
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoding growth inhibitory associated pathways of xenometal–siderophore antibiotic conjugates in S. aureus†
Axia
Marlin
*ab,
Minhua
Cao
ab,
Joelle
El Hamouche
a,
Owen
Glaser
ab and
Eszter
Boros
 *ab
*ab
aDepartment of Chemistry, Stony Brook University, Stony Brook, New York 11790, USA
bDepartment of Chemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA. E-mail: eboros@wisc.edu; amarlin@wisc.edu
First published on 17th March 2025
Abstract
Pathogenic Staphylococcus aureus causes most infectious disease related deaths in the developed world. Continuously evolving resistance to clinically approved antibiotics and combination therapies limits treatment efficacy; new strategies that evade and slow resistance or produce resistant mutants with reduced fitness are needed. We employ antibiotics conjugated to bacterially recognized siderophores to potentiate their efficacy. Acting as a Trojan horse, the siderophore antibiotic conjugates efficiently deliver the antibiotic inside the bacterial cytoplasm by hijacking the iron transport system pathways which are crucial for bacterial survival. Here, we investigated the mechanism of action of gallium xenometallomycins (siderophore antibiotic conjugates incorporating non-endogenous metal ions), Ga–DFO–Cip and Ga–LDFC–Cip, which have demonstrated high potency compared to the parent antibiotic's efficacy in vitro in S. aureus infection. Employing physicochemical, synthetic and transcriptomic analysis studies, this work reveals that kinetically inert, gallium-containing xenometallomycins targeting cytoplasmic bacterial targets impart differential resistance and gene expression profiles when compared to their parent antibiotic in S. aureus bacterial strains. Both Ga–DFO–Cip and Ga–LDFC–Cip effectively disrupt iron–siderophore biosynthesis and uptake machinery. We affirm our results with the radioactive surrogate 67/68Ga–DFO–Cip and demonstrate that the bacterial uptake in Ga–DFO–Cip-resistant S. aureus strains is impaired, leading to diminished compound accumulation in vitro and in vivo.
Introduction
S. aureus (SA) is a Gram-positive pathogen that causes most of the severe and lethal hospital acquired infections in the developed world. This pathogen is responsible for fatal pathologies such as pneumonia, meningitis and toxic shock syndrome.1–4S. aureus infections remain challenging to treat because of the bacterium's ability to persist and adapt. Antibiotic overuse, rapid replication and evolution have promoted the emergence of drug resistant strains of S. aureus worldwide.5,6 Therefore, new antibiotic treatment strategies represent an urgent clinical need.Bacterial virulence is closely associated with the acquisition of essential nutrients such as iron. In human hosts, iron is tightly controlled when sequestered, transported, and stored. Thus, pathogenic bacteria must mobilize iron from their host environment for survival. To do so, most bacterial pathogens produce siderophores, iron-scavenging small molecules for iron sequestration and internalization.7,8 The corresponding iron complexes are internalized via selective and active membrane transport. The siderophore uptake machinery of bacteria is complex and not yet fully understood; a growing number of membrane translocators have been characterized.9–15
The iron–siderophore mediated uptake pathway can provide selective delivery of antimicrobial agents. Antibiotics conjugated to siderophores take advantage of the bacterial iron assimilation pathway, enhancing uptake to potentiate the antibiotic's antibacterial effect (Fig. 1). Potent metal-free (apo-) and pro-drug versions of such siderophore–antibiotic conjugates were introduced by Miller and Nolan.16,17 Cefiderocol, a bidentate catechol–cephalosporin conjugate, was approved by the Federal Drug Administration (FDA) in 2019 (for structures, see Fig. 2).18–21
 | ||
| Fig. 1 Transport internalization pathway for gallium hydroxamate-type siderophore–ciprofloxacin conjugate in wt S. aureus using active uptake machinery. Gallium-mediated mode of action as well as ciprofloxacin-induced toxicity at the molecular level remains unknown and is the subject of this work. | ||
 | ||
| Fig. 2 Chemical structure of cefiderocol, an FDA approved siderophore–antibiotic conjugate, ciprofloxacin (Cip), previously investigated siderophore–antibiotic conjugates, and gallium-based compounds showing growth inhibitory effects on bacteria studied in this work. Red: siderophore, green: linker, and blue: ciprofloxacin or ampicillin. | ||
The recognition and import of siderophore–antibiotic conjugates via selective transmembrane transport requires binding and transposition of a metal-bound species that closely mimics the iron complex. However, an increase in iron concentration in the bacterial growth media results in diminished potency of siderophore–antibiotic conjugates in in vitro assays.22,23 Our group and others have shown that the displacement of Fe(III) by the non-endogenous xenometal Ga(III) not only recovers the growth inhibitory effect but can also significantly potentiate the corresponding siderophore–antibiotic drug conjugate.24–29 Among xenometals of interest, Ga(III) represents the best Fe(III) mimic due to its comparable ionic radius (Ga: 0.76 A; Fe: 0.78 A) and chemical hardness η (Ga: 17; Fe: 12).30 Lack of redox activity under physiological conditions renders Ga(III) unable to recapitulate the biological function of Fe(II/III).
Our group has recently demonstrated that gallium–siderophore–ciprofloxacin (Cip) conjugates Ga–DFO–Cip and Ga–LDFC–Cip (Fig. 2) can outperform the parent antibiotic ciprofloxacin in inhibiting growth of S. aureus in vitro and in vivo.31,32 While this finding is encouraging and motivates further structure–activity relationship (SAR) campaigns, the mechanism of action and source for potentiation of those xenosideromycins in S. aureus are not known (Fig. 1). Previous literature reports hypothesized that the therapeutic effect of Ga(III) compounds arises from Fe(III) substitution in iron-dependent cofactors involved in electron transport cascades and redox-dependent Fe(III) enzymes,24,26,33 while others suggest that Ga(III) selectively alters cellular respiration processes.34
To further improve the in vitro potency and in vivo efficacy for S. aureus infection, a better understanding of the mechanism of action of this unique compound class is required. Specifically, (1) what role do complex inertness and thermodynamic stability play? (2) Is cytoplasmic delivery of Ga(III) required to enhance potency? (3) Does resistance to the parent antibiotic also confer resistance to xenometallomycins? (4) Does the presence of Ga produce unique transcriptomic profiles? (5) How do xenometallomycin-induced mutations impact compound internalization? In this study, we have compiled an extensive set of characterization data to address these questions within the context of S. aureus infection.
Results and discussion
Compound library – design and synthesis
To date, our group has focused on hydroxamate-based siderophores as they confer potency against S. aureus bacterial strains.31,32,35 We selected deferoxamine (DFO) and desferrichrome (DFC) siderophore structures inspired by the natural product sideromycins, salmycin and albomycin; we previously hypothesized that these siderophores retain good affinity for Ga(III) when compared to Fe(III) and are comparatively straightforward to functionalize, using the natural product precedent as a molecular design road map. These conjugates have been shown to effectively retain and enhance the antibiotic potency of Cip in S. aureus. Chemical synthesis of DFO–Cip and LDFC–Cip (Fig. 2), two conjugates incorporating the fluoroquinolone antibiotic ciprofloxacin, was achieved in accordance with previously reported procedures.31,32 Ciprofloxacin and other fluoroquinolone antibiotics rely on cytoplasmic delivery as they target the DNA-gyrase.36–39 To probe the role of the antibiotic, we synthesized the β-lactam containing, deferoxamine-linked ampicillin conjugate DFO–Amp (Fig. 2, for synthesis and characterization, see the ESI†). The chemical synthesis was conducted using sequential, solution-phase amide bond conjugations of building blocks. In our hands, incorporation of the siderophore building block was significantly improved by pre-chelation of Fe(III) or Ga(III), which efficiently masks the otherwise hydrolytically sensitive hydroxamate functional groups and reduces the number of steps to generate the final target construct.Relative stability of Ga(III) and Fe(III) complexes
To form sufficiently inert metal coordination complexes, the chelator must be carefully matched and selected according to the coordinative binding preferences, thermodynamic stability and pH dependent speciation of the corresponding metal ion. In contrast to non-metallated Trojan horse antibiotics such as cefiderocol, which relies on local Fe(III) pools at the site of infection to translocate to the bacterial periplasm,40 xenometal cargos must be pre-chelated and form in vivo compatible, thermodynamically stable complexes. In this regard, hexadentate motifs are preferred over 4- or 2-dentate systems. Deferoxamine (DFO) and desferrichrome (DFC) form thermodynamically stable Fe(III) complexes as evidenced by reported log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K values of 31.3 and 29.07 with DFO and DFC respectively. The gallium complexes have logK values of 28.65 and 27, respectively.41–44 In addition to thermodynamic stability, high relative kinetic inertness is also desirable; as such, rapid dechelation to reach equilibrium would result in immediate loss of the metal payload in vitro and in vivo. To probe the effect of functionalization on the thermodynamic stability and kinetic inertness of the corresponding constructs, we conducted ligand exchange experiments with a 1000-fold excess of ethylenediaminetetraacetate (EDTA). Relative complex half-lives (t1/2), rates of transchelation (k1) and KFe/KGa values are shown in Table 1, as determined by these transchelation experiments and derived from the thermodynamic stability constants for Fe(EDTA) and Ga(EDTA).43
K values of 31.3 and 29.07 with DFO and DFC respectively. The gallium complexes have logK values of 28.65 and 27, respectively.41–44 In addition to thermodynamic stability, high relative kinetic inertness is also desirable; as such, rapid dechelation to reach equilibrium would result in immediate loss of the metal payload in vitro and in vivo. To probe the effect of functionalization on the thermodynamic stability and kinetic inertness of the corresponding constructs, we conducted ligand exchange experiments with a 1000-fold excess of ethylenediaminetetraacetate (EDTA). Relative complex half-lives (t1/2), rates of transchelation (k1) and KFe/KGa values are shown in Table 1, as determined by these transchelation experiments and derived from the thermodynamic stability constants for Fe(EDTA) and Ga(EDTA).43
| t 1/2 (min) | k 1 (min−1) | logKLM |
|
|---|---|---|---|
| Fe–DFO–Cip | 82.66 | 8.39 × 10−3 | 26.89 |
| Ga–DFO–Cip | 60.81 | 1.14 × 10−2 | 26.86 |
| Fe–LDFC–Cip | 10.34 | 6.70 × 10−2 | 28.10 |
| Ga–LDFC–Cip | 57.96 | 1.20 × 10−2 | 27.10 |
In accordance with reported literature values for the corresponding thermodynamic stabilities of DFO and DFC complexes, we observe enhanced thermodynamic stability and kinetic inertness for Fe(III) complexes over Ga(III) complexes. However, functionalization of DFO complexes appears to reduce thermodynamic stability by 4 and 1 orders of magnitude for DFO–Cip and LDFC–Cip respectively. Our data set also demonstrates that kinetic inertness and thermodynamic stability are not correlated, with the Fe–LDFC–Cip complex showing rapid dechelation with half-lives of 10 minutes and below, whereas the M–DFO–Cip series exhibits improved inertness with 60–82 min half-lives. This corroborates previous kinetic inertness and metabolite analysis data obtained with radiochemically tagged analogues and metabolite analyses conducted previously.31,32
Growth inhibition in wt and resistance induced S. aureus
The in vitro potency of the conjugates and their metal complexes was assessed by a conventional microbroth dilution assay, which quantifies the Minimum Inhibitory Concentration (MIC98). The MIC98 was recorded as the lowest compound concentration required to inhibit at least 98% of bacterial growth (OD600 < 0.02) at 16 hours of incubation in comparison to the OD600 of the negative control wells (ESI eqn (1) and Section 3.2†).To determine the impact of the metal chelator and ion separately from the conjugate constructs, we measured the growth inhibitory action of the metal ion citrate salts and siderophore complexes separately (ESI Fig. S23†). In accordance with our previous work on Ga–LDFC and Ga–DFO, Ga–DFC complexes also do not show any significant growth inhibitory activity, indicating that incorporation of an antibiotic small molecule drug is required to harness metal-complex mediated potentiation of growth inhibitory action. Indeed, both Ga–DFO–Cip and Ga–LDFC–Cip show improved MIC98 when compared with the parent antibiotic Cip targeting the cytoplasmic topoisomerase in S. aureus (Fig. 3, Table 2). The corresponding apo- and Fe(III) complexes show comparable or diminished potency, respectively. The DFO–Amp series, which incorporates the cell-wall targeting β-lactam ampicillin, recapitulated previous trends: Ga–DFO–Amp shows a significantly enhanced MIC98 when compared to the corresponding Fe–DFO–Amp complex; however, both compounds remain orders of magnitude less potent when compared to the parent antibiotic.
 | ||
| Fig. 3 Top panel: MIC assay plots of (A) DFO–Cip series, (B) LDFC–Cip series, and (C) M–DFO–Amp in wt SA. Bottom panel: MIC assay plots of (D) DFO–Cip series, (E) LDFC–Cip series and (F) DFO–Amp series in Cip-Res SA. | ||
| S. aureus wt | S. aureus Cip res | S. aureus Ga–DFO–Cip res | S. aureus Ga–LDFC–Cip res | |
|---|---|---|---|---|
| Ciprofloxacin (Cip) | 0.94 | 75 | 3.8 | 3.8 |
| Ampicillin (Amp) | 1.9 | 1.9 | 1.9 | 1.9 |
| DFO–Cip | 3.8 | 15 | >30 | 15 |
| Fe–DFO–Cip | 30 | 15 | >30 | 30 |
| Ga–DFO–Cip | 0.94 | 0.094 | 38 | 1.9 |
| LDFC–Cip | 15 | 7.5 | GP | N/D |
| Fe–LDFC–Cip | 7.5 | GP | GP | N/D |
| Ga–LDFC–Cip | 0.094 | 3.8 | 30 | 150 |
| Fe–DFO–Amp | 30 | GP | >30 | GP |
| Ga–DFO–Amp | 15 | 15 | 15 | 15 |
To demonstrate that the co-delivery of both molecular components must occur via covalent linkage, we also probed the growth inhibitory activity of mixtures of non-functionalized complexes with the native drug. Under these conditions, mixtures of Ga–DFO/Cip and Ga–LDFC/Cip showed no enhanced growth inhibition but reproduced the growth inhibitory capacity of ciprofloxacin (ESI, Fig. S29, and S30†). It is plausible that the siderophore-mediated uptake of the construct increases the total accumulation of ciprofloxacin, resulting in a net increase in delivered antibiotics.
As a first approach to probe if ciprofloxacin and our synthetic constructs exerted growth inhibition using the same mechanisms, we generated Cip, Ga–DFO–Cip, and Ga–LDFC–Cip resistant S. aureus bacterial strains. This was achieved by culturing and repetitive passaging of wt S. aureus in the presence of increasing, sub-MIC amounts of the corresponding antibiotic agent under iron-limiting conditions.45,46 Using this approach, we achieved an increase in the MIC98 of each compound by at least two orders of magnitude within 2–10 passages (ESI Fig. S24–S26†). Ciprofloxacin resistance was established at a MIC98 of 75 μM, compared to a MIC98 of 0.94 μM in the wt S. aureus strain (Table 2 and Fig. S24†). Similarly, the MIC98 increased from 0.94 to 38 μM and 0.094 to 150 μM for Ga–DFO–Cip and Ga–LDFC–Cip, respectively (Table 2, Fig. S25 and S26†). These three strains subsequently provided the basis for the cross-resistance studies. Ciprofloxacin-resistant S. aureus (S. aureus-Cip-res) confers decreased efficacy of Ga–LDFC–Cip (MIC98 = 3.8 μM), when compared to the wt strain, whereas the strain becomes sensitized to Ga–DFO–Cip (MIC98 = 94 nM), indicating non-ciprofloxacin associated mechanisms of growth inhibition. The lack of cross-resistance indicates that resistance mechanisms that inhibit parent drug uptake do not correlate with those of the Ga–DFO–Cip conjugate. Control experiments with DFO–Cip and Fe–DFO–Cip indicate that Ga(III) is required to observe the enhanced potency, while ampicillin and Ga–DFO–Amp show no difference in efficacy. Conversely, the inverse relationship is not observed in cross-resistance experiments with the Ga–DFO–Cip resistant strain (S. aureus GaDFO–Cip-res), with Cip and Ga–LDFC–Cip also showing diminished potency (3.8 and 30 μM, respectively). Finally, the MIC98 of Ga–DFO–Cip and Cip only decreases by 2–4-fold in efficacy in the Ga–LDFC–Cip resistant strain (S. aureus GaLDFC–Cip-res). Finally, the induced resistance mechanisms did not impact the potency of Amp or Ga–DFO–Amp. This further points to resistance mechanisms that do not impact bacterial cell wall composition or emergence of beta-lactamases.
Transcriptomic analysis via RNA sequencing of wt S. aureus under iron-limited conditions
While induction of resistance in bacteria can provide insight into the mechanism of action of novel antibiotic drugs, we also sought to understand what biochemical signature each compound induced in treated S. aureus. To this end, we employed a bottom-up approach via RNA-Sequencing (RNA-Seq). RNA-Seq has become the gold standard tool to study the bacterial transcriptome.47–50 Subjected to antibiotics at sub-MIC concentrations, extensive transcriptional changes can be detected to determine the impact of antibiotics on the expression of trans-membrane transport proteins, structural components of the bacterial cell envelope, DNA damage/repair mechanisms and nutrient homeostasis.One of the challenges in implementing RNA-Seq for our compound class is introduced by iron-depleted assay conditions. This is employed to provide a more host-representative growth environment where metal ions, especially Fe(III), are tightly controlled. Therefore, we first probed changes in gene expression of S. aureus under iron-replete and iron-limited conditions. Differential gene expression analysis plots show the top significant differentially expressed genes with p value < 0.05 experiencing up (red) or down (blue) regulation.
Fig. 4A shows gene expression changes of S. aureus under iron depletion alone. Our results indicate that the heme efflux ATPase system (hrtA/hrtB) and ferritin like protein (ftnA) (Fig. 4A) are downregulated to conserve iron resources within the bacterial cytoplasm.51–53 Accordingly, Fe-dependent nitrate respiration is also downregulated (narGJ). Genes involved in endogenous siderophore synthesis (sbnC) and iron–siderophore uptake (yclQ/yhfQ) are upregulated (Fig. 4A). We next probed the effect of ciprofloxacin on gene expression in iron limited media. Complementary analysis of the differential expression levels further helps to distinguish effects induced by iron insufficiency versus those induced by the presence of ciprofloxacin, which binds DNA gyrase and inhibits topoisomerase IV.36,38,39 As a part of the bacterial SOS responses activated by ciprofloxacin exposure, genes involved in pyrimidine biosynthesis (pyrAB, pyrC, pyrB, pyrF, and pyrP) are upregulated (Fig. 4B),54–56 while hexose transport via the two-component pathway (uhpABC) that regulates the expression of the hexose phosphate transporter uphT is downregulated.57–59 Treatment with ciprofloxacin decreases the uhpT gene transcription, suggesting that the bacteria are unable to maintain survival through hexose phosphate entry. In summary, these control data sets indicate that under iron-depleted conditions, ciprofloxacin down-regulates genes involved in nucleotide synthesis, while iron storage and iron acquisition are up-regulated in the background.
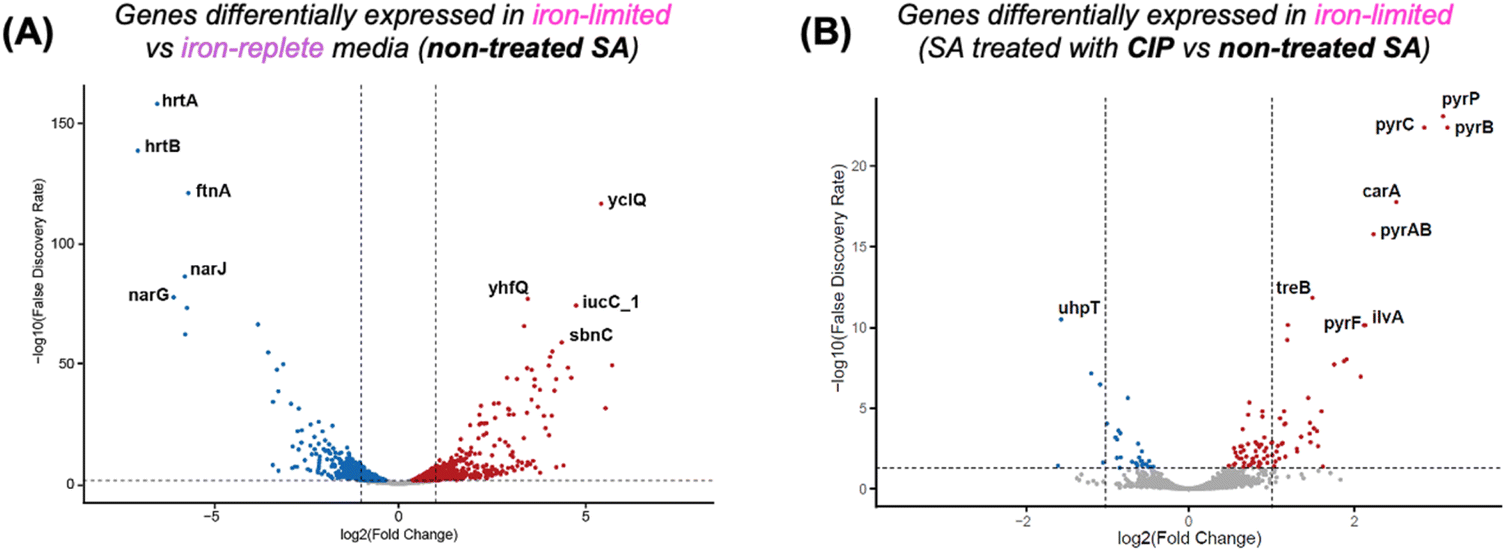 | ||
| Fig. 4 Differential gene expression analysis in non-treated S. aureus (SA) or SA treated with ciprofloxacin. Significantly up- and down-regulated genes (p < 0.05) are shown in red and blue, respectively. Volcano plot of the genes that were differentially expressed in (A) iron limited versus iron replete media in non-treated SA or (B) SA treated with Cip under iron-limited conditions. | ||
Impact of xenometallomycins on S. aureus transcriptomics under iron-limiting conditions
Following our analysis of baseline transcriptomic changes in iron-depleted media, we conducted RNA-Seq analysis of wt S. aureus when exposed to sub-MIC98 concentrations of the sideromycins and metal sideromycins. Following exposure to the DFO–Cip and LDFC–Cip compound series, we observed significant transcriptional changes that can be grouped into 8 broad categories including nucleotide metabolism, ribosomal activity, nitrate and amino acid metabolism, siderophore transport and biosynthesis, heme and saccharide transport. Fig. 5 summarizes our findings, with data grouped separately for the DFO–Cip and LDFC–Cip compound class and in direct comparison with ciprofloxacin. Genes responsible for protein expression that employ heme or other Fe-complexes as cofactors or substrates are highlighted in red. All compounds upregulate genes associated with the mechanism of action of ciprofloxacin under iron-restricted conditions, specifically nucleotide metabolism (pyrB, pyrC, pyrP, pyrAB, and pyrF) including umuC, a low-fidelity DNA polymerase as part of the SOS response commonly observed under fluoroquinolone stress.60–62 All compounds also downregulate NADH-dependent formate dehydrogenase (Fdh), implicated in biofilm formation, and regulated down-stream by Fur under iron-limiting conditions (Fig. 5).63 | ||
| Fig. 5 Heat-map representation of differential gene expression in the presence of sub-MIC98 quantities of sideromycins investigated in this study. Red refers to upregulated genes and blue to down-regulated genes within the heat map. Genes are grouped in accordance with their role within the bacterial organism. Genes in the red box require Fe-containing cofactors or substrates to execute function, such as Fe-heme, or Fe-complexed siderophores. | ||
Major differences are found in the presence of Fe–DFO–Cip and Fe–LDFC–Cip when compared with all other data sets. We observe decreased expression of siderophore transport, biosynthesis and heme import genes (feuC, yusV, ylcQ, fhuD2, isdD, isdC, and isdE), while iron storage genes, nitrogen metabolism (narH, narG, nasD, nasF and narI) and heme-dependent protein expression increase. These changes are notably absent in the presence of the apo-siderophores DFO–Cip and LDFC–Cip and diminished in the presence of Ga–DFO–Cip and Ga–LDFC–Cip. The introduction of the Fe-compounds mimics Fe-sufficient states, which downregulates endogenous siderophore production and uptake, rationalizing why we observe a decrease in efficacy of all Fe-complexes tested to date.
The RNA-Seq data set discussed does not single out differential up/down regulation of genes that arise uniquely in the presence of the gallium complexes. Few genes of interest indicate that the SOS response is activated in the presence of Ga–DFO–Cip (up-regulation of lexA and umuC). Both Ga–DFO–Cip and Ga–LDFC–Cip downregulate rplS (large ribosomal subunit protein bL19), which could indicate diminished efficacy in protein synthesis and thus decreased fitness. Diminished heme import (decrease in expression of isdD, isdC, and isdB), decrease in staphyloferrin synthesis (sbnBCEH) paired with retained expression of the siderophore import machinery and iron storage (fecDC for Ga–DFO–Cip and ftnA for Ga–LDFC–Cip) contributes to the relative success of these compounds in internalization and growth inhibition.
Differentially expressed genes in resistant S. aureus strains
We next pursued differential gene expression analysis in the three-resistance induced S. aureus strains. Fig. 6A displays the corresponding heat-map representation of differential gene expression. Purine biosynthesis (pucG) and amino-acid metabolism related proteins, such as hisB, glnQ, and gltB, are very significantly more down-regulated when compared in the Ga–DFO–Cip and Ga–LDFC–Cip resistant strains, when compared to the ciprofloxacin-resistant background. While none of these proteins contain Fe-cofactors or are known to correlate with iron homeostasis, histidine and glutamate are building blocks of de novo heme biosynthesis in S. aureus.64–67 Further evidence for involvement of heme homeostasis is provided by the strongly upregulated isdG gene in GaDFO–Cip-resistant S. aureus, which encodes for a heme oxygenase that promotes the degradation of heme with subsequent release of iron. Simultaneous downregulation of bacterial ferritin (ftn) further indicates that the organism is using numerous mechanisms to mobilize cytoplasmic Fe(III).68 Additional analysis of the transcriptional profile indicates that each resistant strain shares <40% commonly up- and down-regulated genes with the other two strains (Fig. 6B and C). In addition to well-characterized proteins, approximately 10% of proteins with unknown function were significantly contributing to the compound's unique transcriptomic signature. | ||
| Fig. 6 RNA-Seq analysis of the induced resistant strains. (A) Heat-map representation of differential gene expression in the resistant induced strains (SACip-res, SAGaDFO-res, and SAGaLDFC-res) compared to the wt SA strain. Red refers to upregulated genes and blue to down-regulated genes within the heat map. Genes are grouped in accordance with their role within the bacterial organism. Genes in the red box require Fe-containing cofactors or substrates to execute their function, such as Fe-heme or Fe-complexed siderophores. (B) Venn diagram and a table summarizing up-regulated genes in all three colonies that are above the set threshold of the analysis (p-value ≥ 0.05). (C) Venn diagram and a table summarizing down-regulated genes in all three colonies that are above the set threshold of the analysis (p-value ≥ 0.05). | ||
Mutations observed in ciprofloxacin, GaDFO–Cip and GaLDFC–Cip-resistant S. aureus
In addition to up- and downregulation of genes, we analyzed sequencing data to identify sites of mutations uniquely induced in each of the resistant strains when compared with the parent wt S. aureus strain. Using variant analysis, we identified SNPs (single nucleotide polymorphisms), insertions, deletions or structural variations (INDEL).69Fig. 7A–C list select unique and shared mutations for each resistant strain (for a complete list see also ESI Tables 5–7†). The mutations detected in the ciprofloxacin-resistant strain show numerous point mutations to ribosomal RNA (16S and 23S, entries 8 and 15) and a Snf2 like protein with helicase activity (Fig. 7A, entry 11).70 Additionally, we also detected a single point mutation in an additional protein with sequence homology to helicases (Fig. 7A, entry 13). Of note, we did not observe mutation of topoisomerase which is a more typically observed resistance mechanism to ciprofloxacin in S. aureus;71 however, mutations that affect ribosome assembly have been implicated with exposure to multiple stressors (e.g. Fe-starvation and non-toxic levels of antibiotics) and may contribute to the evolution of high-level multidrug resistance.72 Furthermore, other studies on induced resistance to ciprofloxacin have noted the absence of gyrA or topB mutations and instead report mutations of rpoB, rpoE, and parE, parC genes.71 Indeed, the Ga–LDFC–Cip-resistant S. aureus strain shares a number of mutations with the ciprofloxacin-resistant strain (Fig. 7C, entries highlighted in pink) and shows emergence of rpoB (Fig. 7C, entry 26) and rpoE (Figure7C, entry 38 mutations, underscoring the compound's ciprofloxacin-like character). Other commonly shared mutation sites between the 3 strains encode for genes regulating lipid transport (Fig. 7A, entry 3; Fig. 7C, entry 54), fibrinogen binding protein (Fig. 7A, entry 16; Fig. 7C, entry 31), DNA binding (Fig. 7A, entry 5; Fig. 7C, entry 34) and cell wall biosynthesis (Fig. 7C, entries 29, 35, 39 and 44).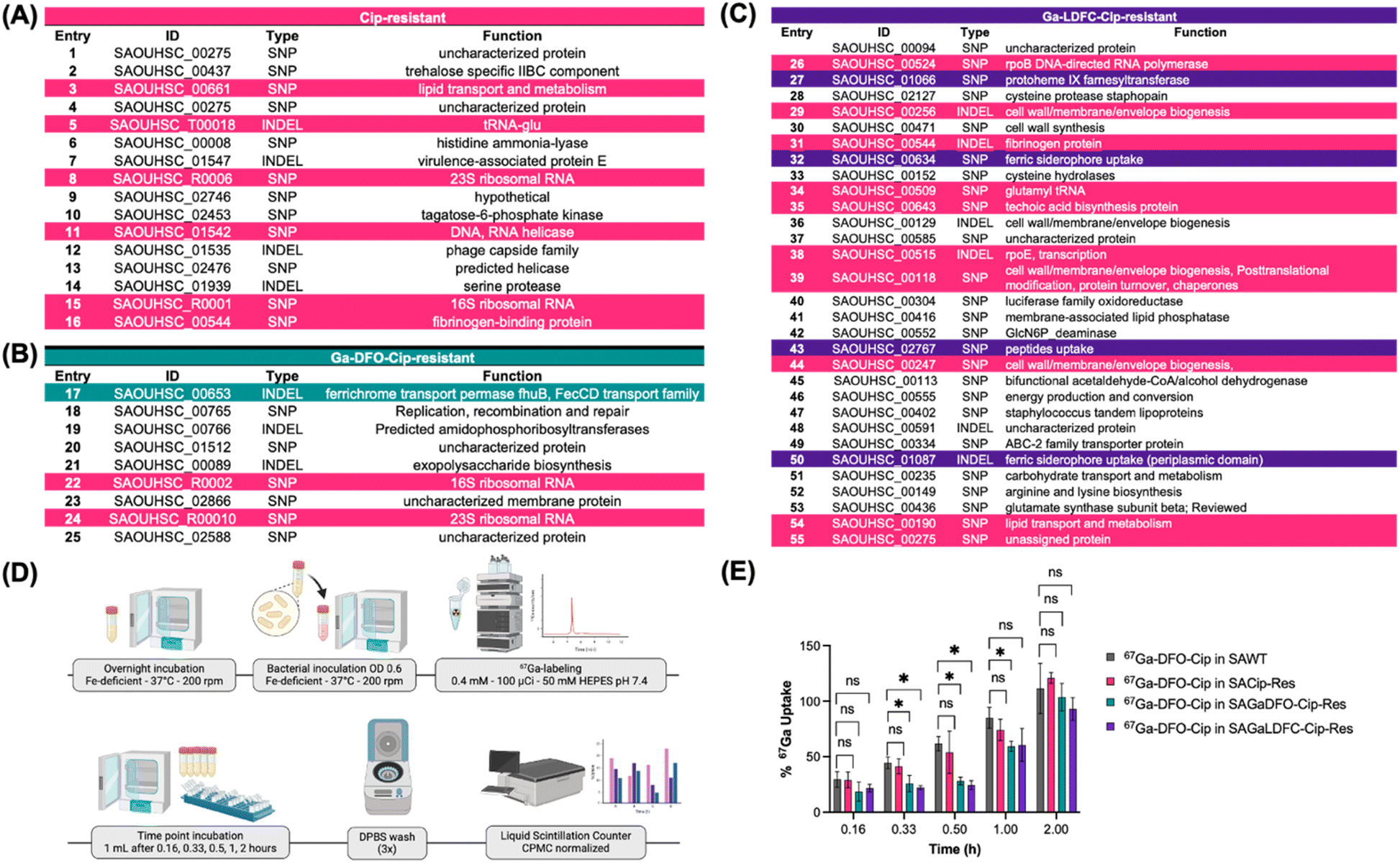 | ||
| Fig. 7 Variants analysis. Top panel: table showing the unique variant in each resistant strain, (A) Cip-resistant, (B) GaDFO–Cip-resistant and (C) GaLDFC–Cip-resistant, that have quality above 100 and allele depth > 30. Bottom panel: (D) radiochemical bacterial uptake assay of 67Ga–DFO–Cip in wt SA and resistant SA under iron deficient conditions. (E) Quantification of radiolabeled 67Ga-DFO-Cip uptake in investigated strains shows decrease of accumulation sideromycin-resistan strains. | ||
The lack of significant cross resistance to Cip, Ga–DFO–Cip and Ga–LDFC–Cip among the resistance-induced strains indicates a significant contribution from differential resistance profiles. Indeed, we identified several unique mutations in the Ga–DFO–Cip resistant and Ga–LDFC–Cip resistant strains. For Ga–DFO–Cip resistant S. aureus, we detected deletion of a 7-nucleotide fragment in a gene that encodes for FhuB (Fig. 7B, entry 17), a previously characterized ferrichrome/hydroxamate siderophore transporter.13 The impaired FhuB transporter effectively decreases import of Ga–DFO–Cip and therefore reduces the compound's efficacy. We also observed the mutation of iron siderophore transport associated genes in the Ga–LDFC–Cip resistant S. aureus strain; however, instead of FhuB, which was unaffected, we observed mutations to two ABC transporter genes (Fig. 7C, entries 32 and 43), both are not yet well characterized, but are implicated in iron siderophore import. The absence of matching iron siderophore transporter mutations in the Ga–DFO–Cip and Ga–LDFC–Cip resistant strains explains the lack of pronounced cross-resistance of these compounds (Table 1). To affirm that these genes play a pivotal role in import of our synthetic sideromycins, we obtained S. aureus strains from the Nebraska Transposon Mutant Library, which contain transposon insertions into S. aureus genes of FhuB (SAOUHSC_00653) and the ferric siderophore uptake transporter (SAOUHSC_01087) to render the corresponding gene non-functional (equivalent to a knock-out mutation).73 As anticipated, Ga–DFO–Cip and Ga–LDFC–Cip lost potency in both tested strains, while Cip retained moderate efficacy. This confirms that siderophore uptake machinery mutations are the bacteria's front-line resistance mechanism to xenometallomycins.
Additional single point mutations were also detected for the gene encoding for cyoE, protoheme IX farnesyltransferase (Fig. 7C, entry 27),74 which is responsible for the incorporation of heme into membrane-associated electron transport cascades.74,75 This mutation is further indicative of the role of Ga(III) in dysregulation of bacterial heme homeostasis and the bacteria's acute need for mobile iron sources within the cytoplasm. Additionally, we also detected a single point mutation in the serine protease gene, which prevents enzymatic release of ciprofloxacin from Ga–LDFC–Cip within the cytoplasm, further contributing to the resistance mechanism of Ga–LDFC–Cip resistant S. aureus. However, we consider neither one of these mutations as pivotal as transporter mutations to establish resistance.76,77
We hypothesized that Ga–DFO–Cip and Ga–LDFC–Cip resistance arise from impeded siderophore transport and therefore should significantly limit the uptake of the corresponding conjugates to the mutant bacterial cells. To probe this hypothesis, we conducted time-dependent radioactive uptake studies in wt and resistant strains using 67Ga–DFO–Cip (Fig. 7D). Quantification of 67Ga–DFO–Cip uptake within the bacterial pellets shows no difference in uptake in wt and Cip-resistant S. aureus; however, 67Ga–DFO–Cip uptake is repressed in Ga–DFO–Cip and Ga–LDFC–Cip resistant S. aureus strains (Fig. 7E). The persistent uptake indicates that the compounds can take advantage of multiple ports of entry, either by FhuB or the alternative ferric siderophore ABC transporter cascade implicated in Ga–LDFC–Cip resistance and mutants may retain a degree of functionality to translocate the conjugates, albeit with significantly reduced efficacy.
In vivo biodistribution in an wt S. aureus infected model
We conducted an in vivo biodistribution assay in an infected mouse model (Fig. 8A) to determine the significance of siderophore transporter mutations in an in vivo environment. Localized soft tissue infections were induced in mice by injecting 108 CFUs of wt SA into the right triceps and 108 CFUs of the SAGaDFO-Cip-resistant bacteria into the contralateral left triceps.31,35,78–81 After 5 hours, mice were intravenously injected with 68Ga–DFO–Cip, and biodistribution was assessed at 1 hour post-injection (Fig. 8A and B), alongside analysis of the metabolites in the urine (Fig. 8B). The biodistribution results revealed approximately 15% uptake of the conjugate in the lung, consistent with findings from previous studies on DFO-based conjugates and the enhanced uptake of ciprofloxacin in pulmonary tissue.32,82 The 68Ga–DFO–Cip complex showed statistically significant, enhanced uptake in the wt SA-infected muscle compared to both the SAGa-DFO-Cip-resistant infected muscle and the control muscle, as shown in Fig. 8C. This selective uptake underscores the importance of active transmembrane transport mediated uptake in live bacteria within the host. With the construct remaining >70%, the 68Ga–DFO–Cip probe dominates biological behavior over degradation and free 68Ga-mediated uptake (Fig. 8B). | ||
| Fig. 8 In vivo biodistribution analysis in an infected model. (A) Workflow scheme, (B) urine metabolite analysis and (C) biodistribution of [68Ga][Ga(DFO–Cip)]3− in the infected model showing predominant accumulation in wt SA infected muscle compared to GaDFO–Cip-resistant infected muscle. *p value = 0.037; **p value = 0.008. | ||
Conclusions
Here, we provide an exhaustive analysis and insight for the mechanism of action of gallium–siderophore conjugates and their ability to potentiate antibacterial activity of clinically employed antibiotic ciprofloxacin in S. aureus bacteria. Differential gene expression analysis of bacteria exposed to sub-MIC concentrations of the gallium–siderophore–ciprofloxacin conjugates reveals signatures characteristic of their parent antibiotic; compound import is facilitated by the bacteria's endogenous iron siderophore machinery, while import of alternative, common iron sources, such as Fe-heme, is repressed. Introduction of the corresponding iron analogues neutralizes Fe–siderophore import and rationalizes their low potency. We affirm the decreased growth inhibitory potency from Ga- to apo to Fe–siderophore–ampicillin conjugate and overall diminished potency relative to the parent antibiotic ampicillin.S. aureus grown in the presence of sub-toxic, progressively increasing concentrations of the sideromycins, elicits mutations that confer enhanced tolerance to the corresponding compounds. Mutations of active, siderophore-complex mediated transport dominate the resistance mechanisms for both Ga-complexes evaluated, which is further affirmed by loss of potency in the corresponding, S. aureus knock-out mutants. In addition to ciprofloxacin-typical signatures, iron import and heme homeostasis become dysregulated; we also uncover that hydroxamate-based siderophores such as deferoxamine and desferrichrome use multiple transmembrane iron transport systems to access the bacterial cytoplasm of S. aureus bacterial strains (Fig. 9). Corresponding in vitro and in vivo experiments with the radioactive analogues 67/68Ga–DFO–Cip affirm that siderophore-specific transmembrane transport is essential to guarantee the delivery and uptake in S. aureus both outside and inside the mammalian host environment.
 | ||
| Fig. 9 Schematic representation of gallium–siderophore–ciprofloxacin conjugate uptake and its downstream effects in S. aureus. | ||
Taken together, our studies also outline design criteria for the future development of sideromycins as antibacterial agents for S. aureus (Fig. 10). Considering that Fe-complexes down-regulate expression of the siderophore uptake machinery, the use of apo- or Ga(III) complexes is preferable. The complex must be sufficiently thermodynamically stable (KML > 25) and kinetically inert (t1/2 > 30 min vs. 1000-fold excess EDTA) to co-deliver metal ions and antibiotic cargo to the cytoplasm. Antibacterial cargos targeting the bacterial cell wall do not benefit significantly from co-delivery of a xenometal. Incorporation of siderophore antibiotic linkers that do not require degradation by endogenous bacterial proteases for activity but employ non-enzymatic release strategies may further prevent or delay emergence of resistance.
 | ||
| Fig. 10 Design criteria for the development of next generation siderophore–fluoroquinolone conjugates for inhibition of S. aureus growth. | ||
Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Specifically, we provide processed NMR spectroscopic information, chromatographic analysis spectra, UV-vis spectra and tabulated biodistribution and stability analysis data as an average of single cohorts of animals as indicated. Tabulated raw RNA-Seq data are provided as supplementary Excel files. Additional raw data are available upon request to the corresponding author.Author contributions
EB, AM designed and planned experiments. AM, OG, JEH, MC conducted experiments and contributed to data analysis. EB and AM conducted data analysis and interpretation. The manuscript and supporting information was written with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Institute of General Medical Sciences (NIGMS) is acknowledged for funding (R35GM142770). The authors utilized the University of Wisconsin-Madison Biotechnology Center Bioinformatics Core Facility (Research Resource Identifier – RRID:SCR_017799) for analysis of differentially expressed genes in the wild-type S. aureus bacteria in response to the compounds and in the induced resistant S. aureus as well the variant analysis of the resistant strains. Dr Mark Berres is acknowledged for his support in RNA sequencing data processing and analysis. Dr Heike Hofstetter is acknowledged for her support in establishing various heteronuclear NMR pulse programs and acquisition methods. The following instrumentation in the Paul Bender Chemical Instrumentation Center at the University of Wisconsin-Madison was supported by NSF CHE-2017891 (Bruker Avance Neo 500); by a generous gift from Paul J. and Margaret M. Bender (Bruker Quazar APEX2 and Bruker 500); NIH S10 OD012245 (Bruker Avance-600). The Electrospray Ionization-Quadrupole-Ion Trap Mass Spectrometer was funded by NIH 1S10 OD020022. Z. Rosenkrans, staff at Small Animal Imaging and Radiotherapy Facility (SAIRF) is acknowledged for tail vein injection during the in vivo study.References
- E. Lappin and A. J. Ferguson, Lancet Infect. Dis., 2009, 9, 281–290 CAS
.
- G. H. Dayan, N. Mohamed, I. L. Scully, D. Cooper, E. Begier, J. Eiden, K. U. Jansen, A. Gurtman and A. S. Anderson, Expert Rev. Vaccines, 2016, 15, 1373–1392 CAS
- V. Pintado, R. Pazos, M. E. Jiménez-Mejías, A. Rodríguez-Guardado, B. Díaz-Pollán, C. Cabellos, J. M. García-Lechuz, J. Lora-Tamayo, P. Domingo, E. Muñez, D. Domingo, F. González-Romo, J. A. Lepe-Jiménez, C. Rodríguez-Lucas, A. Gil, I. Pelegrín, F. Chaves, V. Pomar, A. Ramos, T. Alarcón and E. Pérez-Cecilia, J. Hosp. Infect., 2019, 102, 108–115 CAS
- S. Y. C. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS
- T. J. Foster, FEMS Microbiol. Rev., 2017, 41, 430–449 CrossRef CAS
- Y. Guo, G. Song, M. Sun, J. Wang and Y. Wang, Front. Cell. Infect. Microbiol., 2020, 10 DOI:10.3389/fcimb.2020.00107
- A. M. Timofeeva, M. R. Galyamova and S. E. Sedykh, Plants, 2022, 11, 3065 CrossRef CAS PubMed
- B. Rayner, A. D. Verderosa, V. Ferro and M. A. T. Blaskovich, RSC Med. Chem., 2023, 14, 800–822 CAS
- V. Braun, Front. Biosci., 2003, 8, s1409–s1421 CAS
- J. E. Cassat and E. P. Skaar, Semin. Immunopathol., 2012, 34, 215–235 CAS
- T. E. Clarke, L. W. Tari and H. J. Vogel, Curr. Top. Med. Chem., 2001, 1, 7–30 CAS
- B. S. Conroy, J. C. Grigg, M. Kolesnikov, L. D. Morales and M. E. P. Murphy, BioMetals, 2019, 32, 409–424 CAS
- M. T. Sebulsky, B. H. Shilton, C. D. Speziali and D. E. Heinrichs, J. Biol. Chem., 2003, 278, 49890–49900 CrossRef CAS PubMed
- A. Stintzi, C. Barnes, J. Xu and K. N. Raymond, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10691–10696 CAS
- M. C. van Dijk, R. M. de Kruijff and P.-L. Hagedoorn, Front. Cell Dev. Biol., 2022, 10, 857237 Search PubMed
- T. Zheng, J. L. Bullock and E. M. Nolan, J. Am. Chem. Soc., 2012, 134, 18388–18400 CAS
- C. Ji and M. J. Miller, BioMetals, 2015, 28, 541–551 CAS
- K. S. Kaye, T. Naas, J. M. Pogue and G. M. Rossolini, Infect. Dis. Ther., 2023, 12, 777–806 CrossRef
- K. A. Parsels, K. A. Mastro, J. M. Steele, S. J. Thomas and W. D. Kufel, J. Antimicrob. Chemother., 2021, 76, 1379–1391 Search PubMed
- J. Yao, J. Wang, M. Chen and Y. Cai, Front. Med., 2021, 8 DOI:10.3389/fmed.2021.741940
- A. Sollima, F. Rossini, P. Lanza, C. Pallotto, M. Meschiari, I. Gentile, R. Stellini, A. Lenzi, A. Mulé, F. Castagna, S. Lorenzotti, S. Amadasi, E. Van Hauwermeiren, B. Saccani, B. Fumarola, L. Signorini, F. Castelli and A. Matteelli, Antibiotics, 2024, 13, 453 CAS
- G. J. Anderson and D. M. Frazer, Am. J. Clin. Nutr., 2017, 106, 1559S–1566S CrossRef PubMed
- E. R. Frawley and F. C. Fang, Mol. Microbiol., 2014, 93, 609–616 CAS
- C. R. Chitambar, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2044–2053 CAS
- N. Kircheva and T. Dudev, Inorg. Chem., 2020, 59, 6242–6254 CAS
- S. Hijazi, D. Visaggio, M. Pirolo, E. Frangipani, L. Bernstein and P. Visca, Front. Cell. Infect. Microbiol., 2018, 8, 316 Search PubMed
- K. D. Mjos, J. F. Cawthray, E. Polishchuk, M. J. Abrams and C. Orvig, Dalton Trans., 2016, 45, 13146–13160 CAS
- K. Michalska, M. Rychłowski, M. Krupińska, G. Szewczyk, T. Sarna and J. Nakonieczna, Mol. Pharm., 2022, 19, 1434–1448 CAS
- Z. W. Scott, S. Choi, B. E. Britigan and P. Narayanasamy, ACS Infect. Dis., 2023, 9, 716–738 CAS
- R. G. Pearson, J. Org. Chem., 1989, 54, 1423–1430 CrossRef CAS
- A. Pandey, D. Śmiłowicz and E. Boros, Chem. Sci., 2021, 12, 14546–14556 Search PubMed
- A. Pandey, C. Savino, S. H. Ahn, Z. Yang, S. G. Van Lanen and E. Boros, J. Med. Chem., 2019, 62, 9947–9960 CrossRef CAS PubMed
- V. Vinuesa and M. J. McConnell, Int. J. Mol. Sci., 2021, 22, 2876 CAS
- Y. Wang, B. Han, Y. Xie, H. Wang, R. Wang, W. Xia, H. Li and H. Sun, Chem. Sci., 2019, 10, 6099–6106 CAS
- A. Pandey, M. Cao and E. Boros, ACS Infect. Dis., 2022, 8, 878–888 CAS
- X. S. Pan, J. Ambler, S. Mehtar and L. M. Fisher, Antimicrob. Agents Chemother., 1996, 40, 2321–2326 Search PubMed
- K. A. Gould, X.-S. Pan, R. J. Kerns and L. M. Fisher, Antimicrob. Agents Chemother., 2004, 48, 2108–2115 CrossRef CAS PubMed
- K. M. Evans-Roberts, L. A. Mitchenall, M. K. Wall, J. Leroux, J. S. Mylne and A. Maxwell, J. Biol. Chem., 2016, 291, 3136–3144 CrossRef CAS PubMed
- A. R. Cardoso, L. P. T. Carneiro, G. Cabral-Miranda, M. F. Bachmann and M. G. F. Sales, Chem. Eng. J., 2021, 409, 128135 CrossRef CAS
- A. Ito, T. Nishikawa, S. Matsumoto, H. Yoshizawa, T. Sato, R. Nakamura, M. Tsuji and Y. Yamano, Antimicrob. Agents Chemother., 2016, 60, 7396–7401 CrossRef CAS
- T. Emery and P. B. Hoffer, J. Nucl. Med., 1980, 21, 935–939 CAS
- A. Evers, R. D. Hancock, A. E. Martell and R. J. Motekaitis, Inorg. Chem., 1989, 28, 2189–2195 CrossRef CAS
- N. P. Endicott, E. Lee and T. A. Wencewicz, ACS Infect.
Dis., 2017, 3, 542–553 CrossRef CAS PubMed
- M. I. Tsionou, C. E. Knapp, C. A. Foley, C. R. Munteanu, A. Cakebread, C. Imberti, T. R. Eykyn, J. D. Young, B. M. Paterson, P. J. Blower and M. T. Ma, RSC Adv., 2017, 7, 49586–49599 CAS
- N. Mani, C. H. Gross, J. D. Parsons, B. Hanzelka, U. Müh, S. Mullin, Y. Liao, A.-L. Grillot, D. Stamos, P. S. Charifson and T. H. Grossman, Antimicrob. Agents Chemother., 2006, 50, 1228–1237 CAS
- J. R. Anderson, N. B. Lam, J. L. Jackson, S. M. Dorenkott, T. Ticer, E. Maldosevic, A. Velez, M. R. Camden and T. N. Ellis, Antibiotics, 2023, 12, 887 CAS
- N. Kuptsov, M. Kornienko, D. Bespiatykh, R. Gorodnichev, K. Klimina, V. Veselovsky and E. Shitikov, Viruses, 2022, 14, 567 CAS
- J. Im, D. Lee, O.-J. Park, S. Natarajan, J. Park, C.-H. Yun and S. H. Han, Front. Microbiol., 2022, 13 DOI:10.3389/fmicb.2022.1063650
- L. Bie, M. Zhang, J. Wang, M. Fang, L. Li, H. Xu and M. Wang, Microbiol. Spectr., 2023, 11, e00317–e00323 Search PubMed
- N. J. Croucher and N. R. Thomson, Curr. Opin. Microbiol., 2010, 13, 619–624 CAS
- D. Trivier and R. J. Courcol, FEMS Microbiol. Lett., 1996, 141, 117–127 CAS
- I. Z. Batko, R. S. Flannagan, V. Guariglia-Oropeza, J. R. Sheldon and D. E. Heinrichs, J. Bacteriol., 2021, 203 DOI:10.1128/jb.00458-21
- M. Allard, H. Moisan, É. Brouillette, A. L. Gervais, M. Jacques, P. Lacasse, M. S. Diarra and F. Malouin, Microb. Infect., 2006, 8, 1679–1690 CAS
- K. P. Ha and A. M. Edwards, Microbiol. Mol. Biol. Rev., 2021, 85 DOI:10.1128/MMBR.00091-21
-
L. C. N. da Silva, R. C. Diniz, I. M. d. S. F. Lima, C. I. dos Santos, M. S. Alves, L. I. O. de Souza and A. d. S. Monteiro, in The Rise of Virulence and Antibiotic Resistance in Staphylococcus aureus, IntechOpen, 2017 Search PubMed
- K. H. Maslowska, K. Makiela-Dzbenska and I. J. Fijalkowska, Environ. Mol. Mutagen., 2019, 60, 368–384 CAS
- J. Y. Park, J. W. Kim, B. Y. Moon, J. Lee, Y. J. Fortin, F. W. Austin, S.-J. Yang and K. S. Seo, Infect. Immun., 2015, 83, 1620–1628 CAS
- A. Y. Bhagirath, Y. Li, R. Patidar, K. Yerex, X. Ma, A. Kumar and K. Duan, Int. J. Mol. Sci., 2019, 20, 1781 CAS
- M. G. Bertero, R. A. Rothery, M. Palak, C. Hou, D. Lim, F. Blasco, J. H. Weiner and N. C. J. Strynadka, Nat. Struct. Mol. Biol., 2003, 10, 681–687 CAS
- Y. Dong, X. Miao, Y.-D. Zheng, J. Liu, Q.-Y. He, R. Ge and X. Sun, J. Proteome Res., 2021, 20, 2839–2850 CAS
- L. R. Mesak, V. Miao and J. Davies, Antimicrob. Agents Chemother., 2008, 52, 3394–3397 CrossRef CAS
- S. P. Mattiello, V. C. Barth, J. Scaria, C. a. S. Ferreira and S. D. Oliveira, Sci. Rep., 2023, 13, 18696 CrossRef CAS
- A. A. Pometun, K. M. Boyko, T. S. Yurchenko, A. Y. Nikolaeva, I. S. Kargov, D. L. Atroshenko, S. S. Savin, V. O. Popov and V. I. Tishkov, Biochemistry, 2020, 85, 689–696 CAS
- G. A. Ashniev, N. V. Sernova, A. E. Shevkoplias, I. D. Rodionov, I. A. Rodionova, A. G. Vitreschak, M. S. Gelfand and D. A. Rodionov, BMC Genom., 2022, 23, 558 CrossRef CAS PubMed
- C. M. Beetham, C. F. Schuster, I. Kviatkovski, M. Santiago, S. Walker and A. Gründling, PLoS Pathog., 2024, 20, e1011927 CrossRef CAS PubMed
- T. Saito, D. Duly and R. J. Williams, Eur. J. Biochem., 1991, 197, 39–42 CrossRef CAS PubMed
- H. H. Brewitz, N. Goradia, E. Schubert, K. Galler, T. Kühl, B. Syllwasschy, J. Popp, U. Neugebauer, G. Hagelueken, O. Schiemann, O. Ohlenschläger and D. Imhof, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 1343–1353 CrossRef CAS
- L. J. Lojek, A. J. Farrand, A. Weiss and E. P. Skaar, Int. J. Med. Microbiol., 2018, 308, 582–589 CrossRef CAS PubMed
- N. D. Olson, J. Wagner, N. Dwarshuis, K. H. Miga, F. J. Sedlazeck, M. Salit and J. M. Zook, Nat. Rev. Genet., 2023, 24, 464–483 CAS
- D. P. Ryan and T. Owen-Hughes, Curr. Opin. Chem. Biol., 2011, 15, 649–656 CAS
- D. Ince and D. C. Hooper, J. Bacteriol., 2003, 185, 6883–6892 CrossRef CAS PubMed
- J. E. Gomez, B. B. Kaufmann-Malaga, C. N. Wivagg, P. B. Kim, M. R. Silvis, N. Renedo, T. R. Ioerger, R. Ahmad, J. Livny, S. Fishbein, J. C. Sacchettini, S. A. Carr and D. T. Hung, eLife, 2017, 6, e20420 Search PubMed
- P. D. Fey, J. L. Endres, V. K. Yajjala, T. J. Widhelm, R. J. Boissy, J. L. Bose and K. W. Bayles, mBio, 2013, 4 DOI:10.1128/mbio.00537-12
- E. Stevens, M. Laabei, S. Gardner, G. A. Somerville and R. C. Massey, Sci. Rep., 2017, 7, 13744 Search PubMed
- T. Xu, J. Han, J. Zhang, J. Chen, N. Wu, W. Zhang and Y. Zhang, Front. Microbiol., 2016, 7, 1625 Search PubMed
- K. Stelzner, A. Boyny, T. Hertlein, A. Sroka, A. Moldovan, K. Paprotka, D. Kessie, H. Mehling, J. Potempa, K. Ohlsen, M. J. Fraunholz and T. Rudel, PLoS Pathog., 2021, 17, e1009874 CAS
- K. Rice, R. Peralta, D. Bast, J. de Azavedo and M. J. McGavin, Infect. Immun., 2001, 69, 159–169 CrossRef CAS PubMed
- K. Krasulova, B. Neuzilova, K. D. Bendova, Z. Novy, M. Popper, M. Hajduch and M. Petrik, EJNMMI Radiopharm. Chem., 2024, 9, 20 CrossRef PubMed
- C. Peukert, L. N. B. Langer, S. M. Wegener, A. Tutov, J. P. Bankstahl, B. Karge, F. M. Bengel, T. L. Ross and M. Brönstrup, J. Med. Chem., 2021, 64, 12359–12378 CrossRef CAS PubMed
- M. Petrik, E. Umlaufova, V. Raclavsky, A. Palyzova, V. Havlicek, J. Pfister, C. Mair, Z. Novy, M. Popper, M. Hajduch and C. Decristoforo, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 372–382 CrossRef CAS PubMed
- G. Narayanappa and B. N. Nandeesh, Brain Pathol., 2021, 31, e12950 CrossRef PubMed
- H. X. Ong, D. Traini, M. Bebawy and P. M. Young, Antimicrob. Agents Chemother., 2013, 57, 2535–2540 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08509d |
| This journal is © The Royal Society of Chemistry 2025 |