
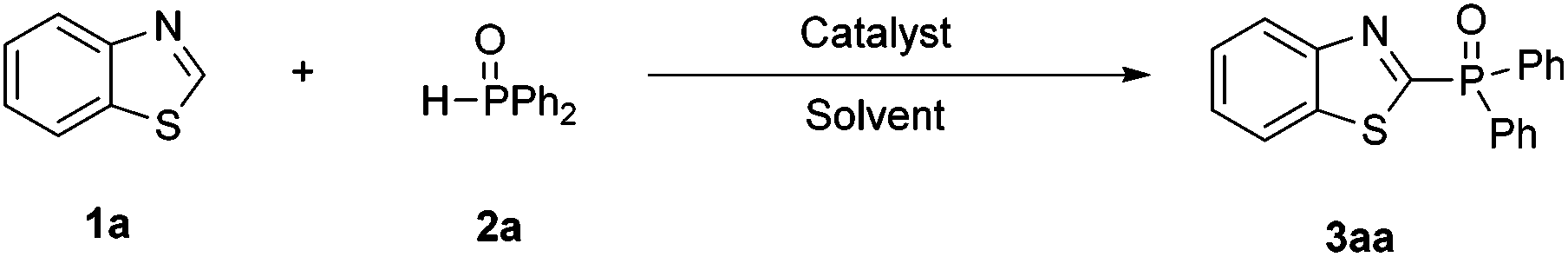
Visible light mediated aerobic radical C–H phosphorization toward arylphosphonates†
Pan
Peng
,
Long
Peng
,
Guangyu
Wang
,
Fangyu
Wang
,
Yi
Luo
and
Aiwen
Lei
*
College of Chemistry and Molecular Sciences, the Institute for Advanced Studies (IAS), Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: aiwenlei@whu.edu.cn
First published on 18th April 2016
Abstract
A visible light mediated radical C–H phosphorization of benzothiazoles with O2 as the sole oxidant was developed towards the synthesis of various 2-diarylphosphoryl benzothiazoles. The phosphonyl radical was identified by EPR experiments. In situ31P NMR experiments showed that Ph2P(O)H mainly converted into the desired product and the oxidative byproduct Ph2P(O)OH.
Arylphosphonates are widely applied in materials science,1 pharmaceutical sciences,2 life sciences3 and catalysis.4 The most common protocols accessing arylphosphonates are transition-metal catalysed cross coupling reactions. Over the past few decades, palladium-,5 nickel-,6 and copper-catalysed7 phosphorization of electrophilic aryl moieties with phosphorus-based nucleophiles has been well investigated. Nevertheless, the pre-functionalization of the electrophilic aryl moieties brings tedious synthetic steps and chemical waste, inevitably restricting their wide application.8 Thus, synthesizing aryl phosphonates by direct C–H phosphorization of arenes with H-phosphonates is desirable. In 2012, Li et al. reported a palladium-catalysed C–H phosphonation of azoles by using persulfate as the oxidant.9 Later on, Zhao's and Wen's group achieved the radical C–H phosphorization of thiazoles by using peroxide or a stoichiometric amount of silver salts respectively.10 However, the use of stoichiometric metal salts or peroxides as the oxidant would result in stoichiometric chemical waste. It is always valuable to achieve direct C–H phosphorization by using a cleaner oxidant to update the existing methods.
As an inexpensive, available and environmentally friendly oxidant, molecular oxygen (O2) has been considered as an ideal oxidant.11 The development of an effective route for C–H functionalization using molecular oxygen as the oxidant under mild conditions is highly desirable. Recently, visible light promoted C–H functionalization using molecular oxygen as the oxidant has also been developed, such as the C–H functionalization of tertiary amines with different nucleophiles,12 intramolecular C–H amination/cyclization,13 C–H thiocyanation of indoles14 and C–H amination of arenes.15 Inspired by these processes, we envisioned that we could achieve aerobic radical C–H phosphorization of arenes using O2 as the sole oxidant via photoredox catalysis (Scheme 1).
 | ||
| Scheme 1 Aerobic radical C–H phosphorization and the plausible mechanism. | ||
Compared to the widely applied iridium and ruthenium photocatalysts, the organic dye Na2Eosin Y is a cheap, environmentally friendly and commercially available photocatalyst.16 On the basis of this, Na2Eosin Y was chosen as the photocatalyst. As illustrated in Scheme 1, Ph2P(O)H could be oxidized with excited Na2Eosin Y* through single-electron transfer to access the Ph2P(O)˙ radical A.17 The excited Na2Eosin Y* was reduced to the Na2Eosin Y˙− radical anion, and then oxidized by O2 to complete the photocatalytic cycle, accompanied by the generation of the O2˙− radical anion. On the other hand, the Ph2P(O)˙ radical A reacted with arenes by radical addition, leading to intermediate B. The rearomatization of intermediate B by oxidation and deprotonation afforded the final arylphosphonates. Besides, the Ph2P(O)˙ radical could react with O2 and convert to the oxidative byproduct Ph2P(O)OH.
To verify the possibility that the Ph2P(O)˙ radical could be accessed through single-electron oxidation with excited Na2Eosin Y* under air, EPR experiments were performed by the addition of the free radical spin trapping agent DMPO (5,5-dimethyl-1-pyrroline N-oxide). As shown in Fig. 1, an EPR signal was identified when DMPO was added to a reaction mixture of diphenylphosphine oxide with Na2Eosin Y under air. Data analysis suggested that the Ph2P(O)˙ radical was generated and then quickly trapped by DMPO, forming a relatively stable radical C (aN = 14.2 G, aH = 19.5 G, aP = 35.6 G).18 This result is consistent with our proposal, indicating that the Ph2P(O)˙ radical could be accessed through single-electron oxidation with excited Na2Eosin Y* under air.
 | ||
| Fig. 1 The electron paramagnetic resonance (EPR) spectra (X band, 9.4 GHz, room temperature). | ||
Based on the above experiments, we chose benzothiazole 1a as the radical acceptor and diphenylphosphine oxide (Ph2P(O)H, 2a) to explore this aerobic C–H phosphorization in the presence of 3 mol% of Na2Eosin Y in CHCl3. To our delight, the desired 2-diphenylphosphinyl benzothiazole 3aa was obtained in 72% yield after 12 h (Table 1, entry 1). As solvents often have a great influence on the radical process, different solvents (such as DMSO, THF, CH3CN, DMF and water) were tested and CHCl3 proved to be the best solvent (Table 1, entries 2–6). It's worth noting that the desired product 3aa was obtained in 63% yield in water (Table 1, entry 6). As previously mentioned, the Ph2P(O)˙ radical would react with O2, which led to extra consumption of Ph2P(O)H. The yield of 3aa was increased to 74% after increasing 2a to 3 equiv. (Table 1, entry 7). Considering that the starting materials remained after the reaction, higher yields were further observed after prolonging the reaction time to 16 h (Table 1, entry 8). Notably, when the photocatalyst was omitted, only 19% yield of the desired product was obtained (Table 1, entry 9). Besides, no reaction occurred (Table 1, entry 10) in the absence of light. And only 5% product was detected when the reaction was performed under a N2 atmosphere (Table 1, entry 11). Those experiments showed that Na2Eosin Y, light and O2 were essential in this transformation.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time | Yieldb |
a Unless otherwise specified, all reactions were carried out using 1a (0.20 mmol), 2a (0.40 mmol) and the catalyst (3 mol%) in solvent (1.0 mL) at rt under 1 atm of air (ballon) and 23 W white light.
b Yield was determined by HPLC analysis using naphthalene as an internal standard, isolated yield in parentheses.
c ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2a (0.6 mmol) was used.
d Reaction was carried out in the dark.
e Under N2 (ballon). 2a (0.6 mmol) was used.
d Reaction was carried out in the dark.
e Under N2 (ballon).
|
||||
| 1 | Na2Eosin Y | CHCl3 | 12 | 72 |
| 2 | Na2Eosin Y | DMSO | 12 | 37 |
| 3 | Na2Eosin Y | THF | 12 | 31 |
| 4 | Na2Eosin Y | CH3CN | 12 | 0 |
| 5 | Na2Eosin Y | DMF | 12 | 9 |
| 6 | Na2Eosin Y | H2O | 12 | 63 |
| 7c | Na2Eosin Y | CHCl3 | 12 | 74 |
| 8c | Na2Eosin Y | CHCl3 | 16 | (85) |
| 9c | — | CHCl3 | 16 | 19 |
| 10c,d | Na2Eosin Y | CHCl3 | 16 | 0 |
| 11c,e | Na2Eosin Y | CHCl3 | 16 | 5 |
To explore the reason why extra two equivalents of Ph2P(O)H were necessary for the transformation, in situ31P NMR spectra were used to monitor the reaction of 1a and 2a under the optimized conditions. As shown in Fig. 2, the target product of 3aa (δ = 21.92 ppm) and Ph2P(O)OH (δ = 27.81 ppm) increased steadily with the decrease of 2a (δ = 23.53 ppm) during the reaction process. These results showed that two extra equivalents of Ph2P(O)H were mainly converted into the oxidative byproduct Ph2P(O)OH.
 | ||
| Fig. 2 In situ NMR spectra of C–H phosphorization. | ||
To gain more insights into the reaction, radical inhibition experiment was carried out. The reaction was completely shut down in the presence of the radical scavenger TEMPO, further indicating that this transformation might go through a radical pathway (for more details, see the ESI†).
The “light/dark” experiments were also performed to explore the importance of light. As shown in Fig. 3, the reaction was totally inhibited in the absence of light and resurrected when the light was on. This result indicated that continuous irradiation with visible light was essential for this photo-catalytic transformation.
 | ||
| Fig. 3 “Light/dark” experiments over the time. | ||
With a better understanding of the reaction mechanism, the compatibility of this transformation was evaluated. As shown in Table 2, benzothiazoles bearing either electron-rich or electron-deficient substituents on the phenyl rings were well tolerated in this protocol, providing the desired products in moderate to excellent yields (3aa–3ha). A range of functional groups, such as methoxy (3ba), methyl (3ca–3da), halogens (3ea–3ga), and nitro substituents (3ha), were well tolerated in this transformation.
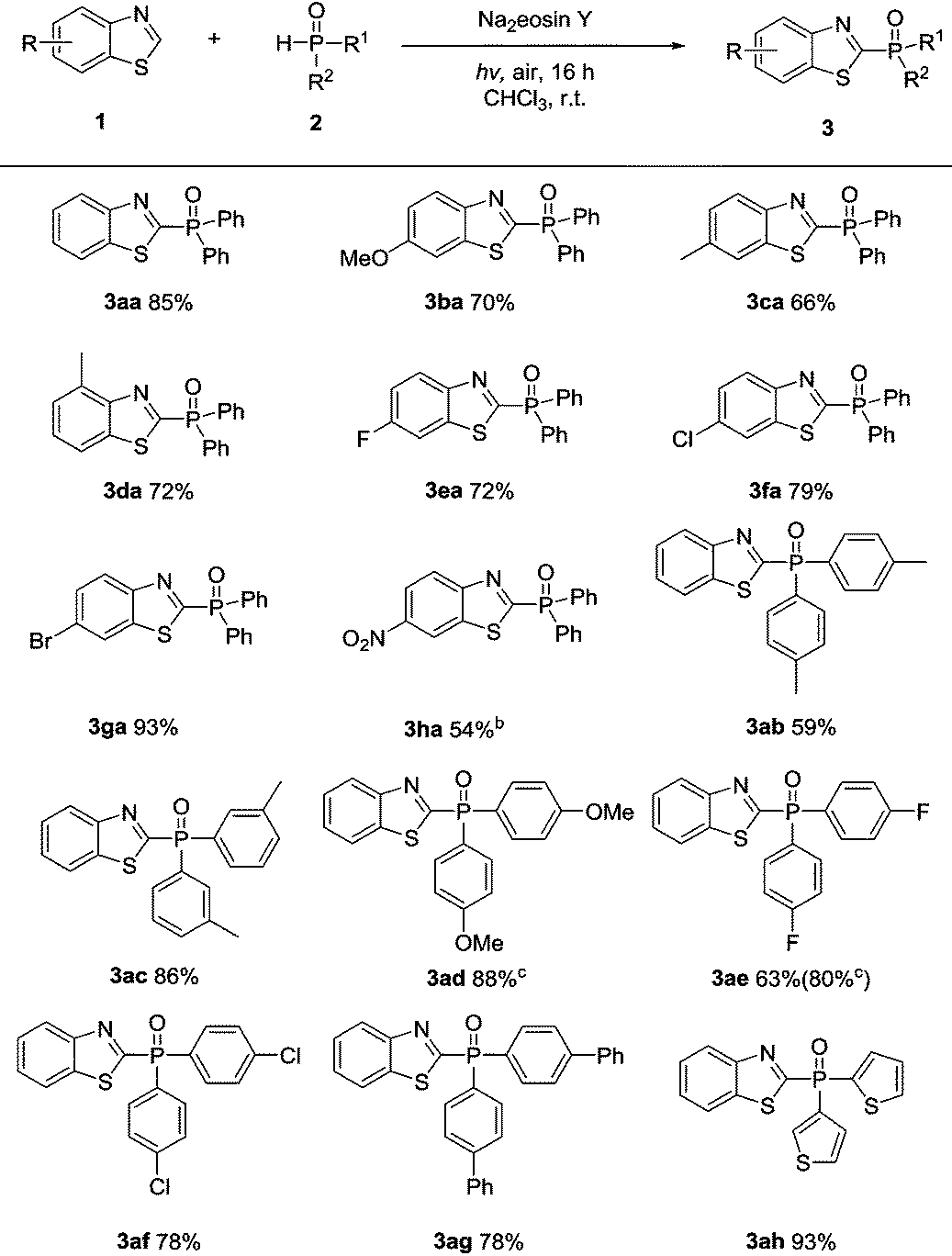
Subsequently, a range of diarylphosphine oxide derivatives were tested. The para- and meta-methyl substituents on the phenyl ring gave the corresponding products 3ab and 3ac in 59% and 86% yields. Treatment of the diarylphosphine oxide involving a para-methoxy group with benzothiazole 1a afforded the desired product 3ad in 88% yield. The halogen groups, including the para-fluoro and para-chloro groups, were also compatible and the corresponding products 3ae and 3af were isolated in 63% and 78% yield respectively. Notably, 80% yield of 3ae could be obtained after increasing the corresponding diarylphosphine oxides to four equivalents. Moreover, diarylphosphine oxide involving para-phenyl reacted with benzothiazole 1a smoothly to afford the corresponding product 3ag in 78% yield. Notably, a heterocyclic phosphine oxide compound was also well tolerated, giving product 3ah in 93% yield.
However, when diethyl phosphite was applied to this protocol, the reaction didn't occur. We speculated that diethyl phosphite might be difficult to be oxidized by excited Na2Eosin Y* due to its higher oxidative potential than diphenylphosphine oxide. Therefore, a much stronger photo-oxidant,19 9-mesityl-10-methylacridinium perchlorate, was selected instead of Na2Eosin Y. As shown in Scheme 2, the desired product could be obtained in 10% HPLC yield.
 | ||
| Scheme 2 Substrate scope of diethyl phosphite. | ||
Conclusions
We have developed a visible light mediated aerobic radical C–H phosphorization of benzothiazoles with H-phosphonates. Various 2-diarylphosphoryl benzothiazoles were synthesized with a wide functional group tolerance including halide, nitryl and thienyl groups. EPR experiments showed that the phosphonyl radical was generated through single-electron oxidation with excited Na2Eosin Y* under air. In situ31P NMR studies indicated that Ph2P(O)H mainly converted into the final product and the oxidative byproduct Ph2P(O)OH. The “light/dark” experiments indicated that continuous irradiation with visible light was essential for this photo-catalytic transformation. Further investigations of the aerobic radical process are ongoing in our laboratory.This work was supported by the 973 Program (2012CB725302), the National Natural Science Foundation of China (21390400, 21520102003, 21272180, 21302148), the Hubei Province Natural Science Foundation of China (2013CFA081), the Research Fund for the Doctoral Program of Higher Education of China (20120141130002), and the Ministry of Science and Technology of China (2012YQ120060). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated.
Notes and references
- (a) J. S. Swensen, E. Polikarpov, A. Von Ruden, L. Wang, L. S. Sapochak and A. B. Padmaperuma, Adv. Funct. Mater., 2011, 21, 3250–3258 CrossRef CAS; (b) C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed; (c) H.-H. Chou and C.-H. Cheng, Adv. Mater., 2010, 22, 2468–2471 CrossRef CAS PubMed; (d) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- (a) R. F. Roush, E. M. Nolan, F. Löhr and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 3603–3609 CrossRef CAS PubMed; (b) P. Lassaux, M. Hamel, M. Gulea, H. Delbrück, P. S. Mercuri, L. Horsfall, D. Dehareng, M. Kupper, J.-M. Frère, K. Hoffmann, M. Galleni and C. Bebrone, J. Med. Chem., 2010, 53, 4862–4876 CrossRef CAS PubMed.
- M. D. Charles, P. Schultz and S. L. Buchwald, Org. Lett., 2005, 7, 3965–3968 CrossRef CAS PubMed.
- (a) Y. Li, L.-Q. Lu, S. Das, S. Pisiewicz, K. Junge and M. Beller, J. Am. Chem. Soc., 2012, 134, 18325–18329 CrossRef CAS PubMed; (b) Y. Guo, H. Fu, H. Chen and X. Li, Catal. Commun., 2008, 9, 1842–1845 CrossRef CAS; (c) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed; (d) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed; (e) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336–345 CrossRef CAS PubMed.
- A. L. Schwan, Chem. Soc. Rev., 2004, 33, 218–224 RSC.
- (a) X. Zhang, H. Liu, X. Hu, G. Tang, J. Zhu and Y. Zhao, Org. Lett., 2011, 13, 3478–3481 CrossRef CAS PubMed; (b) Y.-L. Zhao, G.-J. Wu and F.-S. Han, Chem. Commun., 2012, 48, 5868–5870 RSC; (c) H.-Y. Zhang, M. Sun, Y.-N. Ma, Q.-P. Tian and S.-D. Yang, Org. Biomol. Chem., 2012, 10, 9627–9633 RSC.
- (a) S. Thielges, P. Bisseret and J. Eustache, Org. Lett., 2005, 7, 681–684 CrossRef CAS PubMed; (b) C. Huang, X. Tang, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2006, 71, 5020–5022 CrossRef CAS PubMed.
- C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed.
- C. Hou, Y. Ren, R. Lang, X. Hu, C. Xia and F. Li, Chem. Commun., 2012, 48, 5181–5183 RSC.
- (a) H.-J. Zhang, W. Lin, Z. Wu, W. Ruan and T.-B. Wen, Chem. Commun., 2015, 51, 3450–3453 RSC; (b) X.-L. Chen, X. Li, L.-B. Qu, Y.-C. Tang, W.-P. Mai, D.-H. Wei, W.-Z. Bi, L.-K. Duan, K. Sun, J.-Y. Chen, D.-D. Ke and Y.-F. Zhao, J. Org. Chem., 2014, 79, 8407–8416 CrossRef CAS PubMed.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC.
- (a) J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed; (b) J. Xuan, Y. Cheng, J. An, L.-Q. Lu, X.-X. Zhang and W.-J. Xiao, Chem. Commun., 2011, 47, 8337–8339 RSC; (c) S. Cai, X. Zhao, X. Wang, Q. Liu, Z. Li and D. Z. Wang, Angew. Chem., Int. Ed., 2012, 51, 8050–8053 CrossRef CAS PubMed; (d) Y.-Q. Zou, L.-Q. Lu, L. Fu, N.-J. Chang, J. Rong, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171–7175 CrossRef CAS PubMed; (e) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed.
- (a) S. Maity and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 9562–9566 CrossRef CAS PubMed; (b) Y. Cheng, J. Yang, Y. Qu and P. Li, Org. Lett., 2012, 14, 98–101 CrossRef CAS PubMed.
- W. Fan, Q. Yang, F. Xu and P. Li, J. Org. Chem., 2014, 79, 10588–10592 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688–6699 RSC.
- (a) W.-J. Yoo and S. Kobayashi, Green Chem., 2013, 15, 1844–1848 RSC; (b) J. Xuan, T.-T. Zeng, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. – Eur. J., 2015, 21, 4962–4965 CrossRef CAS PubMed.
- Y. Sueishi and Y. Nishihara, Chem. Res. Chin. Univ., 2000, 16, 313–319 CAS.
- (a) N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed; (b) T. Hering, T. Slanina, A. Hancock, U. Wille and B. Koenig, Chem. Commun., 2015, 51, 6568–6571 RSC; (c) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC; (d) K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715–722 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00049e |
| This journal is © the Partner Organisations 2016 |