Cai, M. Kubicek, J. J. Fleig and B. Yildiz, Chem. Mater., 2012, 24, 1116–1127 CrossRef CAS
- G. P. Summers, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 5275–5279 CrossRef CAS
- M. Abbate, H. Ascolani, F. Prado and A. Caneiro, Solid State Commun., 2004, 129, 113–116 CrossRef CAS
- Y. Chen, W. Jung, Z. Cai, J. J. Kim, H. L. Tuller and B. Yildiz, Energy Environ. Sci., 2012, 5, 7979–7988 CAS
- S. Estradé, J. M. Rebled, J. Arbiol, F. Peiró, I. C. Infante, G. Herranz, F. Sánchez, J. Fontcuberta, R. Córdoba, B. G. Mendis and A. L. Bleloch, Appl. Phys. Lett., 2009, 95, 072507 CrossRef
- J. P. Kemp, D. J. Beal and P. A. Cox, J. Solid State Chem., 1990, 86, 50–58 CrossRef CAS
- S. P. Simner, M. D. Anderson, M. H. Engelhard and J. W. Stevenson, Electrochem. Solid-State Lett., 2006, 9, A478–A481 CrossRef CAS
- A.-K. Huber, M. Falk, M. Rohnke, B. Luerssen, L. Gregoratti, M. Amati and J. Janek, Phys. Chem. Chem. Phys., 2012, 14, 751–758 RSC
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed
- O. Müller and R. Roy, The Major Ternary Structural Families, Springer-Verlag, USA, New York, 1974 Search PubMed
- W. Jung and H. L. Tuller, Energy Environ. Sci., 2012, 5, 5370 CAS
- C. Sun, R. Hui and J. Roller, J. Solid State Electrochem., 2009, 14, 1125–1144 CrossRef
- A. J. Jacobson, Chem. Mater., 2010, 22, 660–674 CrossRef CAS
- Y. Choi, M. C. Lin and M. Liu, J. Power Sources, 2010, 195, 1441–1445 CrossRef CAS
- A. M. Ritzmann, A. B. Muñoz-García, M. Pavone, J. A. Keith and E. A. Carter, Chem. Mater., 2013, 25, 3011–3019 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- D. S. Sholl and J. A. Steckel, Density Functional Theory: A Practical Introduction, John Wiley & Sons, Inc, USA, New Jersey, 2009 Search PubMed
- J. P. Perdew and K. Burke, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- W. C. C. Koehler and E. O. O. Wollan, J. Phys. Chem. Solids, 1957, 2, 100–106 CrossRef CAS
- T. Saitoh, T. Mizokawa, A. Fujimori, M. Abbate, Y. Takeda and M. Takano, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 4257–4266 CrossRef CAS
- P. M. Raccah and J. B. Goodenough, J. Appl. Phys., 1968, 39, 1209–1210 CrossRef CAS
- M. A. Señarís-Rodríguez and J. B. Goodenough, J. Solid State Chem., 1995, 118, 323–336 CrossRef
- K. Sreedhar, J. M. Honig, M. Darwin, M. McElfresh, P. M. Shand, J. Xu, B. C. Crooker and J. Spalek, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6382–6386 CrossRef CAS
- T. Mayeshiba and D. Morgan, Phys. Chem. Chem. Phys., 2015, 17, 2715–2721 RSC
- A. B. Muñoz-García, M. Pavone and E. A. Carter, Chem. Mater., 2011, 23, 4525–4536 CrossRef
- N. Trofimenko and H. Ullmann, Solid State Ionics, 1999, 118, 215–227 CrossRef CAS
- A. B. Muñoz-García and M. Pavone, Chem. Mater., 2016, 28, 490–500 CrossRef
- E. A. Carter, Science, 2008, 321, 800–803 CrossRef CAS PubMed
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef
- G. Makov and M. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014–4022 CrossRef CAS
- Y. Choi, D. S. Mebane, M. C. Lin and M. Liu, Chem. Mater., 2007, 19, 1690–1699 CrossRef CAS
- J. W. Han and B. Yildiz, J. Mater. Chem., 2011, 21, 18983–18990 RSC
- Y. A. Mastrikov, E. Heifets, E. A. Kotomin and J. Maier, Surf. Sci., 2009, 603, 326–335 CrossRef CAS
- J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593–3599 CAS
- J. Druce, T. Ishihara and J. Kilner, Solid State Ionics, 2014, 262, 893–896 CrossRef CAS
- A. Staykov, H. Téllez, T. Akbay, J. Druce, T. Ishihara and J. Kilner, Chem. Mater., 2015, 27, 8273–8281 CrossRef CAS
- S. Piskunov, E. Heifets, T. Jacob, E. A. Kotomin, D. E. Ellis and E. Spohr, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 121406 CrossRef
- V. Sharma, M. K. Mahapatra, P. Singh and R. Ramprasad, J. Mater. Sci., 2015, 50, 3051–3056 CrossRef CAS
- Y.-L. Lee and D. Morgan, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 195430 CrossRef
- V. Sharma, M. K. Mahapatra, S. Krishnan, Z. Thatcher, B. D. Huey, P. Singh and R. Ramprasad, J. Mater. Chem. A, 2016, 4, 5605–5615 CAS
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef
- A. C. Momin, E. B. Mirza and M. D. Mathews, J. Mater. Sci. Lett., 1991, 10, 1246–1248 CrossRef CAS
- S. P. Jiang, J. Mater. Sci., 2008, 43, 6799–6833 CrossRef CAS
- M. Pavone, A. B. Muñoz-García, A. M. Ritzmann and E. A. Carter, J. Phys. Chem. C, 2014, 118, 13346–13356 CAS
- M. Pavone, A. M. Ritzmann and E. A. Carter, Energy Environ. Sci., 2011, 4, 4933–4937 CAS
- A. M. Deml, V. Stevanović, C. L. Muhich, C. B. Musgrave and R. O'Hayre, Energy Environ. Sci., 2014, 7, 1996–2004 CAS
- R. Herger, P. Willmott, C. Schlepütz, M. Björck, S. Pauli, D. Martoccia, B. Patterson, D. Kumah, R. Clarke, Y. Yacoby and M. Döbeli, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 085401 CrossRef
- R. L. Cook and A. F. Sammells, Solid State Ionics, 1991, 45, 311–321 CrossRef CAS
- A. F. Sammells, R. L. Cook, J. H. White, J. J. Osborne and R. C. MacDuff, Solid State Ionics, 1992, 52, 111–123 CrossRef CAS
- L. E. Murr, Interfacial Phenomena in Metals and Alloys, Addison-Wesley Publishing Company, USA, MA, 1975 Search PubMed
- F. F. Abraham, T. Nan-Hsiung and G. M. Pound, Surf. Sci., 1979, 83, 406–422 CrossRef CAS
- P. Wynblatt and R. C. Ku, Surf. Sci., 1977, 65, 511–531 CrossRef CAS
- B. Lezzar, O. Khalfallah, A. Larere, V. Paidar and O. Hardouin Duparc, Acta Mater., 2004, 52, 2809–2818 CrossRef CAS
- J. W. Han, J. R. Kitchin and D. S. Sholl, J. Chem. Phys., 2009, 130, 124710 CrossRef PubMed
- C. C. Battaile, R. Najafabadi and D. J. Srolovitz, J. Am. Ceram. Soc., 1995, 78, 3195–3200 CrossRef CAS
- O. Fukunaga and T. Fujita, J. Solid State Chem., 1973, 8, 331–338 CrossRef CAS
- S. Yoo, A. Jun, Y.-W. Ju, D. Odkhuu, J. Hyodo, H. Y. Jeong, N. Park, J. Shin, T. Ishihara and G. Kim, Angew. Chem., 2014, 126, 13280–13283 CrossRef
- N. Tsvetkov, Q. Lu, L. Sun, E. J. Crumlin and B. Yildiz, Nat. Mater., 2016 DOI:10.1038/nmat4659
- Y.-L. Lee, J. Kleis, J. Rossmeisl and D. Morgan, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224101 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15253h |
| This journal is © The Royal Society of Chemistry 2016 |