
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective Zn-catalyzed hydrophosphinylation of nitrones: an efficient approach for constructing chiral α-hydroxyamino-phosphine oxides†
Shihui
Luo
a,
Xinzhu
Yuan
a,
Jiangtao
Cheng
a,
Zhiping
Yang
a,
Zhongxing
Huang
 b and
Jun (Joelle)
Wang
*a
b and
Jun (Joelle)
Wang
*a
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon, Hong Kong, China. E-mail: junwang@hkbu.edu.hk
bDepartment of Chemistry, The University of Hong Kong, Hong Kong, China
First published on 18th March 2025
Abstract
Although enantioselective hydrofunctionalizations of nitrones are established for the synthesis of various types of chiral hydroxylamines, the asymmetric catalytic hydrophosphinylation of nitrones remains highly challenging. Herein, an efficient asymmetric hydrophosphinylation of nitrones, catalyzed by the dinuclear zinc catalyst derived from ProPhenol, is presented, accommodating a variety of nitrones and phosphine oxides. This approach successfully addresses the long-standing challenge of catalytic hydrophosphinylation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, and offers an efficient and rapid access towards chiral α-hydroxyamino-phosphine oxides. Control experiments suggest that the oxide anion in the nitrone motif is crucial for the enantio-control.
N bond, and offers an efficient and rapid access towards chiral α-hydroxyamino-phosphine oxides. Control experiments suggest that the oxide anion in the nitrone motif is crucial for the enantio-control.
Introduction
Chiral amino phosphorus compounds have garnered significant attention due to their crucial roles in medicinal chemistry and biochemistry,1 as well as their applications as chiral P–N ligands or catalysts in organic synthesis.2 For example, certain α- or β-amino-phosphorus compounds serve as antibacterial agents, antibodies (Fig. 1a), or renin inhibitors (Fig. 1b),2 while other nitrogen-containing phosphorus compounds function as efficient chiral ligands or catalysts in asymmetric catalysis (Fig. 1c).3 Specifically, α-hydroxyaminophosphine oxides are valuable intermediates and building blocks in organic synthesis and drug discovery,3 with some exhibiting notable antitumor activities (Fig. 1d).3b Despite their importance, the effective synthesis of chiral α-hydroxyaminophosphine oxides remains challenging, with no suitable catalytic systems reported to date. Therefore, a practical synthetic route towards enantioselective α-hydroxyaminophosphine oxides is highly desirable. Nitrones are ideal starting materials for constructing chiral α-hydroxyaminophosphine oxides due to their straightforward preparation through simple condensations and crystallizations.4 They are generally more stable and easier to handle than their corresponding imine motifs, and their configurational stability enhances the stereoselectivities in reactions.5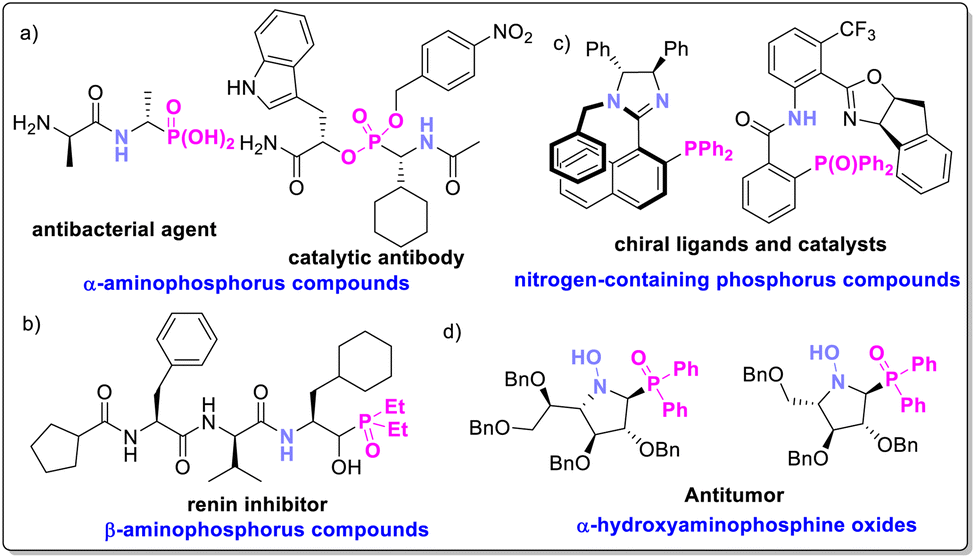 | ||
| Fig. 1 Representative of chiral aminophosphorus compounds. | ||
Asymmetric hydrofunctionalization is a powerful tool for introducing hydrogen and functional groups to CC6 and CN7 double bonds in an enantio- and regioselective manner. Elegant examples of asymmetric hydrofunctionalization of nitrones have been disclosed using carbon nucleophiles with organocatalysts, photocatalysts, Lewis acid catalysts, or transition-metal catalysts (Scheme 1a).8 In 2008, Scheidt's group developed a chiral NHC-catalyzed diastereo- and enantioselective hydrocarbonation of nitrones with homoenolates.8a In 2018, Huang and co-workers discovered an asymmetric hydrocarbonation of nitrones with the combination of photocatalysts and chiral Lewis acids.8b Very recently, Aponick's group reported a copper-catalyzed asymmetric hydrofunctionalization of nitrones with alkyne nucleophiles using tunable axially chiral imidazole-based P,N-ligands.8c Compared to the hydrofunctionalization of CC bonds, reactions involving CN bonds, particularly with heteroatoms nucleophiles, are less developed. While there are limited examples of catalytic enantioselective addition of imine/iminium with phosphonates9 and phosphine oxides10 using Lewis acid or transition-metal catalysts, catalytic hydrophosphinylation of nitrones is rare.
 | ||
| Scheme 1 Overview of asymmetric hydrofunctionalization of nitrones. | ||
Therefore, developing an asymmetric hydrofunctionalization of nitrones with phosphorus nucleophiles addresses an unmet need. As part of our long-term interest in developing asymmetric hydrophosphination reactions,11 we tackle the challenges of asymmetric hydrophosphinylation of nitrones with the dinuclear zinc catalyst derived from ProPhenol (Scheme 1b).
Results and discussion
The nitrogen atom in nitrone cannot directly interact with transition-metal catalysts due to the lack of the lone pair electrons,12 which impairs the enantioselectivity of the reaction. Nevertheless, Lewis acid catalysts, such as the Zn-ProPhenol, may provide metal binding sites and create a chiral pocket to activate both the electrophile and the nucleophile.13 It is hypothesized that the Brønsted basic site binds to phosphine oxide, while the Lewis acidic site coordinates with the oxide anion of the nitrone, thereby promoting enantioselective C–P bond formation. Based on this hypothesis, we started our investigation using nitrone 1a and diphenyl phosphine oxide 2a as model substrates, with diethyl zinc and the (S,S)-ProPhenol ligand as the chiral catalytic system (Table 1). Without additives, the expected product 3aa was obtained in racemic form along with byproduct 4aa (Table 1, entry 1). Pyridine is well known to accelerate reaction rates and enhance enantioselectivities in Lewis acid-catalyzed asymmetric reactions.14 To our delight, the addition of pyridine did increase the ee value from 0% to 63% dramatically (Table 1, entry 2). Further investigation of various solvents revealed that ether solvents consistently gave both good yields and high ee values (Table 1, entries 4–6). Reducing the amount of pyridine slightly decreased the ee values compared to the standard condition (Table 1, entries 6–8). The reaction temperature also played a crucial role in minimizing the side product 4aa, with the ratio of product 3aa to side product 4aa increasing to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 0 °C (Table 1, entry 9 vs. 6). Thus, the optimal conditions were determined to be 10.0 equiv. of pyridine in THF under 0 °C for 3 h (Table 1, entry 6, 80% yield, 93% ee).
:1 at 0 °C (Table 1, entry 9 vs. 6). Thus, the optimal conditions were determined to be 10.0 equiv. of pyridine in THF under 0 °C for 3 h (Table 1, entry 6, 80% yield, 93% ee).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | Yieldb (%) |
3aa:4aac |
ee of 3aad(%) |
|
a Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), Et2Zn (40 mol%), ligand (20 mol%), solvent (2 mL), under N2, 3 h.
b Yields are detected by 31P NMR using P(O)(OMe)3 as the internal standard.
c The ratios are detected by 1H NMR.
d Determined by HPLC analysis with a Chiralcel ID column (hexane/2-propanol 60:40, 1.0 mL min−1, 210 nm).
e No pyridine was added.
f Isolated yield for 3aa.
g 5.0 equiv. of pyridine was added.
h 8.0 equiv. of pyridine was added.
|
|||||
| 1e | Toluene | 0 | 70 | 11:1 |
rac |
| 2 | Toluene | 0 | 73 | 14:1 |
63 |
| 3 | MeCN | 0 | 68 | 8:1 |
91 |
| 4 | 1,4-Dioxane | 0 | 98 | 14:1 |
86 |
| 5 | Anisole | 0 | 81 | 5:1 |
92 |
| 6 | THF | 0 | 87 (80)f | 10:1 |
93 |
| 7g | THF | 0 | 90 | 11:1 |
80 |
| 8h | THF | 0 | 89 | 11:1 |
86 |
| 9 | THF | rt | 59 | 2:1 |
84 |

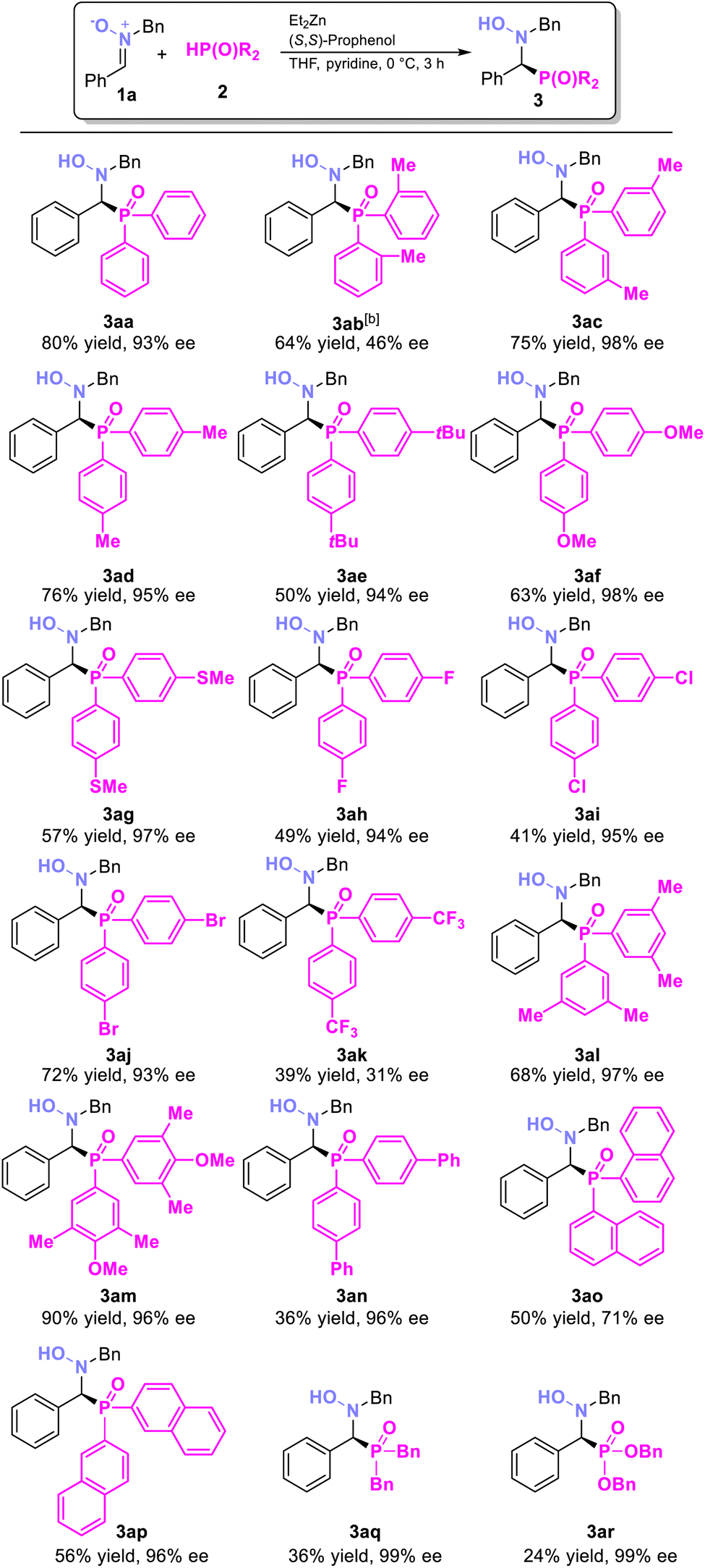
With the optimized conditions in hand, we then investigated the substrate scope of nitrones (Table 2). Generally, nitrones with both electron-withdrawing and electron-donating groups at various positions on the phenyl ring were compatible, yielding the corresponding hydroxyamino phosphine oxides in good yields and high ee values. Nitrone with strong electron-withdrawing substituents (3ea, CF3) on the aromatic ring was also compatible, although with lower conversion. The 1-napthyl nitrone substrate, despite its steric hindrance, also yielded corresponding product in moderate yield and good ee (3ja, 50% yield and 75% ee). After that, we further explored the reaction's compatibility with more valuable and complex substrates (3na–3ra). For example, the substrate 3n, derived from salicylaldehyde and featuring a free hydroxyl group, performed well, giving the expected product 3na in 52% yield with 68% ee. Heteroaromatic compounds were also well tolerated, providing the desired products 3oa and 3pa with good yields and enantioselectivities. Additionally, the derivative of 2,4,5-TMBA, a known COX-2 inhibitor,15 was produced in 48% yield and 62% ee (3qa). The derivative of retinal, an essential chemical for visual phototransduction,16 gave the product in 34% yield, and 69% ee (3ra) under 0 °C for 24 hours, despite its structural complexity. The absolute configuration of 3da was confirmed as (S) by X-ray crystallographic analysis.17

After exploring the nitrone substrates, we turned our attention to various phosphine oxides (Table 3). Both di-m-tolylphosphine oxide and di-p-tolylphosphine oxide gave the desired products in good yield with excellent ee values (3ac, 3ad). Di-o-tolylphosphine oxide, despite its steric hindrance, also yielded the corresponding product in lower yield and ee (3ab 64% yield, 46% ee, 24 h). Phosphine oxides with electron-donating groups on the phenyl ring consistently delivered high yields and excellent ee values (3ac–3ag, 3al–3am, 94–98% ee). In contrast, strong electron-withdrawing groups reduced the ee dramatically (3ak, 39% yield, 31% ee). 3,5-disubstituted and 3,4,5-trisubstituted phenyl phosphine oxides performed remarkably well (3al and 3am, 68–90% yield, 96–97% ee). After that, we further investigated diaryl phosphine oxide with a π-extended conjugated system (3an, 3ao, and 3ap). The 1-napthyl phosphine oxide substrate produces 3ao in 50% yield and 71% ee due to the steric effect, while 3an and 3ap could be generated in very high ee values (96%, 96%). To our delight, dialkyl phosphine oxides and phosphonates also excelled under the standard reaction conditions, achieving extremely high ee values (3aq, 99% ee, 3ar, 99% ee). While the reactivity of the substrates other than the diaryl phosphine oxide is low, this aligns with previous findings that the reactivity order toward electrophiles is Ar2P(O)H > (RO)2P(O)H > R2P(O)H.14b,c
A scale-up reaction using 0.5 mmol of nitrone as the starting material under the standard conditions was performed. The isolated yield remained at 80%, but there was a decrease in enantioselectivity to 81% (Scheme 2a). Additionally, the by-product 4aa was obtained in trace amount with 81% ee. To further elucidate the mechanism and the formation of the by-product 4aa, we conducted several control experiments (Scheme 2b). Diphenylphosphinic acid was detected in the reaction system from HRMS (Fig. S1†) and 31P NMR spectra (Fig. S2†). We hypothesized that product 3aa underwent reduction to form diphenylphosphinic acid and imine 5, which subsequently transformed into 4aa. Therefore, we conducted the reaction using 5 and 2a as starting materials (Scheme 2b, eqn (1)). Surprisingly, the reaction yielded the product in its racemic form, even with or without the addition of pyridine. This suggests that the oxide anion in the nitrone motif is crucial for forming a stronger bond with the zinc atom. Previous reports18 indicated that product 4aa can be generated by the reduction of 3aavia zinc metal present in the catalytic system. However, 3aa did not react with zinc under the reaction conditions (Scheme 2b, eqn (2)). The significant loss of substrate 2a in the reaction reminded us the possibility of 2a as a reducing agent in the catalytic system. We then mixed 3aa with 2a under standard conditions (Scheme 2b, eqn (3)). To our delight, 4aa was produced in 15% yield with 82% ee, with the recovery of 3aa (20% yield, 78% ee). This suggests that the Zn-ProPhenol catalytic system can activate the product 3aa, leading to a nucleophilic attack by diphenyl phosphine oxide, which results in N–O bond cleavage and the formation of 4aa and diphenylphosphinic acid.
 | ||
| Scheme 2 Scale-up reaction and mechanistic study. aReaction conditions: unless indicated, 1a (0.1 mmol), 2a (0.15 mmol), Et2Zn (40 mol%), ligand (20 mol%), pyridine (10.0 equiv.), THF (2 mL), under N2, 3 h. Yields are isolated yields. ee Values are determined by HPLC analysis. | ||
Furthermore, nonlinear effects of the catalyst were observed (Scheme 2c), suggesting the aggregation of the zinc complex.19 Notably, the dimeric assembly 7 was detected by HRMS during the reaction of 1a and 2a under standard conditions for 1 hour (Fig. S3,† the reaction solution was diluted in MeOH before HRMS analysis). Another dimeric assembly 8 was also detected by HRMS when diethylzinc, the (S,S)-ProPhenol ligand and pyridine were mixed in THF (Fig. S4†). Additionally, a Job Plot with UV-vis spectra indicating the complexation20 between zinc and pyridine is presented in Fig. S5.† While the presence of supramolecular species cannot be ruled out, we believed that a dimeric zinc complex 8 is possible to be the active catalyst based on the experimental results and previous reports on dinuclear zinc catalyzed reaction systems.14b,c,21
Conclusions
In summary, we have developed an asymmetric pathway for accessing chiral α-hydroxyaminophosphine oxides using a Lewis acid catalytic system. A wide range of nitrones with various substitutions successfully underwent the reaction, giving the corresponding products in moderate to good yields, and good to excellent ee values. Also, yields of up to 90% and enantioselectivities of up to 99% ee were achieved when employing different types of diaryl phosphine oxides as nucleophiles. The catalytic system also demonstrated compatibility with structurally diverse and complex molecules. Furthermore, the plausible reaction mechanism and the formation of by-product are discussed to understand more about this novel reaction system. Many of the chiral α-hydroxyaminophosphine oxides delivered by this methodology are untouched scaffolds with potential bio-activities, and further studies on their properties are currently underway in our lab.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. Luo performed the experiments and prepared the ESI.† X. Yuan and Dr Z. Yang repeated some experiments and collected some data. J. Cheng prepared some diaryl phosphine oxides. Prof. J. Wang and Prof. Z. Huang conceived and directed the project. S. Luo and Prof. J. Wang wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the National Natural Science Foundation of China (NSFC 22271241), University Grants Committee (Hong Kong) (YCRF C7001-23Y), and HKBU KRPS grant and RC-SFCRG/23-24 for financial support.Notes and references
- (a) J. G. Allen, F. R. Atherton, M. J. Hall, C. H. Hassall, S. W. Holmes, R. W. Lambert, L. J. Nisbet and P. S. Ringrose, Nature, 1978, 272, 56 CAS; (b) P. Kafarski and B. Lejczak, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 63, 193 CAS; (c) A. B. I. I. I. Smith, K. M. Yager and C. M. Taylor, J. Am. Chem. Soc., 1995, 117, 10879 CAS; (d) E. Alonso, E. Alonso, A. Solis and C. D. Pozo, Synlett, 2000, 5, 698 Search PubMed; (e) J. Grembecka, A. Mucha, T. Cierpicki and P. Kafarski, J. Med. Chem., 2003, 46, 2641 CAS; (f) K. Moonen, I. Laureyn and C. V. Stevens, Chem. Rev., 2004, 104, 6177 CAS; (g) S. J. Conway and G. J. Miller, Nat. Prod. Rep., 2007, 24, 687 CAS.
- (a) M. P. Carroll and P. J. Guiry, Chem. Soc. Rev., 2014, 43, 819 CAS; (b) P. H. S. Paioti, K. A. Abboud and A. Aponick, ACS Catal., 2017, 7, 2133 CAS; (c) S. Mishra, J. Liu and A. Aponick, J. Am. Chem. Soc., 2017, 139, 3352 CAS; (d) X.-Y. Dong, Y.-F. Zhang, C.-L. Ma, Q.-S. Gu, F.-L. Wang, Z.-L. Li, S.-P. Jiang and X.-Y. Liu, Nat. Chem., 2019, 11, 1158 CAS; (e) L. Liu, K.-X. Guo, Y. Tian, C.-J. Yang, Q.-S. Gu, Z.-L. Li, L. Ye and X.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 26710 CAS.
- (a) C. De Risi, A. Dondoni, D. Perrone and G. P. Pollini, Tetrahedron Lett., 2001, 42, 3033 CrossRef CAS; (b) P. Zhao, P. Li, J. Xiao and C. Liu, Chem. Commun., 2023, 59, 2624 RSC.
- S.-I. Murahashi and Y. Imada, Chem. Rev., 2019, 119, 4684 CrossRef CAS PubMed.
- (a) S.-I. Murahashi, Y. Imada, T. Kawakami, K. Harada, Y. Yonemushi and N. Tomita, J. Am. Chem. Soc., 2002, 124, 2888 CrossRef CAS PubMed; (b) V. G. Lisnyak, T. Lynch-Colameta and S. A. Snyder, Angew. Chem., Int. Ed., 2018, 57, 15162 CrossRef CAS.
- (a) J. F. Hartwig, Nature, 2008, 455, 314 CrossRef CAS PubMed; (b) X. Zeng, Chem. Rev., 2013, 113, 6864 CrossRef CAS PubMed; (c) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CAS; (d) N. J. Adamson and S. J. Malcolmson, ACS Catal., 2020, 10, 1060 CrossRef CAS; (e) J. Zhou, Q. Yang, C. S. Lee and J. Wang, Angew. Chem., Int. Ed., 2022, 61, e20220216 Search PubMed.
- (a) M. Shibasaki and M. Kanai, Chem. Rev., 2008, 108, 2853 CrossRef CAS PubMed; (b) X. Fu, W.-T. Loh, Y. Zhang, T. Chen, T. Ma, H. Liu, J. Wang and C.-H. Tan, Angew. Chem., Int. Ed., 2009, 48, 7387 CrossRef CAS PubMed; (c) S. Zhang, Z. Feng, C. Jiang, X. Yu, J. Pan, J. Du, Z. Jiang, Y. Chen and T. Wang, Chem.–Eur. J., 2021, 27, 11285 CrossRef CAS PubMed.
- (a) E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2008, 130, 2416 CrossRef CAS PubMed; (b) C.-X. Ye, Y. Y. Melcamu, H.-H. Li, J.-T. Cheng, T.-T. Zhang, Y.-P. Ruan, X. Zheng, X. Lu and P.-Q. Huang, Nat. Commun., 2018, 9, 410 CrossRef PubMed; (c) S. Yin, K. N. Weeks and A. Aponick, J. Am. Chem. Soc., 2024, 146, 7185 CrossRef CAS PubMed.
- (a) P. Merino, E. Marqués-López and R. P. Herrera, Adv. Synth. Catal., 2008, 350, 1195 CrossRef CAS; (b) S. Nakamura, M. Hayashi, Y. Hiramatsu, N. Shibata, Y. Funahashi and T. Toru, J. Am. Chem. Soc., 2009, 131, 18240 CrossRef CAS; (c) L. Yin, Y. Bao, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2013, 135, 10338 CrossRef CAS; (d) K. Yamada, Y. Kondo, A. Kitamura, T. Kadota, H. Morimoto and T. Ohshima, ACS Catal., 2023, 13, 3158 CrossRef CAS.
- (a) K. Yamakoshi, S. J. Harwood, M. Kanai and M. Shibasaki, Tetrahedron Lett., 1999, 40, 2565 CrossRef CAS; (b) G. K. Ingle, Y. Liang, M. G. Mormino, G. Li, F. R. Fronczek and J. C. Antilla, Org. Lett., 2011, 13, 2054 CrossRef CAS PubMed; (c) S. Duan, A. Pan, Y. Du, G. Zhu, X. Tian, H. Zhang, P. J. Walsh and X. Yang, ACS Catal., 2023, 13, 10887 CrossRef CAS.
- (a) Z. Lu, H. Zhang, Z. Yang, N. Ding, L. Meng and J. Wang, ACS Catal., 2019, 9, 1457 CrossRef CAS; (b) Z. Yang, X. Gu, L.-B. Han and J. Wang, Chem. Sci., 2020, 11, 7451 RSC; (c) Z. Yang and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 27288 CrossRef CAS PubMed; (d) Y. Zhang, Y. Jiang, M. Li, Z. Huang and J. Wang, Chem Catal., 2022, 2, 3163 CrossRef CAS; (e) B. Cai, Y. Cui, J. Zhou, Y.-B. Wang, L. Yang, B. Tan and J. Wang, Angew. Chem., Int. Ed., 2023, 135, e202215820 CrossRef; (f) Q. Yang, J. Zhou and J. Wang, Chem. Sci., 2023, 14, 4413 CAS; (g) J. Zhou, L. Meng, S. Lin, B. Cai and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303727 CrossRef CAS PubMed; (h) X.-D. Gu, K. Y. Ngai, W. Wang, B. Li and J. Wang, Chem Catal., 2024, 4, 100903 CrossRef CAS; (i) Y. Jiang, K. W. Cheng, Z. Yang and J. Wang, Chin. Chem. Lett., 2024, 110231 Search PubMed.
- C. Zhao and D. Seidel, J. Am. Chem. Soc., 2015, 137, 4650 CrossRef CAS PubMed.
- (a) B. M. Trost and H. Ito, J. Am. Chem. Soc., 2000, 122, 12003 CAS; (b) B. M. Trost, H. Ito and E. R. Silcoff, J. Am. Chem. Soc., 2001, 123, 3367 CrossRef CAS PubMed; (c) Y. Xiao, Z. Wang and K. Ding, Chem.–Eur. J., 2005, 11, 3668 CAS; (d) B. M. Trost and M. J. Bartlett, Acc. Chem. Res., 2015, 48, 688 CAS; (e) P. Xu and Z. Huang, Nat. Chem., 2021, 13, 634 CAS; (f) Y. Zheng, S. Zhang, K.-H. Low, W. Zi and Z. Huang, J. Am. Chem. Soc., 2022, 144, 1951 CrossRef CAS PubMed.
- (a) B. M. Trost, S. Malhotra and B. A. Fried, J. Am. Chem. Soc., 2009, 131, 1674 CrossRef CAS PubMed; (b) D. Zhao, L. Mao, Y. Wang, D. Yang, Q. Zhang and R. Wang, Org. Lett., 2010, 12, 1880 CrossRef CAS PubMed; (c) D. Zhao, L. Mao, D. Yang and R. Wang, J. Org. Chem., 2010, 75, 6756 CAS; (d) W. Chen, Y. Hui, X. Zhou, J. Jiang, Y. Cai, X. Liu, L. Lin and X. Feng, Tetrahedron Lett., 2010, 51, 4175 CAS; (e) B. M. Trost and A. Quintard, Angew. Chem., Int. Ed., 2012, 51, 6704 CAS; (f) B. M. Trost, S. Malhotra, P. Koschker and P. Ellerbrock, J. Am. Chem. Soc., 2012, 134, 2075 CAS; (g) B. M. Trost, S. Malhotra and P. Ellerbrock, Org. Lett., 2013, 15, 440 CrossRef CAS PubMed.
- (a) C.-C. Chen, C.-C. Chyau and T.-H. Hseu, Lett. Appl. Microbiol., 2007, 44, 387 CAS; (b) M.-R. Wu, M.-H. Hou, Y.-L. Lin and C.-F. Kuo, J. Agric. Food Chem., 2012, 60, 7262 CAS.
- (a) G. Wald, Nature, 1934, 134, 65 CAS; (b) G. Duester, Cell, 2008, 134, 921 CrossRef CAS PubMed; (c) A. Tsin, B. Betts-Obregon and J. Grigsby, J. Biol. Chem., 2018, 293, 13016 CAS.
- CCDC 2408217 [for 3da] contains the ESI crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/data_request/cif.
- (a) P. Merino, A. Lanaspa, F. L. Merchan and T. Tejero, Tetrahedron:Asymmetry, 1997, 8, 2381 CAS; (b) S. Pinet, S. U. Pandya, P. Y. Chavant, A. Ayling and Y. Vallee, Org. Lett., 2002, 4, 1463 CAS; (c) D. Dhavale and S. Jachak, Molecules, 2005, 10, 893 CAS; (d) V. I. Supranovich, V. V. Levin, M. I. Struchkova and A. D. Dilman, Org. Lett., 2018, 20, 840 CAS.
- P. J. Walsh, and M. C. Kozlowski in Fundamentals of Asymmetric Catalysis, University Science Books, Sausalito, 2009, pp. 330–374 Search PubMed.
- (a) C. Y. Huang, in Determination of Binding Stoichiometry by the Continuous Variation Method: The Job Plot, Methods in enzymology, 1982, pp. 509–525 Search PubMed; (b) J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52, 11998 CAS.
- N. Shao, Y.-Y. Luo, H.-J. Lu, Y.-Z. Hu and M.-C. Wang, Tetrahedron, 2018, 74, 2130 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The datasets supporting this article have been uploaded as part of the ESI. CCDC 2408217. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01453k |
| This journal is © The Royal Society of Chemistry 2025 |