
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of oxylipids via a boronic ester cycloetherification approach†
Markus
Tost
 and
Uli
Kazmaier
*
and
Uli
Kazmaier
*
Organic Chemistry, Saarland University, P.O. Box 151150, 66041 Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de
First published on 8th March 2025
Abstract
Deprotonated trimethylsilylethanol can be used as a nucleophile in Matteson homologations. This O-protection group is stable under the usual reaction conditions of the Matteson reaction, but after two further homologation steps it is automatically cleaved off and cycloetherification can take place, giving rise to substituted tetrahydrofurans in a highly stereoselective fashion. This elegant protocol was used in the synthesis of various oxylipids.
Introduction
Substituted tetrahydrofurans (THFs) are frequently occurring secondary metabolites of terrestrial1 as well as marine organisms.2 For example, the large group of amphidinolides3 are rather complex macrolides, many of which show antitumor activities, often in the nanomolar range. In addition to 2,5-disubstituted THF structures, 2,3,5-trisubstituted THFs are also often found in bioactive natural products, that's why new synthesis strategies for their total synthesis are of great interest.4 In addition to methyl groups, the substituents are usually hydroxy groups (acylated and alkylated) (Fig. 1). For example, trans-Kumausin is isolated from the red algae Laurencia nipponica,5 while the lipid diols trans-oxylipid and cis-oxylipid are produced by brown algae on the south coast of Australia.6,7 Both oxylipids show antihelmintic activity in vitro, which prevents larval development in parasitic nematodes. | ||
| Fig. 1 Structures of selected THF-containing natural products. | ||
The pharmacologically interesting properties of these natural products initiated the development of a large number of syntheses towards these unusual structures. A recent review article by Fernandes et al. very nicely compiles the different synthesis routes reported so far.4a For example, intramolecular epoxy openings8 lead to α-oxygenated THF motifs, as in oxylipids.9 Oxa-Michael additions succeed in the formation of cyclic β-oxygenated ketones and carboxylic acid derivatives,10 as they are found in amphidinolides,11 to name just a few methods. In nature, too, these THF motifs are often built up from linear polyketides via cyclization reactions.2a
The highly diastereoselective Matteson homologation (Scheme 1) should also be suitable for the synthesis of substituted tetrahydrofurans, as it allows the formation of arbitrarily substituted and functionalized carbon chains. The reaction of deprotonated dichloromethane with chiral boronic acid esters A yields α-chloroboronic acid ester B in a highly stereoselective fashion, which can then be further reacted with nucleophiles (Nu−), such as Grignard reagents, alkoxides or enolates to yield homologated boronic ester C (Scheme 1a).12 In general, the products are formed as single stereoisomers, based on the double diastereo-differentiating reaction mechanism.13 The repetition of this reaction sequence enables the stereoselective synthesis of highly substituted carbon chains with adjacent stereocenters. As a rule, the carbon chain can be extended without problems, only in case of alkoxy-substituted boronic esters there are sometimes long reaction times and moderate yields. This effect is particularly pronounced for δ-alkoxy-substituted boronic esters, in which the Lewis-basic oxygen very likely coordinates to the Lewis-acid boron atom and thus prevents the addition of another nucleophiles.14 On the other hand, such an interaction should also activate the oxygen as a leaving group, so that the O-substituent can be split off more easily. During studies on the synthesis of leuconolides, Matteson et al. observed the partial cleavage of a benzyl ether, which indicates exactly such an activation.14
 | ||
| Scheme 1 Matteson homologation and new application. | ||
Results and discussion
Our research group has been working for years on the synthesis of natural products, in particular peptides15 and peptide–polyketide conjugates.16 The Matteson homologation is increasingly17 being used, and we were interested to see if this very useful reaction can also be used to generate polyketides and polypropionates with such furan substructures.Therefore, our goal was to build up substituted THF motifs, as in oxylipids, by boronic ester cycloetherification (Scheme 1b). Starting from boronic acid ester D, the α-chloroboronic acid esters G should be accessible via four iterative Matteson homologation steps. Subsequent deprotection of the alcohol and cyclization should lead to THF-derivative H. The method should take advantage of the fact that a 6-ring chelate of G can be formed with a suitable protecting group (PG) who's cleavage is activated by the Lewis acid boron.14 Therefore, it was necessary to find a suitable protecting group, which can be introduced directly via the Matteson reaction, which is stable under the usual reaction conditions, and which can also be split off again, if necessary, under conditions compatible with the boronic acid ester.
In search of a suitable O-protecting group, we turned to the tert-butyldimethylsilyl (TBS) protecting group, which can be split off with fluoride or under acidic conditions. The corresponding TBS-protected boronic ester 1 could be obtained simply by a protecting group switch from the corresponding benzylether. Homologation to the α-chloroboronic ester 1-Cl proceeded without any problems, but the TBS protection group could not be split off with TBAF, even using several equivalents and long reaction times (Scheme 2). Attempts using acidic conditions were also unsuccessful and the use of HF or Py–HF led to the decomposition of the boronic acid ester.
 | ||
| Scheme 2 First attempts of boronic ester cycloetherification. | ||
Since we suspected that the steric demand of the TBS group could prevent coordination towards the boron, we decided to switch to the smaller trimethylsilylethyl (TMSE) protecting group. This functionality can also be split off with fluoride,18 whereby the corresponding alcoholate is formed under fragmentation and ethylene cleavage. In this case, a possible attack of the fluoride takes place further away from the coordinated O-atom and we hoped that steric aspects would not play a role in this case.
As a first example, boronic ester 2 was reacted with deprotonated dichloromethane and subsequently with trimethylsilyl ethanolate, providing the desired boronic ester 3 in good yield (Scheme 3). The two subsequent homologation steps under standard conditions also proceeded without any problems giving rise to boronic ester 5, which we needed for our cyclization experiments. During the further homologation of 5, the corresponding α-chloroboronic ester 5-Cl could be detected in the reaction mixture by NMR spectroscopy, but it underwent the desired cycloetherification to 6 directly within 2 h. Obviously, the chloride present in the reaction mixture is already sufficient to split off TMSCl and ethylene from the oxygen, activated by coordination, whereby the boron alcoholate formed in this process undergoes an SN2-like 1,2-shift to THF derivative 6.
 | ||
| Scheme 3 Successful cycloetherification using cleavable TMSE ethers. | ||
With this successful cycloetherification in our hands, we turned to the application of this method for the synthesis of natural products and comparable structures. We focused on the class of oxylipids, whereby the cis and the trans form differ primarily in the position of the substituents at the 2,5-position of the tetrahydrofuran ring (C-6, C-9) and in the position of the OH group at C-10 (Fig. 1).
To test the reaction sequence on a simple example first, we decided to start with the synthesis of a dia-cis-oxylipid (Scheme 4). In Matteson homologations, neighboring stereogenic centers are always introduced in a 1,2-anti-configuration, which in the case of oxylipids should lead to the dia-cis-oxylipid 14. Although we were recently able to show that the configuration of methyl groups can be reversed in growing chains,19 this is not so easy to do with hydroxyl groups and requires additional steps.
 | ||
| Scheme 4 Synthesis of 7-epi-cis-oxylipid 14via cycloetherification. | ||
In our case, the n-pentyl boronic ester 7 was first homologated with Cl2CHLi and then converted to boronic ester 8 with lithium trimetyhlsilylethanolate under the previously optimized conditions (Scheme 4). Subsequent homologation with deprotonated dibromomethane yielded the α-bromoboronic ester, which was not isolated but directly reacted with NaOPMB to form boronic ester 9. Further homologation with deprotonated dibromomethane and reduction of the α-bromoboronic ester formed allowed the introduction of a methylene group in boronic ester 10,20 which was then subjected to cycloetherification under the previously optimized conditions to the THF derivative 11. The yield achieved was comparable to that of our model reaction (Scheme 3). To complete the synthesis, 11 was reacted with Cl2CHLi at −100 °C and the corresponding α-chloroboronic ester was reacted with freshly produced 8-nonen-1-ylmagnesium bromide in the presence of ZnCl2 to form boronic acid ester 12. Its oxidation to the protected alcohol (13) and subsequent DDQ-mediated cleavage of the PMB ether led to the desired 7-epi-cis-oxylipid 14 as a single diastereomer. By using the enantiomeric boronic ester ent-7, the enantiomeric 7-epi-cis-oxylipid could also be obtained.
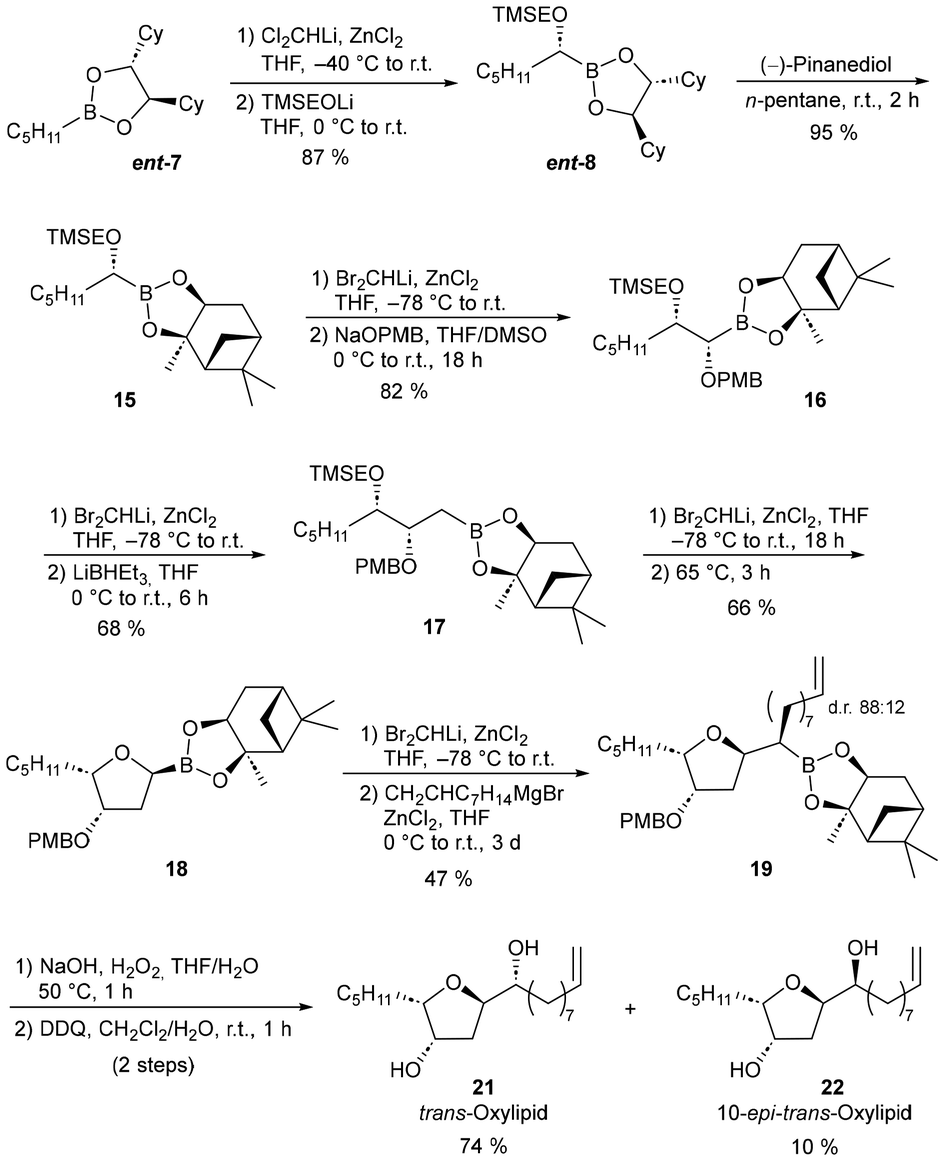
The enantiomeric n-pentyl boronic ester ent-7 was also used to obtain trans-oxylipid (21) (Scheme 5). As already indicated, alkyl groups can be inverted quite easily, but a reversal of the configuration of alkoxy residues is not a trivial issue.21 Unfortunately, some approaches described in the literature22 could not be transferred to the C2-symmetric boronic acid DICHED esters. An alternative approach, however, is the transesterification of a boronic ester with the inversely directing auxiliary, which of course requires additional efforts.23 In the case of DICHED-boronic ester, a transesterification to the inversely directing pinanediol-boronic ester is reported24 as comparably effortless due to the higher thermodynamic stability, which is why we chose this approach (Scheme 5).25Ent-7 was homologated as described before with Cl2CHLi and then converted to ent-8 with TMSEOLi. For transesterification, this boronic ester was dissolved in ether and reacted with 1.1 eq. (−)-Pinanediol, according to a protocol described by Hirschhäuser et al.26 Free (R,R)-DICHED gradually dropped out of the solution, with an equilibrium after 4 d with a conversion rate of 93%. However, if the reaction was not carried out in ether but in pentane, the thermodynamically more stable pinandiol boronic acid ester 15 was quantitatively formed in 2 h, since shortly after the addition of the pinanediol, (R,R)-DICHED precipitated out of the solution and was thus removed from equilibrium. 15 could thus be obtained in a very good yield, easily separated from excess pinanediol and DICHED. In a subsequent homologation with Br2CHLi and substitution with NaOPMB, the 1,2-syn substituted boronic acid ester 16 was obtained in good yield. In the subsequent homologation step Br2CHLi was also chosen in analogy to the last step, as with Cl2CHLi β-elimination was observed as a side reaction. Subsequent substitution with LiBHEt3 resulted in boronic acid ester 17. The subsequent cycloetherification was successful with both the corresponding α-chloroboronic ester and the α-bromoboronic ester, but the ring closure to 18 only took place at an elevated temperature. Interestingly, the subsequent homologation with Cl2CHLi and substitution with the Grignard reagent yielded a significantly worse yield (27%) and poorer selectivity (dr 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32) than the reaction via the corresponding α-bromoboronic ester (dr 88:12). After oxidation to the alcohols (20) and DDQ-mediated cleavage of the PMB protection group, trans-oxylipid 21 as well as the corresponding diastereomer 10-epi-trans-oxylipid 22 were obtained and chromatographically separated.
:32) than the reaction via the corresponding α-bromoboronic ester (dr 88:12). After oxidation to the alcohols (20) and DDQ-mediated cleavage of the PMB protection group, trans-oxylipid 21 as well as the corresponding diastereomer 10-epi-trans-oxylipid 22 were obtained and chromatographically separated.
 | ||
| Scheme 5 Boronic ester cycloetherification in the synthesis of trans-oxylipids. | ||
Conclusions
In summary, Matteson homologation is not only suitable for the synthesis of substituted linear carbon chains in a highly stereoselective manner, but also cyclic tetrahydrofuran motifs. For example, deprotonated trimethylsilylethanol can be used as a nucleophile without any problems, the trimethylsilylethyl protection group is stable under the usual reaction conditions, but after two further homologation steps it is automatically cleaved off and cycloetherification takes place. This elegant protocol was used in the synthesis of various oxylipids.Data availability
Electronic Supplementary Information (ESI) available: Copies of 1H and 13C NMR spectra and experimental details. See https://doi.org/10.1039/D5QO00213C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Saarland University and the DFG (Ka 880/13-1 and Bruker Neo 500–447298507) is gratefully acknowledged. We are grateful to Dr Stefan Boettcher from Pharmaceutical and Medicinal Chemistry department, Saarland University, and Dr. Christine Walt and Alexander Voltz from Helmholtz Institute for Pharmaceutical Research Saarland for support on mass spectrometry.References
- A. Neske, J. Ruiz Hidalgo, N. Cabedo and D. Cortes, Acetogenins from Annonaceae family. Their potential biological applications, Phytochemistry, 2020, 174, 112332 CrossRef CAS PubMed.
- (a) A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Álvarez, Tetrahydrofuran-containing macrolides: a fascinating gift from the deep sea, Chem. Rev., 2013, 113, 4567–4610 CrossRef CAS PubMed; (b) L. Fernández-Peña, C. Díez-Poza, P. González-Andrés and A. Barbero, The tetrahydrofuran motif in polyketide marine drugs, Mar. Drugs, 2022, 20, 120 CrossRef PubMed; (c) P. González-Andrés, L. Fernández-Peña, C. Díez-Poza and A. Barbero, The tetrahydrofuran motif in marine lipids and terpenes, Mar. Drugs, 2022, 20, 642 CrossRef PubMed.
- J. Kobayashi, M. Ishibashi, M. R. Walchli, H. Nakamura, Y. Hirata, T. Sasaki and Y. Ohizumi, Amphidinolide C: the first twenty-five membered macrocyclic lactone with potent antineoplastic activity from the cultured dinoflagellate Amphidinium sp, J. Am. Chem. Soc., 1988, 110, 490–494 CrossRef CAS.
- (a) R. A. Fernandes, R. S. Pathare and D. A. Gorve, advances in total synthesis of some 2, 3, 5-trisubstituted tetrahydrofuran natural products, Chem. – Asian J., 2020, 15, 2815–2837 CrossRef CAS PubMed; (b) R. A. Fernandes, D. A. Gorve and R. S. Pathare, Emergence of 2,3,5-trisubstituted tetrahydrofuran natural products and their synthesis, Org. Biomol. Chem., 2020, 18, 7002–7025 RSC.
- T. Suzuki, K. Koizumi, M. Suzuki and A. Kurosawa, Kumausynes and deacetylkumausynes, four new halogenated c-15 acetylenes from the red alga Laurencia Nipponica Yamada, Chem. Lett., 1983, 12, 1643–1644 CrossRef.
- R. Warren, R. Wells and J. Blount, A novel lipid from the brown alga Notheia anomala, Aust. J. Chem., 1980, 33, 891–898 CrossRef CAS.
- R. J. Capon, R. A. Barrow, S. Rochfort, M. Jobling, C. Skene, E. Lacey, J. H. Gill, T. Friedel and D. Wadsworth, Marine nematocides: Tetrahydrofurans from a southern Australian brown alga, Notheia anomala, Tetrahedron, 1998, 54, 2227–2242 CrossRef CAS.
- U. Koert, Stereoselective synthesis of oligo-tetrahydrofurans, Synthesis, 1995, 115–132 CrossRef CAS.
- (a) R. J. Capon, R. A. Barrow, C. Skene and S. Rochfort, The biomimetic synthesis of marine epoxylipids: Bisepoxides to tetrahydrofurans, Tetrahedron Lett., 1997, 38, 7609–7612 CrossRef CAS; (b) C. Garcia, T. Martin and V. S. Martin, β-Hydroxy-γ-lactones as chiral building blocks for the enantioselective synthesis of marine natural products, J. Org. Chem., 2001, 66, 1420–1428 CrossRef CAS PubMed; (c) Y. Mori, T. Sawada and H. Furukawa, Synthesis of (6S,7S,9R,10R,)-6,9-epoxynonadec-18-ene-7,10-diol, a marine epoxy lipid isolated from the brown alga, Notheia anomala, by an oxiranyl anion strategy, Tetrahedron Lett., 1999, 40, 731–734 CrossRef CAS; (d) R. F. de la Pradilla and A. Castellanos, Katsuki–Jacobsen oxidation–epoxidation of α-silyloxy sulfinyl dienes: application to the formal synthesis of (6S,7S,9R,10R,)-6,9-epoxynonadec-18-ene-7,10-diol, Tetrahedron Lett., 2007, 48, 6500–6504 CrossRef CAS.
- T. Ahmad and N. Ullah, The oxa-Michael reaction in the synthesis of 5-and 6-membered oxygen-containing heterocycles, Org. Chem. Front., 2021, 8, 1329–1344 RSC.
- (a) R. H. Bates, J. B. Shotwell and W. R. Roush, Stereoselective syntheses of the C (1)–C (9) fragment of amphidinolide C, Org. Lett., 2008, 10, 4343–4346 CrossRef CAS PubMed; (b) M. P. Paudyal, N. P. Rath and C. D. Spilling, A formal synthesis of the C1–C9 fragment of amphidinolide C employing the Tamaru reaction, Org. Lett., 2010, 12, 2954–2957 CrossRef CAS PubMed; (c) I. Ciss, M. Seck, B. Figadère and L. Fer, Advances Toward Amphidinolides C, F and U: Isolations, Synthetic Studies and Total Syntheses, Chem. – Eur. J., 2024, 30, e202400471 CrossRef CAS PubMed.
- (a) D. S. Matteson and D. Majumdar, α-Chloro boronic esters from homologation of boronic esters, J. Am. Chem. Soc., 1980, 82, 7588–7590 CrossRef; (b) D. S. Matteson and D. Majumdar, Directed chiral synthesis with pinanediol boronic esters, J. Am. Chem. Soc., 1980, 102, 7590–7591 CrossRef CAS; (c) D. S. Matteson and D. Majumdar, Homologation of boronic esters to α-chloro boronic esters, Organometallics, 1983, 2, 1529–1536 CrossRef CAS.
- P. B. Tripathy and D. S. Matteson, Asymmetric synthesis of the four stereoisomers of 4-methyl-3-heptanol via boronic esters: sequential double stereodifferentiation leads to very high purity, Synthesis, 1990, 200–206 CrossRef CAS.
- D. S. Matteson, R. Soundararajan, O. C. Ho and W. Gatzweiler, (Alkoxyalkyl) boronic ester intermediates for asymmetric synthesis, Organometallics, 1996, 15, 152–163 CrossRef CAS.
- (a) P. Barbie and U. Kazmaier, Total synthesis of cyclomarin A, a marine cycloheptapeptide with anti-tuberculosis and anti-malaria activity, Org. Lett., 2016, 18, 204–207 CrossRef CAS PubMed; (b) L. Junk and U. Kazmaier, Total synthesis of keramamides A and L from a common precursor by late–stage indole synthesis and configurational revision, Angew. Chem., Int. Ed., 2018, 57, 11432–11435 CrossRef CAS PubMed; (c) K. Bauer and U. Kazmaier, Total synthesis of thiamyxins A–C and thiamyxin E, a potent class of RNA–virus–inhibiting (cyclo) depsipeptides, Angew. Chem., Int. Ed., 2023, 62, 202305445 CrossRef PubMed; (d) A. Siebert and U. Kazmaier, Chemical ligation-mediated total synthesis of corramycin, Org. Lett., 2024, 26, 3169–3173 CrossRef CAS PubMed.
- (a) L. Karmann, K. Schulz, J. Herrmann, R. Müller and U. Kazmaier, Total syntheses and biological evaluation of miuraenamides, Angew. Chem., Int. Ed., 2015, 54, 4502–4507 CrossRef CAS PubMed; (b) J. Gorges and U. Kazmaier, Matteson homologation-based total synthesis of lagunamide A, Org. Lett., 2018, 20, 2033–2036 CrossRef CAS PubMed; (c) O. Andler and U. Kazmaier, Total synthesis of apratoxin A and B using Matteson's homologation approach, Org. Biomol. Chem., 2021, 19, 4866–4870 RSC; (d) M. Tost and U. Kazmaier, Synthesis and late-stage modification of (−)-doliculide derivatives Using Matteson's Homologation Approach, Mar. Drugs, 2024, 22, 165 CrossRef CAS PubMed.
- Reviews: (a) D. S. Matteson, Boronic esters in stereodirected synthesis, Tetrahedron, 1989, 45, 1859–1885 CrossRef CAS; (b) D. S. Matteson, Boronic esters in asymmetric synthesis, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed; (c) D. S. Matteson, B. S. L. Collins, V. K. Aggarwal and E. Ciganek, The Matteson reaction, Org. React., 2021, 105, 427–860 Search PubMed.
- S. Fustero, A. G. Sancho, J. L. Acena and J. F. Sanz-Cervera, Fluorous TBAF: a convenient and selective reagent for fluoride-mediated deprotections, J. Org. Chem., 2009, 74, 6398–6401 CrossRef CAS PubMed.
- M. Tost and U. Kazmaier, Stereoselective synthesis of secondary and tertiary boronic esters via Matteson homologation, Org. Lett., 2023, 25, 6835–6839 CrossRef CAS PubMed.
- M. Tost, O. Andler and U. Kazmaier, A Matteson homologation–based synthesis of doliculide and derivatives, Eur. J. Org. Chem., 2021, 6459–6471 CrossRef CAS.
- D. S. Matteson and M. L. Peterson, Synthesis of L-(+)-ribose via (S)-pinanediol (αS)-α-bromoboronic esters, J. Org. Chem., 1987, 52, 5116–5121 CrossRef CAS.
- L. Carmès, F. Carreaux and B. Carboni, Homologation of Boronic Esters with (Dialkoxymethyl)lithiums. Asymmetric Synthesis of α-Alkoxy Boronic Esters, J. Org. Chem., 2000, 65, 5403–5408 CrossRef PubMed.
- (a) D. S. Matteson and H.-W. Man, Hydrolysis of Substituted 1,3,2-Dioxaborolanes and an Asymmetric Synthesis of a Differentially Protected syn,syn-3-Methyl-2,4-hexanediol, J. Org. Chem., 1996, 61, 6047–6051 CrossRef CAS; (b) D. S. Matteson, W. C. Hiscox, L. Fabry-Asztalos, G.-Y. Kim and W. F. Siems, Glass-catalyzed conversion of boronic esters of asymmetric diols to diol sulfites and amine complexes of boron halides, Organometallics, 2001, 20, 2920–2923 CrossRef CAS.
- (a) D. S. Matteson and A. A. Kandil, (S,S)-Diisopropylethanediol (“DIPED”): A new chiral director for the α-chloro boronic ester synthesis, Tetrahedron Lett., 1986, 27, 3831–3834 CrossRef CAS; (b) R. W. Hoffmann and B. Landmann, Stereoselective synthesis of alcohols, XXIII. Transfer of chirality on addition of (α–chloroallyl)boronates to aldehydes, Chem. Ber., 1986, 119, 2013–2024 CrossRef CAS.
- C. D. Roy and H. C. Brown, Stability of boronic esters – Structural effects on the relative rates of transesterification of 2-(phenyl)-1,3,2-dioxaborolane, J. Organomet. Chem., 2007, 692, 764–790 Search PubMed.
- S. Kirupakaran, G. Arago and C. Hirschhäuser, A unified strategy for the synthesis of aldohexoses by boronate assisted assembly of CH2X2 derived C1-building blocks, Chem. Sci., 2023, 14, 9838–9842 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra and experimental details. See DOI: https://doi.org/10.1039/d5qo00213c |
| This journal is © the Partner Organisations 2025 |