
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-water accelerated sulfenylation of indole derivatives under visible light irradiation†
Jun Sup
Lee
ab,
Chulyong
Lee
a,
Jiwon
Jang
a and
Seunghoon
Shin
 *a
*a
aDepartment of Chemistry, Research Institute for Convergence of Basic Science, 222 Wangsimni-ro, Seongdong-gu, Hanyang University, Seoul 04763, Korea. E-mail: sshin@hanyang.ac.kr
bYuhan R&D Institute, 25, Tapsil-ro 35beon-gil, Giheung-gu, Yongin-si, Gyeonggi-do 17084, Korea
First published on 14th March 2025
Abstract
A visible-light promoted sulfenylation of N-carboxyindoles with thiols showed substantially higher rate and selectivity when conducted “on water”. An EDA complex was proposed to form at the water–oil interface, generating thiyl radicals and thus initiating a chain reaction.
Organic chemists have avoided water as the reaction medium, perhaps due to the preconception that substrate dissolution is necessary for reactivity. Consequently, development of organic reactions in pure water has been less common.1 This notion was challenged in 1980 when Rideout and Breslow observed a significant increase in the rate and the selectivity of Diels–Alder reactions in pure water due to the hydrophobic effect.2 In 2005, Sharpless and coworkers demonstrated that the rate of certain pericyclic reactions and epoxide openings in water was faster than in an organic phase.3 For this rate enhancement, reactant heterogeneity is essential, with organic reactants typically forming liquid droplets, assisted by melting point depression.4 This dramatic acceleration in the form of an aqueous suspension was termed an “on-water” reaction.
In connection with green chemistry,5 the application of aqueous medium in visible-light photochemical reactions has recently received increasing attention.6 Potential advantages such as enhanced reductive power of the photocatalysts,7 lowered LUMO of the substrates,8 and facilitated proton transfer9 have stimulated increased interest in exploring the aqueous phase for photocatalytic applications. However, photochemical reactions of water-insoluble substrates that exhibit significant acceleration on water remain limited.10 Recently, the König group reported that cross coupling of cyano heteroarenes can be accelerated in a eutectic mixture of organic reactants, where interfacial hydrogen bonding at the water–oil interface facilitates the formation of a hydrophobic electron donor–acceptor (EDA) complex (Scheme 1A, left).10a The Bae group reported that a (2 + 2) cycloaddition of β-aryl ethenesulfonylfluorides and benzofurans, unreactive in any other organic solvents, uniquely proceeded in water, with the rate surpassing neat conditions.10b Here, on-water acceleration was ascribed to the hydrophobic effect favoring negative volume of activation (ΔV‡) to effectively capture the triplet enthenesulfonyl fluoride with benzofuran (Scheme 1A, right).
 | ||
| Scheme 1 Visible-light-promoted on-water catalysis: sulfenylation of N-carboxyindoles. | ||
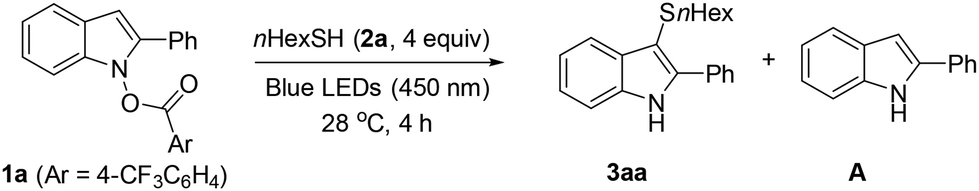
Recently, we11,21,22 and others12 have developed substitutions of N-carboxyindoles as umpoled indole precursors under Brønsted/Lewis acid, Cu, and photocatalytic conditions. Given the biological importance of 3-sulfenylindoles (Scheme 1B),13 substantial efforts have been made via SEAr reactions of indoles with various electrophilic sulfenylating reagents.14 In contrast with previous approaches, we present herein a redox-neutral sulfenylation with thiols using N-carboxyindoles as umpoled indoles. In the pursuit of environmentally sustainable conditions, we discovered that the substitution occurs in water with significantly higher rate and selectivity compared to organic solvents (Scheme 1C). Our findings align with the concept of “on-water” accelerated photocatalysis.
Initially, we noted that previous sulfenylation methods14 had lower yields and a narrower scope for forming alkyl thioethers compared to aryl thioethers. Consequently, our primary optimization goal was to investigate the sulfenylation of N-carboxyindole 1a using aliphatic thiol 2a (Table 1). We first tested photocatalysts in acetonitrile under blue LEDs irradiation (450 nm) (Table S1†). Use of Ru(bpz)3(PF6)2 resulted in extensive decomposition of product 3aa (entry 1), which indicated the photoirradiation-induced instability of 3aa is a significant challenge for optimization. Without a photocatalyst, the reaction was somewhat cleaner, but had low conversion (entry 2). The rate and selectivity were found to be highly solvent-dependent (Table S2†). In ether, the product decomposition was significantly diminished (entry 3). The reaction in an aqueous suspension of 1a resulted in the cleanest conversion (LCMS in Fig. S1†), albeit incomplete (entry 4). The low conversion was due to the insolubility of 1a in water. For example, a mixture of 1a and 2a (4 equiv.) in water forms a heterogeneous mixture at RT (Fig. 1A).15 During the reaction, an orange gummy deposits sat on the magnetic stir bar, encapsulating the unreacted 1a (Fig. 1B). Attempts to improve solubility using a co-solvent (entry 5) or a surfactant (entry 6) resulted in higher conversion, but were accompanied by significant decomposition. Interestingly, the addition of Celite led to complete conversion, yielding 3aa in 83% yield (entry 7). It was assumed that Celite mechanically disrupts solid aggregates, resulting in an even dispersion of reactants. This accelerates full conversion and minimizes over-irradiation-induced decomposition (Fig. 1C). The effect of Celite was not distinct in other solvents, including EtOH, ACN, and ether and Celite was more effective as a dispersant than silica or MS 4 Å (Table S2†). Using shorter or longer wavelength LEDs resulted in a poorer mass balance due to extensive photo-induced decomposition or inactivity, respectively (entries 8 and 9). Testing different leaving groups in 1a revealed that p-CF3 benzoate provided the best yield (Table S3†).
 | ||
| Fig. 1 The effect of Celite as dispersant. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Additiveb | Conv. (%) | 3aa (%) | A (%) |
| a A mixture of 1a (0.05 mmol), nHexSH (2a, 0.2 mmol) and solvents (1 mL) were placed in a vial and was capped under air; crude yields based on 1H NMR spectra. b 50 mg of additives were added. c Ru(bpz)3(PF6)2 (5 mol%). d Cetyltrimethyl ammonium bromide (1.5 equiv.). e λ = 405 nm. f λ = 515 nm. | |||||
| 1 | ACN | [Ru]c | 75 | 39 | 6 |
| 2 | ACN | None | 58 | 33 | 10 |
| 3 | Ether | None | 86 | 66 | 10 |
| 4 | H2O | None | 59 | 48 | 8 |
| 5 | H2O/ACN (1/1) | None | 78 | 57 | 16 |
| 6 | H2O | CTABd | 86 | 14 | 9 |
| 7 | H2O | Celite | >99 | 83 | 15 |
| 8e | H2O | Celite | >99 | 63 | 26 |
| 9f | H2O | Celite | 0 | — | — |
With the optimized conditions established, we examined sulfenylation reaction of 1a starting with aliphatic thiols (Scheme 2). Primary, secondary, and tertiary thiols underwent smooth sulfenylation without issues (entries 1–6). Diverse functional groups, including a silane, an ester, a thiol, and a Boc-protected amine (entries 7–10) were compatible. For less reactive thiols, extended irradiation (entries 7–10) and/or a larger amount of thiol (entries 9 and 10) was required. Notably, N-protected cysteine underwent efficient sulfenylation (entry 11).
 | ||
Scheme 2 Sulfenylation with alkyl (2) and aryl (4) thiols. Reaction conditions: 1 (0.1 mmol), RSH (0.4 mmol), and Celite (100 mg) in water (2 mL) was irradiated with blue LEDs (450 nm, 18 W) for 3–5 h; isolated yields after SiO2 chromatography. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction time: 17 h. bAlkyl thiol (6 equiv.). cDifference from the standard conditions: solvent (CH3CN) and wavelength (405 nm). dThe yield of 3ca and 3ja was determined from the crude 1H NMR spectra. Reaction time: 17 h. bAlkyl thiol (6 equiv.). cDifference from the standard conditions: solvent (CH3CN) and wavelength (405 nm). dThe yield of 3ca and 3ja was determined from the crude 1H NMR spectra. | ||
The conditions developed for aliphatic thiols were directly applied to aromatic thiols (entries 12–27). The sulfenylation proceeded smoothly regardless of the o-, m-, and p-substituents of the aryl thiols (entries 12–23). A furyl thiol gave 5an smoothly (entry 24), but thiazolyl, pyridyl, and pyrimidyl thiols produced unknown byproducts under standard conditions: change of solvent and wavelength of light (CH3CN, 405 nm) produced reasonable yields of 5am–5ap in these cases (entries 25–27). Notably, the synthesis of 5aa can be conducted on a 1 mmol scale, yielding nearly the same high yield (94%), demonstrating the robustness of the current protocol.
Then we tested differently substituted indoles 1 for sulfenylation with both nHexSH and PhSH (entries 28–47). Substrates with a different aryl group at C2 (entries 28–31) and substitutions at C4, C5, C6 positions of the indole core (entries 32–39) were broadly successful. Nonetheless, sterically demanding substrates, such as 1c and 1h proved challenging (entries 30–31 and 40–41), especially in reaction with an aliphatic thiol 2a. Similarly, indoles with alkyl groups at the C2, gave low conversion (entries 42–45). Interestingly, 3-methylindole 1k provided C2-sulfenyl product 5ka in 45% yield (entry 46). However, unsubstituted N-carboxyindole 1l was unreactive under the standard conditions (entry 47). To our delight, the current protocol can be applied to the synthesis of biologically active tubulin polymerase inhibitors 5mq–5oq (Scheme 2).13,16
Subsequently, we investigated the reaction mechanisms. In the presence of TEMPO (2 equiv.), the formation of both 3aa or 5aa was completely suppressed (Fig. S4†). In the reaction with PhSH, PhS-OTMP (m/z = 266.1572 for [M + H]+) was identified, indicating a crucial role of the thiyl radical. We further examined the effects of light (Table S5†). With LEDs off, the reaction of 1a with nHexSH 2a or PhSH 4a did not proceed efficiently, suggesting that a thiyl radical is predominantly formed through photochemical means.17
The distribution of reactants can be monitored by 1H NMR spectroscopy on a heterogeneous mixture of 1a and 2a in water (Fig. 2A and Fig. S9–S12†).10a For instance, 1H NMR spectrum of a immediately agitated mixture of 1a (formal molarity, 0.01 M) and nHexSH (2a, 4 equiv.) in D2O revealed a small amount of 1a and a large quantity of 2a in oil droplets, with a minute quantity of 2a solvated in water (Fig. 2A, up). When filtered through a syringe filter, the oil droplets were removed, leaving only the aqueous solution of 2a in the D2O phase (Fig. 2A, down). Notably, none of 1a remained in the aqueous phase, suggesting that 1a primarily located within the oil droplets alongside the majority of 2a. In addition, it was found that the reaction required a stirring speed of over 300 rpm (Table S4†) to presumably provide sufficient interface between the water and oil droplets.
 | ||
| Fig. 2 Mechanistic experiments and a proposed mechanism. | ||
UV-Vis spectroscopy revealed the interaction of 1a with 2a at the water/oil interface (Fig. S7 and S8†). Addition of 2a into a solution of 1a in CH3CN did not cause any spectral change, but increasing the water ratio in the solvent mixture led to a progressively larger bathochromic shift. This suggests the formation of an electron donor–acceptor (EDA) complex at the water–oil interface.10a The quantum yield was measured to be Φ > 54 for the reaction of 1a with 4a (section 4.6, ESI†), supporting presence of a radical chain.
Based on the above experiments, a plausible mechanism was proposed as shown in Fig. 2B. Visible light absorption of the EDA complex between 1 and 2 (or 4) at the oil–water interface may facilitate a single electron transfer (SET)18 accompanied by a proton transfer (PT).4c Thus generated thiyl radical adds to 1, followed by N–O bond homolysis to form 3 (or 5) and a carboxylate radical. Alternatively, the radical displacement (SRN2′) may be followed,19 in which C–S bond formation and N–O bond cleavage are concerted. The liberated carboxy radical regenerates the thiyl radical through hydrogen atom transfer (HAT). This radical chain appears to be short-lived, as indicated by light on–off experiments conducted with 1a and PhSH in CH3CN (Fig. S5†). In the reaction with aromatic thiols, the mechanism slightly differs: a mixture of 1a and PhSH exhibits a bathochromic shift even in CH3CN, suggesting EDA formation through π–π stacking interaction.20 Similarly to above, this bathochromic shift is further enhanced with increased water composition (Fig. S8†).
One of the practical advantages of aqueous reaction is ability to isolate the product without extractive workup. Exploiting this advantage is demonstrated in the C–S coupling and oxidation into sulfoxide: the crude mixture of 5aa was filtered, washed with a solvent of the next reaction (CH2Cl2) and the filtrate was treated with mCPBA (Fig. S2, section 3.2.2, ESI†). In this fashion, synthesis of sulfoxide 6aa telescoped in good yield (91%) without involving extractive workup of the intermediate 5aa.
In summary, we developed a redox-neutral sulfenylation of umpoled indole derivatives for the synthesis of 3-sulfenylindole. When irradiated with blue LEDs, the sulfenylation occurred in the absence of photocatalyst and the reaction was significantly accelerated by making the reaction mixture heterogeneous in an “on-water” mode. The reaction may proceed through the formation of an EDA complex at the water–oil interface, facilitating the formation of a thiyl radical as a radical chain carrier. For on-water reactions of water-insoluble substrates, Celite was found to be effective as a dispersant to ensure even conversion and to prevent photo-induced decomposition of products.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank National Research Foundation of Korea (NRF-2021R1A2C3010552) and the Korea Drug Development Fund (RS-2023-00259634) for generous support.References
- (a) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- (a) Y.-J. Zuo and J. Qu, J. Org. Chem., 2014, 79, 6832–6839 CAS; (b) K. Karhan, R. Z. Khaliullin and T. D. Kühne, J. Chem. Phys., 2014, 141, 22D528 CrossRef PubMed; (c) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492–5502 CrossRef CAS PubMed; (d) L. L. Thomas, J. Tirado-Rives and W. L. Jorgensen, J. Am. Chem. Soc., 2010, 132, 3097–3104 CrossRef CAS PubMed.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 Search PubMed.
- (a) C. Russo, F. Brunelli, G. C. Tron and M. Giustiniano, J. Org. Chem., 2023, 88, 6284–6293 Search PubMed; (b) K. Sun, Q.-Y. Lv, X.-L. Chen, L.-B. Qu and B. Yu, Green Chem., 2021, 23, 232–248 Search PubMed; (c) S. Barata-Vallejo, D. E. Yerien and A. Postigo, ACS Sustainable Chem. Eng., 2021, 9, 10016–10047 Search PubMed.
- R. Naumann and M. Goez, Green Chem., 2019, 21, 4470–4474 Search PubMed.
- E. Speckmeier, P. J. W. Fuchs and K. Zeitler, Chem. Sci., 2018, 9, 7096–7103 Search PubMed.
- Y. Hou and P. Wan, Photochem. Photobiol. Sci., 2008, 7, 588–596 Search PubMed.
- (a) Y.-M. Tian, W. Silva, R. M. Gschwind and B. König, Science, 2024, 383, 750–756 CrossRef CAS PubMed; (b) S. B. Kim, D. H. Kim and H. Y. Bae, Nat. Commun., 2024, 15, 3876 CrossRef CAS PubMed; (c) N. A. Stini, E. T. Poursaitidis, N. F. Nikitas, M. Kartsinis, N. Spiliopoulou, P. Ananida-Dasenaki and C. G. Kokotos, Org. Biomol. Chem., 2023, 21, 1284–1293 RSC.
- (a) N. H. Nguyen, S. Seo, J. Jang, H. Kim and S. Shin, Org. Lett., 2024, 26, 7149–7154 CrossRef CAS PubMed; (b) N. H. Nguyen, S. M. Oh, C.-M. Park and S. Shin, Chem. Sci., 2022, 13, 1169–1176 RSC; (c) M. Bera, H. S. Hwang, T.-W. Um, S. M. Oh, S. Shin and E. J. Cho, Org. Lett., 2022, 24, 1774–1779 CrossRef CAS PubMed.
- (a) Y. Ye, S.-T. Kim, J. Jeong, M.-H. Baik and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 3901–3909 CrossRef CAS; (b) K. Ali, M. Bera and E. J. Cho, Synlett, 2023, 1019–1022 CAS; (c) Y. Lee, Y. S. Nam, S. Y. Kim, J. E. Ki and H. G. Lee, Chem. Sci., 2023, 14, 7688–7698 Search PubMed.
- (a) G. La Regina, R. Bai, A. Coluccia, V. Famiglini, S. Pelliccia, S. Passacantilli, C. Mazzoccoli, V. Ruggieri, A. Verrico, A. Miele, L. Monti, M. Nalli, R. Alfonsi, L. Di Marcotullio, A. Gulino, B. Ricci, A. Soriani, A. Santoni, M. Caraglia, S. Porto, E. Da Pozzo, C. Martini, A. Brancale, L. Marinelli, E. Novellino, S. Vultaggio, M. Varasi, C. Mercurio, C. Bigogno, G. Dondio, E. Hamel, P. Lavia and R. Silvestri, J. Med. Chem., 2015, 58, 5789–5807 CrossRef CAS PubMed; (b) G. La Regina, R. Bai, W. M. Rensen, E. Di Cesare, A. Coluccia, F. Piscitelli, V. Famiglini, A. Reggio, M. Nalli, S. Pelliccia, E. Da Pozzo, B. Costa, I. Granata, A. Porta, B. Maresca, A. Soriani, M. L. Iannitto, A. Santoni, J. Li, M. M. Cona, F. Chen, Y. Ni, A. Brancale, G. Dondio, S. Vultaggio, M. Varasi, C. Mercurio, C. Martini, E. Hamel, P. Lavia, E. Novellino and R. Silvestri, J. Med. Chem., 2013, 56, 123–149 Search PubMed.
- (a) K. M. Schlosser, A. P. Krasutsky, H. W. Hamilton, J. E. Reed and K. Sexton, Org. Lett., 2004, 6, 819–821 Search PubMed; (b) J. A. Campbell, C. A. Broka, L. Gong, K. A. M. Walker and J.-H. Wang, Tetrahedron Lett., 2004, 45, 4073–4075 CrossRef CAS; (c) M. Chen, Z.-T. Huang and Q.-Y. Zheng, Chem. Commun., 2012, 48, 11686–11688 RSC; (d) Y.-Z. Ji, H.-J. Li, J.-Y. Zhang and Y.-C. Wu, Chem. Commun., 2019, 55, 11864–11867 Search PubMed; (e) F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929–4932 Search PubMed.
- A mixture of 1a and 2a (4 equiv.) have a eutectic melting temperature above 70 °C (Fig. S3†).
- 5mq stimulated the cytotoxic activity of NK cells and arrested the growth of HeLa cells at 10, and 20 nM level, respectively (ref. 13a). 5nq and 5oq showed IC50 = 52 nM and 64 nM, respectively, in MCF-7 cells (ref. 13b).
- For aerobic generation of thiyl radicals, see: J. L. G. Ruano, A. Parra and J. Alemán, Green Chem., 2008, 10, 706–711 Search PubMed.
- (a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2023, 142, 5461–5476 CrossRef; (b) A. K. Wortman and C. R. J. Stephenson, Chemistry, 2023, 9, 2390–2415 CrossRef CAS PubMed.
- This radical displacement was designated as SRN2′ to distinguish them from SRN2 displacements: A. R. Katritzky, G. Z. de Ville and R. C. Patel, Tetrahedron Lett., 1980, 21, 1723–1726 CrossRef CAS.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS PubMed.
- R. Kumar, Q. H. Nguyen, T.-W. Um and S. Shin, Recent Progress in Enolonium Chemistry under Metal-Free Conditions, Chem. Rec., 2022, 22(1), e202100172 Search PubMed.
- D. V. Patil and S. Shin, Organocatalytic Oxidative Functionalization of Alkynes, Asian J. Org. Chem., 2019, 8(1), 63–73 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of all new compounds; reaction development; mechanistic experiments. See DOI: https://doi.org/10.1039/d5ob00429b |
| This journal is © The Royal Society of Chemistry 2025 |