
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanometrology assisted chemical fabrication: direct laser writing of porphyrins onto complex surfaces†
Baptiste Maillota,
Haroon Rashida,
Roxanne Bercyb,
Jean Frédéric Audiberta,
Manuel J. Llansola-Portoles b,
Isabelle Leraya,
Fabien Miomandrea and
Vitor Brasiliense*a
b,
Isabelle Leraya,
Fabien Miomandrea and
Vitor Brasiliense*a
aUniversité Paris-Saclay, ENS Paris-Saclay, CNRS, PPSM, 4 avenue des sciences, 91190 Gif-sur-Yvette, France. E-mail: Vitor.brasiliense@ens-paris-saclay.fr
bUniversité Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91190 Gif-sur-Yvette, France
First published on 24th June 2025
Abstract
The association of operando monitoring methodologies with micro and nanoscale surface modification strategies has recently been shown to enable the preparation of complex yet highly precise organic functional surfaces. While promising, such demonstrations have so far been limited to model systems, consisting on minimally functionalized aryl radicals. With a growing demand for more sophisticated surfaces, bearing multiple functions, a demonstration of the generality of the strategy, including chemically complex moieties and surfaces is deeply needed. In this work, we aim to fill this gap by preparing tetraphenyl porphyrin derivatives modified to become radical precursors that can be activated with light. Operando optical monitoring is used to non-invasively analyze their grafting behavior in different conditions, optimizing the reaction and enabling modification of inert glass surfaces with high precision (30 atto L). We demonstrate that the methodology is compatible with direct laser writing technologies, and use it to prepare photophysically active surfaces with high resolution. We demonstrate that the instrinsic emissive properties of Tetraphenylporphyrin derivatives are well preserved, and that several surface modifing steps can be sequentially stacked, leading to the preparation of surfaces with multiple functions. By controlling the microscale distribution of chemical groups with different photophysical properties, we demonstrate that complex chemical designs can be readily and reliably implemented. This work therefore shows that light activated radical pathways can be broadly used to modify surfaces, opening interesting new perspectives for the implementation of functional materials.
Introduction
The preparation of functional surfaces by microscale surface engineering has captured the imagination of chemists for decades. Indeed, the ability to design chemically active surfaces on a micron-by-micron basis offers exciting new perspectives in a variety of fields such as sensors,1,2 cell biology3 or energy materials.4–6 Among local modification strategies, light-based methodologies are particularly appealing, offering excellent spatio-temporal control over the surface modification process.While lithographic techniques remain the golden standard when it comes to scalability, a strong demand for more flexible methodologies exists, particularly for fast preparation of chemically complex prototypes and proofs-of-concept studies. Indeed, lithography materials are limited, and their use to locally pattern complex surfaces relies on multistep procedures which are prone to generating defects, requiring critical alignments steps that can only be applied to regular surfaces. In such context, microscale additive manufacturing based on direct laser writing methods associated with organic radical reactions has recently emerged as a versatile option for preparing geometrically complex surface distributions of different molecules.7,8 Highly reactive radical intermediates enable wide scope reactions, consequently allowing different chemical functions to be anchored to a variety of surfaces (metals, carbon, glasses, etc.).7–11 Among different radical precursors, diazonium salts are particularly appealing, as diazonium groups are easily activated through several routes (electrochemical reduction, photochemical, temperature, etc.), ultimately generating inert N2 molecules and reactive aryl radicals. Moreover, since radical diazonium salts can be prepared in a single step from widely available aniline substrates, a large array of chemical functionalities can be easily reached with minimal synthetic efforts.
Recently, our team has shown that surface nanometrology can be combined with direct laser writing to implement extremely precise photografting operations,7 paving the way towards preparation of microstructured functional surfaces. While promising, these methodologies have only been demonstrated for aryl-radical intermediates with relatively small substituents, primarily modifying surfaces with only one reactant. It remains to be shown that such photochemical methods can be transposed to larger molecules, bearing more complex electronic structures and solvation properties or to surface patterns containing different chemical groups.
However, as the chemical complexity of the grafting molecules and their environment is increased, the challenge of precisely engineering surfaces grows. For instance, difficulties can arise due to: (i) interaction of the grafting precursors with surface functions, (ii) intrinsic properties of the grafting groups and its intermediates, (iii) hindered ability of radicals to attach to surfaces, etc. For example, the complex electronic and solvation properties found in species used as catalysts and sensors can be accompanied by propensity to aggregate, or by introduction of deexcitation pathways, disfavoring the formation and stability of radical intermediates.
This work therefore aims at demonstrating that nanometrology assisted laser fabrication can indeed enable preparation of multifunctional surfaces, primarily focusing on tetraphenyl porphyrin derivatives containing photoactivable anchoring groups based on aryldiazonium salts. This class of molecules are examples of radical precursors that present complex photochemical structure and functions, finding a wide range of applications in catalysis,12,13 energy conversion,14 sensors,1,15 photodynamic therapy,16,17 etc. Therefore, by demonstrating the direct photografting of porphyrins onto complex surfaces, we directly highlight the potential of our approach to more advanced applications. For instance, local control of the TPP distribution on a surface enables the optimal placement of optical sensors, designed to respond to different concentrations of analytes. Similarly, in contexts where porphyrins are used as catalysts, high-resolution photografting can be used to optimize catalytic coverage and maximize substrate accessibility.
Existing strategies for immobilizing porphyrins onto surfaces, can be broadly categorized in two groups: (i) self-limiting methodologies which rely on the surface properties to control reactivity18–23; or (ii) formulation of composite films, on which porphyrins either associate with different components of the film,24 or constitute the film forming material themselves.16,25,26 In both cases, microscale patterning with high resolution cannot be easily achieved, often requiring multistep lithographic procedures carried out in clean room facilities. Moreover, most traditional methods fail to quantitatively control the layers dimensions when thick yet precise layers are targeted. Therefore, a single step methodology to enable precise direct laser writing of porphyrins would considerably widen the perspectives for preparing functional surfaces, particularly regarding the association with other functional molecules.
In this paper, we show how nanometrology approaches can be implemented to control the photografting of TPPs derivatives on inert surfaces. The methodology is compatible with a wide range of substrates, but we emphasize here the modification of borosilicate glass surfaces to underline the unique optical properties of the grafted TPP. Indeed, owing to their chemical inertness and low absorption in the visible range, glass surfaces only weakly affect the photophysical properties of the grafted molecules, differently from conductive surfaces, which could quench them. After designing a strategy to convert amino-tetraphenylporphyrin (TAPP) precursors into well dispersed photograftable diazonium porphyrins, we identify and optimize their photografting behavior with the help of operando analysis, using quantitative phase imaging as a noninvasive optical nanometrology technique. Finally, the possibility of preparing chemically and geometrically complex structures is explored, associating moieties with different emission properties in a single microscale pattern.
Experimental
Materials, grafting solutions
Porphyrin diazonium salt solutions were prepared from 5,10,15,20-(tetra-4-aminophenyl)-porphyrin (TAPP) (Porphychem), which were dissolved in a mixture of DMSO (Carlo elba reagents) and pH 2.0 Milli-Q water (18.2 MΩ cm−1) most often in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume proportion, or as indicated in the text. The pH of all aqueous solutions used in this paper was adjusted to 2.0 ± 0.1 using a 1 M hydrochloric acid solution (Sigma Aldrich), with the help of a pH meter (Fisher Scientific Accumet, AE150). Dz-TPP Grafting solutions are prepared in situ with a one-pot protocol, by adding 4 equivalents of NaNO2 (Acros Organics) to the TAPP solutions (one equivalent for each amine function). For photosensitized grafting experiments, Rhodamine 6G (Rh6G) was dissolved in the grafting solution after completing the diazotization protocol. Aryldiazonium grafting solutions were prepared from either 4-nitrobenzenediazonium tetrafluoroborate (Sigma Aldrich, 97%), dissolved in pH 2.0 Milli-Q, or by preparing the diazonium in situ from 4-ethynylaniline (Sigma Aldrich, 97%), with addition of 1 eq. NaNO2. In these diazotization experiments, 5 vol.% DMSO was added as a co-solvent to minimize solubility issues. Solutions in all grafting experiments were freshly prepared, and used within 2 h.
:1 volume proportion, or as indicated in the text. The pH of all aqueous solutions used in this paper was adjusted to 2.0 ± 0.1 using a 1 M hydrochloric acid solution (Sigma Aldrich), with the help of a pH meter (Fisher Scientific Accumet, AE150). Dz-TPP Grafting solutions are prepared in situ with a one-pot protocol, by adding 4 equivalents of NaNO2 (Acros Organics) to the TAPP solutions (one equivalent for each amine function). For photosensitized grafting experiments, Rhodamine 6G (Rh6G) was dissolved in the grafting solution after completing the diazotization protocol. Aryldiazonium grafting solutions were prepared from either 4-nitrobenzenediazonium tetrafluoroborate (Sigma Aldrich, 97%), dissolved in pH 2.0 Milli-Q, or by preparing the diazonium in situ from 4-ethynylaniline (Sigma Aldrich, 97%), with addition of 1 eq. NaNO2. In these diazotization experiments, 5 vol.% DMSO was added as a co-solvent to minimize solubility issues. Solutions in all grafting experiments were freshly prepared, and used within 2 h.
Photografting procedure
The grafting reactions were conducted in a home-made setup based on an inverted microscope (Ti2-E Nikon). The grafting reactions are triggered by a 474 nm pulsed laser (LDH-P-C-470, Picoquant, 40 MHz repetition rate) focused on the surface of the substrate with the help of a 100× oil immersion objective (Nikon, NA = 1.49). The laser profile is expanded and collimated to match the back aperture of the objective lens, leading to a focused laser waist in the micrometer range (typ. FWHM = 800 nm). Grafting operations were carried out in a cell of thickness 10 μm, obtained by associating the top part of a thin quartz cell (106-QS, Suprasil, Hellma) and a glass coverslip (VWR international). All glass substrates were used as received. The incident power is systematically measured in the sample focal plane with a hand held powermeter (Ophir NOVA II), and reported along the results below. Unless noted otherwise, total Irradiation powers are kept below 500 nW (≈25 W cm−2) for highly absorbing porphyrin diazonium salt solutions and 200 μW (≈10 kW cm−2) for the other diazoniums, ensuring grafting operations in a reasonable time scale (at most a few minutes), without triggering significant excitation of molecules outside of the beam Rayleigh zone. A precision shutter (Edmund Optics) was used to block the excitation beam during the image acquisition and control the excitation time. The shutter was controlled with the help of an Arduino Mega board, interfaced through Matlab, enabling the control routines to be implemented. Reported grafting experiments were repeated at least 3 times, and both the original data and average grafting curves are reported. Error bars indicate 2 standard deviations from the mean values, representing 95% confidence intervals under the assumption of Gaussian distribution. The standard deviations of mean values from a population consisting of N measurements are estimated as , where σ is the unweighted standard deviation of the measurements. Rinsing operations are systematically carried out after each photografting experiment, using water–DMSO mixtures (ca. 5 mL, with the help of a Pasteur pipette) followed by abundant rinsing with milliQ water (ca. 20–30 ml, dispensed with pissette for a few seconds). The stability of samples was tested by additionally rinsing them with methanol, ethanol and acetonitrile, solvents which easily solubilize any unreacted species, leading no observable changes in the layer morphology, as characterized with atomic force microscopy.
, where σ is the unweighted standard deviation of the measurements. Rinsing operations are systematically carried out after each photografting experiment, using water–DMSO mixtures (ca. 5 mL, with the help of a Pasteur pipette) followed by abundant rinsing with milliQ water (ca. 20–30 ml, dispensed with pissette for a few seconds). The stability of samples was tested by additionally rinsing them with methanol, ethanol and acetonitrile, solvents which easily solubilize any unreacted species, leading no observable changes in the layer morphology, as characterized with atomic force microscopy.
Direct laser writing experiments
Writing experiments are carried out in the same setup by controllably moving the samples at constant speeds with the help of a piezo stage (P-545.3R8H, E727, Physik Instrumente) during continuous irradiation of the sample. Trajectories are created in Matlab software, which was also used to control the sample movement and the shutter.Quantitative phase imaging
Quantitative phase imaging was used as a non-destructive nanometrology tool to monitor the grafting extent, implemented by a diffuser-based phase sensing and imaging (DIPSi) module inserted in the detection path of the microscope.7,8,11,27 As explained in detail elsewhere,27,28 the DIPSi module consisted in the addition of a thin diffuser located 3 mm away from the focal plane of the CCD camera, generating a random interference (speckle) pattern. A Demon Image registration algorithm, implemented in Matlab (imregdemons), was used to track deformations of the speckle pattern, which can be directly related to the optical path difference. In order to ensure good resolution, the total magnification of imaging experiments was increased with the help of its native 1.5× tube lens and a 2.5× relay lens, reaching a total magnification of 375×.Steady state UV Vis and fluorescence measurements
UV Vis absorption spectra of solutions are recorded in a Cary 4000 spectrophotometer (Agilent Technologies), using quartz cuvettes of 1 cm optical path (M&B Thuet). Steady state emission from solutions and the corresponding excitation spectra were collected using a fluorescence emission spectrophotometer (Fluorolog, Horiba). Steady state emission spectra of grafted layers are recorded on the same inverted microscope used for the photografting experiments, using a convergent lens to convert the focused configuration into a widefield illumination. Emission from the layers are collected by a 40× extra-long working distance objective (Nikon, NA = 0.6) and spatially filtered by the optical fibre, leading to a collection zone of 7 μm in the sample referential. The collected photons are then analysed with a fibered spectrometer (SD2000, Ocean Optics).Time resolved fluorescence
Fluorescence emission decay curves are obtained using the time-correlated single-photon counting (TCSPC) method. The setup is made of a titanium sapphire Ti:Sa oscillator (Spectra Physics, Maï Taï) emitting pulses of 100 fs duration at 940 nm (80 MHz frequency). The repetition rate is reduced to 10 MHz by a pulse picker, and non-linear second-harmonic generation crystal generates the desired wavelength at 470 nm (GWU Lasertechnik, UHG-23-PSK). Emitted photons are collected at 90° angle from the excitation beam, through a polarizer at the magic angle (54.7°) with a monochromator centred at the emission wavelength. The incoming photons are collected with a micro-channel plate photomultiplier (MCP-PMT R3809U-50, Hamamatsu) and processed with a TCSPC module (SPC-630, Becker & Hickl). Fluorescence decays are fitted with non-linear least-squares χ2 minimization using Levenberg-Marquardt method under Igor Pro Wavemetrics Inc. Software.Fluorescence lifetime imaging (FLIM)
In FLIM experiments, photografted samples are excited using the same 474 nm laser used in grafting experiments (LDH-P-C-470, Picoquant), with average pulse duration of 250 ps, at 10 MHz repetition rate. Wide field illumination is obtained with CFI Plan Fluor 40× objective (Nikon, NA = 0.75). The signal is collected by a single photon counting camera quadrant anode (MCP-PM, LINCam, Photonscore) based photodetector operating in TCSPC mode. Each photon is digitalized in a 12 bits array in position and Time–Tagged–Time–Resolved (TTTR) with a time resolution of 10 ns and 50 ps respectively.29 The camera is coupled to the optical setup with the help of custom electronic interface and GUI software (Photonscore). Post processing and analysis tools software are carried out using home-built routines implemented in Igor Pro Wavemetrics Inc. Software.Atomic force microscopy (AFM)
Measurements were carried out with a Dimension Icon microscope (Bruker), using PointProbePlus tips (PPP-NCHR, NanoSensors) on tapping mode. All AFM measurements reported in this paper were carried out after copiously rinsing the grafted samples with milliQ water (18.2 MΩ cm−1) and drying the samples under N2 flow.Refractive index estimation
The refractive index of grafted layers (nlayer) are estimated by comparing QPI experiments and ex situ AFM images of the same grafted spots. The ratio between the OPD integrated over the irradiation zone (the optical volume difference, OVD) and the grafted volume V (estimated by integrating the AFM measured object height), yields the refractive index difference between the layer and the grafting medium (nmedium), accordingly to eqn (1):
 | (1) |
nmedium is measured at the observation wavelength (λobs = 633 nm) using an Abbe refractometer (Bausch & Lomb), leading to the value of 1.335 ± 0.001 for aqueous grafting solutions and 1.415 ± 0.001 for the 1:1 DMSO:H2O pH 2.0 ones. A large number (N > 15) of grafting experiments is conducted, leading to the average values and scatter reported in the paper.
Raman spectroscopy
Local Raman spectra from grafted layers are carried out in a home-made Raman microscopy setup based on a TiU2 (Nikon) inverted microscope. The excitation laser (Obis LX/SF, Coherent, λ = 405 nm) is passed through a band pass filter (405/10 Semrock, FF01-405/10) focused on the sample by a 40× extra long working distance objective (ELWD Nikon, NA 0.6), leading to a ≈5 μm spot size. Optical densities are used to limit the incident power in the sample to 4 μW (≈5 W cm−2). Scattered photons are collected by the same objective, and, after elimination of incident wavelength with a long pass filter (409 nm, Semrock, FF02-409), reimaged in the entrance slit plane of a Jobin Yvon HR400 spectrometer equipped with a 300 mm−1 grating, and a N2 cooled CCD sensor (LN/CCD 512 TKB, Princeton Instruments). In order to minimize all risk of sample degradation, experiments were conducted in low temperature (77 K) and low pressure (6 × 10−6 bar) with the help of a liquid nitrogen cooled cryostat (MicrostatN, Oxford Instruments, Andor). We confirmed the stability of the samples by monitoring of the spectra over several minutes identifying no significant spectral changes.X ray photoelectron spectroscopy (XPS)
XPS analysis was performed on a K-Alpha + instrument (ThermoFisher Scientific, East Grinstead, UK) equipped with a monochromatic Al K-Alpha source (1486.6 eV, spot size 400 μm). A flood gun was used to compensate charge due to the insulating nature of the samples, and charging was automatically compensated using the instrument native routine. Survey scans were conducted with a 1 eV step size (2 scans), while detailed analysis of the N 1s and C 1s regions was performed with 0.1 eV step size (9 scans). Data was treated and analysed with the help of the equipment native software (Avantage). A conservative 10% fit error was assumed in the fitting procedures.Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS)
SEM/EDS analysis were performed on a Hitachi S3400N instrument. Data was treated with the instrument native software (EDAX Team) and fit according to standard procedures. In order to make them conductive, a thin Au layer (7 ± 3 nm) was deposited on the sample by sputtering (Emitech K750), operated at a 60 mA current and 10−1 mbar pressure. SEM Images were collected at 5 kV with a 4 mm working distance, while EDS experiments were collected with 8 kV with a 125.4 eV resolution.Results and discussion
Preparation of photoactivable porphyrins
In order to prepare photograftable porphyrins, we start by developing strategies to attach photoactivable anchoring groups to porphyrin precursors. Following our previous work, diazonium moieties constitute ideal candidates as photo anchoring groups, being activatable in the visible range and conveniently prepared in a single step from widely available aromatic amines.7,8,11 However, differently from small aryl derivatives, amino porphyrins such as tetraaminophenylporphyrin (TAPP) cannot be easily solubilized in water, posing a challenge for direct implementation of diazotization reaction, usually carried out in acidic aqueous environments. While diazotization reactions can be conducted in purely organic media, we opted for a co-solvent strategy, easier to implement without substantially modifying the diazotization protocol. DMSO was selected as co-solvent on the basis of a previous demonstration that photografting is possible even in the presence of substantial amounts of DMSO (at least up to 80 vol.%).8 Working in solvent mixtures, however, requires careful consideration of the solution aggregation state, carried out here by analysing the photophysical properties of the different solutions.First, TAPP is dissolved in DMSO. The absorption spectrum of 10 μM TAPP solution is shown in Fig. 1A, showcasing a Soret band at 439 nm and three smaller absorption peaks at 529, 580 and 668 nm (the Q bands), in good agreement with literature data.30 The molar absorptivity of the Soret band, determined by varying the solution concentration, was ε = 70250 M−1 cm−1. This value is notably smaller than the one obtained in a weakly coordinating solvent such as DCM (ε = 93100 M−1 cm−1, ESI S1†). Such variation can be explained by different phenomena, such as incomplete solubilization, interaction between the solvent and TAPP, or presence of small aggregates.31,32 To further characterize TAPP in DMSO, we analysed the fluorescence decay rates obtained from time resolved fluorescence experiments. The presence of supramolecular species is supported by these measurements (ESI S3†): while dissolution in DCM leads to a mono-exponential decay (τ = 7.5 ns, χ2 = 1.26), a biexponential decay is observed in DMSO (τ1 = 3.6 ns, τ2 = 6.3 ns, χ2 = 1.29), suggesting the presence of two emissive populations consistently with the formation of dimers. The solution remains emissive, with a well-defined peak at λem = 701 nm, enabling measurement of an excitation spectra (dashed line). Comparison between the excitation and absorption spectra (Fig. 1A) reveals that emissive species are essentially in a single form in DMSO, as absorption/excitation peaks appear in the same position in both spectra.
 | ||
| Fig. 1 Spectral characterization of photophysical stationary properties of TPP derivatives obtained during the different steps of the diazotization procedure. Absorption spectra are shown in full lines, emission spectra in transparency, and excitation spectra as dashed lines. The excitation and emission wavelengths were adjusted to the respective maxima, as indicated below. Spectra of (A) TAPP in DMSO (λexc = 440 nm, λem = 700 nm), (B) in 1:1 DMSO and H2O (pH 2.0) mixture (λexc = 430 nm, λem = 655 nm) (C) transitorily (t = 1–60 min) after the addition of 4 eq. NaNO2 (λexc = 470 nm, λem = 680 nm), and (D) after 2 h (λexc = 445 nm, λem = 700 nm). In all cases, the spectra have been vertically displaced for clarity. | ||
The situation drastically changes once pH 2.0 water is introduced as a co-solvent, as shown in Fig. 1B, with a split Soret band (λ = 426 nm, εapp ≈ 41 000 M−1cm−1 and λ = 470 nm, εapp ≈ 49000 M−1 cm−1), disappearance of the original Q bands and appearance of a broad NIR Q band centred at 730 nm, associated with hyperporphyrinic behaviour, as previously observed for TAPP.30,33 These spectral changes are probably the result of more than a single phenomenon, as put forth by monitoring the spectra of TAPP DMSO solution during progressive addition of different amounts of water (ESI S2†). At low water concentrations (since 20 μM, or 2 eq.), asymmetric splitting of the Soret band is observed, leading to a small blue-shifted peak (λ = 390 nm) and a larger red-shifted one (λ = 467 nm), concomitantly with the disappearance of the Q bands structure and emergence of a large broad band in the NIR (>800 nm). Similar features are observed if the solution is titrated with an organic proton donor instead of water(ESI S2†), suggesting that protonation of the cycle is responsible for these changes, in good agreement with literature.33,34 At much higher water contents (>100 mM), the blue-shifted Soret satellite increases in intensity and progressively moves towards higher wavelengths, concomitantly with the blueshift of the NIR band, reaching an approximatively symmetrically split Soret band, and a NIR band centered at 730 nm for 50 vol.% H2O. The emission of the porphyrins is also clearly impaired during the process, with almost complete absence of emission bands at 50 vol.% H2O, except for a faint emission band at 654 nm. Using this band to stablish an excitation spectrum leads to the dashed line in Fig. 1B, which bears little resemblance with the absorption spectrum. This discrepancy suggests that several photoactive species are present, leading to complex energy transfer processes, which is consistent with the presence of small aggregates.35 If indeed present, however, aggregates are not sufficiently large to provoke noticeable precipitation, even at much larger concentrations (up to 5 mM). This conclusion is also supported by resonant Rayleigh spectra, a popular methodology to analyse aggregation of porphyrins,36,37 which does not display any clear features (ESI S4†). Small supramolecular species are nevertheless probably present, as photoluminescence decay curves in the presence of 50 vol.% water indicate at least three different decay rates (χ2 = 1.22, ESI S3†).
As the NaNO2 diazotization agent is added to the solution, its spectral properties once again undergo sharp modifications (Fig. 1C), with re-emergence of a well-defined Soret band at 446 nm, disappearance of the broad NIR feature, and reappearance of the Q band structure. These results suggest that the amino phenyl groups are successfully converted into aryldiazoniums (Ph-N2+), forming tetra diazonium phenyl porphyrins (Dz-TPP). The transformation is also accompanied by the reappearance of an emission band at 693 nm, whose excitation spectrum reasonably resembles the absorption, indicating that much less (if any) aggregates are present. Using this band, an excitation spectrum can be obtained and compared to absorption spectrum. As more clearly shown in the final spectra (Fig. 1D) the excitation and absorption bands do not exactly overlap, particularly in the Q bands region, indicating the presence of more than one species. Based on the fact that four charges per molecule are expected to be generated during the diazotization reaction, we hypothesize that this behaviour is related to H2O higher ability to solubilize the newly formed cationic porphyrins. We support this hypothesis by redissolving the Dz-TPP in mixtures of higher water content, leading to the absorption spectra which closely resemble excitation ones (ESI S5†). Determination of the ε of the Soret band in water reveals a value of 95000 cm−1 M−1 (ESI S1†), very close to the value observed in DCM for the TAPP precursor, suggesting that a single species is again present. Taken together, these results clearly indicate that once the diazonium species are formed, a solution containing majorly well dispersed species is obtained.
Surface photografting
A direct laser writing setup was next used to analyse the photografting behaviour of the resulting porphyrin diazonium derivatives onto inert glass surfaces. For that, an optical nanometrology module based on quantitative phase imaging (QPI) is used to analyse the evolution of optical thickness of the layer with irradiation. Following each grafting operation, the surfaces were rinsed and characterized ex situ using atomic force microscopy.Direct photografting
Initial attempts to photograft porphyrin layers were carried out in freshly prepared 5 mM water–DMSO mixtures, confirming that Dz-TPP can graft onto glass surfaces. The reaction is continuously monitored with optical path difference (OPD) images, whose integration around the irradiation zone enable following the evolution of the surface modification operation through the optical volume difference (OVD). A typical OPD image sequence is shown in Fig. 2A (blue), leading to the OVD evolution portrait in Fig. 2C (blue curve, average over N = 5 experiments). Monitoring the OVD over time enables comparison between different grafting conditions by measuring the apparent reaction efficiency (ηapp), i.e. the ratio between the grafting rate and the incident power. For instance, increasing the laser power from nW range to μW range clearly leads to a decrease in the grafting efficiency (ESI S6†), suggesting that almost all molecules in the focal spot are activated at these fluencies, and that the surface modification rate is limited by the diffusive transport of new molecules into the irradiated zone. Increasing the incident power beyond 100 μW leads to homogeneous nucleation of small particles along the laser path, outside of the Rayleigh zone.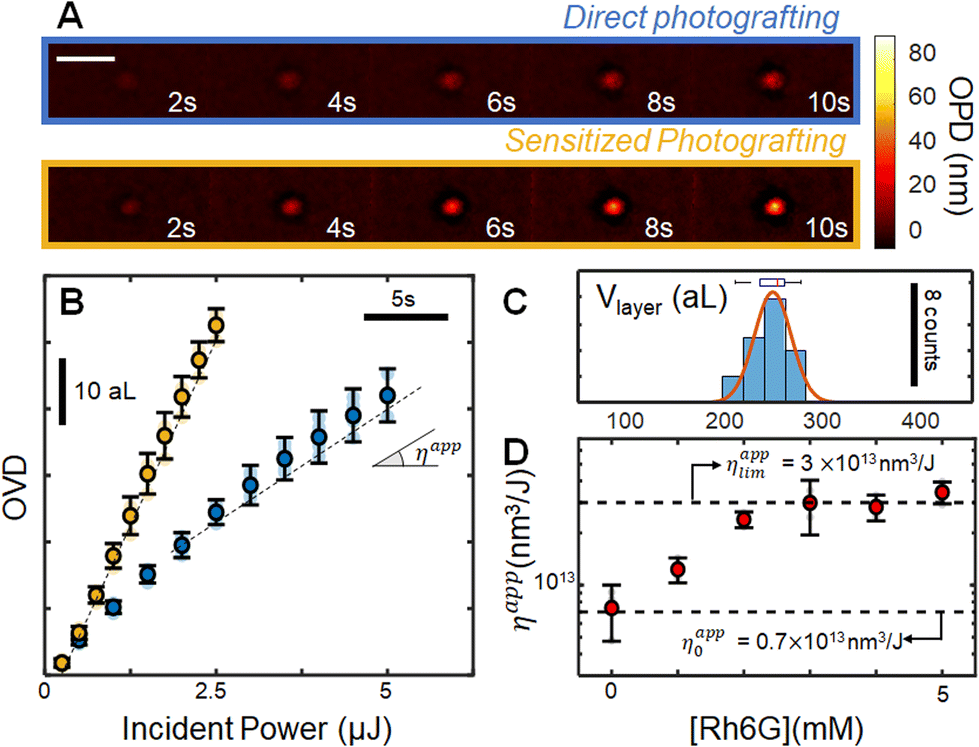 | ||
| Fig. 2 Operando analysis of photografting of 4-DzTPP. (A) OPD images obtained after different irradiation times in the absence (blue, top line), and presence (yellow, bottom line) of 1 eq. Rh6G photosensitizer. The scale bar represents 5 μm. (B) OVD data, obtained by integrating the OPD images over the irradiation zone. The yellow curve shows the mean curve obtained by locally irradiating (∼12.5 W cm−2) a solution of 5 mM Dz-TPP in the presence of 5 mM Rh6G. The markers represent the mean (N = 5, 2σ error) experiments, and raw data points are shown in transparency. (C) Distribution of grafted spots volumes (N = 18, measured with AFM) obtained using an adaptative control algorithm, along with a gaussian fit of the distribution (μ = 249 aL, σ = 18 aL). (D) Evolution of ηapp with the concentration of Rh6G sensitizer. | ||
Comparison between the final OPD image and ex situ atomic force microscopy enables estimation of the average refractive index of the layer, yielding nlayer = 1.455 ± 0.005 (N = 19). Such value is rather low when compared to that of the layers formed with smaller diazonium salts derivatives,7 or to dense porphyrin films,38–40 indicating that the grafted layers are not compact. Low density layers were expected on the basis of the highly reactive radical intermediates generated upon activation of the diazonium groups, associated with the structure of the molecule. Indeed, since several positions can be attacked by radicals during addition of new porphyrin units to the layers, no particular organization is expected, leading to a highly branched structure connected through the phenyl rings. Low refractive indexes suggest that ππ stacking contributions are negligible to the layer structure, as dense stacks would be expected to present optical constants closer to crystalline layers.
We evaluate our ability to control the volume of the grafted layers by using the operando measurement to dynamically adapt the exposition time, as indicated in detail elsewhere.7 After setting the target optical volume to 10 aL (corresponding to a target physical volume V = OVDtarget/Δn of ∼250 aL), we perform several repetitions (N = 18) of the experiment and measure physical volumes ex situ with atomic force microscopy. This data is resumed in the histogram in Fig. 2C (the corresponding AFM images are provided in ESI S7†). The operation precision is estimated by fitting the distribution with a Gaussian function, leading to an average volume of 247 aL, in excellent agreement with the 250 aL target. Defining the operation precision as 2σ (σ being the standard deviation of the gaussian distribution) yields a precision of 36 aL, confirming our ability to control the photografted volumes.
Sensitized grafting
Owing to the existence of low energy excited states, porphyrins frequently present several non-radiative relaxation pathways, and consequently modest emission quantum yields. Since such difficulty is also expected to reduce the photoactivation yield, we have explored the possibility of increasing the photomodification efficiency by introducing a sensitizer. Rhodamine 6G (Rh6G) was selected for this role, owing to the high molar absorptivity and position of its main absorption band, requiring no adaptation of the setup. In the excited state, Rh6G becomes a strong reducing agent (i.e E0(Rh6G+/Rh6G*) = −1.09 V vs. SCE),41 able to intermolecularly activate the diazonium salts through a photo induced electron transfer mechanism.The concentration of Rh6G with respect to the photografting entity was first optimized in a series of experiments using a reaction mimic, p-nitrobenzenediazonium (pNBD). In addition to the structural similarity to the diazonium phenyl groups and Dz-TPP, pNBD was previously shown to optimally graft onto glass, and to present very high refractive index (1.68 ± 0.02),11 facilitating its operando observation. In this experiment series (ESI S8†), we have monitored the grafting rate of 5 mM pNBD solution in the presence of different sensitizer concentrations. Addition of small amounts of Rh6G (<0.5 eq.) leads to a substantial increase in the surface modification efficiency, as measured by the apparent efficiency ηapp. Such tendency saturates as [Rh6G] approaches 1 eq. [4NBD], with almost no increase beyond this threshold. When 4NBD is replaced by the porphyrin derivative, a very similar behaviour is observed: significant acceleration of the grafting reaction at low [Rh6G], which saturates as the sensitizer concentration approaches [Dz-TPP], (Fig. 2D). Typical evolution of the modified zone is indicated in yellow in Fig. 2A and B, revealing a limiting efficiency ηapplim (= 3 × 1013 nm3 J−1) which is roughly 4 times larger than the observed efficiency in the absence of sensitizer (ηapp0 = 0.7 × 1013 nm3 J−1), therefore allowing surface modification at significantly higher rates.
Surface micropatterning and layer characterization
We next demonstrate that such optimized photografting methodology is fully compatible with direct laser writing technology, enabling geometrically complex porphyrin microstructures to be prepared. With the help of a programmable piezoelectric stage, the sample is moved during the grafting operation at controlled speeds (typ. 1 μm s−1, P = 250 nW, or ca. 12.5 W cm−2), drawing arbitrary patterns onto the glass surface. This “writing” strategy is illustrated by printing “ENS Paris-Saclay”, as shown in Fig. 3A. The average resolution was estimated by analysing representative line profiles (N = 24), leading to an average FWHM of 1 μm, only slightly above the excitation laser waist (FWHMlaser = 0.8 μm). | ||
| Fig. 3 Characterization of grafted layer, (A) AFM image of the “ENS Paris-Saclay” pattern, along with FLIM images (inset). (B) Vibrational spectroscopy analysis of the grafted layer, obtained by collecting Raman spectra after excitation at λexc = 405 nm. A total of 20 spectra is collected (gray curves), demonstrating the stability of the measurement. The average Raman spectrum is shown in red, and main vibrational bands are highlighted (assignment in ESI S9†). (C) Emission spectra of the grafted layer, excited in widefield configuration at λ = 474 nm, the average (N = 98) spectrum is shown in red, while the individual spectra are shown in gray (1s acquisition time). (D) Photoluminescecnce decay curves, showing the substrate intrinsic emission (black curve), the layer decay (orange), along with a multiexponential fit (3 terms, dashed lines), and the experiment pulse width (gray). | ||
We confirm the chemical identity of the resulting patterns through a combination of local microscale Raman spectroscopy and surface X-ray photonelectron spectroscopy (XPS) analysis. First, a focused laser is used to extract the Raman signal with high spatial resolution (typ. 1 μm), allowing local characterization of the layer. The excitation wavelength (λexc = 405 nm) is selected to maximize the overlap with the Soret band of the porphyrin, triggering Resonant Raman mechanisms that significantly increase the signal. The resulting spectrum (Fig. 3B) shows several high intensity bands characteristic of tetraphenyl porphyrins, such as the 1553cm−1 (pyrrolic β carbons stretching), and the 1236 cm−1 (meso-carbon-phenyl stretching).42–44 In fact, assignment of all observed bands to normal modes of TPP molecules was possible on the basis of literature data (ESI S9†), unequivocally confirming the chemical identity of the layer. Noteworthy, such excellent agreement between solution data and the grafted layer Raman bands is consistent with the observed refractive index, both suggesting that the grafted layer has a low density.
We further corroborate these findings by performing an extensive surface analysis of the sample combining SEM/EDS and XPS analysis. Using a sacrificial sample, we use SEM/EDS data to show that all organic content of the sample is concentrated on the photomodified area, as EDX spectra collected from the background reveal no N or C contributions. Then, the “ENS Paris-Saclay” sample is analysed using XPS. We observe that (i) that most (ca. 94%) of the nitrogen content of the sample experiences the same chemical environment, showcasing a 399.98 eV binding energy consistently with the TPPs45 and; (ii) that measurements of N to C ratios reveal a value of 0.092 ± 0.01, indicating an average of 4.0 ± 0.4 nitrogen atoms per TPP molecule. These results, fully described in ESI section S9,† demonstrate that the contribution of any eventually unreacted NH2 groups to the chemical can be safely neglected, and that the layer is majorly composed of TPPs.
Photo physical properties
We next focus on characterizing the emission properties of the layer. Using a widefield illumination to excite the sample at 474 nm, we collect the light emitted by the layer using a fibered spectrometer. This leads to the steady state spectra shown in Fig. 3C, which corresponds to the very characteristic red emission of porphyrin, confirming that the photophysical properties of the grafted layers remain intact. Comparatively with the solution spectra of the diazonium precursor (Fig. 1D), two emission bands are now present (λem1 = 670 ± 10 nm, λ2em = 720 ± 10 nm), which correlate very well with emission spectra of tetraphenylporphyrins,38,46,47 as expected. We confirm that the influence of the photosensitizer used in the grafting process are negligible, as emission spectra of layers obtained in the presence of Rh6G sensitizer are identical to the emission of layers prepared by direct grafting (ESI S10†). If present, even small amounts of rhodamine would be readily detectable, owing to the high sensitizer absorption and emission quantum yield at an excitation wavelength.We complete the characterization of the layer emission properties with the help of a fluorescence lifetime imaging camera (FLIM), leading to the average lifetime (τPL) image shown in the inset of Fig. 3A, and to the decay curves shown in Fig. 3D. The resulting emission lifetimes are relatively low, between 0.9 and 1.2 ns, and complex, requiring at least 4 discrete exponential terms to fit the decay curve (fit procedures and results are shown in ESI S11†). Complex emission properties are characteristic of solid layers,48–51 and often arise as a result of complex energy transfer pathways in the solid state, especially for materials with highly overlapping emission and absorption bands within an amorphous structure.
Complex structures
Having demonstrated that pristine surfaces can be precisely microstructured with porphyrin-based layers, we focus next on showing that complex surfaces can also be modified, leading to multifunctional patterns. For that, we sequentially graft layers of different photophysical properties (notably emission), before including the TPP layer. The illustrative target structure consists in three intersecting circles of 10 μm, composed by (i) a post-functionalizable alkyne layer, modified to contain green emitting fluorescein dyes; (ii) a non-emissive nitro-phenylene layer and (iii) the TPP red emitting layer.First, the alkyne layer is prepared from a 5 mM 4-ethynylbenzene diazonium solution. We adjust the incident power (P = 600 μW) to compensate for the weak absorption of the 4-ethynylbenzene radical precursor, leading to an easily observable layer by moving the piezo stage at constant speed of 4 μm s−1. After preparation of the circle, we show that the alkyne functions remain intact by post functionalizing them with azide-modified fluorescein (Az-Fl) fluorophores. For that, a click chemistry procedure was employed, consisting in the incubation of the circle in a 1 mM Az-Fl solution in the presence of catalytic amounts of Cu(I) for 12 h (the full protocol is detailed in ESI S12,† along with the synthesize of Az-Fl). The procedure leads to covalent attachment of fluorescein to the grafted layer, as confirmed by its characteristic green emission upon excitation at 474 nm (control spectra are shown in ESI S13†). Next, we carefully rinse the sample and insert it in a 5 mM 4-nitro-benzenediazonium solution (H2O pH 2.0), grafting a second cycle (P = 600 μW, V = 4 μm s−1), which is not expected to be emissive, as indeed observed. Finally, the rinsing process is re-iterated before using a 5 mM Dz-TPP solution (in 50 vol.% DMSO/H2O), to graft a third cycle containing the porphyrins (P = 250 nW, V = 3 μm s−1), which displays the expected bright red emission, consistently with the data shown in Fig. 3C. The thickness of the final structure is characterized by AFM, as shown in Fig. 4A. Noteworthy, these experiments clearly demonstrate the stability of the grafted layers, able to withstand rinsing with the grafting solutions, its solvents, and several others (ethanol, methanol, acetonitrile, etc.), with no detectable structural changes.
 | ||
| Fig. 4 Photografting chemically complex structures, composed of Dz-TPP (bottom), fluoresceine modified alkyne layer (left), and p-nitro-benzene diazonium (right). (A) AFM characterization of the layers thickness (B) RGB image of the fluorescence emission, (C) FLIM characterization of the layer, along with its associated (D) photoluminesence decay curves, where the incident pulse width is also shown (gray). | ||
Preliminary inspection of the final structure through fluorescence imaging, as shown in the RGB image Fig. 4B, confirm the anticipated trends: the TPP-based layer appears red, while fluorescein-modified can be seen in green and the p-NBD is not visible. Interestingly, in spite of its favourable intrinsic properties, the fluorescein layer emission appears weaker than the TPP. This can be explained by (i) the layers average thickness, (ii) the fact that only a thin layer of fluorophores is grafted by post functionalization, while the whole TPP layer is emissive.
The emission properties are further analysed by FLIM, leading to the image shown Fig. 4C. After spatial filtering of the data on the basis of the layers distribution (ESI S11†), the normalized decay curves shown in Fig. 4D are obtained (see also ESI†), enabling a better observation of decay rates. While it is clear that all three layers present complex decays dominated by small lifetime values (<2 ns), subtle differences can still be spotted, enabling the formation of the lifetime image contrast (Fig. 4C). The pNBD layer has no significant emission, as the detected value in the grafted regions (〈τDZPL〉 <0.7 ns) rather suggests elastic scattering of photons from the excitation pulse. The TPP circle presents a low but significant lifetime, with 〈τTPPPL 〉 ∼1.1 ns. In the regions grafted with fluorescein, a clearer distinction from the other layers is observed, notably due to the presence of a longer component (τFlPL ∼ 1.6 ns), which becomes predominant for t > 3 ns, generating the contrast shown in Fig. 4C.
Conclusions
In this paper, we have shown that geometrically and chemically complex thin organic layers can be prepared by associating controlled photografting and direct laser writing. We have developed porphyrins bearing photoactivable units which photograft onto inert glass surfaces when irradiated at low power (in the 100 nW, or 5 W cm−2 range) with visible wavelengths. Operando nanometrology techniques are used to control the volume of the grafted layers with great precision (ca. 30 attoL for TPP) and to optimize the grafting conditions. In particular, we show that the addition of redox photosensitizers can increase the reaction efficiency by a factor of four. The resulting layers are fully characterized, using a combination of Raman and fluorescence spectroscopy, fluorescence imaging, and optical microscopy, revealing that layers present low compacity and retain many of the precursors photophysical properties. We have shown that geometrically complex porphyrin layers can be prepared, either alone or in association with other, sequentially grafted, chemical groups. As an example of custom microstructure, we prepare surface layers that emit light at different colours, combining up to three different groups in the same motif. This work therefore demonstrates direct micrografting of different functional groups onto surfaces, paving the way for the implementation of multifunctional surfaces in a flexible manner. We anticipate that this proof of concept will find broad applicability in both fundamental and applied research, providing chemists with a methodology to engineer microscale chemical environments, optimally, reliably and quantitatively placing functional molecules at well-defined patterns.Author contributions
BM performed most of the investigation, and contributed to the design of the methodology, HR, RB contributed to the Investigation, performing additional experiments IL, FM, VB provided resources to perform the research, FM, VB, IL, MLP were responsible for research supervision, VB, FM, IL, MLP and BM were responsible for Writing – review & editing; VB was responsible for project administration, methodology design, project conceptualization, validation and writing – original draft.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings described in this paper are available within the article and in its ESI.† Upon reasonable request, raw data can be made available from the corresponding author.Acknowledgements
H.R. thanks CHARMMMAT labex (ANR-11-LABEX-0039) for funding of his M2 internship. B.M. acknowledges financial support from ENS Paris-Saclay through a CDSN fellowship. This project has received financial support from the CNRS through the MITI interdisciplinary programs through its exploratory research program (MITI “Instrumentation, données et analyse in situ ou operando”). This work has been supported as part of France 2030 programme “ANR-11-IDEX-0003” (OI PSiNano). V.B., M.L.P. would like to thank the BrainStormNano Program from OI PSiNano, University Paris-Saclay. M.L.P., and R.B. thanks FISCIENCY grant no. ANR-23-CE50-009. We thank Dr. Robert Pansu for stimulating discussions and suggestions on lifetime experiments and data analysis. This project made use of the materials characterization platform (Atomic force microscopy and Scanning electron microscopy) of the Institut d'Alembert (IDA, CNRS FR3242) of the Ecole Normale Supérieure Paris-Saclay. Rasta Ghasemi is thanked for her assistance in SEM/EDS measurements, and Philippe Decorse for recording the XPS data. This work benefited from the Resonance Raman platforms of I2BC, supported by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05.References
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517–2583 CrossRef PubMed.
- Z. Li and K. S. Suslick, Acc. Chem. Res., 2021, 54, 950–960 CrossRef.
- F. Zeng, Z. Fan, S. Wu, X. Cheng and Y. Tian, RSC Adv., 2019, 9, 924–930 RSC.
- S. McGee, A. Fest, C. Chandler, N. N. Nova, Y. Lei, J. Goff, S. B. Sinnott, I. Dabo, M. Terrones and L. D. Zarzar, ACS Appl. Energy Mater., 2023, 6, 3756–3768 CrossRef CAS.
- A. Hoffmann, P. Jiménez-Calvo, J. Bansmann, V. Strauss and A. J. C. Kuehne, Macromol. Rapid Commun., 2022, 43, 2100731 CrossRef CAS.
- T. Pinheiro, M. Morais, S. Silvestre, E. Carlos, J. Coelho, H. V. Almeida, P. Barquinha, E. Fortunato and R. Martins, Adv. Mater., 2024, 36, 2402014 CrossRef CAS.
- B. Maillot, J. F. Audibert, F. Miomandre and V. Brasiliense, Nanoscale, 2024, 16, 7594–7602 RSC.
- B. Maillot, M. Johnson, J. F. Audibert, F. Miomandre and V. Brasiliense, Nanoscale, 2023, 15, 8754–8761 RSC.
- D. Bélanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- V. Brasiliense, J.-F. Audibert, T. Wu, G. Tessier, P. Berto and F. Miomandre, Small Methods, 2022, 6, 2100737 CrossRef.
- I. Azcarate, C. Costentin, M. Robert and J. M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS.
- K. Rybicka-Jasińska, W. Shan, K. Zawada, K. M. Kadish and D. Gryko, J. Am. Chem. Soc., 2016, 138, 15451–15458 CrossRef.
- F. Werner, J. F. Gnichwitz, R. Marczak, E. Palomares, W. Peukert, A. Hirsch and D. M. Guldi, J. Phys. Chem. B, 2010, 114, 14671–14678 CrossRef CAS.
- I. E. Kolesnikov, A. A. Kalinichev, M. A. Kurochkin, E. Y. Kolesnikov and E. Lähderanta, Mater. Des., 2019, 184(15), 108188–108194 CrossRef CAS.
- D. Xu, Q. Duan, H. Yu and W. Dong, J. Mater. Chem. B., 2023, 11, 5976–5989 RSC.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- Y. Leroux, D. Schaming, L. Ruhlmann and P. Hapiot, Langmuir, 2010, 26, 14983–14989 CrossRef CAS PubMed.
- A. K. D. Dimé, A. Bousfiha and C. H. Devillers, Curr. Opin. Electrochem., 2020, 24, 69–78 CrossRef.
- S. Hebié, A. K. D. Dimé, C. H. Devillers and D. Lucas, Chem. – Eur. J., 2015, 21, 8281–8289 CrossRef.
- D. Dasler, R. A. Schäfer, M. B. Minameyer, J. F. Hitzenberger, F. Hauke, T. Drewello and A. Hirsch, J. Am. Chem. Soc., 2017, 139, 11760–11765 CrossRef CAS PubMed.
- T. Palacin, H. Le Khanh, B. Jousselme, P. Jegou, A. Filoramo, C. Ehli, D. M. Guldi and S. Campidelli, J. Am. Chem. Soc., 2009, 131, 15394–15402 Search PubMed.
- S. Boufi, M. R. Vilar, V. Parra, A. M. Ferraria and A. M. B. Do Rego, Langmuir, 2008, 24, 7309–7315 Search PubMed.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS.
- Z. Liang, H. Y. Wang, H. Zheng, W. Zhang and R. Cao, Chem. Soc. Rev., 2021, 50, 2540–2581 Search PubMed.
- N. U. Day, M. G. Walter and C. C. Wamser, J. Phys. Chem.C., 2015, 119, 17378–17388 CrossRef CAS.
- T. Wu, M. Guillon, C. Gentner, H. Rigneault, G. Tessier, P. Bon and P. Berto, Opt. Lett., 2022, 47, 3079 CrossRef CAS PubMed.
- P. Berto, H. Rigneault and M. Guillon, Opt. Lett., 2017, 42, 5117–5120 CrossRef PubMed.
- Y. Prokazov, E. Turbin, A. Weber, R. Hartig and W. Zuschratter, J. Instrum., 2014, 9, C12015 CrossRef.
- A. B. Rudine, B. D. Delfatti and C. C. Wamser, J. Org. Chem., 2013, 78, 6040–6049 CrossRef CAS.
- R. Fukuda and M. Ehara, J. Chem. Theory Comput., 2013, 9, 470–480 CrossRef CAS.
- R. Shirasawa, I. Takemura, S. Hattori and Y. Nagata, Commun. Chem., 2022, 5, 158 CrossRef CAS PubMed.
- C. C. Wamser and A. Ghosh, JACS Au., 2022, 2, 1543–1560 CrossRef CAS.
- A. B. Rudine, B. D. Delfatti and C. C. Wamser, J. Org. Chem., 2013, 78, 6040–6049 CrossRef CAS PubMed.
- R. F. Khairutdinov and N. Serpone, J. Phys. Chem. B, 1999, 103, 761–769 CrossRef CAS.
- R. F. Pasternack, C. Bustamante, P. J. Ceilings, A. Giannetto and E. J. Gibbs, J. Am. Chem. Soc., 1993, 115, 5393 CrossRef CAS.
- R. F. Pasternack and P. J. Collings, Science, 1995, 935–939 CrossRef CAS.
- M. M. Makhlouf, A. El-Denglawey, H. M. Zeyada and M. M. El-Nahass, J. Lumin., 2014, 147, 202–208 CrossRef CAS.
- M. Morisue, Y. Hoshino, M. Shimizu, S. Tomita, S. Sasaki, S. Sakurai, T. Hikima, A. Kawamura, M. Kohri, J. Matsui and T. Yamao, Chem. Commun., 2017, 53, 10703–10706 RSC.
- N. Kari, M. Zannotti, G. Mamtmin, R. Giovannetti, B. Minofar, D. Řeha, P. Maimaiti, B. Kutilike and A. Yimit, Materials, 2020, 13, 1–20 Search PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- P. Stein, A. Ulman and T. G. Spiro, J. Phys. Chem., 1984, 88, 369–374 CrossRef CAS.
- S. E. J. Bell, A. H. R. Al-Obaidi, M. J. N. Hegarty, J. J. Mcgarvey and R. E. Hester, J. Phys. Chem., 1995, 99, 3959–3964 CrossRef CAS.
- I. H. Wasbotten, J. Conradie and A. Ghosh, J. Phys. Chem. B, 2003, 107, 3613–3623 CrossRef CAS.
- J. P. Macquet, M. M. Millard and T. Theophanides, J. Am. Chem. Soc., 1978, 100, 4741–4746 CrossRef CAS.
- Y. Kobayashi, Y. Yokota, Y. Shoji, S. Sajisha, C. J. Martin, J. Takeya, T. Fukushima and Y. Kim, J. Phys. Chem. C, 2024, 128, 15082–15090 CrossRef CAS.
- M. Ghosh, S. Nath, A. Hajra and S. Sinha, J. Lumin., 2013, 141, 87–92 CrossRef CAS.
- Z. Zhang, A. Brosseau, M. Elie, J. L. Renaud, M. Hamel, S. Gaillard and R. B. Pansu, J. Photochem. Photobiol. A, 2022, 432, 114038 CrossRef CAS.
- R. Métivier, I. Leray, J. P. Lefèvre, M. Roy-Auberger, N. Zanier-Szydlowski and B. Valeur, Phys. Chem. Chem. Phys., 2003, 5, 758–766 RSC.
- Y. S. Liu and W. R. Ware, J. Phys. Chem., 1993, 97, 5980–5986 CrossRef CAS.
- M. N. Berberan-Santos, E. N. Bodunov and B. Valeur, Chem. Phys., 2005, 315, 171–182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: S1: Determination of molar absorptivity coefficients of TAPP in different solvents. S2: Evolution of UV Vis Spectrum of TAPP with the addition of acidic H2O and organic acid. S3: Fluorescence decay of TAPP in different solvents. S4: Resonant Rayleigh spectra of TAPP and Dz TAPP in different solvents. S5: Characterization of TAPP Dz structure in solution by spectroscopy. S6: Effect of laser power on Dz-TAPP grafting efficiency. S7: AFM measurements of grafted TAPP. S8: Optimization of sensitizerto diazonium ratios with 4-nitrobenzenediazonium. S9: Analysis of the chemical properties of the grafted layers. S9.1: Raman Data. S9.2: SEM/EDS data. S9.3: XRay Photoelectron spectroscopy data. S10: Influence of sensitizer on emission spectra of grafted layers. S11: Grafted Layers lifetime analysis and FLIM data treatment. S12: Synthesis of fluorescein azide derivative (Az-Fl) and Click reaction S13: Photophysical properties of polymer layers functionalized by fluorescein derivatives. See DOI: https://doi.org/10.1039/d5nr00765h |
| This journal is © The Royal Society of Chemistry 2025 |