
Machine learning-driven design of single-atom catalysts for carbon dioxide valorization to high-value chemicals: a review of photocatalysis, electrocatalysis, and thermocatalysis
Xiangyu
Wen
 ab,
Xiao
Geng
b,
Guandong
Su
bc,
Yizheng
Li
d,
Qidong
Li
e,
Yuxuan
Yi
f and
Lifen
Liu
*bc
ab,
Xiao
Geng
b,
Guandong
Su
bc,
Yizheng
Li
d,
Qidong
Li
e,
Yuxuan
Yi
f and
Lifen
Liu
*bc
aState Key Laboratory of Chemical Engineering, Zhejiang University, 310027, Hangzhou, China
bKey Laboratory of Industrial Ecology and Environmental Engineering (MOE), Dalian University of Technology, 116024, Liaoning, China. E-mail: lifenliu@dlut.edu.cn
cDepartment of Environment and Food, School of Chemical Engineering, Ocean, and Life Sciences, Dalian University of Technology, Panjin, 124221, China
dDepartment of Building Environment and Energy Engineering, The Hong Kong Polytechnic University, 999077, Hong Kong, China
eSchool of Engineering, The University of British Columbia, BC V1 V 1 V7, 1137 Alumni Ave, Kelowna, Canada
fCollege of Design and Engineering, National University of Singapore, 117575, Singapore
First published on 21st March 2025
Abstract
The pressing need for carbon-neutral technologies has driven extensive research into photocatalytic, electrocatalytic, and thermocatalytic CO2 reduction, with highly efficient single-atom catalysts (SACs) due to their atomically dispersed active sites, tunable coordination environments, and well-defined electronic structures. Recent advances in SACs have demonstrated enhanced activity, selectivity and stability through rational design strategies incorporating transition-metal-based single-atom sites, nitrogen-coordinated frameworks, and perovskite-, graphene-, or MOF-supports. Mechanistically, SACs facilitate CO2 activation via optimized CO2 adsorption, electronic-state modulation and selective stabilization of key intermediates, thus promoting tailored product formation. Despite significant progress, challenges remain in understanding the precise electronic effects governing intermediate binding and selectivity and suppressing metal aggregation under operando conditions. This review systematically integrates experimental findings with machine learning (ML)-assisted first-principles calculations, deep learning (DL) frameworks, and density functional theory (DFT) modeling to refine the performances of SACs. ML-driven Bayesian optimization accelerates catalyst discovery by correlating the synthesis parameters with reaction kinetics and thermodynamics. High-throughput experimental validation combined with multi-technique characterization elucidates the structure–activity relationships, providing insights into the electron transfer dynamics, coordination tuning, and catalytic site evolution. The integration of active learning algorithms enables self-optimizing SACs, dynamically adjusting synthesis and reaction conditions for superior selectivity and faradaic efficiency. By bridging predictive modeling with experimental validation, this review presents a comprehensive framework for the rational design of next-generation SACs, paving the way for high-efficiency conversion of CO2 into valuable chemicals. The synergy between AI-driven catalyst discovery and mechanistic elucidation represents a paradigm shift toward viable and selective CO2 valorization strategies.
 Xiangyu Wen | Mr Xiangyu Wen is currently pursuing a Master's degree in Chemical Engineering (Hydrogen Energy Science and Engineering) at Zhejiang University. His undergraduate research at Dalian University of Technology in 2024 focused on photocatalysis, electrocatalysis, and photo-electrocatalytic CO2 reduction. Presently, his research is centered on the use of machine learning for catalyst design and thermocatalytic CO2 reduction to produce high-value chemicals, with a commitment to advancing CCUS (Carbon Capture, Utilization, and Storage) and clean energy technologies. |
 Guandong Su | Dr Guandong Su is an Assistant Professor in the Department of Environment and Food, School of Chemical Engineering, Ocean, and Life Sciences, Dalian University of Technology. He received his PhD in Environmental Engineering from the National University of Singapore and B.Eng. in Petroleum Engineering from China University of Petroleum-Beijing in 2024 and 2018, respectively. His research interests lie in the interdisciplinary interface of energy and environment to advance sustainable development goals, with an emphasis on providing affordable and clean energy for the future. |
 Yizheng Li | Mr Yizheng LI is a PhD candidate, who is jointly affiliated with the Hong Kong Polytechnic University and the Eastern Institute of Technology (Ningbo) and advised by Prof. Zhangxing CHEN and Prof. Jinyue YAN. He received his M.Eng. and B.Eng. from China University of Petroleum-Beijing. His research interests primarily include interpretable artificial intelligence, machine learning, petroleum engineering, building energy management, clean energy production, energy policy, and sustainable engineering education. |
 Qidong Li | Mr Qidong Li earned his Master's degree in Electrical Engineering through a joint program between the University of British Columbia and the National Research Council Canada, after graduating with a Bachelor's degree in Process Equipment and Control Engineering from China University of Petroleum-Beijing. His research focuses on battery thermal management systems. |
 Yuxuan Yi | Mr Yuxuan Yi is currently pursuing a Master's degree in Environmental Engineering at the National University of Singapore. His undergraduate research at Dalian University of Technology focused on photocatalysis. Presently, his research interests encompass seawater reverse osmosis and environmental data analytics. |
 Lifen Liu | Lifen Liu is currently a Full Professor in the School of Chemical Engineering, Marine and Life Science, Dalian University of Technology. She received her Bachelor, Master and Ph.D. degrees from DUT. She was a Visiting Scholar and Collaborator at the Sydney University, Australia from 1997–1999, a Research Associate at HKUST, HK, and a Senior Research Fellow at the Georgia Institute of Technology, USA (2010.10–2011.05). Her current and main scientific interests include environmental nanotechnology and pollution control engineering, specifically single-atom catalysts, functional electrode and fuel cell technologies for cost-effective processes. She has published more than 200 international peer-reviewed papers. |
Green foundation1. This review discusses advancements in green chemistry, particularly in the CO2 reduction reaction (CO2RR) using single-atom catalysts (SACs). Key developments include the use of SACs for photocatalysis, electrocatalysis, and thermocatalysis, with improved CO2 activation, enhanced selectivity, and increased efficiency through atomic-level precision.2. The integration of experimental techniques with machine learning (ML), deep learning models (DL), and DFT establishes a data-driven framework for optimizing the design of SACs. This approach facilitates the iterative refinement of the catalyst properties, prediction of reaction pathways, and enhancement of efficiency and selectivity, thereby generating wider interest. 3. The future of this field lies in achieving a comprehensive understanding of mechanisms and refining SAC design through ML/DL/DFT and experimental validation. These insights will guide the rational development of SACs with enhanced performance, aiding the scalability of CO2 reduction technologies and contributing to sustainability. |
1 Introduction
Rapid industrialization has greatly increased CO2 emissions, intensifying the greenhouse effect and creating various social, ecological and environmental challenges. China's rising share in global emissions has spurred efforts for “dual carbon” goals, emphasizing the need for effective solutions like catalytic CO2 reduction technologies. These technologies enable CO2 conversion into valuable small organic molecules via thermocatalysis, electrocatalysis,1–4 and photocatalysis,5 offering promising pathways for CO2 capture, storage (CCS), and utilization (CCU).6,7 Thermocatalysis at high temperatures can activate CO2, breaking C–O bonds and thereby producing C1 and C2 products. Meanwhile, in electrocatalysis,8,9 CO2 gets electrochemically reduced to form intermediates like formate, CO and methane;10 in photocatalysis,11 which mimics photosynthesis, light energy is used to generate charge carriers to activate and reduce CO2.12 These last two methods offer efficient solutions under mild conditions to reduce CO2 emissions.13 Converting CO2 into high-value chemicals, such as CO, CH4, HCOOH and C2 compounds, is both a carbon utilization strategy and a focus area of CO2RR for academic and industrial researchers.14 The related progress is shown in the pivotal role of catalytic science and technology in driving advances in energy, environmental and industrial sectors, enhancing reaction efficiency and selectivity and lowering activation energies.15 In recent years, the rapid progress of nanotechnology has propelled “nanocatalysts” to the forefront of energy, chemical, and environmental engineering owing to their high surface areas and unique catalytic properties.16,17 Catalysts are designed to reduce the size of active metal components and to unveil the distinct catalytic effects at the nano-, sub-nano- and SAC-scale.18 The concept of SACs was introduced while creating a Pt1/FeOx catalyst, where a single platinum atom was anchored on a ferrite nanocrystal.SACs offer distinct advantages over traditional nanocatalysts, including exceptionally high atomic efficiency, large surface areas and well-defined, uniform active sites. These properties make SACs highly effective in catalytic applications such as organocatalysis,19,20 photocatalysis21,22 and electrocatalysis.23,24 In CO2RR, SACs, especially those incorporating Co, N and C (e.g., M–N–C), exhibit superior performance due to their tailored coordination environments, enhanced electronic properties and precise control over the active site geometry. The high reactivity of these materials stems from their ability to fine-tune the electronic structure and optimize the catalytic pathway, facilitating efficient CO2 activation and reduction to small organic molecules such as CO, formate, and methane. The mechanism involving CO2 adsorption, activation via metal sites, electron transfer and bond cleavage, and formation of specific products, is determined by the catalyst's electronic structure and the reaction conditions.25 Despite significant progress in the design of high-density, high-stability SACs, the underlying mechanisms and their transformation during reactions remain poorly understood. While SACs theoretically maximize atomic efficiency, the increased surface energy of the isolated metal atoms, due to their higher surface area, can lead to aggregation under reaction conditions. As the metal loading increases, SACs tend to cluster, leading to a loss of catalytic performance. Therefore, achieving both high stability and large substrate loading in SACs remains a major challenge. This issue is particularly relevant in CO2RR, where maintaining the integrity of the isolated metal sites is crucial for efficient CO2 activation and selective reduction to organic molecules. Hence, solutions for optimizing support materials, such as nitrogen-doped carbon frameworks, to stabilize isolated metal sites and prevent aggregation while maintaining their high reactivity and selectivity for CO2RR26 are highly desirable.
Prediction and optimization of SACs using ML have become key research directions, with recent major advancements in ML offering powerful tools for accelerating catalyst discovery,27 fine-tuning reaction parameters, and unveiling complex reaction pathways.28,29 A key advancement in photocatalysis is the development of NiNi heterobimetallic SACs, which outperform traditional catalysts by utilizing a redox-active ligand for CO2 activation, highlighting the importance of dual-metal cooperation in catalyst design.30 In electrocatalysis, ML-guided DFT calculations have optimized SACs, achieving low overpotentials and high CH4 selectivity. Cu–Al catalysts, optimized via ML and DFT, show improved faradaic efficiency and selectivity, with active learning refining the reaction conditions such as the temperature and pressure.31 Thermocatalysis has benefited from AI platforms like Carbon Copilot (CARCO), which accelerated the discovery of SACs for CO2 conversion, achieving high precision in catalyst design within weeks.32 Mechanistic studies using ML and ab initio calculations, such as with CuPt/TiO2 catalysts, reveal that interface design is crucial for stabilizing CO2 intermediates, further guiding the rational design of SACs.33
While existing reviews34–36 have extensively covered the preparation methods, characterization techniques, auxiliary agents, and catalyst supports for SACs, there is a notable paucity of discussions on the synergistic optimization of ML/DL/DFT approaches alongside experimental strategies in the design of SACs, the identification of optimal reaction conditions, and the fine-tuning of SACs preparation parameters. In this context, our review offers a comprehensive analysis of the preparation methods, characterization techniques, mechanistic insights, reaction kinetics, and the integration of ML predictions for SACs in CO2RR. We emphasize the pivotal role of these approaches in optimizing the reaction pathways and enhancing the catalytic performance. Additionally, by employing machine learning to elucidate and validate the CO2 reduction mechanisms, we aim to predict previously unexplored reduction pathways and intermediates, thereby providing a robust theoretical and technical foundation for the tailored production of advanced chemicals in industrial applications.
2 Single-atom catalysts: design, preparation, characterization and prediction
2.1 Formation and optimization of single-atom catalysts
The preparation of SACs involves dispersing isolated metal atoms onto a stable support to prevent aggregation into larger clusters or nanoparticles.37 Single-atom dispersion requires strong metal–support interactions to prevent aggregation during synthesis or post-synthesis processes.38,39 In wet-chemical methods, aggregation is inevitable unless the metal atoms or complexes strongly bind to the support. SACs consist of isolated metal atomic sites dispersed on a support, where metal atoms are not arranged in continuous lattice structures as in metal nanoparticles, nor embedded in the surface lattice of metal oxides or other non-oxide compounds. These active sites typically consist of the metal atom and its first coordination shell, composed of oxygen or other non-metallic atoms. The active sites interact with nearby surface structures, such as metal atoms and oxygen vacancies. These sites adsorb and activate reactant molecules, forming surface intermediates, and ultimately generate products. The structural complexity of SACs arises from the diversity of the ligand shells surrounding the dispersed metal atoms. These metal atoms are often located at defect sites on the support, such as step edges, corners, and voids. Although the metal atoms are isolated, their chemical and coordination environments vary significantly depending on their specific positions. This variation in ligand coordination induces different electronic perturbations in the metal atoms’ valence orbitals, altering their electronic structure and ultimately affecting the catalytic performance (Fig. 1). Furthermore, the addition of two or more metal atoms to a single dispersed metal atom can form dimers or trimers, which often exhibit distinct an sometimes enhanced catalytic properties.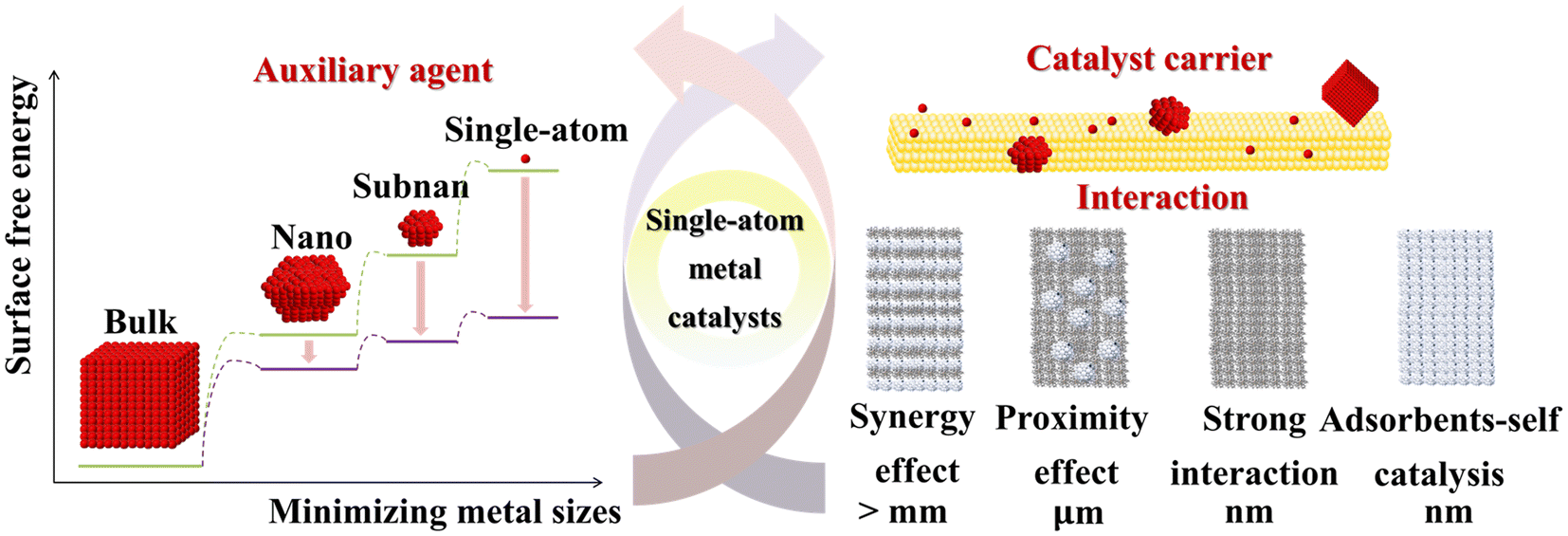 | ||
| Fig. 1 Effects of metal-size reduction on the surface free energy of auxiliary agents, and the interactions between auxiliary agents and catalyst supports in single-atom catalysts. | ||
CO2RR has attracted considerable attention in recent years, especially in the fields of photocatalysis, electrocatalysis, and Fischer–Tropsch synthesis (FTS).40,41 SACs have gained prominence due to their exceptional catalytic properties. When supported on materials such as carbon-based substrates, SACs can mimic the activation and reduction capabilities of homogeneous catalysts, facilitating CO2 conversion into small organic molecules.42 Despite their potential, challenges persist, including the strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation energy, competition with the hydrogen evolution reaction (HER), low product selectivity, and catalyst stability issues.43 Recent advancements have focused on optimizing SACs to enhance CO2 activation, tune electronic properties, and improve reaction pathways. For example, supporting SACs on nitrogen-doped carbon materials has been shown to significantly improve the CO2RR efficiency by enhancing the charge transfer and stabilizing the key intermediates. Understanding and controlling the metal–support interaction is critical for improving the SACs performance, as it modulates the CO2 binding energy and the formation of the desired products.
O bond dissociation energy, competition with the hydrogen evolution reaction (HER), low product selectivity, and catalyst stability issues.43 Recent advancements have focused on optimizing SACs to enhance CO2 activation, tune electronic properties, and improve reaction pathways. For example, supporting SACs on nitrogen-doped carbon materials has been shown to significantly improve the CO2RR efficiency by enhancing the charge transfer and stabilizing the key intermediates. Understanding and controlling the metal–support interaction is critical for improving the SACs performance, as it modulates the CO2 binding energy and the formation of the desired products.
The use of a single-atom iron catalyst supported on carbon nitride has been demonstrated, where metal sites dispersed on a nitrogen-doped carbon matrix serve as active centers for the adsorption and activation of CO2,44 enabling its reduction to CO and hydrocarbons. Similarly, ultra-thin nitrogen-doped carbon nanosheets have been employed as supports for single-atom nickel catalysts, significantly enhancing the efficiency of CO2RR. By integrating principles from homogeneous catalysis with quantum computational chemistry, these SACs have been engineered to mimic the CO2 activation processes observed in homogeneous systems. Co-doping with carbon and nitrogen further boosts the catalytic efficiency by modifying the electronic environment of the SACs, optimizing the CO2 adsorption, and facilitating the formation of reaction intermediates.
To further enhance the SACs catalytic performance, future development should focus on the following key aspects:
1. Optimization of metal–support interactions: tuning the interactions between metal active sites and the support is essential for improving the SACs performance. By modifying these interactions, the electronic structure of the metal atoms can be adjusted, thereby improving the CO2 adsorption and facilitating the formation of reaction intermediates.
2. Synergistic effect of multi-metallic sites: incorporating two or more metal atoms into SACs can enhance its catalytic performance. This synergistic effect provides new avenues for SACs optimization.
3. Regulation of surface defects: introducing specific defects, such as oxygen vacancies or nitrogen doping, can effectively modulate the electronic environment of SACs, optimizing the catalytic pathways and enhancing the catalytic efficiency and selectivity.
4. Catalyst stability and reusability: improving the stability of SACs and preventing the loss or aggregation of metal atoms during prolonged reactions is essential for the industrial application of SACs.
2.2 Performance predictions by DFT and machine learning
The electronic structure of SACs is linked to bonding between the metal centers and reactants, with key factors such as the d-band center, electronegativity, and ionization energy playing a crucial role. The position of the d-band center controls electron transfer to adsorbed species, enhancing CO2RR.49 Electronegativity and ionization energy govern the catalyst's ability to donate or withdraw electrons, affecting interactions with CO2 and products. Promoters such as alkali metals can further modify the electronic structure, adjusting the electron density and enhancing the adsorption of intermediates. The support material affects the metal dispersion, catalyst stability, and geometric configuration, influencing the electronic interactions and CO2RR efficiency.
ML models are increasingly used to predict SAC performance in CO2RR, leveraging high-dimensional data to uncover structure–activity relationships. Parameters such as the coordination number, bond length, d-band center, and electronegativity are key input features in these models. Computational methods like DFT predict the binding and adsorption energies, which are essential for understanding the CO2RR mechanisms. These properties are fundamental in the rational design of optimized catalysts.50
Key structural features vary for different CO2RR pathways, including photocatalysis, electrocatalysis, and thermocatalysis (Table 1). In photocatalysis, the band gap and electronic affinity govern light absorption and electron transfer efficiency. Electrocatalysis emphasizes charge transfer and the d-band center for efficient electron flow at the electrode surface. In thermocatalysis, geometric descriptors like the coordination number and bond lengths are critical for the reaction rate and product distribution, particularly at high temperatures.
| Descriptors | Photocatalysis | Electrocatalysis | Thermocatalysis |
|---|---|---|---|
| Structural and geometrical properties | |||
| Bond lengths | ✓ | ✓ | ✓ |
| Bond angles | ✓ | ✓ | ✓ |
| Atomic radius | ✓ | ✓ | ✓ |
| Coordination numbers | ✓ | ✓ | ✓ |
| Atomic positions (one-hot encoding) | ✓ | ✓ | ✓ |
| Number of specific element atoms | ✓ | ✓ | ✓ |
| Surface hollow, bridge, and top sites | ✓ | ✓ | ✓ |
| Chemical and elemental properties | |||
| Electronegativity | ✓ | ✓ | ✓ |
| Ionization energy | ✓ | ✓ | ✓ |
| Atomic mass | ✓ | ✓ | ✓ |
| d-electron count | ✓ | ✓ | ✓ |
| d-band center | ✓ | ✓ | ✓ |
| Valence electron number | ✓ | ✓ | ✓ |
| Electron affinity | ✓ | ✓ | ✓ |
| Physics-informed descriptors | |||
| SOAP (smooth overlap of atomic positions) | ✓ | ✓ | ✓ |
| MBTR (many-body tensor representation) | ✓ | ✓ | ✓ |
| ACSF (atom-centered symmetry functions) | ✓ | ✓ | ✓ |
| Synthesis and experimental parameters | |||
| Calcination temperature (°C) | ✓ | ✓ | |
| Calcination time (h) | ✓ | ✓ | |
| Reduction temperature (°C) | ✓ | ✓ | |
| Reduction time (hours) | ✓ | ✓ | |
| Reaction temperature (°C) | ✓ | ✓ | ✓ |
| Reaction pressure (MPa) | ✓ | ✓ | ✓ |
| GHSV | ✓ | ✓ | ✓ |
| Time on stream (h) | ✓ | ✓ | ✓ |
| Experimental data and metrics | |||
| Tafel slope (η) | ✓ | ||
| Band gap (eV) | ✓ | ||
| Catalyst per volume (mg mL−1) | ✓ | ||
| Sacrificial agent | ✓ | ||
Machine learning models, particularly those integrated with DFT calculations, have shown significant potential in predicting the electronic structure and energetics of SACs. For instance, the first-coordination sphere–support interaction (FCSSI) has emerged as a key descriptor for regulating the catalyst performance in CO2RR. ML models, such as extreme gradient boosting regression (XGBR), utilize DFT-derived data to predict limiting potentials (UL), which directly influence reaction pathways and selectivity toward specific products.53 Moreover, ML has proven effective in identifying intermediate species and elucidating reaction pathways. For example, the use of ML models to predict product selectivity in SACs supported on modified graphene (e.g., MoO4 on H-doped graphene and FeO4 on N,O-doped graphene) demonstrated the ability to distinguish between products like formate and CO.54 This highlights the potential of ML to identify SACs that minimize side reactions, such as HER,55 and favor target products such as formic acid or CO. In addition to predicting known pathways, ML models can uncover previously unobserved reaction mechanisms. For example, the prediction of new C–C coupling processes in graphdiyne-based SACs for C3 product formation offers new possibilities for CO2RR, potentially enhancing the overall CO2 conversion efficiency.56 These models provide an integrated framework to validate existing hypotheses and predict novel catalytic routes, which is critical for developing more selective and efficient CO2RR catalysts.
The application of graph neural networks (GNNs) and transformer-based models, such as CatBERTa, has further strengthened the predictive capabilities of ML models. These hybrid models can predict adsorption energies by utilizing spatial coordinates and textual representations of catalytic systems, thus allowing for the discovery of unknown intermediates and reaction pathways.57 Additionally, ML models can identify optimal reaction conditions, such as metal centers (Mn, Co, Ru, Os) and support interactions, which are crucial for product selectivity, as seen in the case of CH4 production.58 By integrating ML with computational studies, the exploration of CO2RR mechanisms becomes more systematic and accurate. This approach not only allows for the identification of new intermediates and reaction steps, but also enables experimental validation, which accelerates the discovery of high-performance SACs. The synergy between ML predictions and experimental data fosters an iterative process that refines reaction mechanism models, facilitating the rational design of SACs for efficient CO2RR.
ML-based methods, when coupled with quantum-level simulations, offer valuable insights into CO2RR mechanisms on SACs. These approaches facilitate the prediction of intermediates, reaction pathways, and optimal conditions, leading to the design of SACs with tailored properties for enhanced CO2RR performance.
Active ML methods, including reinforcement and self-supervised learning, enable models to autonomously search for optimal SAC structures and reaction conditions. These models iteratively refine predictions through continuous feedback from experimental data, accelerating the discovery of high-performance SACs. Moreover, ML facilitates the design of catalyst supports and surface modifications by considering local coordination environments and the electronic effects induced by dopants.54 In practical applications, ML has already contributed to the discovery of SACs such as Zn-NP3 and Sn-P4, which exhibit promising CO2RR activity and selectivity.60 By predicting the optimal catalyst structures and conditions before synthesis, these models minimize trial-and-error processes and accelerate the development of efficient SACs for CO2RR. Furthermore, ML-driven frameworks enable the high-throughput screening of catalyst materials and reaction parameters, expediting the discovery of novel SACs. The iterative refinement of SAC structures and reaction conditions continuously enhances the catalyst performance and CO2 utilization strategies.
In summary, integrating ML with SAC design and optimization offers a transformative approach to developing high-performance catalysts. By predicting the optimal catalyst structures and reaction conditions, ML accelerates the discovery and optimization of SACs for more efficient CO2RR.
To address these computational limitations, various ML models, including Random Forest (RF), Support Vector Machines (SVM), Neural Networks (NN), and Graph Neural Networks (GNNs), have been employed to predict the catalytic performance of SACs and gain insights into their underlying mechanisms. These models utilize the structural and electronic properties of catalysts to predict the catalytic activity, stability, selectivity, and key reaction parameters such as activation energy and charge transfer.
From a mechanistic perspective, Zhu et al.61 identified eight key descriptors that characterize the electronic and structural properties of catalysts: Nd (the number of d-shell valence electrons of metal atoms), which influences reactivity; EA (electron affinity), representing the metal's ability to accept electrons; CT (charge transfer from metal-zeolites to intermediates), quantifying electron redistribution during the catalytic step; Hc (hydrogen count in carbon atoms) and Hs (hydrogen in H2O, CH3OH, CH4), which influence the reactant activation; Nc (coordination number of carbon to metal), capturing the interaction strength between the carbon-based intermediates and the metal; Ps (number of proton/electron transfers), offering insights into the proton or electron transfer mechanisms; and NHB (hydrogen bonds), which stabilize the reaction intermediates.
To predict CT (e)—the charge transfer between the metal-zeolites and intermediates—ML models are trained using three key features: C (coordination number for intermediate to metal), No (coordination number for oxygen to metal), and NI (number of atoms in intermediates). These features capture the strength of the interaction between intermediates and the metal, the interaction of oxygen-containing species with the metal, and the complexity of the intermediates. By analyzing the relationships between these features and charge transfer, ML models help identify active sites and understand the critical electronic redistributions necessary for catalytic activity (Fig. 2).
 | ||
| Fig. 2 Application of DFT and machine learning/deep learning in predicting reaction pathways (a) Machine learning model with eight features, and the ability the XGBoost algorithm in predicting the free energy change in CO2 reduction process, (b) Machine learning model with six features for CT prediction on metal-zeolites, (c) 10-feature scheme, using three additional descriptors to replace the CT values, for free energy change prediction in the CO2 reduction process using XGBoost, (d) flow chart of reactivity and product calculation of CO2 reduction reactions on 26 metal-zeolites, (e) external test in the CO2 reduction process using ExtraTree. Reproduced from ref. 61, with permission of ACS Catalyst, copyright 2022. | ||
Once trained, ML models can predict key DFT-derived quantities, such as CT (e), activation barriers, and intermediate energies, at a significantly reduced computational cost. This accelerates the screening of catalysts and intermediates, allowing DFT calculations to focus on the most promising candidates. Additionally, ML enables efficient exploration of complex parameter spaces, uncovering insights into the reaction mechanisms that DFT alone might not fully resolve.
From a mechanistic perspective, ML provides deeper insights into catalytic processes by identifying key descriptors and elucidating their influence on reactivity. It offers a fundamental understanding of how electronic structures drive charge transfer and how intermediate coordination impacts catalytic efficiency. The integration of ML with DFT and experimental data accelerates SACs discovery and optimization, providing a powerful framework for designing next-generation catalytic materials with tailored properties.
2.3 Efficient and innovative methods for the preparation of single-atom catalysts
The preparation methods for SACs impact their performance in CO2 reduction, with each method offering distinct advantages and limitations (Fig. 3). Co-precipitation is cost-effective and scalable, yielding SACs with high surface area and catalytic activity.62 However, high metal loading can cause aggregation, reducing catalyst efficiency. It is suitable for large-scale production, but struggles with metal dispersion at high loadings.63 | ||
| Fig. 3 Synthesis, purification, and application of single-atom catalysts for CO2 reduction: from precursor preparation to catalytic reactor implementation. | ||
Ion exchange enables precise metal deposition, increasing active site density and surface area,64,65 but suffers from high reagent consumption and catalyst instability66 due to site degradation.67 Chemical vapor deposition (CVD) and atomic layer deposition (ALD) achieve atomic-level metal dispersion, yet remain cost-intensive and complex, limiting their scalability. The sol–gel method, though solvent-free with good dispersion, risks metal aggregation at high temperatures. Electrodeposition, often following substrate cleaning and electropolishing, facilitates nanostructure formation, while spin-coating efficiently deposits functional layers like perovskites, followed by annealing. Hydrothermal synthesis grows metal oxide nanowires, and photolithography with etching fabricates precise substrate patterns.68
For large-scale applications, co-precipitation offers cost efficiency and scalability, whereas CVD and ALD provide superior atomic precision but face economic and scalability constraints. The optimal method depends on balancing the cost, precision, and industrial feasibility.
2.4 Comparison in the characterization of single-atom catalysts
Accurate characterization is crucial for understanding the structure–property relationships of SACs, which exhibit unique catalytic behaviors due to their atomically dispersed active sites. The complexity of SACs, including their coordination environment, electronic structure, and metal–support interactions, requires the use of complementary techniques that each address specific aspects of their structure and performance.Microscopic methods, such as transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) combined with energy-dispersive X-ray spectroscopy (EDS), provide spatial insights into the dispersion and localization of single atoms. TEM reveals the distribution of active sites, while STEM-EDS achieves atomic-scale resolution, providing detailed insights into the elemental composition and structural features. Spectroscopic techniques, including X-ray absorption fine structure (XAFS) and X-ray photoelectron spectroscopy (XPS), play a crucial role in probing the local coordination environment, oxidation state, and chemical bonding of single atoms. These methods reveal the critical interactions between the active sites and the support, while also characterizing the surface's elemental composition and chemical states.
In situ techniques, such as Fourier transform infrared spectroscopy (FTIR) and Raman spectroscopy, allow for real-time monitoring of active sites and surface chemical environments during reactions. FTIR monitors the adsorption behavior and vibrational modes of reaction intermediates, while Raman spectroscopy tracks dynamic changes in catalytic sites under reaction conditions, elucidating the reaction mechanisms. Thermal characterization methods, such as temperature-programmed reduction (TPR) and thermogravimetric analysis (TGA), assess metal–support interaction strength, reducibility, and thermal stability, all of which are essential for evaluating the catalyst durability under operational conditions.
The integration of these techniques offers a comprehensive understanding of SACs, connecting their atomic structure, electronic properties, and reaction mechanisms to the catalytic performance and selectivity. This holistic approach not only advances fundamental knowledge, but also informs the rational design of next-generation catalysts for CO2 hydrogenation and other critical reactions.
3 Photocatalytic CO2 reduction
3.1 Mechanism of photocatalytic reduction
The photocatalytic reduction of CO2 with H2 utilizes light energy, typically from ultraviolet or visible light, to drive the reaction, converting CO2 and H2 into valuable products such as methane, methanol, and other organic compounds. This process involves several critical steps. First, photon absorption occurs when the semiconductor catalyst absorbs light, exciting electrons from the valence band to the conduction band, thereby generating electron–hole pairs (e−/H+). The electrons and holes are then separated and migrate within the catalyst's conduction and valence bands, with electrons (e−) participating in the CO2RR, while holes (H+) are involved in the oxidation of H2. During CO2RR, the electrons reduce CO2 into products such as carbon monoxide, methanol, or methane, depending on the catalyst and reaction conditions. This reaction proceeds through e− and H+ transfer, with different intermediates forming based on the targeted product. The oxidation process involves H+ at the catalyst surface, which oxidize H2, generating H+ that provide the protons necessary for CO2RR. These key steps are crucial for optimizing the catalyst performance and guiding the development of efficient photocatalysts for CO2 hydrogenation (Table 2).| Product | CO2 reduction pathways | E eq (V) | ΔH (kJ mol−1) |
|---|---|---|---|
| CO | CO2 + 2e− + 2H+ → CO + H2O | –0.10 | –41.2 |
| CH4 | CO2 + 8e− + 8H+ → CH4 + 2H2O | 0.17 | –253.0 |
| C2H6 | 2CO2 + 14e− + 14H+ → C2H6 + 4H2O | 0.14 | 264.3 |
| C3H8 | 3CO2 + 20e− + 20H+ → C3H8 + 6H2O | 0.09 | 374.2 |
| C2H4 | 2CO2 + 12e− + 12H+ → C2H4 + 4H2O | 0.08 | 127.8 |
| CH3OH | CO2 + 6e− + 6H+ → CH3OH + H2O | 0.03 | –131.0 |
| C2H5OH | 2CO2 + 12e− + 12H+ → C2H5OH + 3H2O | 0.09 | 216.1 |
| C3H7OH | 3CO2 + 18e− + 18H+ → C3H7OH + 5H2O | 0.1 | 331.2 |
| CH3CHO | 2CO2 + 10e− + 10H+ → CH3CHO + 3H2O | 0.06 | 130.1 |
| HCOOH | CO2 + 2e− + 2H+ → HCOOH | –0.12 | –31.2 |
| CH3COOH | 2CO2 + 10e− + 10H+ → CH3COOH + 2H2O | 0.11 | 122.1 |
In photocatalytic CO2 hydrogenation, the use of SACs is particularly effective. SACs, with their atomic-level dispersion on supports, create well-defined active sites that facilitate efficient electron and proton transfer, thereby optimizing the reduction steps. These catalysts enhance the reaction rates, selectivity, and stability by minimizing energy barriers, stabilizing reaction intermediates, and promoting specific electron transfer processes. The preparation of SACs—including metal choice, support material, and synthesis method—significantly influences their catalytic performance. By fine-tuning the electronic properties of metal atoms and their interaction with the support, SACs can be optimized to enhance CO2 activation, improve product selectivity, and boost the overall efficiency of the photocatalytic reduction process (Fig. 4).
 | ||
| Fig. 4 Engineering single-atom catalysts for efficient photocatalytic CO2 reduction: modulation of the electron pair alignment and recombination dynamics using auxiliary agents and catalyst supports to enhance the catalyst stability. | ||
Inspired by photosynthesis, photocatalysis leverages light-driven redox reactions for CO2 fixation. In this process, photogenerated charge carriers are separated in the semiconductor69 and transferred to the catalyst surface, where they react with adsorbed CO2. This results in the conversion of CO2 into valuable organic compounds such as formic acid, CO, alcohols, and hydrocarbons.70 Compared to traditional CO2 purification methods, photocatalysis offers advantages such as mild reaction conditions, low energy consumption, and minimal environmental impact, positioning it as a promising technology for sustainable CO2 utilization and carbon management.71
3.2 Reaction kinetics of photocatalytic reduction
The reaction kinetics of photocatalytic CO2RR, particularly when catalyzed by SACs, begins with photon absorption, which excites electrons and generates active sites on the catalyst surface. The efficiency of this process is largely influenced by the metal's ability to facilitate electron transfer and the support material's role in stabilizing the excited states. Early research on photocatalytic CO2RR dates back to the 1970s, initially focusing on semiconductor materials for water splitting applications. In the 1980s, research shifted towards using solar energy to drive CO2RR into organic chemicals, with early studies concentrating on methane and carbon monoxide production. Initial reaction models were oversimplified, typically assuming surface adsorption and electron transfer as the primary steps. However, with advancements in the catalyst materials and reaction conditions, the kinetics of the process became increasingly complex, involving multiple steps, various intermediates, and charge carrier participation.As catalyst materials diversified, selectivity and product distribution in photocatalytic CO2RR emerged as critical research themes. Different catalysts not only affect the separation efficiency of electron–hole pairs, but also play a decisive role in the formation pathways of intermediates, thus determining the final product. The reaction kinetics of photocatalysis involve several complex stages, including catalyst excitation, electron–hole pair separation and migration, and the formation and transformation of intermediates.
Enhancements in reaction kinetics are often achieved through orbital coupling between metal atoms and adjacent ligands, especially in bimetallic sites. For instance, Pt–Ru dimers on g-C3N4 demonstrate significantly improved catalytic performance for CO2RR and HER by weakening the binding strength with intermediates and adjusting the orbital energy levels.72 This strategy allows for more efficient catalytic processes by optimizing the interaction between the catalyst and the reaction intermediates.

The rate of a photocatalytic reaction (rPC) is generally linearly dependent on the light intensity (Ilight). However, under high light conditions, the reaction rate may become limited by the catalyst's electron carrier capacity, resulting in kinetic saturation. The rate is also influenced by the concentrations of CO2 (CCO2) and H2 (CH2), as well as by the surface adsorption characteristics of the catalyst, illumination conditions, and reactant concentrations (e.g., nPC, aPC, bPC). Moreover, the lifetime of the photogenerated electrons and the efficiency of the charge carrier separation are critical factors that impact the overall reaction efficiency. The separation efficiency of charge carriers is closely related to the catalyst surface structure and the light intensity. If photogenerated electrons recombine with holes on the catalyst surface, the reaction efficiency will be significantly reduced. Therefore, optimizing the charge carrier migration and separation efficiency is crucial for improving the performance of photocatalytic CO2 reduction.
3.3 Photocatalytic reduction of CO2 using single-atom catalysts
Optimizing CO2 adsorption and activation on the catalyst surface is key to improving the photocatalytic performance. However, a detailed understanding of surface site dynamics is still lacking. Feng et al.79 introduced an innovative strategy by constructing Mn single-atom sites on TiO2 nanostructures, which modified the local coordination environment, enhancing CO2 adsorption and the localization of photogenerated electrons, thus improving the catalytic efficiency. Similarly, Zhang et al.80 developed a dual-site system combining Au/Co double-monoatom-supported CdS for CO2RR. In this system, Co sites promote CO2 adsorption and reduction, while Au sites facilitate the collection of photogenerated electrons, selectively producing CO and CH4 with 82% selectivity. This design underscores the importance of effectively distributing photogenerated charges across different single-atom sites to enhance the photocatalytic selectivity.
Auxiliary agents play a vital role in optimizing SACs for photocatalytic CO2RR by modulating their electronic structures, stabilizing active sites, and facilitating charge transfer. Strategies such as defect engineering, heteroatom doping, and co-catalyst integration significantly influence reaction pathways, improving the catalytic activity and product selectivity.81–83 One effective approach involves introducing oxygen vacancies or structural distortions in the catalyst support to enhance the CO2 adsorption and charge separation. For instance, in Pd-SA/TiO2, oxygen vacancies created under a hydrogen-rich atmosphere serve as strong anchoring sites for Pd single atoms, preventing agglomeration and tuning their electronic structure. This modification increases the CO selectivity to 92.51% and catalytic activity to 56.84 μmol g−1 h−1 by facilitating charge transfer and minimizing electron–hole recombination.84
Heteroatom doping further refines the electronic properties of SACs by modifying charge distribution at the active sites. Doping with elements such as nitrogen, sulfur, or phosphorus stabilizes the reaction intermediates and enhances CO2 activation. In Cu-based SACs, nitrogen-doped carbon supports strengthen metal–support interactions, promoting C–C coupling and enabling selective C2+ product formation. The dopant atoms shift the d-band center of the Cu sites, reducing the activation energy barriers and optimizing multi-electron transfer processes.85
Co-catalyst incorporation provides another effective strategy for improving SACs performance by introducing additional charge transfer pathways or cooperative catalytic mechanisms.86 Overall, the integration of defect engineering, heteroatom doping, and co-catalysts significantly enhances SACs performance in photocatalytic CO2RR. These strategies collectively improve charge transfer, stabilize reaction intermediates, and suppress recombination losses, leading to higher catalytic efficiency and selectivity.
MOFs have attracted considerable interest for their ability to modulate electron transport in photocatalysis, owing to their tunable porosity, high surface area, and the potential to tailor active sites. Porphyrins and their derivatives, known for their excellent photosensitivity, can create conductive pathways through their stacked units. The incorporation of metals further enhances electron transport, although precise control of these pathways remains a challenge. Huang et al.88 employed a thermal-induced strategy, using thorium ions as unstable coordination nodes in porphyrin-based MOFs to control electron transport, thereby improving carrier separation and enhancing photocatalytic efficiency. This study provides valuable insights into carrier dynamics within photocatalytic systems. MOFs provide high surface area, tunable porosity, and defined coordination sites, ensuring uniform metal dispersion and electronic modulation. Their confinement effect prevents metal aggregation, while enhancing CO2 activation. MOF-derived carbons retain structural integrity and improve conductivity, facilitating charge transfer and reaction kinetics.
Recent developments in SACs have demonstrated their potential for CO2RR, driven by their unique electronic properties, high dispersion, and enhanced catalytic activity through defect engineering. SACs offer additional active sites at the atomic scale, which significantly enhance catalytic performance. Cheng et al.89 utilized the large surface area and planar conjugate structure of g-C3N4 to anchor Ni single atoms through a self-limiting method.90 This approach not only stabilized the Ni single atoms, but also promoted efficient CO2 adsorption and electron transfer from g-C3N4 to Ni via Ni–N coordination, resulting in a CO2 conversion rate that is 7.8 times higher than that of pure g-C3N4 under visible light. This highlights the critical role of tuning active sites for improving CO2RR efficiency.91
Zhang et al.92 developed Co-SA@SP by anchoring single-atom cobalt on commercial superconducting carbon black. The Co–N4 coordination sites in this material reduced the CO˙ desorption energy barrier, enhancing the catalytic reactivity, CO selectivity (84.2%), and stability under UV light. The suppression of HER further promoted CO2RR, demonstrating the potential of SACs to inhibit competitive reactions and enhance selectivity. This simple and cost-effective catalyst offers a promising pathway for scalable photocatalytic CO2RR in industrial applications. Catalyst carriers critically influence SACs’ photocatalytic CO2RR by modulating the electronic structure, charge transfer, and active site stability. The carrier dictates metal–support interactions, adsorption strength, and reaction selectivity.
Semiconductor supports with oxygen vacancies effectively anchor single metal atoms, preventing aggregation while tuning their electronic environment. In Pd-SA/TiO2, oxygen vacancies enhance metal–support interactions, optimizing Pd's oxidation state and CO2 activation. This defect engineering improves the charge separation and suppresses recombination, boosting the catalytic efficiency.84
Carbon-based supports, such as graphene, CNTs, and nitrogen-doped carbon (N–C), offer high conductivity and π-conjugation networks that enhance charge transport. In Cu-SACs, N–C stabilizes Cu atoms, improving C–C coupling and favoring C2+ products. Strong metal–support interactions (SMSI) optimize Cu's electronic state, lowering activation barriers and increasing the CO2 conversion selectivity.85 Hybrid architectures incorporating plasmonic nanostructures or co-catalysts further enhance the SACs performance. Plasmonic-metal-SACs hybrids use localized surface plasmon resonance (LSPR) to extend the carrier lifetimes and improve charge transfer, lowering the activation barriers. Co-catalyst integration facilitates charge separation and directional electron flow, optimizing the reaction selectivity.86
In summary, defect engineering, conductivity tuning, and confinement effects are key to improving the SACs stability and reactivity. Future research should focus on optimizing the metal–support interactions and charge transfer pathways to enhance SAC-based CO2RR.
The perovskite structure, characterized by a corner-sharing octahedral arrangement of metal cations and halide anions, provides a versatile platform for modulating the electronic properties, optimizing the charge carrier dynamics, and facilitating enhanced photocatalytic activity. In Cs3Sb0.5Bi1.5Cl4Br5 perovskite catalysts, the formation of a built-in electric field (IEF) within heterojunctions facilitates efficient electron–hole separation, a key factor in improving the photocatalytic CO2RR efficiency. The coupling of these materials with other catalysts, such as Bi-BTC frameworks, has shown that the synergistic effects between the two components can significantly boost photocatalytic performance, leading to high CO and CH4 yields.93
In addition, engineering the electronic structure of perovskites through doping or phase transition can further enhance their catalytic properties. For example, doping perovskite structures with metal ions or using phase transitions to generate piezoelectricity can increase the adsorption capacity for CO2 and reduce the energy barrier for its conversion.94 The piezoelectric effect, induced by phase transition under external stimuli, can produce a polarized electric field that accelerates CO2 activation, contributing to improved catalytic efficiency. In the case of Cs3Bi2Br9-based perovskites, the introduction of Cu atoms into the perovskite lattice has been shown to promote charge separation and enhance the electronic structure of the material, which further lowers the energy barriers for CO2RR. The atomically dispersed Cu acts as a critical site for charge transfer, improving the selectivity for CO production while maintaining stability over multiple cycles.95
The engineering of heterojunctions by combining perovskites with other semiconductors, such as Co3O4, has proven highly effective for promoting efficient charge separation. The formation of built-in electric fields at the heterojunction interface enhances the photocatalytic CO2RR performance, with Na-ion incorporation further strengthening these electric fields to boost CO2 adsorption and activation.96 Thus, perovskite SACs possess unique properties, such as their tunable band structures, efficient charge separation, and enhanced CO2 adsorption, all of which play crucial roles in improving the directional selectivity of photocatalytic CO2RR.97 These structural features, coupled with innovative doping strategies and heterojunction formation, position perovskite-based SACs as promising candidates for the efficient conversion of CO2 into valuable chemicals (Fig. 5).
 | ||
| Fig. 5 Lead-free halide-perovskite-based heterojunction: (a) charge density difference (isosurface level, 0.04 e Å−3) over the CSBX/BOF heterojunction. The yellow and cyan regions represent the positive and negative electron density isosurfaces, respectively, KPFM surface potential images (middle), and the corresponding topography and surface potential line profiles (bottom) for (b) CSBX and (c) BOF three-dimensional topographical maps (top). (d) Schematic of the band structures of CSBX and BOF (left), interfacial charge transfer and the formation of an IEF upon hybridisation (middle), and CSBX/BOF S-scheme heterojunction under light irradiation (right). Reproduced from ref. 93, with permission of Applied Catalysis B: Environment and Energy, copyright 2025. | ||
4 Electrocatalytic CO2 reduction
4.1 Mechanism of electrocatalytic reduction
CO2RR converts electrical energy into chemical energy, producing valuable small molecules via electrochemical reduction.92 However, in aqueous systems, the HER often competes with CO2RR, significantly reducing selectivity and efficiency. To address this, the development of highly efficient electrocatalysts with high activity, selectivity, and stability is crucial for advancing CO2RR technology. SACs have emerged as promising candidates due to their well-defined active sites, maximizing atom utilization. The performance of SACs is primarily determined by the electronic structure and coordination environment of the single metal atoms, which can effectively modulate CO2 adsorption, activation, and reduction. SACs promote the reduction process by facilitating the formation of key intermediates such as ˙CO2, ˙COOH, and ˙CHO through unique reaction pathways, while minimizing HER by optimizing the binding energies of hydrogen and CO2 intermediates. The enhanced selectivity and efficiency of SACs arise from the atomic-level control over the reaction mechanisms, enabling precise tuning of the product distribution. Ongoing efforts to improve SACs for CO2RR have focused on stabilizing the metal sites, increasing conductivity, and optimizing the local electronic environment to achieve scalable, practical performance.CO2RR is a complex multi-electron, multi-proton process where intermediates like ˙CO are crucial for C2 product formation. Strong interactions between ˙CO and metal catalysts (e.g., Pt, Fe, Ni) can poison the active sites, favoring HER and diminishing the CO2RR efficiency.98 On the other hand, metals with weak ˙CO binding (e.g., Au, Ag, Zn) facilitate ˙CO desorption, which prevents efficient C–C coupling and results in CO as the final product.99 However, metals with moderate ˙CO binding strengths, such as Cu, Ni, Co, and Zn, are among the few catalysts capable of selectively producing C2 and other organic carbon products,100 striking a balance between ˙CO binding and desorption, thereby enabling effective C–C coupling.
Electrocatalytic CO2RR typically occurs in an electrochemical cell, where an electrode (a metal or alloy electrode) catalyzes the reaction between CO2 and H2, converting them into useful chemicals. During this process, CO2 is adsorbed onto the catalyst surface, where it is activated through electron transfer, generating reactive intermediates like ˙CO2˙− (a radical anion) or ˙CO2H (a hydrogenated intermediate). This step requires overcoming the adsorption energy barrier between CO2 and the electrode, typically occurring on metal electrodes (e.g., copper, silver, gold). The reduction reaction involves the transfer of electrons and protons; electrons are delivered via the electrode, while protons come from the electrolyte. The electrode surface reduces CO2 intermediates, generating species such as CO2˙− or CO2H.
The electrocatalytic reaction proceeds through the formation of intermediates, which are progressively reduced to final products like methanol, methane, and other hydrocarbons (Fig. 6). The overpotential, defined by the current density and voltage applied, plays a critical role in determining the product selectivity. Different electrocatalysts significantly affect the distribution of products. For instance, copper-based catalysts are often superior to others, promoting a range of carbon-containing organic products such as alkanes, alkenes, and alcohols, while silver and gold catalysts typically favor the formation of CO.
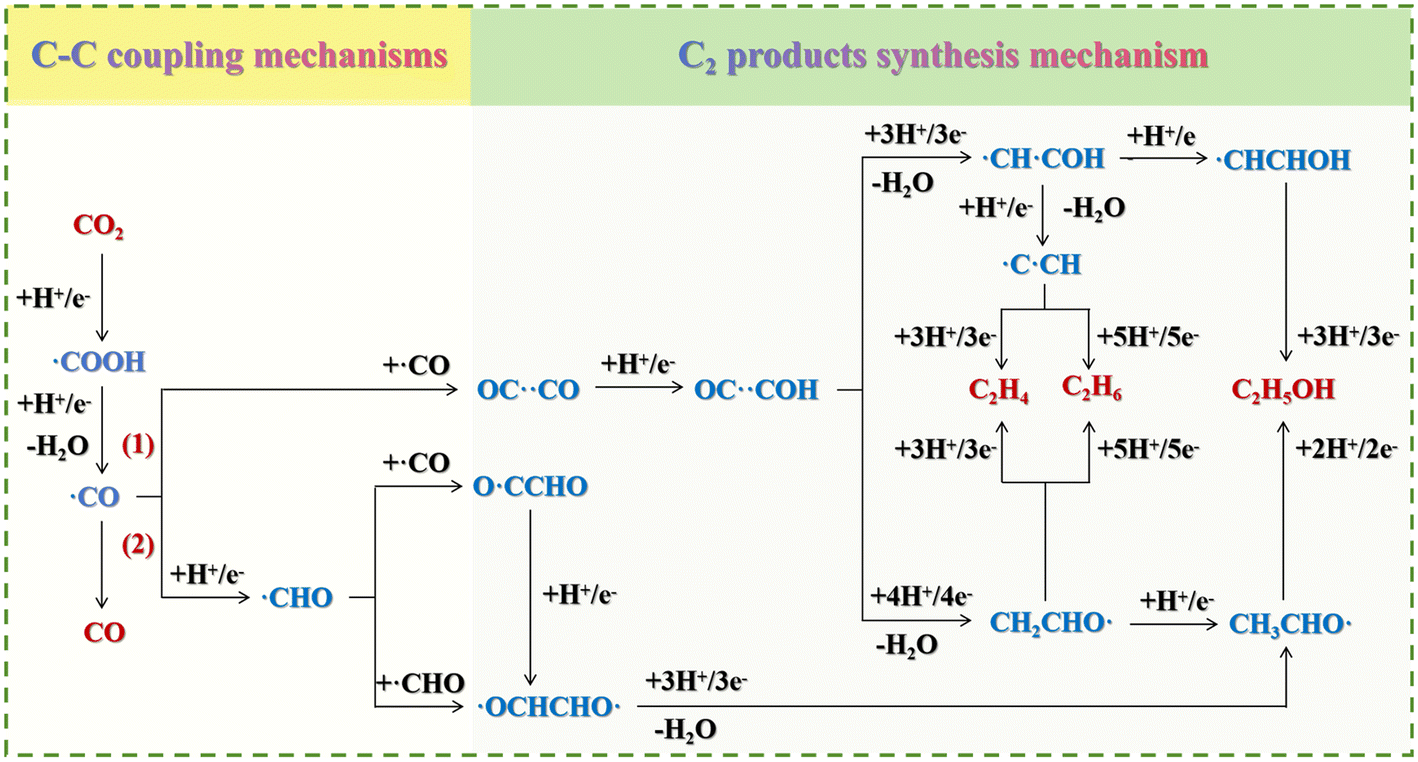 | ||
| Fig. 6 Reaction pathways of CO2 reduction to produce value-added C2 chemicals (C2H4, C2H6, and C2H5OH) via electrocatalysis by adding (1) ˙CO before H+/e− and (2) H+/e− before ˙CO. | ||
Thus, understanding and optimizing the interaction between CO2 intermediates, the electronic properties of the metal catalysts, and reaction conditions are key to enhancing the efficiency and selectivity of electrocatalytic CO2RR.
4.2 Reaction kinetics of electrocatalytic reduction
In recent years, significant progress has been made in understanding the electrocatalytic CO2RR using SACs, leading to improved catalytic performance. SACs are particularly appealing due to their high metal atom utilization, distinct electronic properties, and excellent catalytic efficiency. However, the isolated active sites in SACs can limit their effectiveness in reactions that require the simultaneous activation of multiple reactants or intermediates. To address this limitation, strategies have been developed to combine the advantages of SACs with multi-atomic sites, resulting in “fully exposed metal cluster catalysts” (FECCs).101 These FECCs can simultaneously activate different reactants, enhancing the reaction kinetics and improving the overall electrocatalytic performance.Recent studies102 have demonstrated that tuning the atomic dispersion of metal species within SACs can significantly enhance the catalytic efficiency. For instance, by adjusting the pyrolysis temperature, researchers have successfully transitioned nickel (Ni) species from clusters to single-atom scales, resulting in a Ni FECC with superior CO2RR performance. The optimized Ni FECC achieved a FE of up to 99% for CO production, with a CO partial current density of 347.2 mA cm−2, and exhibited stable performance over 20 hours of electrolysis. Theoretical calculations revealed that sulfur doping plays a crucial role in regulating the charge distribution across multiple Ni sites, thus accelerating the reaction kinetics and facilitating efficient CO2RR to CO.
Another study103 focused on the development of a model Ni SAC with well-defined Ni–N4 coordination on a conductive carbon support, which was used to explore the CO2RR mechanism. Using advanced operando techniques, such as X-ray absorption spectroscopy, Raman spectroscopy, and near-ambient XPS, it was found that the Ni+ sites in the Ni SAC were highly active for CO2 activation, acting as the actual catalytic sites for the CO2RR. Electrochemical kinetics studies identified the rate-determining step of the CO2RR as the conversion of ˙CO2@ + H+ → ˙COOH, providing key insights into the mechanistic details of CO2RR.
Despite the outstanding performance of SACs in CO2RR, the relationship between their electronic and geometric structures and their electrocatalytic properties remains an area of ongoing investigation. Factors such as the coordination environment of metal atoms, the nature of the conductive support, and the interactions with secondary catalysts all significantly influence the efficiency and selectivity of SACs in CO2RR. Understanding these structure–function correlations is critical for the rational design of SACs that exhibit high activity, selectivity, and stability.104
The kinetics of electrocatalytic CO2RR are influenced by several key factors, including current density, applied voltage, catalyst surface properties, and the concentrations of CO2 and H2. Typically, the reaction rate is proportional to the current density (j, A cm−2), with the reaction rate increasing as the current density increases, until a saturation point is reached. The overpotential (η) represents the difference between the required voltage for the reaction and the theoretical voltage, reflecting the activation barrier on the catalyst surface and significantly affecting the reaction rate. This relationship is often described by the Tafel equation, which characterizes the logarithmic dependence of the reaction rate on the overpotential:
η = aEC + bEC·log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j j |
Additionally, the reaction rate (rEC) is affected by the concentrations of CO2 and H2. The rate for CO2RR typically follows the equation:
| rEC = k·j |
| rEC = kEC·(CCO2)aEC·(CH2)bEC |
Overall, SACs have demonstrated remarkable potential in CO2RR, with atomically dispersed active sites, tunable electronic structures, and enhanced reaction kinetics. However, challenges remain in the catalyst design, synthesis, and characterization. Future research should focus on developing novel synthetic strategies to precisely control the atomic dispersion, improving the catalyst stability under practical conditions, and further elucidating the structure–activity relationships.
4.3 Electrocatalytic reduction of CO2 using single-atom catalysts
The CO2RR involves complex intermediates and competing reaction pathways, making optimization challenging. Jin et al.111 addressed this by constructing atomically dispersed heterodiatomic pairs, where ˙COOH intermediates bridge across heteroatomic sites, lowering their formation energy. This modification enhances electron delocalization via Mo–Fe d–d orbital coupling, which promotes ˙CO desorption and improves CO selectivity. The introduction of dual- and multi-active centers has also gained attention for its ability to activate multiple reactants, improving selectivity and product diversity. For CO2RR, dual-site catalysts often feature separate sites for CO2 activation and subsequent hydrogenation or C–C coupling. The spatial arrangement of these sites influences the migration of intermediates, and optimizing ˙H coverage can help mitigate HER, improving the selectivity for CO2RR over hydrogen evolution.
Single-atom alloy catalysts (SAAs) are another promising approach for CO2RR to C2+ products, although challenges remain in achieving selective C–C coupling. Cao et al.112 reported on a Bi–Cu SAA catalyst that achieved 73.4% FE for C2+ products. The interaction between Bi and Cu modulated the electronic structure of Cu, lowering the CO2 adsorption and activation energy, enhancing intermediate formation, and improving the C2+ selectivity. This study highlights the potential of SAAs in regulating active sites, and provides a new strategy for catalyst design. In a different approach, Cai et al.113 used Pd atoms for CO2RR to formic acid with a Pd/C–Pt/C composite electrode, demonstrating a new method for low-temperature aqueous-phase electrocatalysis. Additionally, Quan et al.114 developed nitrogen-coordinated Cu SACs (Cu/NC) for CO2 electrocarboxylation with styrene, achieving 92% FE and nearly 100% product selectivity for phenylsuccinic acid.
Metals such as Fe, Co, Cu, Zn, and Sn are increasingly used in CO2RR due to their distinct product selectivity. Sn, In, and Pb primarily produce formic acid while effectively inhibiting HER,115,116 whereas Zn, Au, and Ag favor CO production due to their strong interaction with the CO˙ intermediate, which is crucial for large-scale CO production.117–119 However, the high cost of Au and Ag limits their practical application. Cu, as the only metal capable of producing C2+ products, holds significant potential, as C2 products (ethanol and ethylene) offer greater commercial value compared to C1 products (CO and methane).120 Despite this, the structural sensitivity of copper single atoms and their role in CC coupling remain areas of ongoing research. It has been assumed that CO2RR to both CO and C2 products occurs at the same Cu site, but recent studies suggest that distinct Cu sites may be involved in these steps.121 Further mechanistic investigations are required to clarify these processes and optimize the catalyst performance. Cu-based catalysts have been extensively studied for CO2RR, with various modifications improving their selectivity and efficiency. This strategy can be extended to other Lewis-acid metals to further enhance the CO2RR performance. Wang et al.122 developed an InBi bimetallic catalyst supported on In2O3 nanodots on Bi2O2CO3 nanosheets, where the synergistic effect between the metals and the enhanced CO2 adsorption led to 97.17% FE in an H-type electrolyzer.
Heteroatom doping, particularly with nitrogen, sulfur, or phosphorus, alters the electronic properties of the support material and enhances the SACs activity. Nitrogen-doped carbon supports improve the interaction between metal centers and CO2, facilitating CO2 adsorption and reduction. This doping stabilizes the metal sites and prevents agglomeration, thus improving the catalyst's stability over prolonged reactions. The coordination environment of the metal center is further optimized by the inclusion of multiple heteroatoms in the support structure. In Cu SACs, nitrogen-doped carbon support adjusts the binding strength of ˙CO and ˙COOH intermediates, enhancing CO2RR to C2H4. This synergy between the metal and support is crucial for maximizing the CO2RR performance. Co-catalysts, often in the form of small nanoparticles or sub-nanoclusters, have been integrated with SACs to promote cooperative catalysis. In these tandem systems, one site activates CO2, while the other facilitates CO coupling, leading to higher-order products such as C2H4 and CH4. Cu SACs coupled with copper nanoclusters exhibit enhanced selectivity for C2+ products by promoting CO2RR to CO, followed by coupling at the NP site.123
Moreover, modifying the coordination environment around the metal site further enhances the SACs performance. For instance, Cu SACs supported by N2-bidentate sites in a graphdiyne structure show higher selectivity for methane production, with reduced side reactions. This design achieves a FE of 80.6% for methane and a current density of 160 mA cm−2.124
The support curvature also affects the SACs activity. Ni SACs supported on spherical carbon materials exhibit optimized nanocurvatures, which influences the interfacial electric field, modulating intermediate adsorption during CO2RR. This results in a CO partial current density of 400 mA cm−2 at 99% FE.125
Cu SACs supported by nitrogen-doped carbon quantum dots (CQDs) exhibit enhanced catalytic performance, with a FE of over 80% for ethanol production and stable performance over 50 hours. This improvement stems from the atomic dispersion of Cu sites and the interaction with the nitrogen-doped support.126
While precious metals such as Ru, Pd, and Pt have demonstrated high efficiency in CO2RR, their high cost and limited scalability prompt the exploration of more affordable and scalable SACs.129–131 Liu et al.132 developed a Sn–Cu diatomic catalyst by introducing Sn into isolated Cu sites, which achieved a FE of 99.1% for CO production.133 The high efficiency of this catalyst is attributed to its efficient atom utilization, SMSI, and the high unsaturation of the coordination environment around the metal atoms. Additionally, the nitrogen-doped porous carbon support improves mass transfer, ensuring that the catalyst can maintain high performance over extended periods.
Su et al.134 reported a Cu/Zn hierarchical porous catalyst that inhibits HER, while favoring alcohol formation. This catalyst enhances intermediate interactions with the surface. Furthermore, it features a porous structure that increases the density of surface active sites and improves the surface-to-volume ratio, promoting the formation of C2 products. In another study, Chen et al.135 employed a Cu2+ electrolyte to balance Cu ion dissolution and deposition, achieving 83.3% ethylene selectivity after 4 hours of electrolysis.136 The periodic removal of accumulated bicarbonate during the reaction further stabilized the catalyst, maintaining high performance and selectivity. The combination of ultrafast thermal shock synthesis and pore engineering enhanced the electrode's reactive surface area, facilitated charge transfer, and improved selectivity, ultimately boosting the catalyst's overall efficiency. Fan et al.137 addressed stability concerns in acidic systems by modifying the electrode surface with benzimidazole-functionalized ionomer groups. This modification activated CO2 and enhanced C–C coupling, achieving over 80% multi-carbon selectivity. The control of the microscopic water environment and the improvement of the catalyst stability under acidic conditions further contributed to the high performance.
Oxygen or nitrogen vacancies in the support material can improve metal–support interactions and increase the number of available active sites. Supports like TiO2 or carbon materials with oxygen vacancies can stabilize metal sites and facilitate electron transfer, lowering the activation energy for CO2RR. This enhances the reaction rates and improves selectivity for valuable products such as C2+ compounds. Using Ti3C2O2-based MXene supports showed that the asymmetric coordination of the metal center improved CO2RR and product selectivity. The support's ability to modulate the metal's electronic properties and interaction with intermediates contributed to enhanced catalytic activity, such as improved formic acid production.138
The chemical composition of the carrier is also important. Nitrogen-doped carbon supports, for instance, can enhance the electronic environment of SACs, improving CO2 adsorption and the formation of C2+ products. Supports with high surface area and tunable porosity, such as MOFs, are ideal for dispersing metal atoms uniformly, improving accessibility to reactants and boosting the catalyst efficiency.139
Tandem catalysis, where SACs are combined with nanoclusters or sub-nanometric catalysts, can further enhance performance. These hybrid systems allow for cooperative interactions, where one component activates CO2 and the other facilitates CO coupling to form higher-order products. Cu-based SACs combined with copper nanoclusters show improved selectivity for C2+ products due to this synergy.123 The stability of SACs is also influenced by the support. Conductive and stable supports, like graphene or carbon nanotubes, help maintain the high dispersion of metal atoms and ensure efficient electron transfer, preventing aggregation and preserving catalytic activity. A study of CuFONC, a carbon composite with Cu single atoms supported by F, O, and N-doped motifs, found that the carrier stabilized the Cu sites, enhancing CO–C coupling and selectivity for C2 products. The carrier's chemical environment strengthened the interaction between Cu and CO intermediates, leading to an FE of 80.5%.140 The curvature of the carrier also plays a role. Ni SACs on spherical carbon exhibited superior CO2 reduction performance due to the stronger electric fields generated by the nanocurvature. These fields help optimize the adsorption of intermediates, enhancing the SACs activity.125
Overall, these studies demonstrate the effectiveness of SACs in CO2RR, highlighting various strategies for improving the efficiency, selectivity, and stability. The use of different metal combinations, such as Cu–Sn, Cu–Bi, and Cu–Zn, as well as modifications to support structures and electrolyte conditions, plays a critical role in tailoring the catalytic properties and enhancing the overall CO2RR. The transition to more affordable, scalable SACs is essential for advancing CO2RR technologies, with significant potential for commercial applications in producing valuable carbon-based products.141
The modification of the graphene matrix plays a critical role in improving the selectivity and efficiency of CO2RR. For example, nitrogen doping and the introduction of vacancy defects have been shown to modulate the electronic environment, optimizing the charge distribution at the active sites. In iron-based SACs supported on nitrogen-doped graphene (Fe–N–C), nitrogen plays a key role in lowering the CO2RR onset potential and widening the potential range for achieving high CO FE. This is due to the enhanced charge capacity of the ˙COOH intermediate, which facilitates selective CO2RR over competing reactions like hydrogen evolution. Additionally, the introduction of defects provides additional catalytic sites, improving reactivity while maintaining the material's structural integrity (Fig. 7).142,143
 | ||
| Fig. 7 Illustration and benchmark of the local embedding for modeling single-atom catalysts, along with the synthesis procedure and morphology characterization: (a) schematic of the synthesis process of CoPc/C3N4/G,142 (b) binding energies of CO on FeN4 predicted using DFT with four functionals (BEEF-vdW, PBE, RPBE+U, and HSE06) and CCSD (T) local embedding. Experimental TPD measurements of the CO binding energy are 0.93–1.18 eV and 1.04 eV,143 (c) schematic showing fragment selection in local embedding calculations (left) and electron density of the FeN4C10-embedded fragment (right), (d–f) SEM, TEM, and HR-TEM images of CoPc/C3N4/G.143 Reproduced from ref. 142 and 143, with permission of ACS Catalysis, copyright 2024. | ||
Another significant aspect of graphene-based SACs is the tailored design of the interfacial microenvironment (Fig. 7). For instance, in CoPc catalysts supported on a graphitic carbon nitride/graphene (C3N4/G) matrix, the graphene matrix enriches CO2 and dissociates H2O to produce active hydrogen species, further enhancing the catalytic performance. The interaction between C3N4 and graphene also optimizes the electronic properties of the active sites, leading to a higher turnover frequency (TOF) and exceptional CO selectivity, with a FE consistently above 98%.144 These findings highlight the importance of both structural and compositional engineering in graphene-based SACs, demonstrating that the manipulation of defects, doping, and microenvironment tuning are essential for improving the CO2RR performance.
Thus, the combination of hierarchical graphene structures, defects, heteroatom doping, and interfacial microenvironment design results in SACs with enhanced catalytic stability, product selectivity, and CO2RR efficiency. These strategies are crucial for advancing the development of efficient and high-performance catalysts for CO2 conversion.
5 Thermocatalytic CO2 reduction based on Fischer–Tropsch synthesis
5.1 Reaction kinetics and mechanism of Fischer–Tropsch synthesis
The FTS mechanism for CO2 hydrogenation on SACs involves several key stages: adsorption of CO2, activation via hydrogenation, and product formation through well-defined pathways. The reaction starts with the adsorption of CO2, which is then hydrogenated to form active intermediates such as ˙CO, methanol, or methane. This process typically follows a surface adsorption–reaction–desorption cycle. Temperature plays a crucial role in these reactions; FTS generally occurs at elevated temperatures (300–500 °C), where the reaction heavily relies on thermal energy to overcome activation barriers. CO2 hydrogenation to olefins and other FTS products is an exothermic reaction, with low temperatures favoring product formation, while higher temperatures enhance the reaction kinetics. In practice, unreacted syngas is often recycled to optimize conversion. High pressure conditions also favor olefin formation. The reaction equations of CO2 hydrogenation to alkane, olefin and alcohol are shown in the following formulas:| CO2 + H2 ↔ CO + H2O |
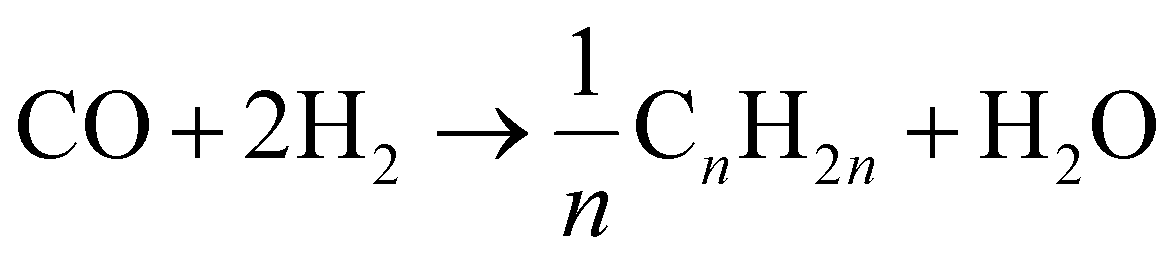


The reaction rates in FTS follow the Arrhenius equation, where the rate is exponentially dependent on temperature:

Here, rFTS is the reaction rate, A is the pre-exponential factor, Ea is the activation energy, R is the gas constant, and T is the absolute temperature. The reaction rate also depends on the concentrations of CO2 and H2, with the dependence often described by a power-law expression:
| rFTS = kFTS·(CCO2)aFTS·(CH2)bFTS |
where kFTS is the rate constant, and aFTS and bFTS are the reaction orders with respect to CO2 and H2, respectively. Higher reactant concentrations typically increase the reaction rate, particularly at elevated temperatures.
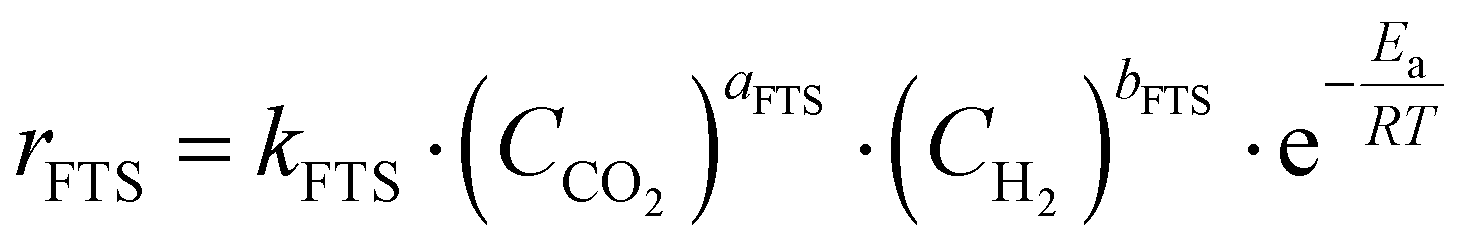
At lower temperatures, the reaction is often limited by electron transfer and hydrogenation steps. At higher temperatures, the rate is more influenced by the activation energy. Temperature adjustments allow for selective production of the desired products: lower temperatures favor methane, while higher temperatures promote olefin and alcohol formation.
The reaction mechanism varies depending on the catalyst and temperature. For Fe-based catalysts, the active phase has shifted from metallic iron to the FeCx phase in recent studies,145 following the Mars-van Krevelen (MvK) mechanism. This mechanism divides the reaction into the surface and outer layers of the catalyst, with fast reactions following the Langmuir–Hinshelwood (L–H) pathway, where most of the FTS activity occurs at a small fraction of active sites. The slow pathway, governed by the MvK mechanism, occurs predominantly at the majority of active sites.146
Brübach et al.147 explored the kinetics of CO2 hydrogenation over Fe-based catalysts, identifying key inhibitory factors such as the strong adsorption of oxygen-containing species and the effects of shifting the equilibrium to lower CO partial pressures. They proposed new kinetic expressions based on the L–H model, which incorporate these dynamics.


Product distribution in CO2 hydrogenation typically follows the Anderson–Schulz–Flory (ASF) model:
| Wn = n(1 + α)2αn−1 |
The integration of reaction kinetics models with ML and DL is an emerging direction for uncovering unknown mechanisms. Sun et al.148 employed a combined approach of artificial neural networks (ANN) and response surface methodology (RSM) to simulate the product distribution of FTS in a microchannel reactor. The model demonstrated the ability to predict the formation rates of hydrocarbon products in the C2–C15 range, as well as the olefin-to-paraffin ratio (OPR), showing advantages in rapid convergence and high predictive accuracy. This approach offers a novel perspective for studying the kinetics of complex catalytic processes. Wang et al.149 developed a radial basis function neural network (RBFNN), which offers fast convergence and reduced computation time, to generate data matrices with a limited set of experimental data. A comprehensive kinetic model, coupled with a genetic algorithm (CKGA), was used to select mechanisms and infer potential reaction pathways. The RSM-based RBFNN, constructed via central composite design, processed responses and subsequent singular/multiple optimizations to produce the data matrix. This approach provides a highly practical and valuable tool for process engineering design and practice, especially for understanding product distribution during FTS (Fig. 8).
 | ||
| Fig. 8 Machine learning assists in unveiling the reaction kinetics process: (a) integrating machine learning models, (b) schematic of the genetic algorithm (GA) for mechanism discrimination and data regression (CLD refers to chain-length dependent), (c) schematic of RSM for significance analysis (NPR refers to a normal plot of residues, RVP refers to residues versus predictions, BCP refers to the Box-Cox plot for power transform, and CD refers to Cook's distance), (d) schematic of RBFNN formulated from the Gaussian function. Reproduced from ref. 149, with permission of ACS OMEGA, copyright 2021. | ||
5.2 Single-atom catalysts in CO2 conversion via Fischer–Tropsch synthesis
The synthesis of SACs for CO2 hydrogenation typically involves the reduction of metal precursors to isolated metal atoms, which are subsequently dispersed onto supports through thermal or chemical reduction. The catalytic performance of SACs is strongly influenced by strong metal–support interactions (SMSI), which stabilize metal atoms and facilitate the activation of CO2 and H2. These interactions play a crucial role in determining the reaction pathway and product selectivity. Notably, the metal's d-band structure modulates the adsorption and activation of CO, ultimately affecting the product distribution. For instance, Co1/W6S8 favors the formation of C2+ hydrocarbons,150 whereas Ni1/W6S8 exhibits higher selectivity toward methanol, highlighting the critical role of metal–support interactions in controlling the reactivity and selectivity.Iron-based SACs, particularly those supported on CeO2, further exemplify the influence of support interactions and preparation conditions on catalytic performance. Wang et al.151 demonstrated that iron species can be effectively dispersed on CeO2via the deposition–precipitation method, forming Feδ+ single atoms under CO2 hydrogenation conditions (300 °C, 2 MPa). These single atoms are stabilized through Fe–O–Ce bonds, which play a key role in CO2 activation. Additionally, co-doping with Ru enhances the iron dispersion and prevents metal aggregation under reaction conditions, thereby stabilizing the catalyst and modulating the electronic environment of the active sites.
The CO2 hydrogenation reaction on SACs proceeds through the adsorption of CO2 and H2 onto metal atoms, leading to protonation and electron transfer, forming intermediates such as ˙CO and ˙HCOOH. The binding strength of CO2 and its intermediates is a critical factor in determining the reaction mechanism and product distribution. Metal–support interactions, particularly those involving reducible oxides such as CeO2, facilitate electron transfer and optimize the binding energies of the reaction intermediates, thereby influencing the catalytic selectivity.
In conclusion, the rational design of SACs for CO2 hydrogenation necessitates precise control over metal dispersion, oxidation state, and metal–support interactions. Co-doping strategies, such as incorporating Ru, improve metal dispersion and enhance the stability of active sites under reaction conditions. The optimization of metal–support interactions, particularly by tuning the metal's d-band structure, is essential for achieving superior catalytic activity and selectivity.
6 Advanced strategies for single-atom catalysts innovation in CO2 reduction
Recent advancements in SACs have demonstrated their significant potential in enhancing CO2RR, offering exceptional atomic efficiency and tunable catalytic sites. By maximizing atomic utilization, SACs ensure that each active site corresponds to a single metal atom, thereby substantially enhancing catalytic efficiency compared to conventional nanoparticle-based catalysts. This atomic-level precision, combined with the ability to finely tune the electronic structure and coordination environment of active sites, facilitates efficient CO2 activation and reduction. SACs have been shown to catalyze the formation of key small organic molecules, such as formate, carbon monoxide, and methane, through mechanisms involving CO2 adsorption, electron transfer, and bond cleavage.Despite these promising advancements, a comprehensive mechanistic understanding of SAC-mediated CO2RR remains incomplete. Current knowledge of precise reaction pathways, the identification of key intermediates, and the role of atomic coordination in determining catalytic activity remains limited. Gaining deeper insights into these mechanistic aspects is crucial for optimizing SACs for large-scale applications. A fundamental understanding of the reaction mechanisms will enable the rational design of SACs with enhanced stability, selectivity, and efficiency-critical attributes for achieving sustainable CO2RR and contributing to global carbon neutrality objectives.
Furthermore, SACs represent a paradigm shift from conventional catalysts, which typically consist of supported metal or metal oxide nanoparticles. The unique atomic-scale structure of SACs facilitates novel reaction pathways that differ significantly from those of traditional catalysts. These distinct reaction dynamics are crucial for advancing CO2RR, enabling more efficient conversion and improved product selectivity.
This integrated framework, as shown in Fig. 9, enables the precise design and optimization of SACs, paving the way for scalable and efficient CO2 conversion.
 | ||
| Fig. 9 Holistic iterative optimization framework for CO2 reduction using single-atom catalysts: integrating advanced machine learning, deep learning, DFT, reaction kinetics and thermodynamic models, high-throughput experimental feedback, and Bayesian optimization for the rational design and enhancement of catalytic performance and mechanistic insights. | ||
The performance of SACs is fundamentally governed by their structural design, which directly impacts the catalytic activity and selectivity. This review introduces an innovative approach for CO2RR, where experimental data guide the selection of reaction conditions, preparation methods of SACs, and key microstructural features. These factors serve as critical parameters in ML models and DFT calculations. By developing mechanistic models based on reaction kinetics and thermodynamics, we aim to iteratively refine active learning models, self-supervised learning frameworks, and large-scale predictive models. In combination with multimodal models, high-throughput experimental data and material characterization results are used to uncover the relationship between the catalyst structure, catalytic properties, reaction pathways, and intermediate selectivity. These efforts reveal previously unexplored CO2RR mechanisms, providing valuable insights into the rational design of SACs for the highly selective production of target chemicals. Furthermore, by applying Bayesian optimization principles, we propose a strategy to enhance the experimental outcomes, enabling continuous iteration between model predictions and experimental validation.
6.1 Optimization of catalyst synthesis in structure and conditions
The catalytic hydrogenation of CO2 encompasses multiple pathways, including thermocatalytic, electrocatalytic, photocatalytic, and photo-electrocatalytic reduction. Among these, thermocatalysis has reached a relatively advanced stage of industrial application, while electrocatalytic and photocatalytic systems remain largely confined to laboratory research.Photocatalysis, inspired by natural photosynthesis, provides an alternative approach for CO2RR, offering advantages such as low energy consumption and high flexibility. However, photocatalytic systems currently face limitations, including low conversion efficiency and high costs associated with photovoltaic systems. Challenges such as poor interfaces, crystal lattice mismatches, and dangling bonds still limit the performance of photocatalytic and electrocatalytic materials.
Electrocatalysis presents several advantages, including the ability to operate under mild reaction conditions (ambient temperature and pressure) and the flexibility to modulate intermediate species. These features make electrocatalysis a promising option for scalable CO2RR. However, high costs associated with electrocatalytic systems, particularly expensive materials used in catalysts and electrodes, pose significant barriers to commercialization.
Future research should focus on developing advanced materials to address the fundamental limitations of these systems. For example, low-dimensional semiconducting materials are promising for constructing high-quality interfaces with well-aligned band gaps, facilitating efficient light absorption and charge transfer. The integration of van der Waals heterostructures into photoelectrodes can enhance exciton dissociation, enabling more efficient electron and hole transfer to the catalyst's active sites, thereby improving the overall catalytic performance. Optimizing reaction conditions—such as the temperature, pressure, and CO/H2 ratio—will be crucial for enhancing the catalytic efficiency of SACs. Particular attention should be paid to the design of new support materials, such as perovskite-based structures, to regulate metal–support interactions. These innovations will enable better control over the electronic properties and coordination environments of SACs, improving the selectivity and efficiency of CO2 reduction.
Overall, while SACs have shown significant promise in CO2 hydrogenation and other critical reactions, addressing the remaining challenges related to the cost, reaction conditions, and mechanistic understanding is essential for large-scale implementation of SACs in sustainable CO2 reduction. Continued improvement in both the fundamental understanding of SACs and the development of new materials will help achieve more efficient, cost-effective, and scalable solutions for CO2 utilization, advancing scientific innovation and global sustainability goals.
6.2 Machine learning-driven design and pathways of single-atom catalysts for CO2 reduction
The use of SACs for CO2 reduction into valuable chemicals has attracted significant attention due to their potential to reduce carbon emissions and store renewable energy. Achieving high selectivity and efficiency in CO2RR necessitates a comprehensive understanding of the reaction mechanisms, active sites, and intermediate species involved. By integrating ML with DFT calculations, this approach has emerged as a powerful tool for elucidating the reaction mechanisms, optimizing the catalyst design, and predicting reaction pathways.ML techniques, including DL, active learning, and self-supervised learning, facilitate the prediction of key catalytic properties, such as intermediate adsorption energies, binding affinities, and the electronic structures of SACs. Analyzing large datasets enables ML models to uncover correlations between the catalyst composition, support materials, and catalytic performance, offering valuable insights into designing highly selective SACs for specific high-value CO2RR products. Notably, ML can guide the design of SACs with tailored properties, such as optimized metal–support interactions and atomic arrangements, thereby improving the reaction pathways and catalytic efficiency.
Integrating ML models with multi-modal data, such as structural and operational information derived from techniques like electron microscopy, further advances the comprehension of catalytic systems. This integration aids in designing SACs with specific properties that optimize the reaction mechanisms and enhance the catalytic performance. For instance, in Cu-based SACs, ML models have been employed to control the morphology of metal nanoparticles, which, in turn, influences the reaction mechanism and product distribution, thereby improving the CO2RR performance.
Despite the promise of ML in catalyst design, several challenges remain. A major hurdle is the lack of high-quality, annotated datasets for effective ML model training. Data scarcity and experimental noise often lead to inaccurate predictions, hindering the development of optimal catalysts. Furthermore, ML models struggle to capture the full complexity of catalytic systems, especially non-equilibrium effects that influence the SACs stability and reactivity. To overcome these challenges, future research should focus on expanding and refining datasets through high-throughput experimental techniques and advanced simulations. Hybrid approaches combining data-driven ML methods with first-principles calculations, such as ML-DFT, could further enhance the prediction accuracy and reliability. Another limitation of the current ML models is their limited interpretability. While ML can accurately predict the catalytic properties, understanding the underlying physical mechanisms remains a significant challenge. Enhancing the transparency of ML models through explainable AI techniques is crucial for gaining deeper insights into the catalytic processes and guiding the rational design of SACs with improved performance.
Coupling ML with operando techniques, such as X-ray spectroscopy and electron microscopy, facilitates the real-time monitoring of active sites and structural dynamics during reactions. This real-time analysis provides valuable insights into the formation of reaction intermediates and the evolution of active sites throughout the CO2RR process, offering a deeper understanding of the catalytic behavior of SACs.
While ML models hold great promise for advancing the design and optimization of SACs for CO2RR, addressing challenges related to the data quality, model interpretability, and system complexity is essential for realizing their full potential. Integrating ML with high-throughput experimentation, advanced simulations, and operando techniques will pave the way for the development of more efficient and selective SACs, ultimately advancing CO2RR technologies and contributing to global sustainability goals.
6.3 Synergistic optimization of catalysts via machine learning and experimental validation
Developing SACs for CO2RR requires integrating advanced ML, DFT, and experimental validation to enhance the catalyst performance and optimize the reaction pathways. The synergy between predictive models and experimental data provides deeper mechanistic insights, guiding the rational design of SACs with high selectivity and efficiency.To elucidate the reaction mechanisms of SACs and identify the active phases in CO2RR, both computational and experimental approaches must be integrated. Computational techniques such as DFT predict the electronic structure of active sites, adsorption energies, and reaction intermediates. By incorporating ML models (e.g., RF, DL, and transformer models), these predictions enable the accurate identification of key catalytic properties and reaction pathways. Optimization of SACs can be achieved by selecting the appropriate descriptors for catalysts, auxiliary agents, and support materials, with a focus on tailoring the spatial structure to enhance the catalytic performance.
Bayesian theory provides a robust framework for constructing thermodynamic and kinetic models based on experimental data, facilitating the prediction of highly selective, efficient, and stable SACs. Integrating these predictions with experimental validation enables iterative model refinement. Incorporating new experimental data enhances the predictive accuracy, facilitating the development of more precise models. This continuous feedback loop between the computational predictions and experimental testing is crucial for uncovering previously unknown reaction pathways and optimizing SACs for CO2RR. Furthermore, by continuously updating models with new data and refining reaction mechanisms, optimal reaction conditions for specific products can be predicted. The iterative optimization process enables tailored SACs design, adjusting the catalyst structures and reaction environments based on the desired product distribution.
The dynamic interplay between the ML predictions, DFT simulations, and experimental validation accelerates SACs development, ensuring more efficient and selective CO2RR. The continuous refinement of computational models and experimental methodologies deepens mechanistic understanding, facilitates the design of highly selective SACs, and optimizes reaction conditions, ultimately advancing scalable catalytic applications.
7 Conclusion
SACs have shown great potential in CO2RR due to atomically dispersed active sites, which allow for tunable catalytic properties. However, further development is necessary to fully understand the reaction mechanisms, optimize conditions, and scale up the catalysts. The integration of DFT, ML, and experimental validation holds immense promise for designing next-generation SACs with improved efficiency, selectivity, and stability.1. The synergy between DFT and ML has greatly advanced the SACs design for CO2RR. DFT provides insights into the electronic structure and reaction intermediates, while ML optimizes the catalyst properties, predicts the reaction pathways, and tailors the SACs functionalities to enhance both efficiency and selectivity.
2. Iterative ML refinement through continuous experimental validation creates a dynamic feedback loop, progressively improving SACs. Integrating experimental data enhances the prediction accuracy, enabling the design of SACs with superior stability and selectivity for specific products.
3. Achieving deeper mechanistic understanding requires precise identification of active sites. Combining ML, DFT, and experimental techniques helps elucidate complex reaction pathways, optimizing the SACs coordination environments and improving selectivity.
4. The SACs design must account for the different reaction conditions, such as temperature, pressure, and CO/H2 ratios. Fine-tuning these parameters alongside well-designed SACs enhances the catalytic performance and stability under practical conditions.
5. Multimodal modeling, combining DFT structural descriptors with experimental data, allows for precise control over the SACs active sites and configurations, facilitating the development of highly active and scalable catalysts for industrial CO2RR applications.
These advancements are crucial for realizing practical CO2RR technologies and achieving global sustainability goals.
Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We ackowledge the financial support from China NSFC No.22276026 received by Prof. Lifen Liu.References
- T. J. Wang, W. S. Fang, Y. M. Liu, F. M. Li, P. Chen and Y. Chen, Heterostructured Pd/PdO nanowires for selective and efficient CO2 electroreduction to CO, Energy Chem., 2022, 70, 407–413 CAS.
- W. Wang, Z. Ma, X. Fei, X. Wang, Z. Yang, Y. Wang, J. Q. Zhang, H. Ning, N. Tsubaki and M. B. Wu, Joint tuning the morphology and oxygen vacancy of Cu2O by ionic liquid enables high-efficient CO2 reduction to C2 products, Chem. Eng. J., 2022, 436, 135029 Search PubMed.
- W. Wang, X. Wang, Z. Ma, Y. Wang, Z. Yang, J. Zhu, L. Lv, H. Ning, N. Tsubaki and M. Wu, Carburized In2O3 Nanorods Endow CO2 Electroreduction to Formate at 1 A cm−2, ACS Catal., 2022, 13, 796–802 Search PubMed.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman and J. Feijóo, et al., Operando studies reveal active Cu nanograins for CO2 electroreduction, Nature, 2023, 614, 262–269 Search PubMed.
- Z. Feng, X. Zhu, J. Yang, K. Zhong, Z. Jiang, Q. Yu, Y. Song, Y. Hua, H. Li and H. Xu, Inherent Facet-Dominant effect for cobalt oxide nanosheets to enhance photocatalytic CO2 reduction, Appl. Surf. Sci., 2022, 578, 151848 CrossRef CAS.
- R. Murakami, K. Shiota, A. Uchida and F. Inagaki, Light-swing CO2 capture: photoirradiation-based chemical CO2 release based on photoisomerization of azobenzene-amine/guanidine derivatives, Green Chem., 2024, 26, 7406 CAS.
- S. S. Satter, J. S. Lopez, M. L. Hubbard, Y. Jiang, R. A. Dagle and J. Kothandaraman, Reactive direct air capture of CO2 to C-C coupled products using multifunctional materials, Green Chem., 2024, 26, 8242–8255 CAS.
- J. X. Gu, X. Zhao, Y. Sun, J. Zhou, C. Y. Sun, X. L. Wang, Z. H. Kang and Z. M. Su, A photo-activated process cascaded electrocatalysis for the highly efficient CO2 reduction over a core–shell ZIF-8@Co/C, J. Mater. Chem. A, 2020, 8, 16616–16623 CAS.
- L. Ye, Y. Ying, D. Sun, Z. Zhang, L. Fei, Z. Wen, J. Qiao and H. Huang, Highly Efficient Porous Carbon Electrocatalyst with Controllable N–Species Content for Selective CO2 Reduction, Angew. Chem., Int. Ed., 2020, 59, 3244–3251 Search PubMed.
- C. X. Zhao, J. N. Liu, B. Q. Li, D. Ren, X. Chen, J. Yu and Q. Zhang, Multiscale Construction of Bifunctional Electrocatalysts for Long-Lifespan Rechargeable Zinc-Air Batteries, Adv. Funct. Mater., 2020, 36, 2003619 Search PubMed.
- Y. Zhou, Z. Wang, L. Huang, S. Zaman, K. Lei, T. Yue, Z. Li, B. You and B. Y. Xia, Engineering 2D Photocatalysts toward Carbon Dioxide Reduction, Adv. Energy Mater., 2021, 11, 2003159 CAS.
- O. S. Bushuyev, P. D. Luna, C. T. Dinh, J. Lagemaat, S. O. Kelley and E. H. Sargent, What Should We Make with CO2 and How Can We Make It?, Joule, 2018, 2, 825–832 CAS.
- J. M. Spurgeon and B. A. Kumar, comparative technoeconomic analysis of pathways for commercial electrochemical CO2 reduction to liquid products, Energy Environ. Sci., 2018, 11, 1536–1551 CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 CAS.
- C. Wang, Z. Sun, Y. Zheng and Y. H. Hu, Recent progress in visible light photocatalytic conversion of carbon dioxide, J. Mater. Chem. A, 2019, 7, 865–887 CAS.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Photoelectrochemical devices for solar water splitting-materials and challenges, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Low-dimensional catalysts for hydrogen evolution and CO2 reduction, Nat. Rev. Chem., 2018, 2, 0105 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Y. Xiong, W. Sun, Y. Han, P. Xin, X. Zheng, W. Yan, J. Dong, J. Zhang, D. Wang and Y. Li, Cobalt single atom site catalysts with ultrahigh metal loading for enhanced aerobic oxidation of ethylbenzene, Nano Res., 2021, 14, 2418–2423 CrossRef CAS.
- Y. Xiong, W. Sun, P. Xin, W. Chen, X. Zheng, W. Yan, L. Zheng, J. Dong, J. Zhang, D. Wang and Y. Li, Gram-Scale Synthesis of High-Loading Single-Atomic-Site Fe Catalysts for Effective Epoxidation of Styrene, Adv. Mater., 2020, 32, 2000896 CrossRef CAS PubMed.
- B. H. Lee, S. Park and M. Kim, et al., Reversible and cooperative photoactivation of single-atom Cu/TiO2 photocatalysts, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed.
- J. H. Wang, M. S. Yin, Q. Zhang, F. Cao, Y. Xing, Q. Zhao, Y. Wang, W. Xu, W. Wu and M. Wu, Single-Atom Fe-N4 sites promote the triplet-energy transfer process of g-C3N4 for the photooxidation, J. Catal., 2021, 404, 89–95 CrossRef CAS.
- H. Ou, D. Wang and Y. Li, How to select effective electrocatalysts: Nano or single atom?, Nano Select, 2021, 2, 492–511 CAS.
- Q. N. Zhan, T. Y. Shuai, H. M. Xu, C. J. Huang, Z. J. Zhang and G. R. Li, Syntheses and applications of single-atom catalysts for electrochemical energy conversion reactions, Chin. J. Catal., 2023, 47, 32–66 CrossRef CAS.
- Q. Sun, C. Jia, Y. Zhao and C. Zhao, Single atom-based catalysts for electrochemical CO2 reduction, Chin. J. Catal., 2022, 43, 1547–1597 CrossRef CAS.
- L. Wang, S. Lyu and S. Li, In situ observation of structural evolution of single-atom catalysts: From synthesis to catalysis, ChemPhysMater, 2024, 3, 24–35 CrossRef.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C. T. Dinh, P. D. Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Accelerated discovery of CO2 electrocatalysts using active machine learning, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P. C. Chen, H. Wang, C. J. Pollock, X. Huang, Y. T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Operando studies reveal active Cu nanograins for CO2 electroreduction, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- W. Fang, W. Guo, R. Lu, Y. Yan, X. Liu, D. Wu, F. M. Li, Y. Zhou, C. He, C. Xia, H. Niu, S. Wang, Y. Liu, Y. Mao, C. Zhang, B. You, Y. Pang, L. Duan, X. Yang, F. Song, T. Zhai, G. Wang, X. Guo, B. Tan, T. Yao, Z. Wang and B. Y. Xia, Durable CO2 conversion in the proton-exchange membrane system, Nature, 2024, 626, 86–91 CrossRef CAS PubMed.
- Y. Xiao, H. T. Zhang and M. T. Zhang, Heterobimetallic NiFe Complex for Photocatalytic CO2 Reduction: United Efforts of NiFe Dual Sites, J. Am. Chem. Soc., 2024, 146, 28832–28844 CrossRef CAS PubMed.
- Z. Wu, J. Liu, B. Mu, X. Xu, W. Sheng, W. Tao and Z. Li, Machine-Learning assisted screening of double metal catalysts for CO2 electroreduction to CH4, Appl. Surf. Sci., 2024, 648, 159027 CrossRef CAS.
- Y. Li, S. Wang, Z. Lv, Z. Wang, Y. Zhao, Y. Xie, Y. Xu, L. Qian, Y. Yang, Z. Zhao and J. Zhang, Transforming the synthesis of carbon nanotubes with machine learning models and automation, Matter, 2025, 8, 101913 CrossRef CAS.
- V. Sumaria, T. B. Rawal, Y. F. Li, D. Sommer, J. Vikoren, R. J. Bondi, M. Rupp, A. Prasad and D. Prasad, Machine Learning, Density Functional Theory, and Experiments to Understand the Photocatalytic Reduction of CO2 on CuPt/TiO2, J. Phys. Chem. C, 2024, 128, 14247–14258 CrossRef CAS.
- J. Liu, Y. Cai, R. Song, S. Ding, Z. Lyu, Y.-C. Chang, H. Tian, X. Zhang, D. Du, W. Zhu, Y. Zhou and Y. Lin, Recent progress on single-atom catalysts for CO2 electroreduction, Mater. Today, 2021, 48, 95–114 CrossRef CAS.
- X. F. Ziang Shang, G. Chen, R. Qin and Y. Han, Recent Advances on Single-Atom Catalysts for Photocatalytic CO2 Reduction, Small, 2023, 19, 2304975 CrossRef PubMed.
- W. Hu, H. Yang and C. Wang, Progress in photocatalytic CO2 reduction based on single-atom catalysts, RSC Adv., 2023, 13, 20889–20908 RSC.
- J. Lin, B. Qiao, J. Liu, Y. Huang, A. Wang, L. Li, W. Zhang, L. F. Allard, X. Wang and T. Zhang, Design of a Highly Active Ir/Fe(OH)x Catalyst: Versatile Application of Pt-Group Metals for the Preferential Oxidation of Carbon Monoxide, Angew. Chem., Int. Ed., 2012, 51, 2920–2924 CAS.
- B. J. O'Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, Catalyst Design with Atomic Layer Deposition, ACS Catal., 2015, 5, 1804–1825 Search PubMed.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng and D. Geng, et al., Single-atom catalysis using Pt/graphene achieved through atomic layer deposition, Sci. Rep., 2013, 3, 1775 CrossRef.
- L. Zhang, Z. J. Zhao and J. Gong, Nanostructured Materials for Heterogeneous Electrocatalytic CO2 Reduction and their Related Reaction Mechanisms, Angew. Chem., Int. Ed., 2017, 56, 11326 CrossRef CAS PubMed.
- T. Ouyang, H. H. Huang, J. W. Wang, D. C. Zhong and T. B. Lu, A Dinuclear Cobalt Cryptate as a Homogeneous Photocatalyst for Highly Selective and Efficient Visible-Light Driven CO2 Reduction to CO in CH3CN/H2O Solution, Angew. Chem., 2017, 129, 756–761 CrossRef.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Single-atom electrocatalysts, Angew. Chem., Int. Ed., 2017, 56, 13944 CrossRef CAS PubMed.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Defect and Interface Engineering for Aqueous Electrocatalytic CO2 Reduction, Joule, 2018, 12, 2551–2582 CrossRef.
- N. Corbin, J. Zeng, K. Williams and K. Manthiram, Heterogeneous molecular catalysts for electrocatalytic CO2 reduction, Nano Res., 2019, 12, 2093–2125 CrossRef CAS.
- Z. Yang, W. Gao and Q. Jiang, A machine learning scheme for the catalytic activity of alloys with intrinsic descriptors, J. Mater. Chem. A, 2020, 8, 17507–17515 RSC.
- K. Tran and Z. W. Ulissi, Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution, Nat. Catal., 2018, 1, 696–703 CrossRef CAS.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C. T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S. C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Accelerated discovery of CO2 electrocatalysts using active machine learning, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- W. Gao, Y. Chen, B. Li, S. P. Liu, X. Liu and Q. Jiang, Determining the adsorption energies of small molecules with the intrinsic properties of adsorbates and substrates, Nat. Commun., 2020, 11, 1196 CrossRef CAS PubMed.
- X. Ma, Z. Li, L. E. Achenie and H. Xin, Machine-Learning-Augmented Chemisorption Model for CO2 Electroreduction Catalyst Screening, J. Phys. Chem. Lett., 2015, 6, 3528–3533 CrossRef CAS PubMed.
- X. Wan, Z. Zhang, H. Niu, Y. Yin, C. Kuai, J. Wang, C. Shao and Y. Guo, Machine-Learning-Accelerated Catalytic Activity Predictions of Transition Metal Phthalocyanine Dual-Metal-Site Catalysts for CO2 Reduction, J. Phys. Chem. Lett., 2021, 12, 6111–6118 CrossRef CAS PubMed.
- S. Özsoysal, B. Oral and R. Yıldırım, Analysis of photocatalytic CO2 reduction over MOFs using machine learning, J. Mater. Chem. A, 2024, 12, 5748–5759 RSC.
- Q. Yang, L. Zhao, R. Bao, Y. Fan, J. Zhou, D. Rong, H. Zhou and D. Zhang, Interpretable machine learning-assisted advanced exergy optimization for carbon-neutral olefins production, Renewable Sustainable Energy Rev., 2025, 208, 115027 CrossRef CAS.
- H. Sun and J. Y. Liu, Advancing CO2RR with O-Coordinated Single-Atom Nanozymes: A DFT and Machine Learning Exploration, ACS Catal., 2024, 14, 14021–14030 CrossRef CAS.
- L.-H. Mou, J. Du, Y. Li, J. Jiang and L. Chen, Effective Screening Descriptors of Metal-Organic Framework-Supported Single-Atom Catalysts for Electrochemical CO2 Reduction Reactions: A Computational Study, ACS Catal., 2024, 14, 12947–12955 CrossRef CAS.
- R. Ding, J. Chen, Y. Chen, J. Liu, Y. Bando and X. Wang, Unlocking the potential: machine learning applications in electrocatalyst design for electrochemical hydrogen energy transformation, Chem. Soc. Rev., 2024, 53, 11390–11461 RSC.
- M. Sun and B. Huang, Direct Machine Learning Predictions of C3 Pathways, Adv. Energy Mater., 2024, 14, 2400152 CrossRef CAS.
- J. Ock, S. Badrinarayanan, R. Magar, A. Antony and A. B. Farimani, Multimodal language and graph learning of adsorption configuration in catalysis, Nat. Mach. Intell., 2024, 6, 1501–1511 CrossRef.
- M. Ren, X. Guo, S. Zhang and S. Huang, Design of Graphdiyne and Holey Graphyne-Based Single Atom Catalysts for CO2 Reduction With Interpretable Machine Learning, Adv. Funct. Mater., 2023, 33, 2213543 CrossRef CAS.
- X. Bai, Y. Li, Y. Xie, Q. Chen, X. Zhang and J. R. Li, High-throughput screening of CO2 cycloaddition MOF catalyst with an explainable machine learning model, Green Energy Environ., 2025, 10, 132–138 CrossRef CAS.
- L. Wang, H. Chen, L. Yang, J. Li, Y. Li and X. Wang, Single-atom catalysts property prediction via Supervised and Self-Supervised pre-training models, Chem. Eng. J., 2024, 487, 150626 Search PubMed.
- Q. Zhu, Y. Gu, X. Liang, X. Wang and J. Ma, A Machine Learning Model to Predict CO2 Reduction Reactivity and Products Transferred from Metal-Zeolites, ACS Catal., 2022, 12, 12336–12348 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Metal-Doped Nitrogenated Carbon as an Efficient Catalyst for Direct CO2 Electroreduction to CO and Hydrocarbons, Angew. Chem., 2015, 127, 10908–10912 CrossRef.
- R. Guo, C. Guo, Z. Bi, H. Zhang, N. Lv, B. Xi, G. Hu and J. Xu, The single atom Fe loaded catalytic membrane for effective peroxymonosulfate activation and pollution degradation, Appl. Catal., B, 2024, 356, 124243 CAS.
- A. V. Bukhtiyarov, M. A. Panafidin, I. P. Prosvirin, N. S. Smirnova, P. V. Markov, G. N. Baeva, I. S. Mashkovsky, G. O. Bragina, C. Rameshan, E. Yu. Gerasimov, Y. V. Zubavichus, V. I. Bukhtiyarov and A. Yu. Stakheev, Deliberate control of the structure-specific active sites in PdIn bimetallic catalysts using adsorbate induced segregation effects, Appl. Surf. Sci., 2023, 608, 155–086 Search PubMed.
- G. A. Naikoo, F. Arshad, I. U. Hassan, F. B. Omar, M. M. Tambuwala, M. Mustaqeem and T. A. Saleh, Trends in bimetallic nanomaterials and methods for fourth-generation glucose sensors, Trends Anal. Chem., 2023, 162, 117042 CAS.
- S. K. Lahiri, C. Zhang, M. Sillanpää and L. Liu, NiO@SiO2 photo-catalyst prepared by ion-exchange method for fast elimination of reactive dyes from wastewater, Mater. Today Chem., 2022, 23, 100–677 Search PubMed.
- A. Kowalczyk, A. Borcuch, M. Michalik, M. Rutkowska, B. Gil, Z. Sojka, P. Indyka and L. Chmielarz, MCM-41 modified with transition metals by template ion-exchange method as catalysts for selective catalytic oxidation of ammonia to dinitrogen, Microporous Mesoporous Mater., 2017, 240, 9–21 CAS.
- V. Andrei, I. Roh, J. A. Lin, J. Lee, Y. Shan, C. K. Lin, S. Shelton, E. Reisner and P. Yang, Perovskite-driven solar C2 hydrocarbon synthesis from CO2, Nat. Catal., 2025, 137–146 CrossRef CAS.
- K. Maeda, D. An, R. Kuriki, D. Lu and O. Ishitani, Graphitic carbon nitride prepared from urea as a photocatalyst for visible-light carbon dioxide reduction with the aid of a mononuclear ruthenium(II) complex, J. Org. Chem., 2018, 14, 1806–1812 CAS.
- W. Zhang, Z. Jin and Z. Chen, Rational-Designed Principles for Electrochemical and Photoelectrochemical Upgrading of CO2 to Value-Added Chemicals, Adv. Sci., 2022, 9, 2105204 CrossRef CAS PubMed.
- X. Tao, Y. Wang, J. Qu, Y. Zhao, R. Li and C. Li, Achieving selective photocatalytic CO2 reduction to CO on bismuth tantalum oxyhalogen nanoplates, J. Mater. Chem. A, 2021, 9, 19631–19636 Search PubMed.
- X. M. You, B. Xu, H. Zhou, H. Qiao, X. Lv, Z. Huang, J. Pang, L. Yang, P. F. Liu, X. Guan, H. G. Yang, X. Wang and Y. F. Yao, Ultrahigh Bifunctional Photocatalytic CO2 Reduction and H2 Evolution by Synergistic Interaction of Heteroatomic Pt-Ru Dimerization Sites, ACS Nano, 2024, 18, 9403–9412 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Electrochemical photolysis of water at a semiconductor electrode, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. Halmann, Photoelectrochemical reduction of aqueous carbon dioxide on p-type gallium phosphide in liquid junction solar cells, Nature, 1978, 275, 115–116 CrossRef CAS.
- J. Xia, N. Karjule, B. Mondal, J. Qin, M. Volokh, L. Xing and M. Shalom, Design of melem-based supramolecular assemblies for the synthesis of polymeric carbon nitrides with enhanced photocatalytic activity, J. Mater. Chem. A, 2021, 9, 17855–17864 RSC.
- J. Wu, Y. Huang, W. Ye and Y. Li, CO2 Reduction: From the Electrochemical to Photochemical Approach, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed.
- H. Zhang, Y. Wang, S. Zuo, W. Zhou, J. Zhang and X. W. D. Lou, Isolated Cobalt Centers on W18O49 Nanowires Perform as a Reaction Switch for Efficient CO2 Photoreduction, Chem. Soc., 2021, 143, 2173–2177 Search PubMed.
- Y. Li, S. Wang, X. Wang, Y. He, Q. Wang, Y. Li, M. Li, G. Yang, J. Yi, H. Lin, D. Huang, L. Li, H. Chen and J. Ye, Facile top-down strategy for direct metal atomization and coordination achieving a high turnover number in CO2 Photoreduction, J. Am. Chem. Soc., 2020, 142, 19259–19267 Search PubMed.
- C. Feng, T. T. Bo, P. Maity, S. W. Zuo, W. Zhou, K. W. Kuang, O. F. Mohammed and H. Zhang, Regulating Photocatalytic CO2 Reduction Kinetics through Modification of Surface Coordination Sphere, Adv. Funct. Mater., 2024, 34, 2309761 CAS.
- Y. Zhang, B. Johannessen, P. Zhang, J. Gong, J. Ran and S. Z. Qiao, Reversed Electron Transfer in Dual Single Atom Catalyst for Boosted Photoreduction of CO2, Adv. Mater., 2023, 35, 2306923 Search PubMed.
- H. Haroon and Q. Xiang, Single-Atom based Metal-Organic Framework Photocatalysts for Solar-Fuel Generation, Small, 2024, 20, 2401389 CAS.
- Y. Wang, L. Liao, G. Zhu, W. Xie, Q. Zhou, F. Yu, H. Zhou and H. Zhou, Metal-nitrogen coordinated single atomic photocatalysts for solar energy conversion, Coord. Chem. Rev., 2025, 523, 216254 CrossRef CAS.
- Q. Yang, H. Liu, Y. Lin, D. Su, Y. Tang and L. Chen, Atomically Dispersed Metal Catalysts for the Conversion of CO2 into High-Value C2+ Chemicals, Adv. Mater., 2024, 36, 2310912 CrossRef CAS PubMed.
- Y. Zheng, W. Li, J. Ju, J. Jiang, L. Zhang, H. Jiang, Y. Hu and C. Li, Oxygen vacancy mediated Pd-SA/TiO2 single-atom catalyst created via ultra-fast one-step synthesis for enhanced CO2 photoreduction, J. Colloid Interface Sci., 2025, 683, 280–290 CrossRef CAS PubMed.
- B. Rhimi, M. Zhou, Z. Yan, X. Cai and Z. Jiang, Cu-Based Materials for Enhanced C2+ Product Selectivity in Photo-/Electro-Catalytic CO2 Reduction: Challenges and Prospects, Nano-Micro Lett., 2024, 16, 64 CrossRef CAS PubMed.
- Y. Luo, X. Wang, F. Gao, L. Jiang, D. Wang and H. Pan, From Single Atom Photocatalysts to Synergistic Photocatalysts: Design Principles and Applications, Adv. Funct. Mater., 2024, 2418427 CrossRef.
- W. Lyu, Y. Liu, J. Zhou, D. Chen, X. Zhao, R. Fang, F. Wang and Y. Li, Modulating the Reaction Configuration by Breaking the Structural Symmetry of Active Sites for Efficient Photocatalytic Reduction of Low-concentration CO2, Angew. Chem., Int. Ed., 2023, 62, e202310733 CrossRef CAS PubMed.
- Z. W. Huang, K. Q. Hu, X. B. Li, Z. N. Bin, Q. Y. Wu, Z. H. Zhang, Z. J. Guo, W. S. Wu, Z. F. Chai, L. Mei and W. Q. Shi, Thermally Induced Orderly Alignment of Porphyrin Photoactive Motifs in Metal–Organic Frameworks for Boosting Photocatalytic CO2 Reduction, J. Am. Chem. Soc., 2023, 145, 18148–18159 CAS.
- L. Cheng, H. Yin, C. Cai, J. Fan and Q. Xiang, Single Ni Atoms Anchored on Porous Few–Layer g–C3N4 for Photocatalytic CO2 Reduction: The Role of Edge Confinement, Small, 2020, 16, 2002411 CAS.
- P. Zhang, X. Zhan, L. Xu, X. Fu, T. Zheng, X. Yang, Q. Xu, D. Wang, D. Qi, T. Sun and J. Jiang, Mass production of a single-atom cobalt photocatalyst for high-performance visible-light photocatalytic CO2 reduction, J. Mater. Chem. A, 2021, 9, 26286–26297 CAS.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem., 2018, 2, 65–81 CAS.
- P. Zhang, X. Zhan, L. Xu, X. Fu, T. Zheng, X. Yang, Q. Xu, D. Wang, D. Qi, T. Sun and J. Jiang, Mass production of a single-atom cobalt photocatalyst for high-performance visible-light photocatalytic CO2 reduction, J. Mater. Chem. A, 2021, 9, 26286–26297 CAS.
- J. Lee, G. I. N. Waterhouse, Y. Mao, Z. Wang, E. Zhu, J. Low, S. P. Chai and L. L. Tan, Lead-free halide perovskites and Bi-BTC frameworks: Engineering S-scheme heterojunctions for photocatalytic CO2 conversion, Appl. Catal., B, 2025, 365, 124942 CAS.
- J. He, X. Wang, P. Feng, Y. Zhou, K. Wang, B. Zou and M. Zhu, Isostructural phase transition-induced piezoelectricity in all-inorganic perovskite CsPbBr3 for catalytic CO2 reduction, Appl. Catal., B, 2024, 355, 124186 CrossRef CAS.
- Y. Feng, D. Chen, M. Niu, Y. Zhong, Z. He, S. Ma, K. Yuan, H. Ding, K. Lv, L. Guo, W. Zhang and M. Ma, Ligand free synthesis of atomically dispersed Cu doping ultrathin Cs3Bi2Br9 for efficient photoreduction CO2 with high CO selectivity, Appl. Catal., B, 2025, 365, 124931 CrossRef CAS.
- Z. L. Liu, Y. F. Mu, X. R. Li, Y. X. Feng, M. Zhang and T. B. Lu, Constructing strong built-in electric field in lead-free halide-perovskite-based heterojunction to boost charge separation for efficient CO2 photoreduction, Appl. Catal., B, 2025, 366, 125012 CrossRef CAS.
- H. Sun, Z. Lin, R. Tang, Y. Liang, S. Zou, X. Zhang, K. Chen, R. Zheng and J. Huang, Enhanced solar urea synthesis from CO2 and nitrate waste via oxygen vacancy mediated-TiOx support lead-free perovskite, Appl. Catal., B, 2025, 360, 124511 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, A selective and efficient electrocatalyst for carbon dioxide reduction, Nat. Commun., 2014, 5, 32–42 Search PubMed.
- B. Zhang and J. Zhang, Rational design of Cu-based electrocatalysts for electrochemical reduction of carbon dioxide, Energy Chem., 2017, 26, 1050–1066 Search PubMed.
- J. He, N. J. J. Johnson, A. Huang and C. P. Berlinguette, Electrocatalytic Alloys for CO2 Reduction, ChemSusChem, 2018, 11, 48–57 CAS.
- Q. Hao, Q. Tang, H. X. Zhong, J. Z. Wang, D. X. Liu and X. B. Zhang, Fully exposed nickel clusters with electron-rich centers for high-performance electrocatalytic CO2 reduction to CO, Sci. Bull., 2022, 67, 1477–1485 CAS.
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Elucidating the Electrocatalytic CO2 Reduction Reaction over a Model Single–Atom Nickel Catalyst, Angew. Chem., Int. Ed., 2019, 59, 798–803 Search PubMed.
- H. Gu, J. Wu and L. Zhang, Recent advances in the rational design of single-atom catalysts for electrochemical CO2 reduction, Nano Res., 2022, 15, 9747–9763 CAS.
- N. Qiu, J. Li, H. Wang and Z. Zhang, Emerging dual-atomic-site catalysts for electrocatalytic CO2 reduction, Sci. China Mater., 2022, 65, 3302–3323 CAS.
- Y. Li, Z. He, F. Wu, S. Wang, Y. Cheng and S. Jiang, Defect engineering of high-loading single-atom catalysts for electrochemical carbon dioxide reduction, Mater. Rep. Energy, 2023, 3, 100197 CAS.
- D. Gao, T. Liu, G. Wang and X. Bao, Structure Sensitivity in Single-Atom Catalysis toward CO2 Electroreduction, ACS Energy Lett., 2021, 6, 713–727 CAS.
- Y. E. Kim, Y. N. Ko, B. S. An, J. Hong, Y. E. Jeon, H. J. Kim, S. Lee, J. Lee and W. Lee, Atomically Dispersed Nickel Coordinated with Nitrogen on Carbon Nanotubes to Boost Electrochemical CO2 Reduction, Energy Lett., 2023, 8, 3288–3296 CAS.
- Y. Wang, Y. Liu, W. Liu, J. Wu, Q. Li, Q. Feng, Z. Chen, X. Xiong, D. Wang and Y. Lei, Regulating the coordination structure of metal single atoms for efficient electrocatalytic CO2 reduction, Energy Environ. Sci., 2020, 13, 4609–4624 RSC.
- S. Yang, H. An, S. Arnouts, H. Wang, X. Yu, J. de. Ruiter, S. Bals, T. Altantzis, B. M. Weckhuysen and W. V. D. Stam, Halide-guided active site exposure in bismuth electrocatalysts for selective CO2 conversion into formic acid, Nat. Catal., 2023, 6, 796–806 CrossRef CAS.
- J. Feng, L. Zhang, S. Liu, L. Xu, X. Ma, X. Tan, L. Wu, Q. Qian, T. Wu, J. Zhang, X. Sun and B. Han, Modulating adsorbed hydrogen drives electrochemical CO2-to-C2 products, Nat. Commun., 2023, 14, 4615 CrossRef CAS PubMed.
- Z. Jin, M. Yang, Y. Dong, X. Ma, Y. Wang, J. Wu, J. Fan, D. Wang, R. Xi, X. Zhao, T. Xu and J. Zhao, et al., Atomic Dispersed Hetero-Pairs for Enhanced Electrocatalytic CO2 Reduction, Nano-Micro Lett., 2024, 16, 4 CrossRef CAS PubMed.
- Y. Cao, S. Chen, S. Bo, W. Fan, J. Li, C. Jia, Z. Zhou, Q. Liu, L. Zheng and F. Zhang, Single Atom Bi Decorated Copper Alloy Enables C-C Coupling for Electrocatalytic Reduction of CO2 into C2+ Products, Angew. Chem., 2023, 135, e202303048 CrossRef.
- F. Cai, D. Gao, H. Zhou, G. Wang, T. He, H. Gong, S. Miao, F. Yang, J. Wang and X. Bao, Electrochemical promotion of catalysis over Pd nanoparticles for CO2 reduction, Chem. Sci., 2017, 8, 2569–2573 RSC.
- Y. Quan, R. Yu, J. Zhu, A. Guan, X. Lv, C. Yang, S. Li, J. Wu and G. Zheng, Efficient carboxylation of styrene and carbon dioxide by single-atomic copper electrocatalyst, J. Colloid Interface Sci., 2021, 601, 378–384 CrossRef CAS PubMed.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and J. Feng, Electrodeposited Zn Dendrites with Enhanced CO Selectivity for Electrocatalytic CO2 Reduction, ACS Catal., 2015, 5, 4586–4591 CAS.
- Y. Chen and M. W. Kanan, Tin Oxide Dependence of the CO2 Reduction Efficiency on Tin Electrodes and Enhanced Activity for Tin/Tin Oxide Thin-Film Catalysts, J. Am. Chem. Soc., 2012, 134, 1986–1989 CAS.
- H. Mistry, R. Reske, Z. Zeng, Z. J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, Exceptional Size-Dependent Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles, Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, Insight into Electrochemical CO2 Reduction on Surface-Molecule-Mediated Ag Nanoparticles, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- J. M. Spurgeon and B. Kumar, A comparative technoeconomic analysis of pathways for commercial electrochemical CO2 reduction to liquid products, Energy Environ. Sci., 2018, 11, 1536 RSC.
- C. Kim, F. Dionigi, V. Beermann, X. Wang, T. Möller and P. Strasser, Alloy Nanocatalysts for the Electrochemical Oxygen Reduction (ORR) and the Direct Electrochemical Carbon Dioxide Reduction Reaction (CO2RR), Adv. Mater., 2019, 31, 1805617 Search PubMed.
- W. Gao, Y. Xu, L. Fu, X. Chang and B. Xu, Experimental evidence of distinct sites for CO2-to-CO and CO conversion on Cu in the electrochemical CO2 reduction reaction, Nat. Catal., 2023, 6, 885–894 CrossRef CAS.
- Q. Wang, X. Yang, H. Zang, C. Liu, J. Wang, N. Yu, L. Kuai, Q. Qin and B. Geng, InBi Bimetallic Sites for Efficient Electrochemical Reduction of CO2 to HCOOH, Small, 2023, 19, 2303172 CrossRef CAS PubMed.
- A. R. Woldu, A. G. Yohannes, Z. Huang, P. Kennepohl, D. Astruc, L. Hu and X. C. Huang, Experimental and Theoretical Insights into Single Atoms, Dual Atoms, and Sub-Nanocluster Catalysts for Electrochemical CO2 Reduction (CO2RR) to High-Value Products, Adv. Mater., 2024, 36, 2414169 CrossRef CAS PubMed.
- H. Dai, T. Song, X. Yue, S. Wei, F. Li, Y. Xu, S. Shu, Z. Cui, C. Wang, J. Gu and L. Duan, Cu single-atom electrocatalyst on nitrogen-containing graphdiyne for CO2 electroreduction to CH4, Chin. J. Catal., 2024, 64, 123–132 Search PubMed.
- B. Wang, M. Wang, Z. Fan, C. Ma, S. Xi, L. Y. Chang, M. Zhang, N. Ling, Z. Mi, S. Chen, W. R. Leow, J. Zhang, D. Wang and Y. Lum, Nanocurvature-induced field effects enable control over the activity of single-atom electrocatalysts, Nat. Commun., 2024, 15, 1719 Search PubMed.
- R. Purbia, S. Y. Choi, C. H. Woo, J. Jeon, C. Lim, D. K. Lee, J. Y. Choi, H.-S. Oh and J. M. Baik, Highly selective and low-overpotential electrocatalytic CO2 reduction to ethanol by Cu-single atoms decorated N-doped carbon dots, Appl. Catal., B, 2024, 345, 123694 CrossRef CAS.
- S. Cui, C. Yu, X. Tan, W. Li, Y. Zhang and J. Qiu, A tandem catalyst with high CO2 capture capability to achieve a promoted CO2-to-CH4 electrochemical conversion, Chem. Eng. J., 2023, 470, 144083 CrossRef CAS.
- J. Ding, B. H. Yang, X. L. Ma, S. Liu, W. Liu, Q. Mao, Y. Huang, J. Li, T. Zhang and B. Liu, A tin-based tandem electrocatalyst for CO2 reduction to ethanol with 80% selectivity, Nat. Energy, 2023, 8, 1386–1394 CrossRef CAS.
- Y. Wang, S. L. Marquard, D. Wang, C. Dares and T. J. Meyer, Single-Site, Heterogeneous Electrocatalytic Reduction of CO2 in water as the Solvent, ACS Energy Lett., 2017, 2, 1395–1399 CrossRef CAS.
- S. Baskaran and Y. Jung, Mo2CS2-MXene supported single-atom catalysts for efficient and selective CO2 electrochemical reduction, Appl. Surf. Sci., 2022, 592, 153339 CrossRef CAS.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.C. Cheong, L. Zheng, F. Ren and G. Ying, et al., MXene (Ti3C2) vacancy-confined single-atom catalyst for efficient functionalization of CO2, J. Am. Chem. Soc., 2019, 141, 4086–4093 Search PubMed.
- W. Liu, H. Li, P. Ou, J. Mao, L. Han, J. Song, J. Luo and H. L. Xin, Isolated Cu-Sn diatomic sites for enhanced electroreduction of CO2 to CO, Nano Res., 2023, 16, 8729–8736 CrossRef CAS.
- L. Zhang, J. Feng, L. Wu, X. Ma, X. Song, S. Jia, X. Tan, X. Jin, Q. Zhu, X. Kang and J. Ma, et al., Oxophilicity-controlled CO2 electroreduction to C2+ alcohols over Lewis acid metal-doped Cuδ+ catalysts, J. Am. Chem. Soc., 2023, 145, 21945–21954 CrossRef CAS PubMed.
- X. Su, Y. Sun, L. Jin, L. Zhang, Y. Yang, P. Kerns, B. Liu, S. Li and J. He, Hierarchically porous Cu/Zn bimetallic catalysts for highly selective CO2 electroreduction to liquid C2 products, Appl. Catal., B, 2020, 269, 118800 CrossRef CAS.
- G. Chen, J. Fu, B. Liu, C. Cai, H. Li, Z. Zhang, K. Liu, Z. Lin and M. Liu, Passivation of Cu nanosheet dissolution with Cu2+-containing electrolytes for selective electroreduction of CO2 to CH4, Environ. Sci.:Nano, 2022, 9, 3312–3317 RSC.
- Z. Xu, Y. Xie and Y. Wang, Pause electrolysis for acidic CO2 reduction on 3-dimensional Cu, Mater. Rep. Energy, 2023, 3, 100173 CAS.
- M. Fan, J. E. Huang, R. K. Miao, Y. Mao, P. Ou, F. Li, X. Y. Li, Y. Cao, Z. Zhang and J. Zhang, et al., Cationic-group-functionalized electrocatalysts enable stable acidic CO2 electrolysis, Nat. Catal., 2023, 6, 763–772 CrossRef CAS.
- S. Cao, H. Chen, X. Wei, J. Li, C. Yang, Z. Chen, S. Wei, S. Liu, Z. Wang and X. Lu, Asymmetric coordination engineering of active centers in MXene-based single atom catalysts for high-performance ECO2RR, Carbon, 2024, 225, 119094 CrossRef CAS.
- S. Chen, T. Luo, X. Li, K. Chen, Q. Wang, J. Fu, K. Liu, C. Ma, Y.-R. Lu, H. Li, K. S. Menghrajani, C. Liu, S. A. Maier, T.-S. Chan and M. Liu, Design of reaction-driven active configuration for enhanced CO2 electroreduction, Nano Energy, 2024, 128, 109873 CrossRef CAS.
- Z. Lv, C. Wang, Y. Liu, R. Liu, F. Zhang, X. Feng, W. Yang and B. Wang, Improving CO2-to-C2 Conversion of Atomic CuFONC Electrocatalysts through F, O-Codrived Optimization of Local Coordination Environment, Adv. Energy Mater., 2024, 14, 2400057 CrossRef CAS.
- S. Yang, H. Wang, Y. Xiong, M. Zhu, J. Sun, M. Jiang, P. Zhang, J. Wei, Y. Xing, Z. Tie and Z. Jin, Ultrafast Thermal Shock Synthesis and Porosity Engineering of 3D Hierarchical Cu–Bi Nanofoam Electrodes for Highly Selective Electrochemical CO2, Nano Lett., 2023, 23, 10140–10147 CrossRef CAS PubMed.
- Z. Wang, J. Yang, Z. Song, M. Lu, W. Wang, Z. Ren and Z. Chen, Reprogramming the Microenvironment of Cobalt Phthalocyanine by a Targeted Multifunctional π-Conjugated Modulator Enables Concerted CO2 Electroreduction, ACS Catal., 2024, 14, 8138–8147 CrossRef CAS.
- J. Lei and T. Zhu, Impact of Potential and Active-Site Environment on Single-Iron-Atom-Catalyzed Electrochemical CO2 Reduction from Accurate Quantum Many-Body Simulations, ACS Catal., 2024, 14, 3933–3942 CAS.
- A. Kumar, M. Ubaidullah, P. V. Pham and R. K. Gupta, Three-dimensional macro-structures of graphene-based materials for emerging heterogeneous electrocatalysis: Concepts, syntheses, principles, applications, and perspectives, Chem. Eng. J., 2024, 499, 156664 CrossRef CAS.
- J. M. Gracia, F. F. Prinsloo and J. W. Niemantsverdriet, Mars-van Krevelen-like Mechanism of CO Hydrogenation on an Iron Carbide Surface, Catal. Lett., 2009, 133, 257–261 CAS.
- J. Chai, R. Pestman, W. Chen, N. Donkervoet, A. I. Dugulan, Z. Men, P. Wang and E. J. M. Hensen, Isotopic Exchange Study on the Kinetics of Fe Carburization and the Mechanism of the Fischer-Tropsch Reaction, ACS Catal., 2022, 12, 2877–2887 CAS.
- L. Brübach, D. Hodonj and P. Pfeifer, Kinetic Analysis of CO2 Hydrogenation to Long-Chain Hydrocarbons on a Supported Iron Catalyst, Ind. Eng. Chem. Res., 2022, 61, 1644–1654 Search PubMed.
- Y. Sun, G. Yang, M. Xu, J. Xu and Z. Sun, A simple coupled ANNs–RSM approach in modeling product distribution of Fischer–Tropsch synthesis using a microchannel reactor with Ru–promoted Co/Al2O3 catalyst, Int. J. Energy Res., 2020, 44, 1046–1061 CAS.
- Y. Wang, J. Hu, X. Zhang, A. Yusuf, B. Qi, H. Jin, Y. Liu, J. He, Y. Wang, G. Yang and Y. Sun, Kinetic Study of Product Distribution Using Various Data-Driven and Statistical Models for Fischer-Tropsch Synthesis, ACS Omega, 2021, 6, 27183–27199 CAS.
- Q. Zhang, L. Guo and Z. Hao, CO hydrogenation on M1/W6S8 (M = Co and Ni) single-atom catalysts: Competition between C2 hydrocarbons and methanol synthesis pathways, Mol. Catal., 2019, 464, 10–21 CAS.
- X. Wang, X. P. Fu, W. Z. Yu, C. Ma, C. J. Jia and R. Si, Synthesis of a ceria-supported iron–ruthenium oxide catalyst and its structural transformation from subnanometer clusters to single atoms during the Fischer–Tropsch synthesis reaction, Inorg. Chem. Front., 2017, 4, 2059–2067 CAS.
| This journal is © The Royal Society of Chemistry 2025 |