
Oxetane as a part of modern medicinal chemistry toolbox: the advanced synthesis of 3,3-disubstituted building blocks†
Eduard V.
Litskan
ab,
Oleksandr V.
Semenchenko
ab,
Serhii V.
Lynnyk
ac,
Dmitry S.
Granat
ab,
Bohdan V.
Vashchenko
ab,
Anastasiia Ye.
Hoida
ab,
Daria A.
Tolmachova
a,
Dmytro O.
Leha
 ab,
Oleksandr O.
Grygorenko
ab,
Dmytro M.
Volochnyuk
*abd and
Serhiy V.
Ryabukhin
*abd
ab,
Oleksandr O.
Grygorenko
ab,
Dmytro M.
Volochnyuk
*abd and
Serhiy V.
Ryabukhin
*abd
aEnamine Ltd, 78 Winston Churchill str., 02094 Kyiv, Ukraine. E-mail: s.v.ryabukhin@gmail.com; d.volochnyuk@gmail.com
bTaras Shevchenko National University of Kyiv, 60 Volodymyrska str., 01601 Kyiv, Ukraine
cV. N. Karazin Kharkiv National University, 4 Svobody Square, Kharkiv, Ukraine
dInstitute of Organic Chemistry, National Academy of Sciences of Ukraine, 5 Akademik Kuhar str., 02660 Kyiv, Ukraine
First published on 22nd April 2025
Abstract
Despite numerous potential advantages of oxetanes, their restricted synthetic accessibility and propensity to ring-opening hamper their wide application in drug design. In this work, we disclose our experimental achievements and 10 years of experience in oxetane chemistry and provide a comprehensive study on the oxetane core tolerance towards the reaction conditions of the typical toolbox of organic and medicinal chemists, which is based on our original, unpublished developments. The scope of examined reactions includes oxidation, reduction, alkylation, acylation, nucleophilic substitution, C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C
C/C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond formation, hydrolysis, and protecting groups cleavage, and demonstrates the stability of the oxetane's moiety towards acidic and basic conditions. Over 40 transformations were applied to generate the oxetane chemical stability profile, which could direct the design of other synthetic approaches for the synthesis and incorporation of oxetanes in the future. The additional aim of the work included the optimization of the synthetic protocols with the possibility of scaling up to 1 kg in a single run. Newly designed synthetic protocols in our hands allowed for the preparation of novel 3,3-disubstituted oxetanes as small building blocks (over 100 examples, almost 90% of which were not reported previously in the literature). These results benefit the development of oxetanes as a part of the toolbox of modern medicinal chemistry, as well as their incorporation into drug development programs.
C bond formation, hydrolysis, and protecting groups cleavage, and demonstrates the stability of the oxetane's moiety towards acidic and basic conditions. Over 40 transformations were applied to generate the oxetane chemical stability profile, which could direct the design of other synthetic approaches for the synthesis and incorporation of oxetanes in the future. The additional aim of the work included the optimization of the synthetic protocols with the possibility of scaling up to 1 kg in a single run. Newly designed synthetic protocols in our hands allowed for the preparation of novel 3,3-disubstituted oxetanes as small building blocks (over 100 examples, almost 90% of which were not reported previously in the literature). These results benefit the development of oxetanes as a part of the toolbox of modern medicinal chemistry, as well as their incorporation into drug development programs.
Introduction
Since the development of the first preparative methods for the parent oxetane (originally called trimethylene oxide) in 19161 and 1949,2 this simple heterocyclic core has remained exotic for the chemical community for many years, mainly due to its uncertain reactivity. According to classical heterocyclic chemistry textbooks, oxetane's ability to undergo ring-opening reactions lies in the reactivity of oxiranes and tetrahydrofurans. It is well-known that oxiranes have found applications as useful reactive intermediates with a thoroughly characterized reactivity profile. Meanwhile, the tetrahydrofuran backbone is regarded as a stable moiety, highlighted by THF as one of the most popular solvents in organic chemistry. In a chapter of “PATAI'S Chemistry of Functional Groups” published in 1967,3 one can find practical guidance underlining that the oxetane fragment generally tolerates alkali conditions excepting Grignard reagents and complex hydrides. At the same time, this ring is rather unstable in an acidic environment and is easily cleaved by external nucleophiles available in the reaction medium. Additionally, ring-opening reactions were indicated to be possible under thermal and transition metal-catalyzed conditions. However, all the available data were scarce and inconsistent for providing a comprehensive grasp of the oxetane's reactivity profile, thus leading to limited applications of this class of compounds as either reactive intermediates in organic synthesis or “stable” fragments in functional-oriented design for many years. Nevertheless, the continuous discovery of oxetane-bearing natural products4 paved the way for the development of the chemotherapy medication paclitaxel, primarily isolated in 1971 from Taxus brevifolia and approved by the FDA for medical use in 1993. Commonly known by its brand name Taxol®, it shares the title of the only oxetane-containing FDA-approved drug with its semi-synthetic analogs Docetaxel (Taxorete®) and Cabazitaxel (Jevtana®). This fact called for further seminal insightful studies by the Carreira's group in collaboration with Hoffmann-La Roche (Roger-Evans, Müller),5–7 which uncovered distinct patterns of oxetane's core drug-like properties and isosteric relationships with carbonyl and gem-dimethyl groups. Particularly, structural variations revealed improved metabolic stability and lower lipophilicity of the oxetane fragment compared to groups commonly exploited in MedChem projects, thus promoting it to an “emerging motif” in drug discovery.8 Nowadays, oxetanes have entered routine medicinal chemistry, as pointed out by the explosive increase in the number of oxetane-based substances used in drug discovery (Fig. 1, box A). The collection and analysis of such an impressive data array allow for a precise formulation of the oxetane role in medicinal chemistry, which is discussed in several review articles.9–11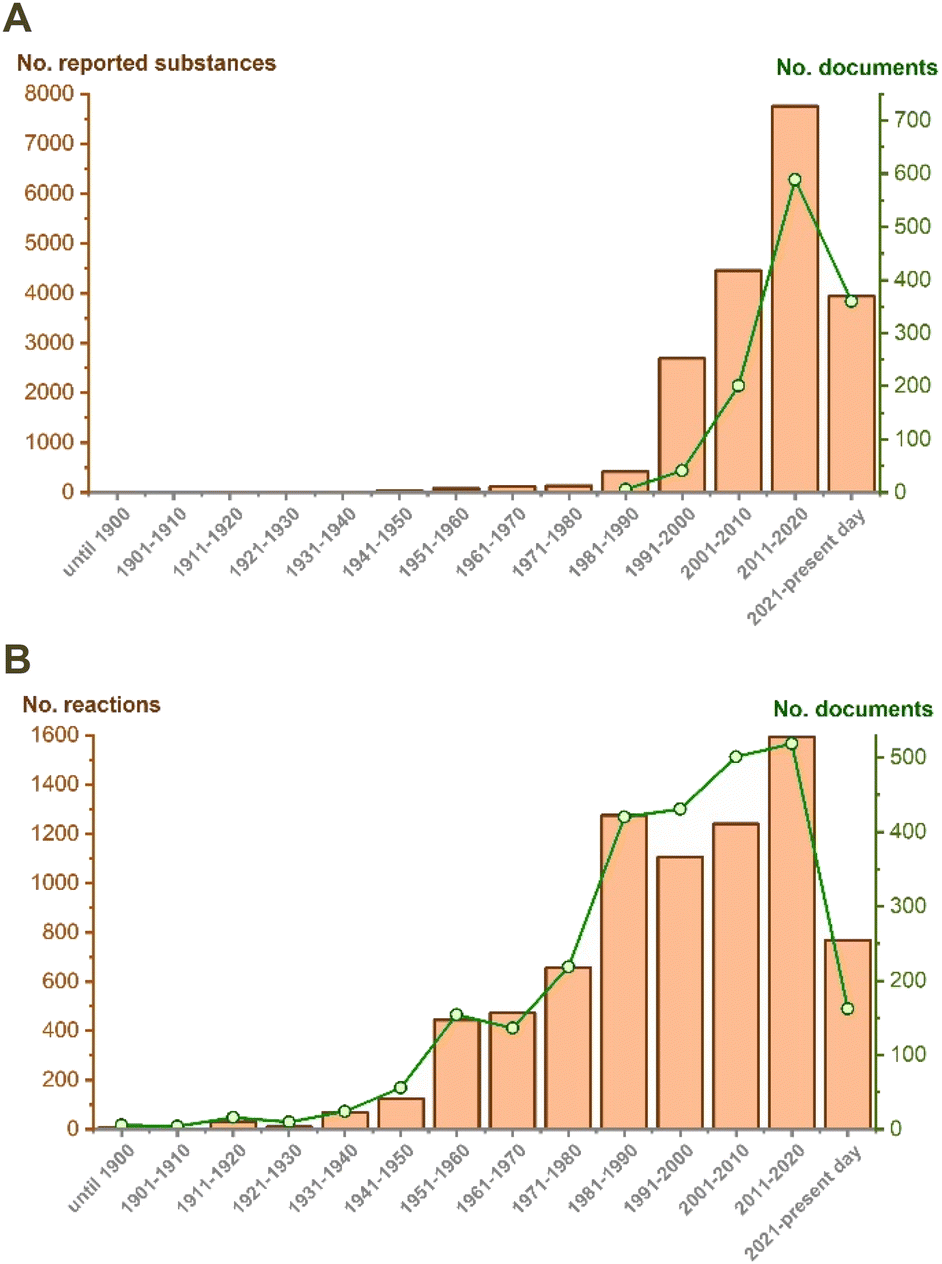 | ||
| Fig. 1 (A) The number of biologically active oxetanes deposited in the Reaxys database along with the number of the corresponding references; (B) the number of oxetane ring cleavage reactions deposited in the Reaxys database along with the number of the corresponding references. | ||
Meanwhile, the number of oxetane ring cleavage examples observed in synthetic practice is increasing as well (Fig. 1, box B, and Scheme 1). Those included the Brønsted11 or Lewis12,13 acid-catalyzed reactions, reactions with hydrides14 and organometallic reagents,1,15,16etc. Apparently, there were attempts to systematize all the information on the issue. However, as far as we know, only one review article discussed the possible synthetic application of oxetane cleavage reactions.17
 | ||
| Scheme 1 Selected representative examples of the ring-opening reactions of oxetanes. | ||
Still, detailed data regarding the chemical stability of the oxetane ring remains unavailable, impeding further advancements in this area and hindering the growth of diversity and nomenclature of commercially available oxetane-derived building blocks. Our recent analysis of commercially accessible building blocks revealed only 891 examples of those containing oxetane rings.18 This value is extremely low compared to other MedChem-relevant classes of compounds.
As mentioned above, it is well-known that the oxetane ring is prone to ring-opening reactions mostly in acidic conditions, even upon isolation of oxetanes from the reaction mixtures or their storage.19 Our research is intended to share our synthetic experience gained while performing in-house programs directed to enriching the stock with 3,3-disubstituted oxetane-derived building blocks (Fig. 2) using 3-oxetanone as a common starting compound available on a kilogram scale.
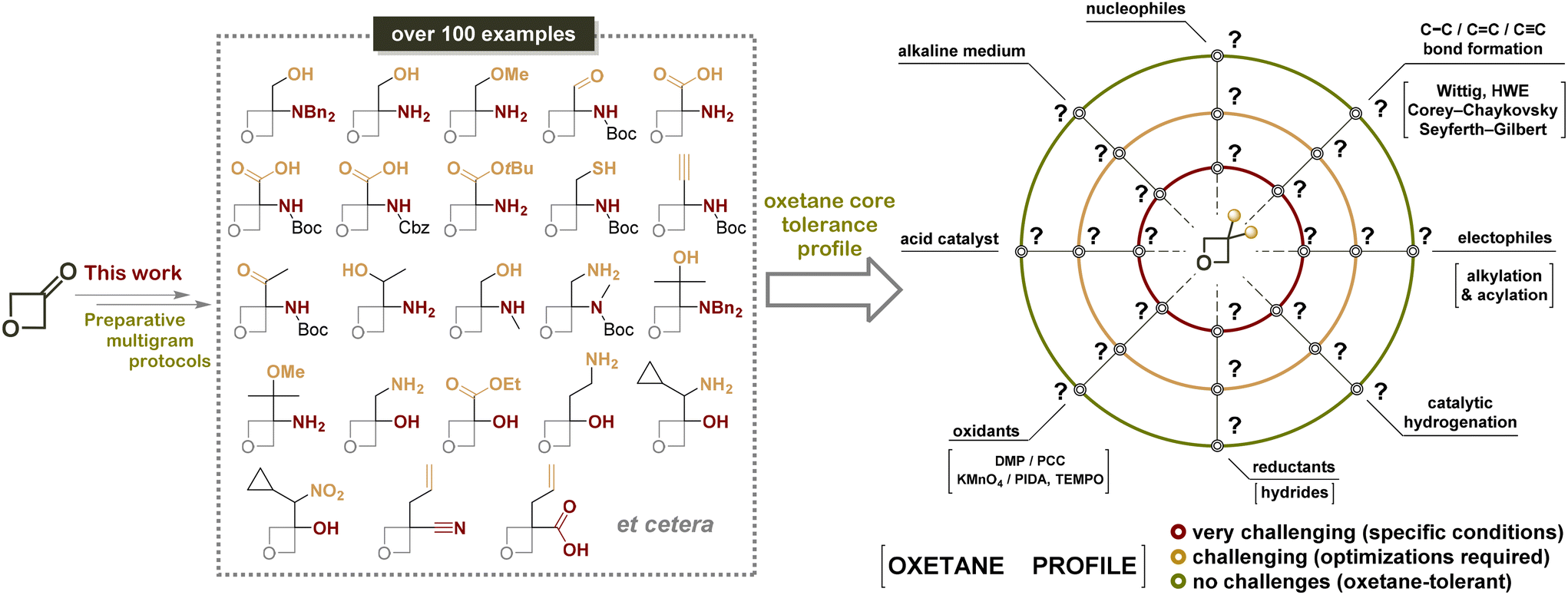 | ||
| Fig. 2 Synopsis of the research: the synthesis of the title compounds and further analysis of the oxetane reaction profile. | ||
The performed transformations cover a series of functional group interconversions, including oxidation, reduction, alkylation, acylation, nucleophilic substitution, hydrolysis, and protecting group cleavage, as well as C–C/CC/CC bond formation. Among others, the evaluated protocols include the application of acidic or basic reaction conditions. If the oxetane core was unstable under the model reaction conditions, we modified and/or optimized them to make the required transformations possible.
This paper summarizes our many years of experimental findings and efforts in the discussed research area, including hundreds of test experiments (both successful and unfruitful). The proposed optimized and efficient transformations can give rise to a series of novel small sp3-enriched building blocks, promising for modern practice-oriented organic and medicinal chemistry. Notably, among 107 compounds obtained, the synthesis of only 12 examples was previously reported. Additionally, this work offers an outline of oxetane core tolerance toward different reaction conditions by analogy with the “robustness screen” approach applied earlier for the catalytic reactions,20 which could be a helpful comprehensive manual for synthetic and medicinal chemists working in the field (Fig. 2).
Results and discussion
First, we have aimed to obtain a series of common oxetane-containing precursors with a 3,3-disubstituted pattern. For this purpose, we utilized the commercially available bulk oxetan-3-one (1) as the starting material. Initial transformations (T) involved a series of well-established ketone functionalization reactions (Fig. 3). In particular, the Strecker synthesis with TMSCN as the cyanide source and dialkylamine yielded 3-cyano-3-dibenzylamino oxetane. It opened pathways to a diverse scope of oxetane amino acids and their derivatives. The oxetane-tolerant introduction of the aminomethyl group alongside a tertiary hydroxyl group was achieved via the Henry synthesis with nitromethane and other nitroalkanes. In turn, the Horner–Wadsworth–Emmons (HWE) reaction transformed oxetan-3-one into α,β-unsaturated ester derivatives, facilitating further functionalization through Michael addition and dihydroxylation (Scheme 2).21–24 | ||
| Fig. 3 Toolbox of transformations (T) in the chemistry of oxetanes designed and elaborated in this investigation (green– suitable method; yellow – despite the method generally could be used, the conditions are not optimized, therefore providing low yields and/or purity of products; brown – the formation of products was not observed, the starting materials were regenerated; red – the set of conditions led to the decomposition of the starting materials). Detailed procedures and reaction schemes are given in the ESI.† | ||
 | ||
| Scheme 2 The utility of oxetanone for the preparation of functionalized derivatives tolerating the oxetane moiety. | ||
Next, we have systematically tested a wide range of reactions and conditions from the common toolbox of organic chemists, aiming to maximize the synthetic utility of this fragment. Following the aforementioned Strecker synthesis, the subsequent transformation was the hydrolysis of the nitrile fragment. This step was crucial as it enabled the formation of a series of amino acid derivatives, significantly expanding the range of accessible compounds.
The hydrolysis was performed under the oxetane-tolerant basic conditions following the previously described strategies.19 This approach proved highly efficient and provided the desired products in high purity and yield. The acidic catalysis facilitated the ring-opening of oxetane and the formation of unwanted byproducts, which is the particular reaction limiting the use of oxetanes. Notably, the treatment with acids is also standard during the isolation step of the product according to the common protocols. This has inspired us to conduct a detailed study and optimize the methods of further reactions in this work to develop efficient procedures that are tolerant to the oxetane core.
A set of other essential transformations included the Corey–Chaykovsky cyclopropanation with trimethyl sulfoxonium-derived ylide, which provided a pathway to spirocyclic 5-oxaspiro[2.3]hexane-1-carboxylate, shown for the case of the acrylate mentioned above (Scheme 2, T31).
Recognizing the success of the use of the basic conditions, we applied the same strategy for ester hydrolysis in subsequent stages, again preventing ring-opening reactions and producing high-purity products on the multigram scale (Scheme 3, T7).
 | ||
| Scheme 3 Synthesis of oxetane-3-carboxylic acids. | ||
The presence of the acidic carboxylic group itself is compatible with the oxetane core; the ring-opening reaction is observed only in the case of treatment with strong acids. This limited the esterification reaction to basic conditions, including the use of alkyl halides. The mild reaction conditions included the treatment with Hunig's base (Scheme 4, T9). In contrast, attempts to perform the esterification using the corresponding alcohol in the presence of HCl expectedly led to decomposition. The synthesis of tert-butyl esters involved a reaction with isobutylene in the presence of a catalytic amount of TsOH, with the oxetane ring remaining intact.
 | ||
| Scheme 4 Synthesis of amino ester derivatives via esterification. | ||
As expected, esters facilitated a straightforward pathway to the corresponding primary alcohols. However, initial attempts to use aluminum and boron hydride reagents proved suboptimal. Our first experiments with LiAlH4 at temperatures above 0 °C resulted in the decomposition of oxetane carboxylates, contrary to results reported in the literature.23,25 Performing the reaction at temperatures between −30 and −10 °C was successful in most cases. The reaction was relatively slow in the case of derivatives with the N-Boc-protected group and provided moderate yields of the target hydroxymethyl derivatives. Alternatively, using NaBH4 at 0 °C was more fruitful (Scheme 5, T1). Despite being uncommon and unsuitable for most of the existing esters, this approach minimized the side decomposition reaction while maintaining reasonable reaction rates, leading to higher yields of the desired alcohols.
 | ||
| Scheme 5 Reduction of oxetane-3-carboxylates to hydroxyalkyl-substituted derivatives. | ||
It should be noted that the direct reduction of carboxylic acids to primary alcohols was challenging and low-yielding. The treatment with LiAlH4 did not show signs of any reaction below 0 °C, and rapid decomposition was observed at elevated temperatures, which agrees with the literature data.26
An attempt to use mixed anhydride in the reaction with NaBH4 resulted in limited success.27 The treatment of oxetane-3-carboxylate with a Grignard reagent (MeMgBr), at −20 °C to rt, resulted in the formation of ca. 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 mixture of the mono- and double addition products, i.e., ketones and tertiary alcohols, that were separated by column chromatography (Scheme 6, T20).24,28–31 Further reduction of the ketone to secondary alcohol was performed successfully using the conditions described above for esters, i.e., LiAlH4 at −30 °C or NaBH4 at 0 °C (Scheme 6, T16).
:7 mixture of the mono- and double addition products, i.e., ketones and tertiary alcohols, that were separated by column chromatography (Scheme 6, T20).24,28–31 Further reduction of the ketone to secondary alcohol was performed successfully using the conditions described above for esters, i.e., LiAlH4 at −30 °C or NaBH4 at 0 °C (Scheme 6, T16).
 | ||
| Scheme 6 The reaction with Grignard reagents and the reduction of ketones. | ||
The obtained hydroxy groups allowed for a wide range of diversification providing oxetane-containing building blocks. The corresponding ethers were synthesized via Williamson alkylation in the presence of NaH or t-BuOK at temperatures varying from 0 to 80 °C (depending on the reaction, Scheme 7, T21).32–35 The mesylation reaction proceeded smoothly with MsCl and Et3N in CH2Cl2 at 0 °C (Scheme 7, T34).36–39 Several successful strategies were developed for the synthesis of fluorine-containing oxetanes. For this purpose, we utilized oxygen-containing functional groups in direct deoxyfluorination (Scheme 7, T13). Notably, DAST and morph-DAST were successfully used to transform alcohols to fluorides at −78 to 0 °C with the oxetane ring left intact. However, the deoxyfluorination attempts were unsuccessful for the carboxylic acids due to insufficient reactivity even at elevated temperatures up to 100 °C. The use of more robust SF4 and HF resulted in substrate decomposition rather than the desired trifluorination.
 | ||
| Scheme 7 Transformations of oxetan-3-yl methanols. | ||
The oxidation of hydroxymethyl-substituted oxetanes with Dess–Martin reagent (DMP) or PCC was an efficient approach to the synthesis of the corresponding aldehydes when additional protected functional groups were present. It was found that DMP was most suitable in the case of the presence of α-NHBoc substituent, while chromium(VI) reagents worked well in the case of alkyl-substituted heterocycles (Scheme 8, T8, and T28). Similarly, oxidation to carboxylic acids was substrate-specific. The radical pathway using TEMPO with PIDA, despite being most suitable for α-NHBoc-substituted substances, sometimes led to partial decomposition. Less reactive α-alkyl-substituted compounds were successfully converted to the corresponding carboxylic acid via the robust treatment with KMnO4; no decomposition was observed in this case (Scheme 8, T12).27,40
 | ||
| Scheme 8 Oxidation of alcohols to aldehydes and carboxylic acids. | ||
The carbonyl group of aldehydes and ketones could be used to prepare other classes of oxetanes. Model reactions included the deoxofluorination of aldehydes (Scheme 9, T38) as an important reaction for the incorporation of the CHF2 substituent into the oxetane core. The Seyferth–Gilbert reaction using the Ohira–Bestmann reagent is suitable for introducing the highly valuable acetylene fragment into the oxetane core (Scheme 9, T24).
 | ||
| Scheme 9 Reactions of oxetane-derived aldehydes with nucleophilic reagents. | ||
An alternative route for the incorporation of fluorine atoms involved mesylation followed by the nucleophilic substitution reaction with a fluoride source at 60 °C (Scheme 10, T14).
 | ||
| Scheme 10 A set of nucleophilic substitution reactions for the preparation of halides, azides, nitriles, and benzoates. | ||
In the case of N-Boc protected oxetane-3-amines, the nucleophilic substitution of mesylate with the azide anion failed, showing no reaction below 40 °C. Upon further heating, cyclization products were identified (Scheme 10, T46).41 Other substrates, i.e. 3-fluorooxetanes, proved to be suitable for the nucleophilic substitution of MsO-group, providing azides after reaction with NaN3 at 80 °C in DMF (Scheme 10, T46), or bromides when treated with LiBr at 60 °C for 5 h (Scheme 10, T14).
Sulfur-containing derivatives were obtained from the mesylates. The reaction proceeded with reasonable yields within a narrow temperature range (ca. 40 °C) with KSAc or NaSMe as the sulfur sources (Scheme 11, T11).42
 | ||
| Scheme 11 Results of the use of S-nucleophiles for the substitution of bromide. | ||
Significantly, no conversion was observed at 20 °C, while raising the temperature to 60 °C led to partial or complete rearrangements of starting materials. The use of hydrazine monohydrate was successful in cleaving the S-acetyl moiety to give thiol from the corresponding thioacetate.
Since most of the aforementioned compounds were bifunctional, the critical task was related to the selective installation and cleavage of the corresponding protecting groups. In particular, quantitative oxetane-tolerant cleavage of the N-Bn group of 3-aminooxetnes was achieved using an enriched 20% Pearlman's catalyst under 80 bar of H2 at 60 °C (Scheme 12, T18).43,44
 | ||
| Scheme 12 Catalytic debenzylation for the preparation of primary and secondary amines (higher pressure up to 80 atm only provided more rapid conversion; no changes in yields or purity were observed); reductive amination. | ||
Some oxetan-3-amines could be purified and stored as hydrochlorides, i.e. via the dilution of amines in a basic form with t-BuOMe, followed by the titration with 2 M HCl–Et2O until pH 7 was reached (shown for the case of compounds 63, 74, and 83). Due to the presence of a basic amino group, the oxetane core remained intact in these cases, at least in the case of the calculated amount of acid used to give the neutral media. Using the saturated solution of HCl in 1,4-dioxane is less fruitful in such a case due to the harsher conditions required to obtain a pure amine hydrochloride.
Introducing N-Boc protection using Boc2O at 50 °C was straightforward, mild, and relatively successful in all cases (Scheme 13, T22). However, its removal proved more challenging due to the commonly required use of acids (mostly HCl or TFA). In contrast to the previous cases when the basic center is attached to the oxetane core, thus allowing the transformation of oxetane-3-amines in hydrochloride forms, the standard direct treatment with HCl in Et2O or 1,4-dioxane resulted in partial or complete decomposition of the substrate at rt after 1 h.
 | ||
| Scheme 13 Incorporation and removal of N-Boc group. | ||
In contrast, the reaction with TFA in CH2Cl2 proceeded smoothly, providing a reliable method for N-Boc removal and tolerating the oxetane fragments present in the molecule (Scheme 13, T6). Further treatment of the obtained amine trifluoroacetates with 2 M HCl–Et2O was applied to give the corresponding hydrochlorides, which are typically more crystalline.
For the Cbz removal, fruitful results were obtained in the presence of Et3N under 60 bar of H2 (Scheme 14, T19).40 These methods proved more effective and oxetane-tolerant than the procedures applied for related compounds previously and could be applied to multigram syntheses.45
 | ||
| Scheme 14 Incorporation and removal of N-Cbz and N-Fmoc groups. | ||
The oxetane-tolerant reduction with cobalt boride (obtained in situ from CoCl2 and NaBH4) of nitriles in the presence of Boc2O was rapid and well-suited for the efficient preparation of primary amines on a multigram scale (Scheme 15, T4). We also studied two more common methods for this reaction. In particular, LiAlH4 decomposed the oxetane core at any temperature in the range above 0 °C. However, Ra–Ni at 60 °C under 80 atmospheres of H2 was an oxetane-tolerant successful approach; no undesirable side reactions were observed.
 | ||
| Scheme 15 Synthesis of aminomethyl- and aminoethyl-substituted oxetanes. | ||
However, these conditions were inappropriate for reducing the nitro group to amine, as they required prolonged reaction times and provided products with low purity. The milder conditions, specifically, hydrogenation under 50 atm in the presence of Pd(OH)2/C, solved the latter issue (Scheme 15, T5). This method proved to be a preparative approach for obtaining 3-hydroxy-3-methylamino oxetanes via the Henry reaction.
The transformations of azides to the corresponding primary amine relied on the standard Staudinger procedure with PPh3 and H2O (Scheme 15, T10). Attempts to perform this transformation with Pd/C catalysis yielded a low amount of product with unsatisfactory purity.46
Yet another way to access amines involves amide reduction using AlH3 at −78 °C to −50 °C. Attempts to use NaBH4 or LiAlH4 procedure under various conditions resulted in the decomposition of the starting oxetane-containing amides.
A strategy developed for the construction of spirocyclic oxetane derivatives involved ring-closing metathesis of two allyl fragments attached to the oxetane core using the Grubbs catalyst at 120 °C (Scheme 16, T25).6 The corresponding diallyl fragment was accessed via the Petasis reaction with the corresponding allyl amine.47 The trifluoroacetyl protecting group proved to be the most suitable for the reaction of interest. Its removal posed no significant issues and was achieved through basic hydrolysis at 60 °C (Scheme 16, T30). The CC double bond of the side chain can be easily reduced by catalytic hydrogenation (Scheme 16, T17).
 | ||
| Scheme 16 Synthesis of spirocyclic derivatives via the metathesis or iodocyclization reaction. | ||
The other type of spirocyclic scaffold construction relied on the iodocyclization reaction that also tolerated the oxetane fragment (Scheme 16, T15).
Conclusion
Our experimental findings disclosed in this study significantly expand the synthetic toolkit for 3,3-disubstituted oxetanes, highlighting challenging assumptions about their instability in ring-opening reactions. The findings demonstrate a broader tolerance of oxetanes to diverse conditions than previously considered (Fig. 2).The oxetane reactivity profile briefly summarizes all efforts and attempted syntheses (Fig. 4). Starting from widespread bulky oxetan-3-one, common ketone transformations (e.g., Strecker, Horner–Wadsworth–Emmons, Grignard) enable functionalization of the latter up to a hundred-gram scale. Basic hydrolysis of nitriles and esters provides amino acids efficiently, avoiding acid-mediated ring opening.
 | ||
| Fig. 4 The resulting profile of the reactivity of oxetanes according to all synthetic transformations performed in this work. | ||
As observed in numerous attempted syntheses, the reduction methods required precise optimization, proving crucial for modifying functional groups adjacent to the oxetane core.
Optimized conditions enabled selective carbonyl reductions of esters, amides, and ketones at lower temperatures to minimize by-products. Several strategies were advised for introducing amino and aminoalkyl groups into oxetanes. In contrast to heterogeneous reduction conditions, the azide transformations via the Staudinger reaction proceeded smoothly. Additional optimization was required for the reduction of nitro and nitrile groups.
Protection and deprotection strategies included Pearlman's catalyst for N-Bn/N-Cbz removal. The introduction of N-Boc protection was relatively straightforward, although its removal required significant optimization with satisfactory results only in the case of TFA as the reagent.
Oxidation of oxetanes to aldehydes was efficiently achieved using DMP or PCC, while carboxylation was possible via TEMPO/PIDA or KMnO4 oxidation. Fluorination involved deoxy-/deoxofluorination and mesylate substitution, while alkylation, mesylation, and nucleophilic substitution facilitated sulfur and halogen derivative synthesis. Additional transformations included the Seyferth–Gilbert reaction (acetylene introduction), Corey–Chaykovsky cyclopropanation, and Grubbs-catalyzed ring-closing metathesis. Some challenges remained, particularly with nucleophilic substitutions, which led to the rearrangement or elimination products, and trifluorination of carboxylic acids, which proved ineffective.
This work compiles newly designed optimized methods for synthesizing and functionalizing 3,3-disubstituted oxetanes, disproving conventional concerns about their fragility. Many reactions were successfully scaled up, enhancing practical applications in medicinal and synthetic organic chemistry, affording a library of accessible oxetanes with almost 100 novel compounds (see the ESI, Scheme S27†). These findings should support drug discovery by expanding the accessible chemical space and improving the physicochemical properties of novel compounds.
Abbreviations
| DAST | Diethylaminosulfur trifluoride |
| DIPEA | N,N-Diisopropylethylamine |
| DMP | Dess–Martin periodinane |
| HWE | Horner–Wadsworth–Emmons |
| PCC | Pyridinium chlorochromate |
| PIDA | Phenyliodine(III) diacetate ((diacetoxyiodo)benzene) |
| RCM | Ring-closing metathesis |
| TBAF | Tetra-n-butylammonium fluoride |
Author contributions
Conceptualization, S. V. R., D. M. V. and D. S. G.; methodology, E. V. L., D. S. G. and O. O. G.; investigation, E. V. L., O. V. S., S. V. L., D. S. G., B. V. V., A. Y. H., D. A. T. and D. O. L.; formal analysis, B. V. V., A. Y. H. and O. O. G.; validation, E. V. L., D. S. G., B. V. V. and D. O. L.; visualization, B. V. V. and D. O. L.; writing – original draft, E. V. L. and B. V. V.; writing – review & editing, B. V. V., D. M. V., O. O. G. and S. V. R.; resources, D. M. V. and O. O. G.; supervision, D. M. V. and S. V. R.; funding acquisition, project administration, S. V. R.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by Enamine Ltd and Ministry of Education and Science of Ukraine (Grant No. 0123U102102). The authors thank Mr Bohdan S. Sosunovych, and Mr Illia O. Doroshenko, for their help with manuscript preparation, Prof. Andriy O. Tolmachov for his encouragement and support, and all the brave people of Ukraine for making this publication possible.References
- C. G. Derick and D. W. Bissell, Studies of trimethylene oxide. I. Preparation and characterization, J. Am. Chem. Soc., 1916, 38(11), 2478–2486, DOI:10.1021/ja02268a023.
- C. R. Noller, Trimethylene oxide, Org. Synth., 1949, 29, 92, DOI:10.15227/orgsyn.029.0092.
- R. J. Gritter, Reaction of Cyclic Esters, PATAI'S Chemistry of Functional Groups, ch. 9, 11967, pp. 373–443, DOI:10.1002/9780470771075.ch9.
- V. M. Dembitsky, Highly Oxygenated Cyclobutane Ring in Biomolecules: Insights into Structure and Activity, Oxygen, 2024, 4(2), 181–235, DOI:10.3390/oxygen4020012.
- G. Wuitschik, M. Rogers-Evans, K. Müller, H. Fischer, B. Wagner, F. Schuler, L. Polonchuk and E. M. Carreira, Oxetanes as Promising Modules in Drug Discovery, Angew. Chem., Int. Ed., 2006, 45(46), 7736–7739, DOI:10.1002/anie.200602343.
- G. Wuitschik, M. Rogers-Evans, A. Buckl, M. Bernasconi, M. Märki, T. Godel, H. Fischer, B. Wagner, I. Parrilla, F. Schuler, J. Schneider, A. Alker, W. B. Schweizer, K. Müller and E. M. Carreira, Spirocyclic Oxetanes: Synthesis and Properties, Angew. Chem., Int. Ed., 2008, 47(24), 4512–4515, DOI:10.1002/anie.200800450.
- G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, Oxetanes in Drug Discovery: Structural and Synthetic Insights, J. Med. Chem., 2010, 53(8), 3227–3246, DOI:10.1021/jm9018788.
- O. O. Grygorenko, D. M. Volochnyuk and B. V. Vashchenko, Emerging Building Blocks for Medicinal Chemistry: Recent Synthetic Advances, Eur. J. Org. Chem., 2021, 2021(47), 6478–6510, DOI:10.1002/ejoc.202100857.
- G. Huang, D. Hucek, T. Cierpicki and J. Grembecka, Applications of Oxetanes in Drug Discovery and Medicinal Chemistry, Eur. J. Med. Chem., 2023, 261, 115802, DOI:10.1016/j.ejmech.2023.115802.
- J. J. Rojas and J. A. Bull, Oxetanes in Drug Discovery Campaigns, J. Med. Chem., 2023, 66(18), 12697–12709, DOI:10.1021/acs.jmedchem.3c01101.
- J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry, Chem. Rev., 2016, 116(19), 12150–12233, DOI:10.1021/acs.chemrev.6b00274.
- J. C. Mullis and W. P. Weber, Regiospecificity of Reactions of Epoxides and Oxetanes with Trimethylsilyl Cyanide, J. Org. Chem., 1982, 47(15), 2873–2875, DOI:10.1021/jo00136a011.
- S. A. Carr and W. P. Weber, Titanium Tetrachloride Promoted Reactions of Allylic Trimethylsilanes and Oxetane, J. Org. Chem., 1985, 50(15), 2782–2785, DOI:10.1021/jo00215a038.
- S. Searles, K. A. Pollart and E. F. Lutz, Oxetanes. VI.1 Reductive Cleavage and Substituent Effects 2, J. Am. Chem. Soc., 1957, 79(4), 948–951, DOI:10.1021/ja01561a046.
- S. Searles, The Reaction of Trimethylene Oxide with Grignard Reagents and Organolithium Compounds, J. Am. Chem. Soc., 1951, 73(1), 124–125, DOI:10.1021/ja01145a045.
- C. Huynh, F. Derguini-Boumechal and G. Linstrumelle, Copper-Catalysed Reactions of Grignard Reagents with Epoxides and Oxetane, Tetrahedron Lett., 1979, 20(17), 1503–1506, DOI:10.1016/S0040-4039(01)86190-6.
- S. Ahmad, M. Yousaf, A. Mansha, N. Rasool, A. F. Zahoor, F. Hafeez and S. M. A. Rizvi, Ring-Opening Reactions of Oxetanes: A Review of Methodology Development and Synthetic Applications, Synth. Commun., 2016, 46(17), 1397–1416, DOI:10.1080/00397911.2016.1208245.
- Y. Zabolotna, D. M. Volochnyuk, S. V. Ryabukhin, D. Horvath, K. S. Gavrilenko, G. Marcou, Y. S. Moroz, O. Oksiuta and A. Varnek, A Close-up Look at the Chemical Space of Commercially Available Building Blocks for Medicinal Chemistry, J. Chem. Inf. Model., 2022, 62(9), 2171–2185, DOI:10.1021/acs.jcim.1c00811.
- B. Chalyk, A. Grynyova, K. Filimonova, T. V. Rudenko, D. Dibchak and P. K. Mykhailiuk, Unexpected Isomerization of Oxetane-Carboxylic Acids, Org. Lett., 2022, 24(26), 4722–4728, DOI:10.1021/acs.orglett.2c01402.
- L. Pitzer, F. Schäfers and F. Glorius, Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations, Angew. Chem., Int. Ed., 2019, 58(25), 8572–8576, DOI:10.1002/anie.201901935.
- B. Jones, M. Proud and V. Sridharan, Synthesis of Oxetane/Azetidine Containing Spirocycles via the 1,3-Dipolar Cycloaddition Reaction, Tetrahedron Lett., 2016, 57(25), 2811–2813, DOI:10.1016/j.tetlet.2016.05.053.
- J. D. Beadle, N. H. Powell, P. Raubo, G. J. Clarkson and M. Shipman, Synthesis of Oxetane- and Azetidine-Containing Spirocycles Related to the 2,5-Diketopiperazine Framework, Synlett, 2016,(1), 169–172, DOI:10.1055/s-0035-1560593/id/jr000-2001/bib.
- T. Fujishima, T. Suenaga and T. Nozaki, Concise Synthesis and Characterization of Novel Seco-Steroids Bearing a Spiro-Oxetane Instead of a Metabolically Labile C3-Hydroxy Group, Tetrahedron Lett., 2014, 55(28), 3805–3808, DOI:10.1016/j.tetlet.2014.05.060.
- T. Suenega and T. Fujishima, The C4-Functionalized 9,10-Seco-5,7,10(19)-Cholestatriene Derivatives: Concise Synthesis and Characterization of Novel Vitamin D Analogues with a Four-Membered Heterocyclic Ether, Tetrahedron, 2018, 74(13), 1461–1467, DOI:10.1016/j.tet.2018.02.001.
- R. Zhang, M. Sun, Q. Yan, X. Lin, X. Li, X. Fang, H. H. Y. Sung, I. D. Williams and J. Sun, Asymmetric Synthesis of Pyrrolidines via Oxetane Desymmetrization, Org. Lett., 2022, 24(12), 2359–2364, DOI:10.1021/acs.orglett.2c00564.
- E. G. Tse, S. D. Houston, C. M. Williams, G. P. Savage, L. M. Rendina, I. Hallyburton, M. Anderson, R. Sharma, G. S. Walker, R. S. Obach and M. H. Todd, Nonclassical Phenyl Bioisosteres as Effective Replacements in a Series of Novel Open-Source Antimalarials, J. Med. Chem., 2020, 63(20), 11585–11601, DOI:10.1021/acs.jmedchem.0c00746.
- D. Li, D. L. Sloman, A. Achab, H. Zhou, M. A. McGowan, C. White, C. Gibeau, H. Zhang, Q. Pu, I. Bharathan, B. Hopkins, K. Liu, H. Ferguson, X. Fradera, C. A. Lesburg, T. A. Martinot, J. Qi, Z. J. Song, J. Yin, H. Zhang, L. Song, B. Wan, S. Daddio, N. Solban, J. R. Miller, B. Zamlynny, A. Bass, E. Freeland, B. Ykoruk, C. Hilliard, J. Ferraro, J. Zhai, I. Knemeyer, K. M. Otte, S. Vincent, N. Sciammetta, A. Pasternak, D. J. Bennett and Y. Han, Oxetane Promise Delivered: Discovery of Long-Acting IDO1 Inhibitors Suitable for Q3 W Oral or Parenteral Dosing, J. Med. Chem., 2022, 65(8), 6001–6016, DOI:10.1021/acs.jmedchem.1c01670.
- R. A. Croft, J. J. Mousseau, C. Choi and J. A. Bull, Structurally Divergent Lithium Catalyzed Friedel–Crafts Reactions on Oxetan-3-Ols: Synthesis of 3,3-Diaryloxetanes and 2,3-Dihydrobenzofurans, Chem. – Eur. J., 2016, 22(45), 16271–16276, DOI:10.1002/chem.201604031.
- S. E. Kephart, L. R. Zehnder, B. Huang and S. C. Sutton, Synthesis of Oxetane-3-Carboxaldehyde and Methyl Oxetane-3-Carboxylate via Homologation of Oxetane-3-One, Tetrahedron, 2016, 72(26), 3641–3646, DOI:10.1016/j.tet.2016.03.078.
- V. A. Bhosale, M. Nigríni, M. Dračínský, I. Císařová and J. Veselý, Enantioselective Desymmetrization of 3-Substituted Oxetanes: An Efficient Access to Chiral 3,4-Dihydro-2H-1,4-Benzoxazines, Org. Lett., 2021, 23(24), 9376–9381, DOI:10.1021/acs.orglett.1c03419.
- Z. Wang, Z. Chen and J. Sun, Catalytic Enantioselective Intermolecular Desymmetrization of 3-Substituted Oxetanes, Angew. Chem., Int. Ed., 2013, 52(26), 6685–6688, DOI:10.1002/anie.201300188.
- M. Motoi, H. Suda, K. Shimamura, S. Nagahara, M. Takei and S. Kanoh, A Facile Synthesis of Oxetane Derivatives for Preparing Cross-Linked Polyoxetane Resins Bearing the Bromide at the Spacer End, Bull. Chem. Soc. Jpn., 1988, 61(5), 1653–1659, DOI:10.1246/bcsj.61.1653.
- R. Hamill, B. Jones, C. M. Pask and V. Sridharan, Synthesis of Oxetane/Azetidine Containing Spirocycles, Tetrahedron Lett., 2019, 60(16), 1126–1129, DOI:10.1016/j.tetlet.2019.03.042.
- C. H. Lin, Synthesis and Liquid Crystalline Behavior of Photoreactive Side Chain Liquid Crystalline Polyoxetanes Containing Cinnamoyl Biphenyl Mesogen, Asian J. Chem., 2015, 27(4), 1495–1500, DOI:10.14233/ajchem.2015.18540.
- B. Schulte, C. A. Dannenberg, H. Keul and M. Möller, Formation of Linear and Cyclic Polyoxetanes in the Cationic Ring-Opening Polymerization of 3-Allyloxymethyl-3-Ethyloxetane and Subsequent Postpolymerization Modification of Poly(3-Allyloxymethyl-3-Ethyloxetane), J. Polym. Sci., Part A:Polym. Chem., 2013, 51(5), 1243–1254, DOI:10.1002/pola.26494.
- F. Toselli, M. Fredenwall, P. Svensson, X. Q. Li, A. Johansson, L. Weidolf and M. A. Hayes, Hip to Be Square: Oxetanes as Design Elements to Alter Metabolic Pathways, J. Med. Chem., 2019, 62(16), 7383–7399, DOI:10.1021/acs.jmedchem.9b00030.
- X. Fang, Y. M. Zhang, K. Chang, Z. Liu, X. Su, H. Chen, S. X. A. Zhang, Y. Liu and C. Wu, Facile Synthesis, Macroscopic Separation, E/Z Isomerization, and Distinct AIE Properties of Pure Stereoisomers of an Oxetane-Substituted Tetraphenylethene Luminogen, Chem. Mater., 2016, 28(18), 6628–6636, DOI:10.1021/acs.chemmater.6b02746.
- N. G. W. Rose, M. A. Blaskovich, A. Wong and G. A. Lajoie, Synthesis of Enantiomerically Enriched β,γ-Unsaturated-α-Amino Acids, Tetrahedron, 2001, 57(8), 1497–1507, DOI:10.1016/S0040-4020(00)01146-7.
- M. A. Blaskovich, G. Evindar, N. G. W. Rose, S. Wilkinson, Y. Luo and G. A. Lajoie, Stereoselective Synthesis of Threo and Erythro β-Hydroxy and β-Disubstituted-β-Hydroxy α-Amino Acids, J. Org. Chem., 1998, 63(11), 3631–3646, DOI:10.1021/jo972294l.
- D. Vigo, L. Stasi and S. Gagliardi, Synthesis of 3,3-Disubstituted Oxetane Building Blocks, Tetrahedron Lett., 2011, 52(5), 565–567, DOI:10.1016/j.tetlet.2010.11.118.
- T. I. Mukhametshin, A. I. Petrov, N. V. Kuznetsova, V. A. Petrov, N. V. Averianova, I. K. Garaev, A. V. Kostochko, A. T. Gubaidullin, D. B. Vinogradov and P. V. Bulatov, Synthesis and Copolymerization of Azidomethyl-Substituted Oxetanes: The Morphology of Statistical Block Copolymers, Chem. Heterocycl. Compd., 2017, 53(6–7), 811–821, DOI:10.1007/s10593-017-2128-3/metrics.
- E. M. Skoda, J. R. Sacher, M. Z. Kazancioglu, J. Saha and P. Wipf, An Uncharged Oxetanyl Sulfoxide as a Covalent Modifier for Improving Aqueous Solubility, ACS Med. Chem. Lett., 2014, 5(8), 900–904, DOI:10.1021/ml5001504.
- T. Fujishima, T. Nozaki and T. Suenaga, Design and Synthesis of Novel 1,25-Dihydroxyvitamin D3 Analogues Having a Spiro-Oxetane Fused at the C2 Position in the A-Ring, Bioorg. Med. Chem., 2013, 21(17), 5209–5217, DOI:10.1016/j.bmc.2013.06.032.
- S. Wu, C. Xu, K. Xia, Y. Lin, S. Tian, H. Ma, Y. Ji, F. Zhu, S. He and X. Zhang, Ring Closure Strategy Leads to Potent RIPK3 Inhibitors, Eur. J. Med. Chem., 2021, 217, 113327, DOI:10.1016/j.ejmech.2021.113327.
- G. P. Möller, S. Müller, B. T. Wolfstädter, S. Wolfrum, D. Schepmann, B. Wünsch and E. M. Carreira, Oxetanyl Amino Acids for Peptidomimetics, Org. Lett., 2017, 19(10), 2510–2513, DOI:10.1021/acs.orglett.7b00745.
- M. Kumar, V. K. Sharma, R. Kumar and A. K. Prasad, Biocatalytic Route to C-3′-Azido/-Hydroxy-C-4′-Spiro-Oxetanoribonucleosides, Carbohydr. Res., 2015, 417, 19–26, DOI:10.1016/j.carres.2015.08.015.
- L. Gazzard, K. Williams, H. Chen, L. Axford, E. Blackwood, B. Burton, K. Chapman, P. Crackett, J. Drobnick, C. Ellwood, J. Epler, M. Flagella, E. Gancia, M. Gill, S. Goodacre, J. Halladay, J. Hewitt, H. Hunt, S. Kintz, J. Lyssikatos, C. Macleod, S. Major, G. Médard, R. Narukulla, J. Ramiscal, S. Schmidt, E. Seward, C. Wiesmann, P. Wu, S. Yee, I. Yen and S. Malek, Mitigation of Acetylcholine Esterase Activity in the 1,7-Diazacarbazole Series of Inhibitors of Checkpoint Kinase 1, J. Med. Chem., 2015, 58(12), 5053–5074, DOI:10.1021/acs.jmedchem.5b00464.
Footnote |
| † Electronic supplementary information (ESI) available: Sequential synthetic schemes, details on experiments and syntheses; spectral and analytical data for the synthesized compounds; and copies of NMR spectra. See DOI: https://doi.org/10.1039/d5qo00572h |
| This journal is © the Partner Organisations 2025 |