
Orthogonal cleavage of the 2-naphthylmethyl group in the presence of the p-methoxy phenyl-protected anomeric position and its use in carbohydrate synthesis†
Vittorio
Cattaneo
,
Davide
Oldrini
,
Alessio
Corrado
,
Francesco
Berti
and
Roberto
Adamo
*
GSK Vaccines, Via Fiorentina 1, 53100 Siena, Italy. E-mail: roberto.x.adamo@gsk.com
First published on 29th April 2016
Abstract
Orthogonal removal of naphthylmethyl (NAP) and anomeric O-p-methoxyphenyl (PMP) ethers using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone and cerium(IV) ammonium nitrate, respectively, is described. These reactions were tested in the oligosaccharide assembly of biologically relevant motifs, such as α-D-Man-(1,3)-[α-D-(1,6)-Man]-α-D-Man, α-D-Fuc-(1,2)-α-D-Fuc and α-D-NeuNAc-(2,3)-β-D-Gal. The usefulness of these chemoselective deprotections was proven in the synthesis of high mannoses.
Introduction
Aromatic protecting groups are largely used in carbohydrate chemistry, where typically building blocks need temporary protections to mask hydroxyls which are not involved in the formation of the glycosidic bond.1 Protecting groups can aid the control of stereoselectivity and modulate the reactivity of the coupling partners during this step. The choice of the protecting groups is, therefore, crucial in these multi-step syntheses.2 Aromatic ethers can be removed at the end of the oligosaccharide assembly by catalytic hydrogenation, which makes their use very popular.2Electron-rich aromatic ethers are preferable to ester protecting groups as arm building blocks utilized in carbohydrate synthesis or when the control of stereoselectivity needs to exclude the anchimeric assistance, as in the formation of 1,2 cis-glycosidic linkages.2
In particular the NAP (2-naphthylmethyl) group is becoming very attractive for the possibility of easy removal in the presence of other aromatic protections, such as benzyl groups, by oxidative cleavage with DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone),3,4 catalytic hydrogenation5 or under acidic conditions.6
NAP and PMB (p-methoxybenzyl) can be orthogonally removed when used for the protection of non-anomeric hydroxyls by means of DDQ and CAN (cerium(IV) ammonium nitrate), respectively.7,8 NAP can be cleaved while leaving PMB intact by fine tuning Pd/C-catalyzed hydrogenolysis.5 Conversely, taking advantage of PMB liability under acidic conditions, this group can be removed without impacting NAP by means of HCl and HFIP (hexafluoro-2-propanol).6
Due to its acid sensitivity, PMB is rarely used to protect the anomeric positions, and PMP (p-methoxyphenyl) is generally preferred. After the release of the PMP protection, the anomeric hydroxyl is usually transformed into an appropriate leaving group, such as imidate or phosphate (Scheme 1), to obtain a glycosyl donor ready for glycosylation.9 Direct conversion of glycosyl p-methoxyphenol into the corresponding thiol donor has also been described.10 Hence, PMP is generally used to protect the C-1 position in acceptors that are converted into donors at a later stage of the synthetic strategy.
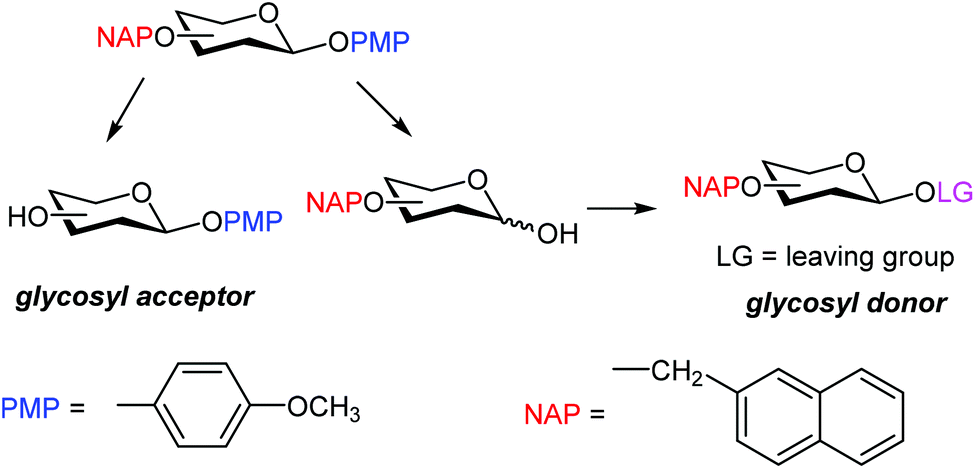 | ||
| Scheme 1 Usefulness of orthogonal removal of NAP and MP protections in carbohydrate synthesis. | ||
Here we show that NAP can be selectively removed in the presence of the PMP-protected anomeric position, and vice versa. Next, this feature was proven useful in oligosaccharide assembly.
Results and discussion
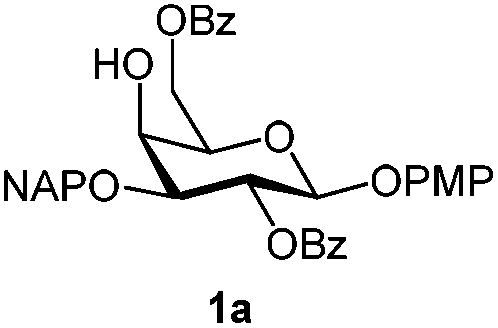
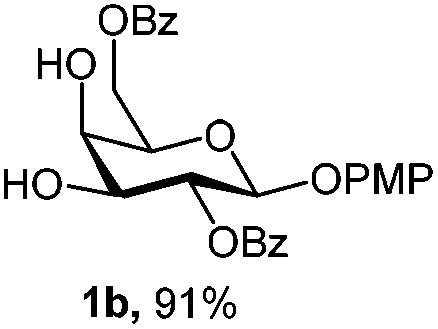
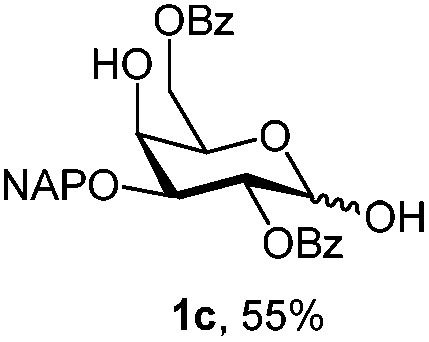
Initially we observed that oxidative cleavage of NAP with DDQ in galactopyranoside 1a gave compound 1b without loss of the PMP anomeric protection, as demonstrated by the presence in the 1H NMR spectrum of a singlet at 3.77 ppm assigned to the OCH3 signal.Conversely, treatment of 1a with CAN provided the product 1c, where the two α- and β-anomeric positions appeared in the 1H NMR spectrum at 5.66 (J1,2 3.6 Hz) and 4.78 (J1,2 8.0 Hz), respectively.
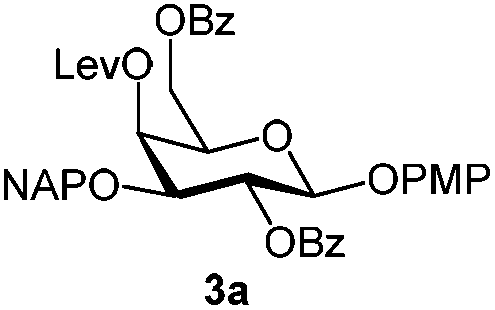
To better explore these orthogonal deprotections, we synthesized the set of monosaccharides depicted in Table 1 (procedures are reported in the ESI†). Typically, treatment of the substrate with 5 equiv. of DDQ in a CH2Cl2–MeOH mixture for 5–6 h gave selective cleavage of the NAP protection in good yield. Of note, more prolonged reaction time (<6 h) for NAP removal in compound 7a led to the loss of the 4-O-benzyl group. This was confirmed by acetylation of the formed product and appearance in the 1H NMR spectrum of a triplet (J 9.8 Hz) at 5.48 ppm, which was unambiguously assigned to H-4.
| Entry | Substrate | NAP removal | PMP removal |
|---|---|---|---|
| a For procedures see the Experimental section and the ESI. | |||
| 1 |
|
|
|
| 2 |
|
|
|
| 3 |
|
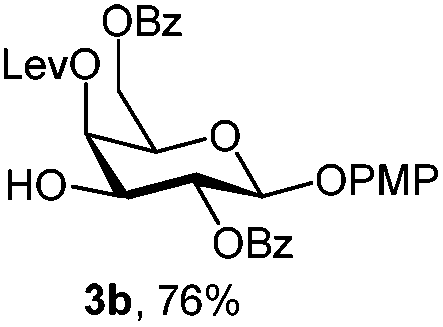
|
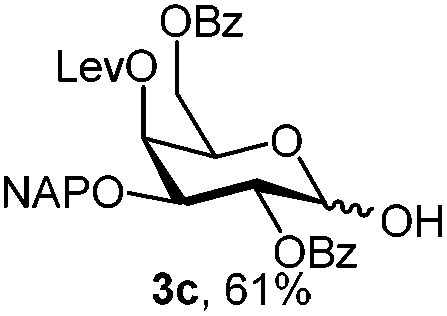
|
| 4 |
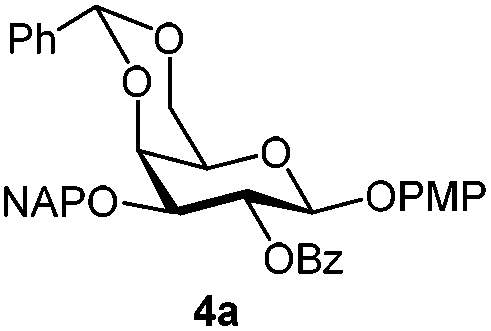
|
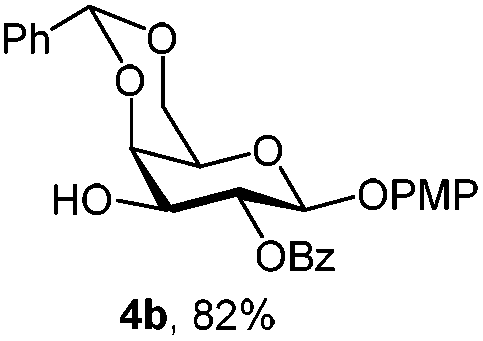
|
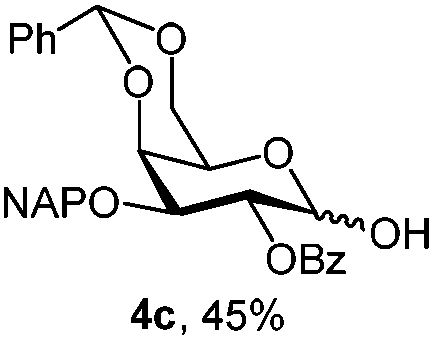
|
| 5 |
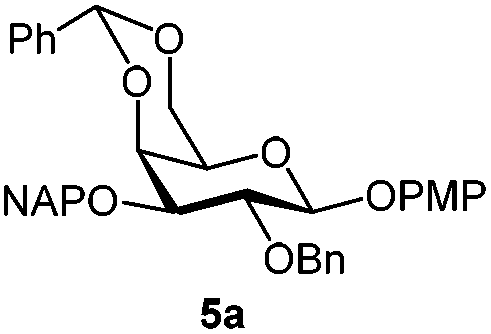
|
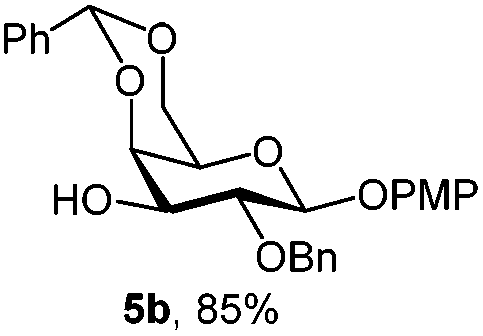
|
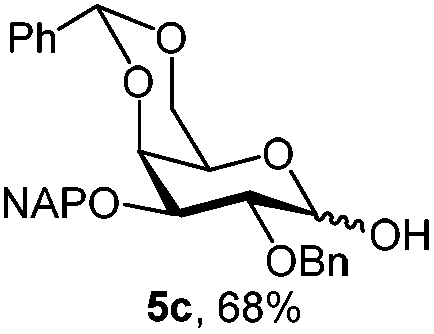
|
| 6 |
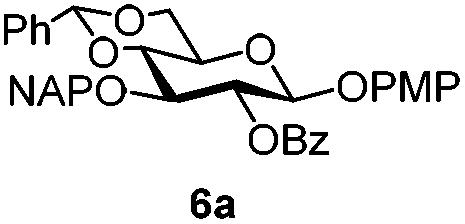
|
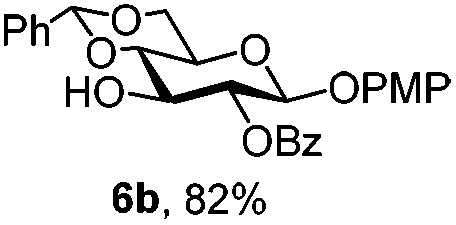
|
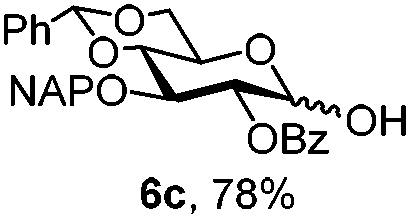
|
| 7 |
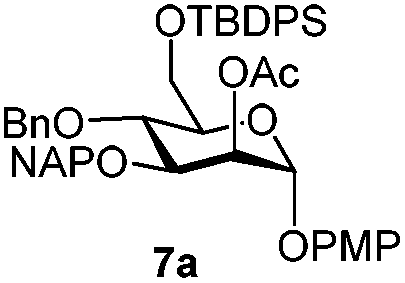
|
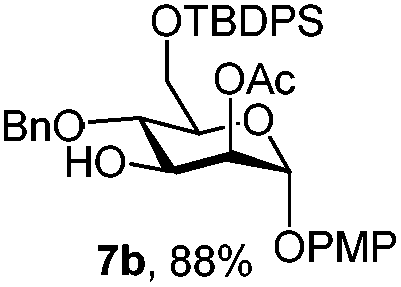
|
|
| 8 |
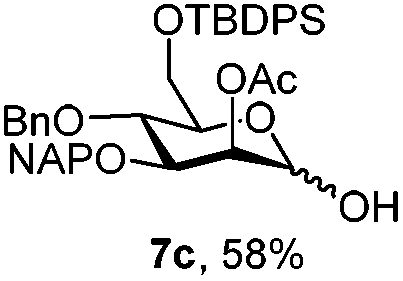
|
|
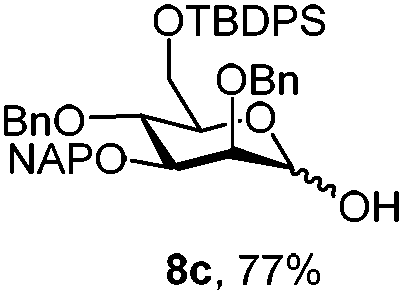
|
| 9 |
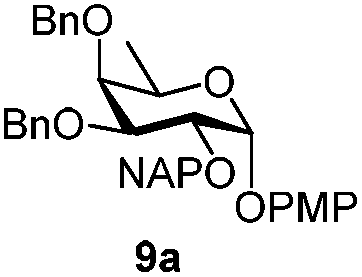
|
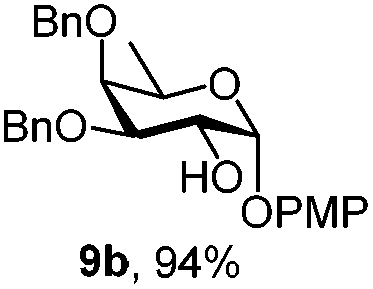
|
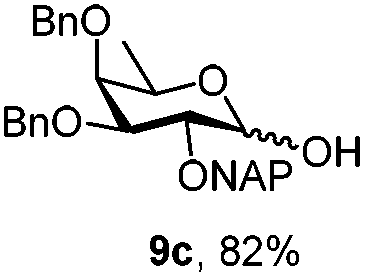
|
| 10 |
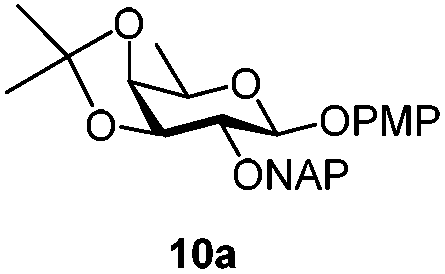
|
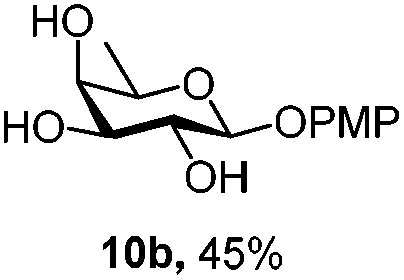
|
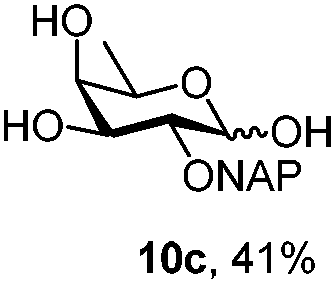
|
Occurrence of debenzylation with DDQ either in the presence or absence of water has been well documented,11 hence it needs to be taken into account as a possible side reaction.
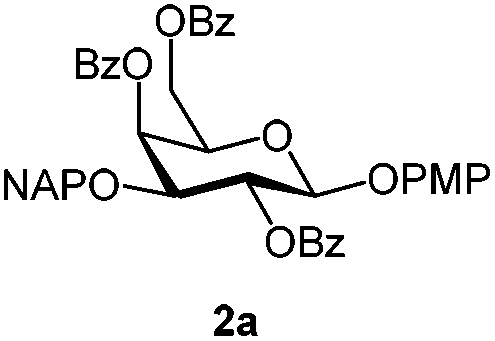
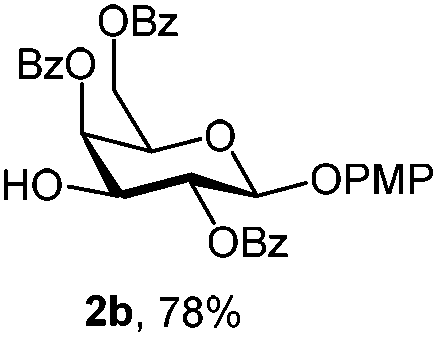
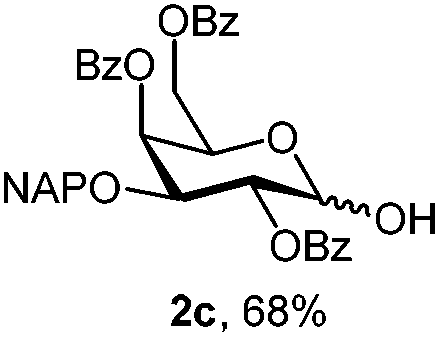
For PMP removal, the use of 5–6 equiv. of CAN in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile–water for 6 h provided the 1-OH products in moderate to good yields for all the tested compounds (Table 1),12 without impairing the NAP protection. The moderate yield of some examples could be ascribed to the formation of a quinone adduct as a by-product, which has been reported in this reaction.13 As shown in Table 2 for compound 2a, the reaction occurred giving a higher yield when acetonitrile–water was used as the solvent mixture instead of acetone–water.
:1 acetonitrile–water for 6 h provided the 1-OH products in moderate to good yields for all the tested compounds (Table 1),12 without impairing the NAP protection. The moderate yield of some examples could be ascribed to the formation of a quinone adduct as a by-product, which has been reported in this reaction.13 As shown in Table 2 for compound 2a, the reaction occurred giving a higher yield when acetonitrile–water was used as the solvent mixture instead of acetone–water.
| Entry | Substrate | Conditions | Product | Yield |
|---|---|---|---|---|
| 1 | 2a | DDQ (3 eq.), CH2Cl2–MeOH, 6 h, r.t. | 2b | 35% |
| 2 | 2a | DDQ (5 eq.), CH2Cl2–MeOH, 6 h, r.t. | 2b | 78% |
| 3 | 2a | CAN (3 eq.), CH3CN–H2O, 6 h, r.t. | 2c | 28% |
| 4 | 2a | CAN (5 eq.), (CH3)2CO–H2O, 6 h, r.t. | 2c | 45% |
| 5 | 2a | CAN (5 eq.), CH3CN–H2O, 12 h, r.t. | 2c | 53% |
| 6 | 2a | CAN (5 eq.), CH3CN–H2O, 6 h, 0 °C | 2c | 68% |
| 7 | 7a | CAN (5 eq.), CH3CN–H2O, 6 h, r.t. | 7c | 15% |
| 8 | 7a | CAN (5 eq.), CH3CN–H2O, 6 h, 0 °C | 7c | 58% |
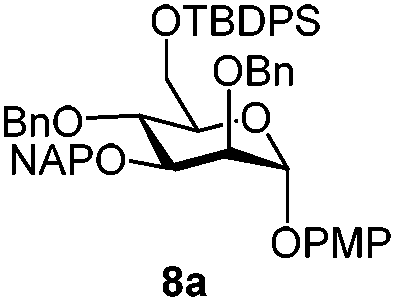
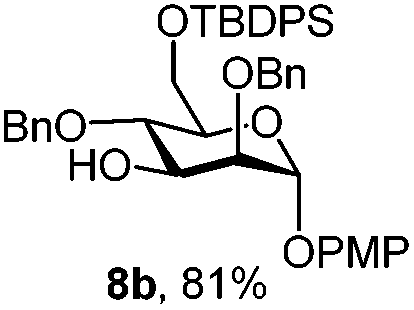
| 9 | 8a | CAN (5 eq.), CH3CN–H2O, 6 h, r.t. | 8c | 28% |
| 10 | 8a | CAN (5 eq.), CH3CN–H2O, 6 h, 0 °C | 8c | 77% |
Furthermore a reaction temperature of 0 °C avoided the formation of by-products. Particularly for substrates 7a and 8a concomitant loss of the NAP protection was observed as a side reaction (Table 2, entries 7–10). After acetylation of the by-products the 1H NMR signals related to the H-3 positions were shifted to a region between 5.45 and 5.30 ppm, clearly indicating that NAP deprotection had taken place.
Under the optimized conditions esters (Ac, Lev, Table 1, entries 1–4), benzyl ethers (Table 1, entries 5–9), benzylidene (Table 1, entries 4–6) and silyl groups (Table 1, entries 7 and 8) were untouched. In contrast, isopropylidene protection was proven labile with both DDQ and CAN because of the intrinsic acidity of the reagents (Table 1, entry 10), as manifested in the 1H NMR spectrum by the disappearance of the two signals related to the CH3 groups at 1.32 and 1.31 ppm, respectively.
Having established the orthogonality of the two protective groups, we assessed the applicability of the method to generate useful glycosyl donors.
We focused our attention to the 3-ONAP mannoside 7a, which was used as the acceptor to form the α-D-Man-(1,3)-[α-D-(1,6)-Man]-α-D-Man trisaccharide motif overexpressed in cancer cells,14–16 and which is contained in high mannoses, a family of oligomannose containing glycans highly expressed in mammalian cells and HIV.17–19 To obtain acceptor 11 with the two OH groups at positions 2 and 6 available for glycosylation, compound 7a was first subjected to TBDPS removal (Scheme 2) and then to NAP cleavage. The attempt to remove the TBDPS group from 7b caused migration of the 2-O-acetyl group to position 3 during the treatment with TBAF. Glycosylation of the acceptor with trichloroacetimidate 1220 in the presence of TMSOTf as the promoter afforded the desired trisaccharide 13 uneventfully.
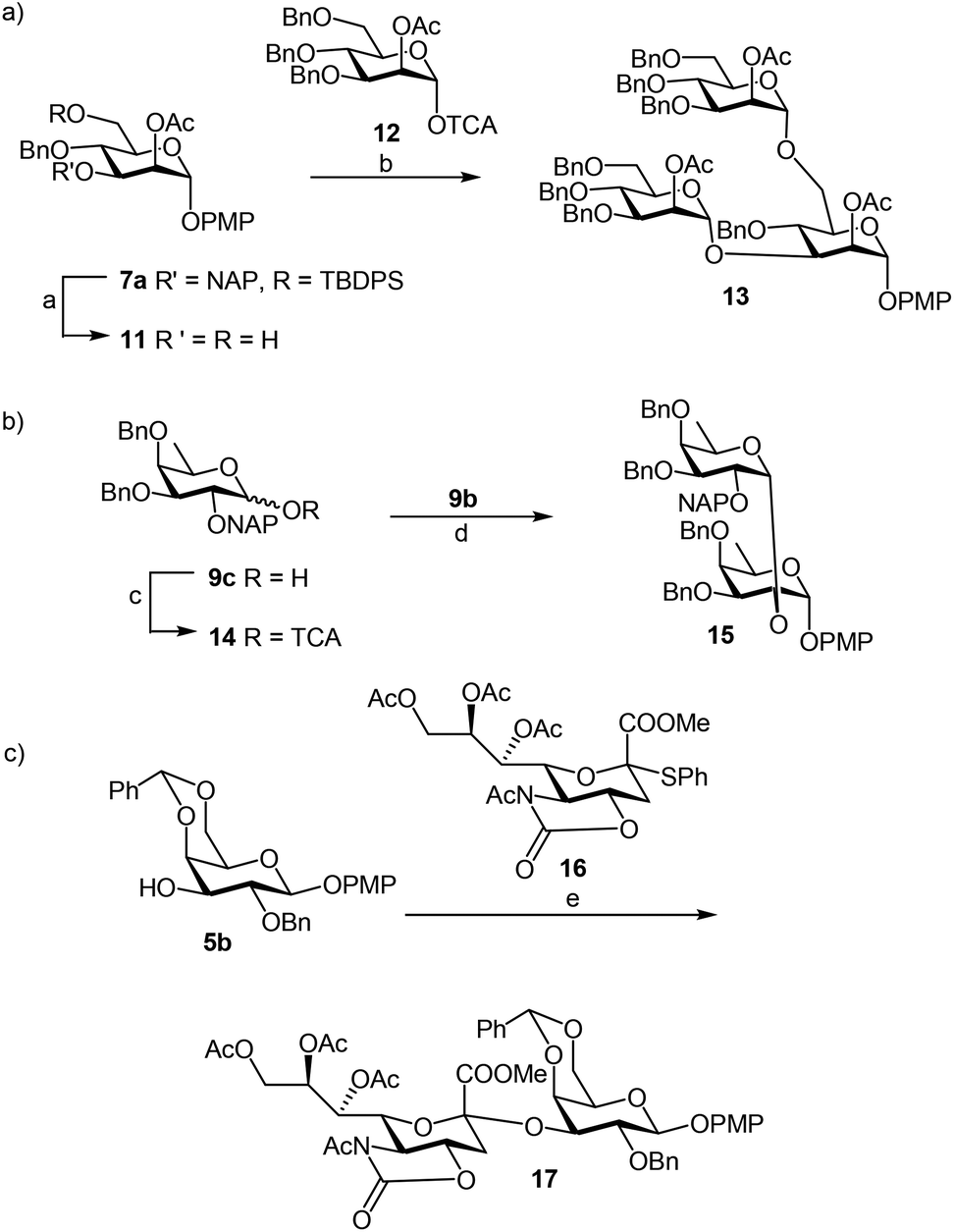 | ||
| Scheme 2 Application of PMP protected sugars as acceptors. Reaction conditions: a. TBAF, THF; DDQ, MeOH–CH2Cl2, 71%; b. TMSOTf, CH2Cl2, −20 °C, 78%; c. Cl3CCN, DBU, CH2Cl2, 91%; d. CH2Cl2, TMSOTf, CH2Cl2, −20 °C, 82%; e. NIS–TfOH, CH2Cl2, −40 °C, 65%. | ||
Next, we utilized the fucoside building block 9c to generate the α-D-Fuc-(1,2)-α-D-Fuc motif, found in the Schistosoma mansoni complex glycan.21 After removing the PMP protection with CAN, trichloroacetimidate 14 was prepared in high yield (Scheme 2) by a classic reaction with Cl3CCN and DBU. TMSOTf promoted glycosylation of PMP protected acceptor 9b with the attained donor 14 provided disaccharide 15 with exclusive α-stereoselectivity, as proved by the J1,2 = 3.6 Hz at 4.89 ppm for the newly generated glycosidic linkage in the 1H NMR spectrum.22 The non-participating NAP group exhibited a favourable effect on the stereocontrol of the reaction.
In another example, 3-OH galactoside 5b prepared from 5a (Table 1) was used as the acceptor of the N-acetyloxazolidinone sialic donor 16 to synthesize the α-D-NeuNAc-(2,3)-β-D-Gal disaccharide 17 which is present in a variety of mammalian and microbial glycans (Scheme 2). The NIS–TfOH promoted reaction gave the α-anomer stereoselectively. The H-3e signal at 2.89 ppm in the 1H NMR spectrum was compared with a similar disaccharide synthesized by Hsu et al.,23 where H-3e was at 2.89 ppm. Furthermore, the C-1 chemical shift of 169 ppm and 3J(C-1, H-3eq) of 6.0 were in perfect agreement with the assigned configuration.
Finally we tested the deprotection in the context of the multistep synthesis of high mannoses (Scheme 3). First, the PMP deprotected mannoside 8c was converted into the corresponding trichloroacetimidate donor 18 by treatment of the 1-OH sugar with Cl3CCN and DBU. After coupling the linker through a TMSOTf promoted reaction in good yield (→19), removal of the NAP protection gave the acceptor 20 ready for glycosylation with donor 12.20
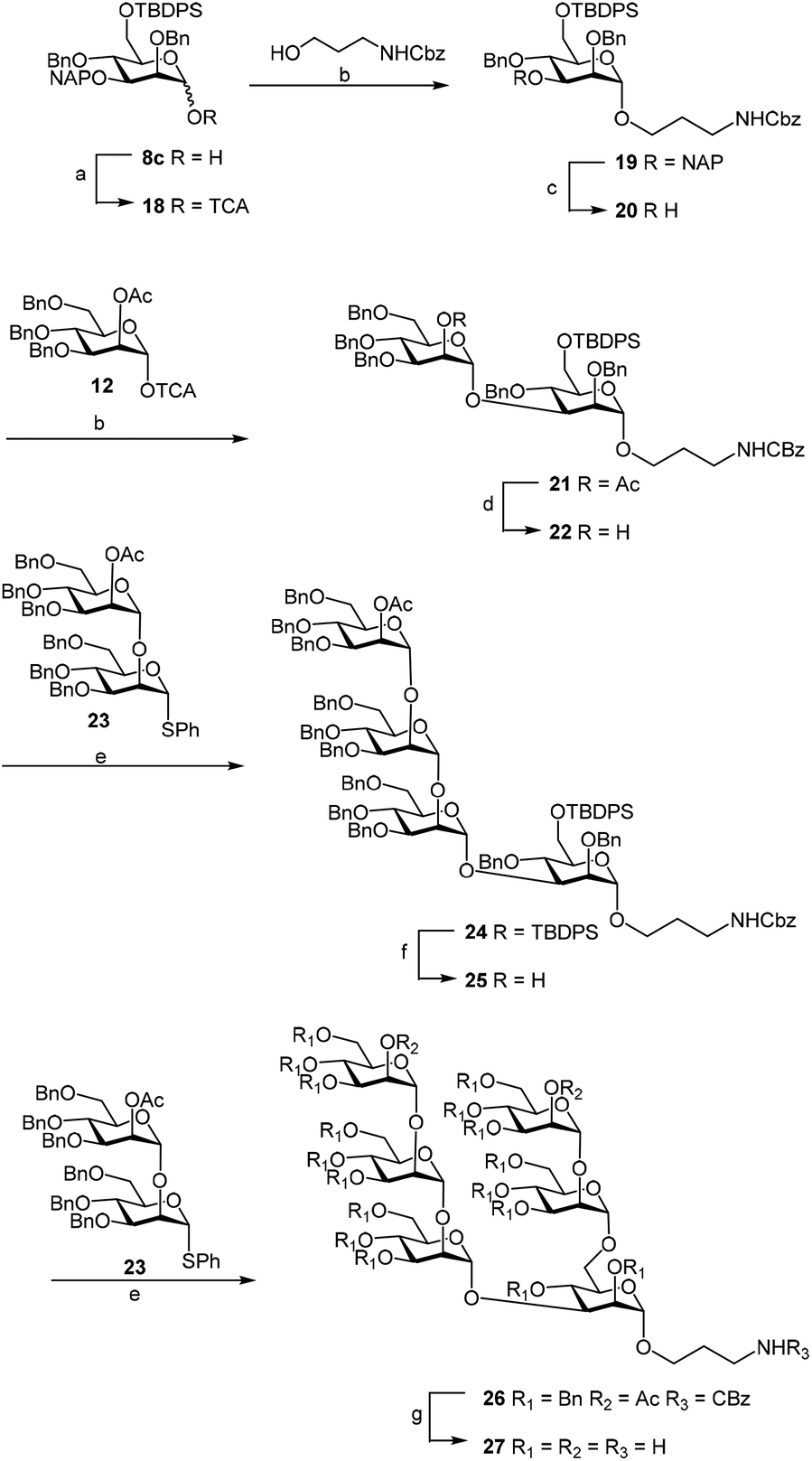 | ||
| Scheme 3 Application of NAP deprotection in multistep synthesis of Man6. Reaction conditions: a. Cl3CCN, DBU, CH2Cl2, 67%; b. TMSOTf, CH2Cl2, −30 °C, 73% from linker, 91% from 12; c. DDQ, MeOH–CH2Cl2, 78%; d. NaOMe, MeOH, 94%; e. NIS–TfOH, CH2Cl2, −30 °C, 88% from 22, 81% from 25; f. TBAF, THF, 91%; g. NaOMe, MeOH, then H2, Pd–C, 74%. | ||
Subsequent methanolysis of the acetyl ester from the synthesized disaccharide 21 made the 2-OH position of 22 available for further elongation. Glycosylation of 22 with the known donor 23,24 using NIS–TfOH as the promoter, gave the tetramannoside 24. Desilylation of the latter compound with TBAF in THF gave acceptor 25, which was again glycosylated with 2324 under the aforementioned conditions to afford the hexamannosides 26. Deacetylation under Zemplen conditions and Pd–C catalyzed hydrogenolysis yielded the so called Man6 glycan 27. The NMR spectra of the final compound were in agreement with those reported in the literature.25
Conclusions
In summary, we developed a general protocol for orthogonal removal of the NAP protection in the presence of an anomeric PMP group using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone and cerium(IV) ammonium nitrate, respectively. We demonstrated that these chemoselective reactions are useful for sequential oligosaccharide assembly of biologically relevant motifs, such as α-D-Man-(1,3)-[α-D-(1,6)-Man]-α-D-Man, α-D-Fuc-(1,2)-α-D-Fuc and α-D-NeuNAc-(2,3)-β-D-Gal. In the case of Fuc-α-(1,2)-Fuc disaccharide the 2-ONAP protection governed the stereocontrol of the reaction. The usefulness of orthogonal cleavage of NAP and PMP protection was proved in the synthesis of a hexamannose (Man6), belonging to high mannose glycans.17–19 We expect that the reported finding will find applications in synthetic strategies leading to variously substituted glycans.Experimental
General procedure for NAP removal
To a solution of sugar (0.1 mmol) in 4:1 CH2Cl2–MeOH (5 ml) DDQ (5 equiv.) was added. The mixture was stirred for 6 h, when TLC (cyclohexane–EtOAc) showed disappearance of the starting material. The crude mixture was diluted with CH2Cl2 (15 ml) and extracted three times with aq. NaHCO3. Combined organic layers were evaporated under vacuum and purified on silica gel (cyclohexane–EtOAc) to provide the deprotected compound.
General procedure for PMP removal
To a solution of sugar (0.1 mmol) in 4:1 (or 9:1) CH3CN–H2O (5 ml), CAN (5 equiv.) was added. The mixture was stirred for 6 h at 0 °C or room temperature, then TLC (cyclohexane–EtOAc) showed the completion of the reaction. The crude mixture was diluted with CH2Cl2 (15 ml) and extracted three times with aq. NaCl. Combined organic layers were evaporated under vacuum and purified on silica gel (cyclohexane–EtOAc) to provide the deprotected compound.
4-Methoxyphenyl 2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,3)-[2-O-acetyl-3,4,6-tri-O-benzyl-α-D-(1,6)-mannopyranosyl]-2-O-acetyl-4-O-benzyl-α-D-mannopyranoside 13
To a solution of mannoside donor 12 (365 mg, 2.5 mmol) and acceptor 11 (100 mg, 0.23 mmol) in dry CH2Cl2 (5 ml) containing 4 Å MS (250 mg), TMSOTf (2 μl, 0.01 mmol) was added at −20 °C under nitrogen. The mixture was stirred for 30 min, and then TLC (4:1 cyclohexane–EtOAc) showed the completion of the reaction. The crude was quenched with TEA, concentrated and chromatographed on silica gel to give 13 (245 mg, 78%) as a transparent oil. [α]25D = +48.7° (c 0.33, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.41–6.80 (m, 39H, H–Ar), 5.54 (t, J = 1.7 Hz, 1H, H-2C), 5.47 (dd, J = 1.6, 2.4 Hz, 1H, H-2B), 5.45 (d, 1H, H-1B), 5.38 (dd, J = 1.4, 3.1 Hz, 1H, H-2A), 5.29 (d, 1H, H-1A), 5.00 (d, 1H, H-1C), 4.93–4.51 (m, 14H, 7 × CH2Ph), 4.48–4.47 (m, 1H), 4.05–3.98 (m, 3H), 3.95–3.83 (m, 6H), 3.82–3.71 (m, 8H), 3.66 (s, 3H, OCH3), 2.22, 2.20 (2 s, 3H each, 2 × CH3CO). 13C NMR (CDCl3): δ 170.4, 170.3, 170.2 (3 × CO), 155.1–114.5 (C–Ar), 102.0 (C-1A), 97.9 (C-1B), 95.8 (C-1C), 80.0, 77.9, 77.7, 75.3, 75.2, 75.0, 74.3, 74.0, 74.5, 73.4, 72.5, 71.8, 71.7, 71.6, 71.5, 68.9, 68.7, 68.5, 68.4, 55.5 (OCH3), 21.1, 21.0 (3 × CH3CO). HR ESI MS C80H86O20: [M + H]+ calc. 1367.5642; found 1367.5791.
4-Methoxyphenyl 3,4-O-benzyl-α-D-fucopyranosyl-(1,2)-3,4-O-benzyl-2-O-naphtylmethyl-α-D-fucopyranoside 15
Fucoside donor 14 (85 mg, 0.13 mmol) and acceptor 9b (50 mg, 0.1 mmol) were dissolved in dry CH2Cl2 (5 ml) containing 4 Å MS (250 mg). After stirring for 15 min under nitrogen at −20 °C, TMSOTf (1 μl, 0.005 mmol) was added. Stirring was prolonged for 30 min, and then TLC (9:1 cyclohexane–EtOAc) showed the completion of the reaction. The crude was quenched with TEA, concentrated and chromatographed on silica gel to give 15 (79 mg, 85%) as a syrup. [α]25D = +72.5° (c 0.60, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.39–6.63 (m, 32H, H–Ar), 5.51 (d, J = 3.6 Hz, 1H, H-1A), 4.99 (d, J = 3.3 Hz, 1H, H-1B), 4.96–4.50 (m, 10H, 5 × CH2Ar), 4.56 (dd, J = 3.4, 10.5 Hz, H-2A), 4.15 (dd, J = 2.7, 9.9 Hz, 1H, H-3A), 4.11–4.07 (m, 1H, H-5B), 4.02–3.98 (m, 1H, H-5A), 4.02 (dd, J = 2.3, 9.3 Hz, H-2B), 3.91 (dd, J = 7.0, 8.9 Hz, 1H, H-3B), 3.70–3.55 (d, 1H, H-4B), 3.63 (s, 3H, OCH3), 1.09 (d, J = 6.0 Hz, 3H, H-6B), 0.90 (d, J = 6.3 Hz, 3H, H-6A). 13C NMR (CDCl3, 100 MHz): δ 155.8–114.4 (C–Ar), 95.4 (C-1A), 94.4 (C-1B), 83.8 (C-5A,B), 82.2 (C-3B), 82.5 (C-3A), 78.2 (C-4A), 77.4 (C-4B), 76.0, 74.7, 73.0, 72.6, 70.7 (CH2Ar), 67.0 (C-2B), 63.3 (C-2A), 55.6 (OCH3), 16.6, 16.3 (C-6A,B). HR ESI MS C58H60O10: [M + H]+ calc. 939.4084; found 939.4056.
4-Methoxyphenyl 2-O-benzyl-4,6-O-benzylidene-3-O-(methyl 5 acetamido-7,8,9-tetra-5-N,4-O-carbonyl-3,5-dideoxy-D-glycero-α-D-galacto-non-2-ulopyranosylonate)-D-galactopyranoside 17
Galactoside acceptor 5b (40 mg, 0.08 mmol) and silayl donor 16 (57 mg, 0.1 mmol) were stirred in CH2Cl2 (4 ml) containing 4 Å MS (200 mg) at −60 °C for 15 min. NIS (23 mg, 0.1 mmol) and TfOH (7 μl, 0.08 mmol) were added, and stirring was continued for 30 min at −30 °C then TLC (1:1 toluene–EtOAc) showed the completion of the reaction. The mixture was neutralized with TEA, filtered and concentrated. Chromatography of the residue (cyclohexane–EtOAc) gave 50 mg of product 17 (65%). [α]25D = +34.0° (c 0.22, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.54–6.82 (m, 14H, H–Ar), 5.70 (s, 1H, PhCH), 5.58–5.54 (m, 1H, H-8B), 4.96–4.89 (m, 3H, PhCHa, H-6B, incl. d, 4.88, J = 8.6 Hz, H-1A), 4.80–4.55 (m, 3H, PhCHb, H-6aA, H-4B), 4.49–4.43 (m, 1H, H-7B), 4.39–4.19 (m, 2H, H-3A, 6bA), 4.39–4.19 (dd, J = 2.7, 9.9 Hz, 1H, H-3A), 4.11–4.07 (m, 1H, H-5B), 3.98–3.78 (m, 1H, H-2A,9B, incl. s, 3.79, OCH3), 3.66–3.30 (m, 2H, H-5A,B), 2.89 (dd, J = 3.3, 12.1, H-3eB), 2.20–2.05 (m, 14H, H-3aB, 4 × CH3CO). 13C NMR (CDCl3): δ 171.9, 171.4, 171.0, 169.8, 165.7 (5 × CO), 138.5–126.2 (C–Ar), 102.9 (C-1B), 101.8 (PhCH), 76.9, 76.4, 76.0, 75.5 (PhCH2), 76.4, 74.2, 73.5, 70.4, 69.2 (C-6A), 66.3, 63.2 (C-9B), 60.4, 59.2 (OCH3), 53.7 (C-5C), 52.8 (COOCH3), 36.6 (C-3B), 24.5, 21.1, 20.8, 20.7 (4 × CH3CO). HR ESI MS C46H51NO19: [M + H]+ calc. 922.3134; found 922.3166.
3-(Benzyloxycarbonyl)aminopropyl 2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,3)-2,4-di-O-benzyl-6-O-tertbutyldiphenyl silyl-α-D-mannopyranoside 21
To a solution of acceptor 20 (1 g, 1.26 mmol) and donor 12 (1.04 g, 1.64 mmol) in CH2Cl2 (20 ml) containing preactivated 4 Å MS, TMSOTf (14 μl, 0.08 mmol) was added at −30 °C. The mixture was stirred for 30 min, when TLC (7:3 cyclohexane–EtOAc) showed the completion of the reaction. The mixture was neutralized with TEA, filtered and concentrated. Purification of the residue on silica gel (cyclohexane–EtOAc) gave the product 21 (2.1 g, 94%). [α]25D = +30.0° (c 2.4, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.87–6.25 (m, 39H, H–Ar), 5.52 (br. s, 1H, H-2B), 5.25 (s, 1H, H-1B), 5.13, 5.09 (2 d, J = 11.7 Hz, 2H, CH2Cbz), 4.95–4.51 (m, 11H, 5 × CH2Ph, incl. s, 4.88, H-1A), 4.16–3.77 (m, 8H), 3.76–3.66 (m, 4H), 3.47–3.42 (s, 1H, H-1′b), 3.27–3.19 (m, 2H, H-3′), 2.11 (s, 3H, CH3CO), 1.77–1.72 (m, 2H, H-2′), 1.12 (s, 9H, tBuSi). 13C NMR (CDCl3): δ 163.5, 156.3 (2 × CO), 138.6–127.8 (C–Ar), 99.7 (C-1B), 97.3 (C-1A), 78.8, 78.0, 75.1, 74.9, 74.8, 74.3, 73.7, 73.2, 72.9, 72.3, 72.1, 69.2, 68.9, 66.5 (C-6A), 65.1 (C-6B), 63.1 (C-1′), 38.5 (C-3′), 29.5 (C-2′), 26.8 (tBuSi), 21.0 (CH3CO). HR ESI MS C76H83NO16Si: [M + Na]+ calc. 1268.5531; found 1268.5543.
3-(Benzyloxycarbonyl)aminopropyl 3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,3)-2,4-di-O-benzyl-6-O-tertbutyldiphenylsilyl-α-D-mannopyranoside 22
Compound 21 (1 g, 0.8 mmol) was dissolved in MeOH (10 ml) and NaOMe was added until the pH was 9–10. The mixture was stirred overnight, then neutralized with Dowex H+ and filtered. The filtrate was concentrated and purified on silica gel (cyclohexane–EtOAc) to provide the 2-OH compound 22 (910 mg, 84%). [α]25D = +15.8° (c 2.05, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.85–6.27 (m, 39H, H–Ar), 5.32 (s, 1H, H-1B), 5.20, 5.11 (2 d, J = 11.7 Hz, 2H, CH2Cbz), 4.97–4.57 (m, 11H, 5 × CH2Ph, incl. s, 4.91, H-1A), 4.19–3.90 (m, 9H), 3.80–3.71 (m, 4H), 3.47–3.41 (s, 1H, H-1′b), 3.31–3.21 (m, 2H, H-3′), 2.56 (br. s, 1H, OH), 1.80–1.73 (m, 2H, H-2′), 1.17 (s, 9H, tBuSi).13C NMR (CDCl3): δ 156.4 (CO), 138.6–126.5 (C–Ar), 101.6 (C-1B), 97.5 (C-1A), 80.4, 78.2, 75.0, 74.9, 74.8, 74.5, 73.4, 73.3, 72.4, 72.0, 71.9, 69.4, 68.8, 66.5 (C-6A), 65.3 (C-6B), 63.1 (C-1′), 38.6 (C-3′), 29.6 (C-2′), 26.8 (tBuSi). HR ESI MS C74H81NO12Si: [M + Na]+ calc. 1226.5461; found 1226.5433.3-(Benzyloxycarbonyl)aminopropyl 2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,2)-3,4,6-tri-O-benzyl-α-D-manno pyranosyl-(1,2)-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,3)-2,4-di-O-benzyl-6-O-tertbutyldiphenylsilyl-α-D-mannopyranoside 24
Disaccharide acceptor 22 (1 g, 0.98 mmol) and donor 23 (1 g, 0.81 mmol) were stirred in CH2Cl2 (40 ml) containing 4 Å MS (2 g) at −40 °C for 15 min. After addition of NIS (275 mg, 1.2 mmol) and TfOH (10 μl, 0.11 mmol) stirring was continued for 30 min at −30 °C when TLC (7:3 cyclohexane–EtOAc) showed the completion of the reaction. The mixture was neutralized with TEA, filtered and concentrated. Chromatography of the residue (cyclohexane–EtOAc) gave 1.5 g of 24 (88%). [α]25D = +18.9° (c 2.15, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.77–7.03 (m, 70H, H–Ar), 5.63 (br. s, 1H, H-2D), 5.34 (s, 1H, H-1B), 5.28 (s, 1H, H-2C), 5.23–5.11 (m, 3H, CH2Cbz, NH), 4.98–4.32 (m, 22H, 11 × CH2Ph, incl. s, 4.86, H-1A), 4.16–3.47 (m, 21H), 3.47–3.41 (s, 1H, H-1′b), 3.24–3.09 (m, 2H, H-3′), 2.21 (s, 3H, CH3CO), 1.77–1.71 (m, 2H, H-2′), 1.12 (s, 9H, tBuSi). 13C NMR (CDCl3): δ 170.2, 156.3 (2 × CO), 138.8–126.5 (C–Ar), 101.2 (C-1B), 100.5 (C-1C), 99.4 C-1D), 97.2 (C-1A), 80.3, 79.4, 78.3, 75.2, 75.0, 74.5, 74.7, 74.2, 73.4, 73.0, 72.7, 72.4, 72.0, 71.9, 69.8, 69.2, 68.7, 66.5, 65.2, 63.2 (C-1′), 38.4 (C-3′), 29.6 (C-2′), 26.8 (tBuSi), 21.2 (CH3CO). HR ESI MS C130H141NO24Si: [M + Na]+ calc. 2150.9511; found 2150.9525.
3-(Benzyloxycarbonyl)aminopropyl 2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,2)-3,4,6-tri-O-benzyl-α-D-manno pyranosyl-(1,2)-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,3)-2,4-di-O-benzyl-α-D-mannopyranoside 25
Tetrasaccharide 24 (1.5 g, 0.7 mmol) was dissolved in 30 ml of 1:4 pyridine–dioxane and 70% HF·pyridine was added. After stirring overnight, the mixture was concentrated and purified on silica gel (cyclohexane–EtOAc) to give 25 (1.17 g, 91%). [α]25D = +29.4° (c 0.46, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.44–7.17 (m, 60H, H–Ar), 5.64 (br. s, 1H, H-2D), 5.33 (s, 1H, H-1B), 5.28 (s, 1H, H-2C), 5.19–5.13 (m, 3H, CH2Cbz, NH), 4.92–4.32 (m, 22H, 11 × CH2Ph, incl. s, 4.88, H-1A), 4.18–3.51 (m, 21H), 3.39–3.39 (s, 1H, H-1′b), 3.24–3.13 (m, 2H, H-3′), 2.20 (s, 3H, CH3CO), 1.74–1.60 (m, 2H, H-2′). 13C NMR (CDCl3): δ 170.1, 156.4 (2 × CO), 138.8–126.5 (C–Ar), 101.0 (C-1B), 100.6 (C-1C), 99.5 C-1D), 97.6 (C-1A), 79.4, 79.3, 78.3, 77.9, 75.2, 74.7, 74.2, 73.4, 73.1, 72.8, 72.5, 72.3, 72.2, 72.0, 69.7, 69.9, 68.5, 66.6, 65.2, 62.2 (C-1′), 39.3 (C-3′), 29.6 (C-2′), 21.1 (CH3CO). HR ESI MS C114H123NO24: [M + Na]+ calc. 1912.8333; found 1912.8351.
3-(Benzyloxycarbonyl)aminopropyl 2-O-acetyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,2)-3,4,6-tri-O-benzyl-α-D-manno pyranosyl-(1,2)-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,3)-6-O-[2-O-acetyl-3,4,6-tri-O-benzyl-α-D-manno pyranosyl-(1,2)-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1,2)]-2,4-di-O-benzyl-α-D-mannopyranoside 26
Glycosylation of acceptor 25 (430 mg, 0.35 mmol) and donor 23 (507 mg, 0.46 mmol) as described for 24 yielded hexasaccharide 26 (490 mg, 81%). The product was similar to that described in the literature,25 except for the signals of the linker 3.75–3.72 (m, 1H, H-1′a), 3.47–3.41 (s, 1H, H-1′b), 3.17–3.04 (m, 2H, H-3′), 1.71–1.68 (m, 2H, H-2′). HR ESI MS C170H131NO35: [M + Na]+ calc. 2819.2312; found 2819.2331.Man6
NMR data for this compound were in agreement with those reported in the literature for a similar hexamannoside.25Acknowledgements
We gratefully acknowledge the support of Dr Daniela Proietti with ESI MS analysis.Notes and references
- (a) P. G. M. Wuts and T. W. Greene, Protective groups in organic synthesis, John Wiley and Sons Inc., Hoboken, New Jersey, 2007, p. 370 Search PubMed; (b) K. Jarowicki and P. Kocienski, J. Chem. Soc., Perkin Trans. 1, 1999, 1589 RSC.
- (a) S. Crotti and R. Adamo, Curr. Org. Synth., 2013, 10, 501 CrossRef CAS; (b) X. Zhu and R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900 CrossRef CAS PubMed; (c) J. T. Smooth and A. Demchenko, Adv. Carbohydr. Chem. Biochem., 2009, 62, 161 CrossRef; (d) P. H. Seeberger, Chem. Soc. Rev., 2008, 37, 19 RSC; (e) T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611 CrossRef CAS PubMed.
- J. Xia, J. L. Alderfer, C. F. Piskorz and K. L. Matta, Chem. – Eur. J., 2001, 7, 356 CrossRef CAS.
- V. Wittmann, Angew. Chem., Int. Ed., 2006, 45, 3399 CrossRef CAS.
- L. Lázár, L. Jánossy, M. Csávás, A. B. Herczeg and S. Antusa, ARKIVOC, 2012,(v), 312 Search PubMed.
- A. G. Volbeda, H. A. V. Kistemaker, H. S. Overkleeft, G. A. van der Marel, D. V. Filippov and J. D. C. Codée, J. Org. Chem., 2015, 80, 8796 CrossRef CAS PubMed.
- J. A. Wright, J. Yu and J. B. Spencer, Tetrahedron Lett., 2001, 42, 4033 CrossRef CAS.
- Y. Li, B. Roy and X. Liu, Chem. Commun., 2011, 47, 8952 RSC.
- S. Iacobucci, N. Filippova and M. d'Alaraco, Carbohydr. Res., 1995, 277, 321 CrossRef CAS PubMed.
- Z. Zhang and G. Magnusson, Carbohydr. Res., 1996, 13, 41 CrossRef.
- (a) T. Tanaka, Y. Oikawa, N. Nakajima, T. Hamada and O. Yonemitsu, Chem. Pharm. Bull., 1987, 35, 2203 CrossRef CAS; (b) A. F. Sviridov, M. S. Ermolenko, D. V. Yashunsky, V. S. Borodkin and N. K. Kochetkov, Tetrahedron Lett., 1987, 28, 3839 CrossRef CAS; (c) E. Vedejs, R. A. Buchanan and Y. Watanabe, J. Am. Chem. Soc., 1989, 111, 8430 CrossRef CAS.
- D. B. Werz and P. H. Seeberger, Angew. Chem., Int. Ed., 2005, 44, 6315 CrossRef CAS PubMed.
- S. Nie, W. Li and B. Yu, J. Am. Chem. Soc., 2014, 136, 4157 CrossRef CAS PubMed.
- H. Schachter, Carbohydr. Res., 2009, 244, 1391 CrossRef PubMed.
- S. Goč and M. Janković, Dis. Markers, 2013, 36, 847 CrossRef PubMed.
- L. Y. Lee, M. Thaysen-Andersen, M. S. Baker, N. H. Packer, W. S. Hancock and S. Fanayan, J. Proteome Res., 2014, 13, 4783 CrossRef CAS PubMed.
- V. Y. D. M. Mandal, X. Geng and S. J. Danishefsky, Angew. Chem., Int. Ed., 2004, 43, 2557–2561 CrossRef PubMed.
- M. O. V. Y. Dudkin, X. Geng, M. Mandal, W. C. Olson and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 9560 CrossRef PubMed.
- V. Y. D. X. Geng, M. Mandal and S. J. Danishefsky, Angew. Chem., Int. Ed., 2004, 43, 2562 CrossRef PubMed.
- F. Yamazaki, S. Sato, T. Nukada, Y. Ito and T. Ogawa, Carbohydr. Res., 1990, 201, 31 CrossRef CAS PubMed.
- K.-H. Koo, S. Sarda, X. Xu, J. P. Caulfield, M. R. McNeil, S. W. Homans, H. R. Morris and A. Dell, J. Biol. Chem., 1995, 270, 17114 CrossRef.
- P. I. Abronina, A. I. Zinin, D. A. Romashin, N. N. Malysheva, A. O. Chizhov and L. O. Kononov, Synlett, 2015, 2267 CrossRef CAS.
- C.-H. Hsu, K.-C. Chu, Y.-S. Lin, J.-L. Han, Y.-S. Peng, C.-T. Ren, C.-Y. Wu and C.-H. Wong, Chem. – Eur. J., 2010, 16, 1754 CrossRef CAS PubMed.
- C. T. Tanifum and C.-W. T. Chang, J. Org. Chem., 2009, 74, 634 CrossRef CAS PubMed.
- H.-K. Lee, C. N. Scanlan, C.-Y. Huang, A. Y. Chang, D. A. Calarese, R. A. Dwek, P. M. Rudd, D. R. Burton, I. A. Wilson and C.-H. Wong, Angew. Chem., Int. Ed., 2004, 43, 1000 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00144k |
| This journal is © the Partner Organisations 2016 |