DOI:
10.1039/D0RA09041G
(Paper)
RSC Adv., 2021,
11, 203-209
Simple primary β-amino alcohols as organocatalysts for the asymmetric Michael addition of β-keto esters to nitroalkenes†
Received
23rd October 2020
, Accepted 3rd December 2020
First published on 22nd December 2020
Abstract
Simple primary β-amino alcohols act as an efficient organocatalysts in the asymmetric Michael addition of β-keto esters with nitroalkenes affording highly pure chiral Michael adducts. Also, both enantiomers of the adducts were obtained, depending on the specific catalyst used and reaction temperature.
1. Introduction
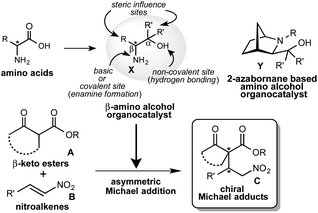
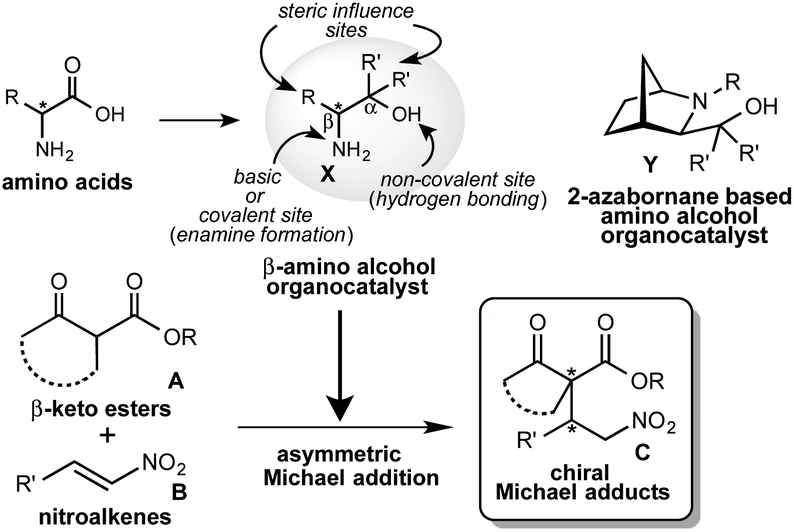
From the last decade, the development of new optically active multifunctional organocatalysts and their use in asymmetric synthesis as independent chiral sources have drawn considerable interest in the scientific community.1 A lot of excellent covalent and non-covalent organocatalysts have been developed for use in a wide range of asymmetric reactions.2 However, it is always a challenging task to design and synthesize a new class of multifunctional organocatalysts and to explore them as self-determining and eco-friendly catalysts in asymmetric synthesis. In recent years, we are continuously exploring the multifunctional β-amino alcohol organocatalysts X (Scheme 1),3 that are easily derived from commercially available amino acids in one or two steps and are less sensitive to air, show low toxicity and are eco-friendly. This amino alcohol X contains an amino group acting as a basic or covalent enamine formation site, a non-covalent hydroxyl group acting as a hydrogen bonding site in a single molecule and also the substituents at α- and β-positions, which might also be effective in controlling the enantioselective reaction course (Scheme 1). In our previous study, this catalyst has worked as an efficient organocatalyst in 1,3-dipolar cycloaddition,4 Diels–Alder reactions5 and aldol reactions.6 Asymmetric Michael addition is recognized as a versatile and powerful key carbon–carbon bond and also a powerful tool for constructing carbon–heteroatom bond providing an effective route for the synthesis of chiral compounds which act as precursor for a range of biologically and pharmaceutically important compounds. Furthermore, this addition is also useful for the construction of chiral building blocks containing quaternary carbon stereocenters, which act as a key synthetic intermediates for the complex compounds and synthetic drug candidates.7 Consequently, several efforts have been made in recent years to develop an efficient organocatalyst for this reaction.8 Our group has reported that the cage type 2-azanorbornane-based amino alcohols Y act as an efficient organocatalyst in this addition.10 However, this catalyst Y has a complex structure and requires a multistep route for the synthesis, although efficient catalytic activity is observed. Based on these backgrounds, we have planned to try the asymmetric Michael addition of β-keto esters A with nitroalkenes B using more simplified β-amino alcohol organocatalyst X (Scheme 1).
 |
| | Scheme 1 Asymmetric Michael addition of β-keto esters with nitroalkenes. | |
In this paper, we describe an efficient catalytic activity displayed by β-amino alcohol organocatalyst catalyst X in the Michael addition of A with B to afford chiral Michael adducts C at satisfactory chemical yields and stereoselectivities (up to 80%, up to dr = 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, up to 99% ee). In addition, an interesting property was observed that both enantiomers of the adducts were obtained depending on used specific catalyst and a reaction temperature with excellent stereoselectivities (up to 99:1, up to 99% ee).
:1, up to 99% ee). In addition, an interesting property was observed that both enantiomers of the adducts were obtained depending on used specific catalyst and a reaction temperature with excellent stereoselectivities (up to 99:1, up to 99% ee).
2. Results and discussion
2.1. Preparation and screening of catalysts
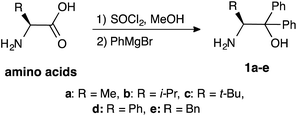
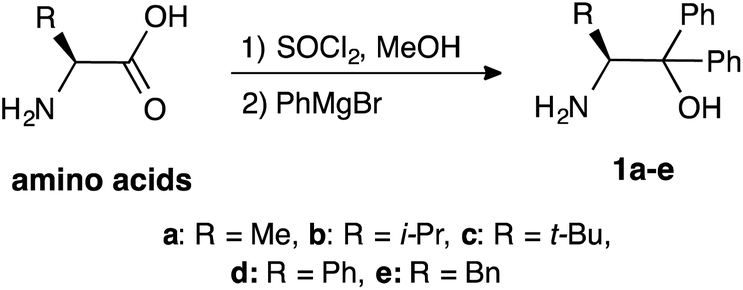
β-Amino alcohol organocatalysts 1a–e with different substituents at the α- and β-position were prepared from the corresponding amino acids (Scheme 2). The reaction of amino acids with thionyl chloride in methanol, followed by Grignard reaction of the obtained methyl esters afforded the desired 1a–e.5a
 |
| | Scheme 2 Preparations of β-amino alcohols 1a–e. | |
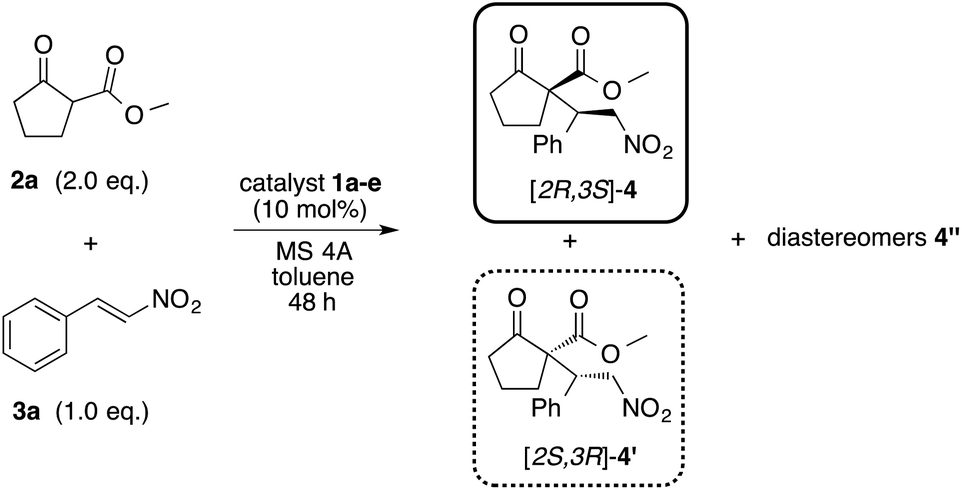
In order to investigate the asymmetric catalytic activity of the obtained amino alcohols 1a–e, model reaction was carried out using methyl-2-oxocyclopentanecarboxylate 2a and nitrostyrene 3a in toluene at 0 °C and −30 °C for 48 h (Table 1) respectively. All catalysts showed a catalytic activity in this reaction (entries 1–5). When the reaction was carried out at 0 °C using catalyst 1a with methyl group at β-position, the desired Michael adduct [2S,3R]-4 was obtained with moderate chemical yield, diastereoselectivity and low enantioselectivity (62%, dr = 83:17, 45% ee) (entry 1). Similar reaction using catalyst 1b with isopropyl group did not show a much change in the chemical yield and diastereoselectivity, but enantioselectivity was quite increased (64%, dr = 88:12, 56% ee) (entry 2). Interestingly, the use of catalyst 1c with bulky tert-butyl group afforded the enantiomer adduct [2S,3R]-4′ of 4 in moderate chemical yield, diastereoselectivity and good enantioselectivity (68%, dr = 91:9, 88% ee) (entry 3). In addition, catalyst 1d with more sterically influential phenyl group provided adduct 4 with moderate chemical yield, good diastereoselectivity and low enantioselectivity (65%, dr = 83:17, 11% ee) (entry 4). Furthermore, catalyst 1e with benzyl group also did not show satisfactory enantioselectivity, although chemical yield and diastereoselectivity were moderate (55%, dr = 86:14, 10% ee) (entry 5). On the other hand, the decrease of temperature to −30 °C substantially improved diastereoselectivity and enantioselectivities (entries 1–5). Catalyst 1a afforded adduct 4 with good chemical yield, excellent diastereoselectivity and enantioselectivity (75%, dr = 99:1, 99% ee) (entry 1). Catalyst 1b also afforded 4 with good chemical yield and in excellent diastereoselectivity and enantioselectivity (70%, dr = 96:4, 98% ee) (entry 2). Interestingly, the enantiomer adduct 4′ of 4 was obtained at 0 °C using 1b, but the decrease to −30 °C afforded 4, although the reason is not clear. Similarly, the use of catalyst 1c gave 4′ in good chemical yield and diastereoselectivity with excellent enantioselectivity (80%, dr = 98:2, 99% ee) (entry 3). The use of bulky catalyst 1d significantly increased the enantioselectivity with good chemical yield and diastereoselectivity (65%, dr = 96:4, 98% ee) (entry 4). Bulkier catalyst 1e brought about the increase of enantioselectivity to afford 4′, but satisfactory chemical yield and stereoselectivities were not obtained (50%, dr = 85:15, 60% ee) (entry 5). However, similarly to the adduct from catalyst 1b, the use of catalyst 1e also afforded the enantiomer adduct 4′ of 4, although the reason is not clear. Each catalyst showed satisfactory asymmetric catalytic activity at −30 °C. It might be for a reason that the conformation of the transition state of this Michael addition reaction using catalyst 1a with substrates 2a, 3a was fixed at the decrease of reaction temperature at −30 °C affording high enantioselectivity, although the reason is not clear. Especially, catalyst 1a with methyl group and 1c with bulky tert-butyl group at β-position showed better catalytic activity than others. The determination of absolute configuration and stereoselectivity of 4, 4′ were confirmed on comparision with previous data.10
Table 1 Asymmetric Michael addition of β-keto ester 2a with nitrostyrene 3a using amino alcohol organocatalysts 1a–e
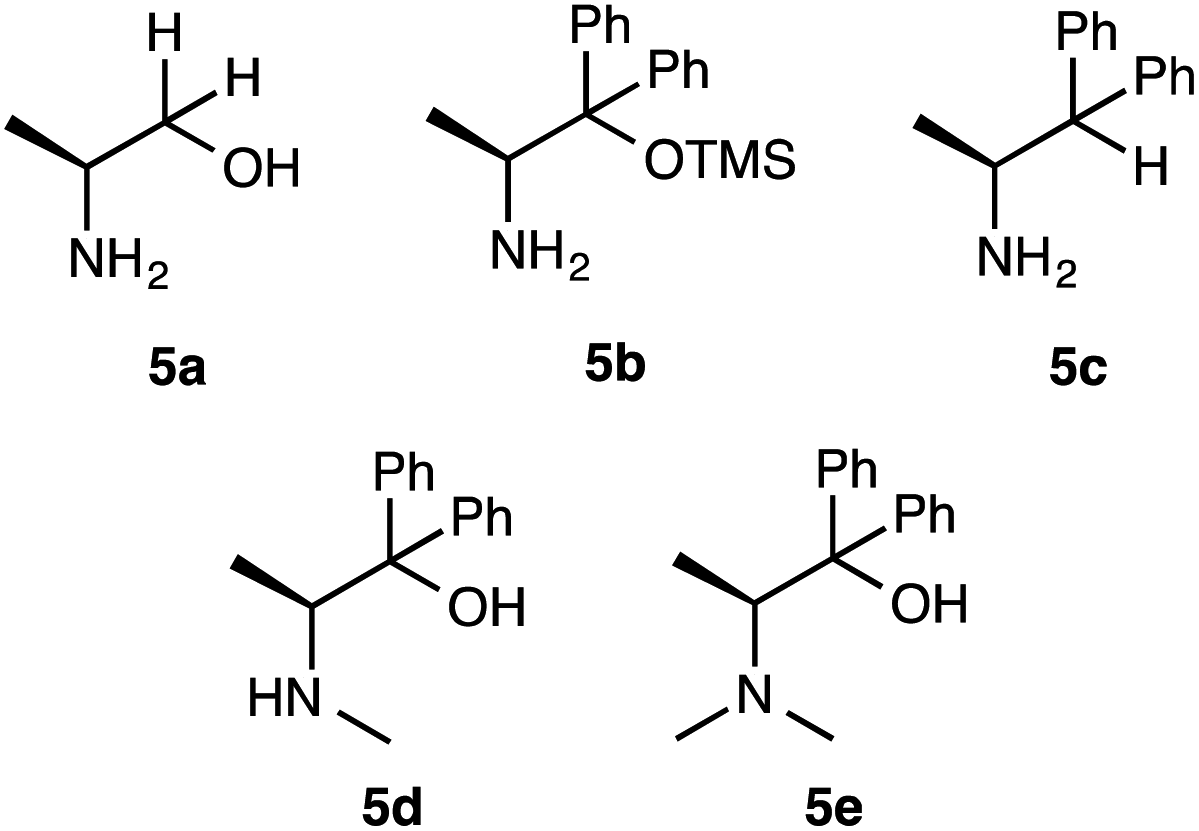
To further improve the results, we evaluated the effect of solvents, molar ratio of catalyst or substrates, and reaction time using the simplest superior catalyst 1a (Table 2). Initially, solvent effect was examined (entries 1–9). The reaction of 2a (2 eq.) with 3a (1 eq.) using 10 mol% of catalyst 1a was performed at −30 °C for 48 h in polar solvents (CH3CN, DMSO and MeOH), respectively (entries 1–3). However, catalyst 1a did not show satisfactory catalytic activity in those solvents (CH3CN: 26%, dr = 88:19, 38% ee) (DMSO: 40%, dr = 75:25, 9% ee) (MeOH: 45%, dr = 83:17, 9% ee) (entries 1–3). In addition, non-polar hexane also did not work as better solvent for this reaction (hexane, 40%, dr = 90:10, 46% ee) (entry 4). On the other hand, catalyst 1a showed enough catalytic activity in ether solvents (Et2O, i-Pr2O) (Et2O: 60%, dr = 95:5, 98% ee) (i-Pr2O: 73%, dr = 98:2, 98% ee) (entries 5 and 6). However, cyclic etherate THF did not work as a good solvent (28%, dr = 85:5, 20% ee) (entry 7). Furthermore, halogenated CH2Cl2 also was not effective solvent (45%, dr = 83:17, 64% ee) (entry 8). From these results, toluene was observed to be the best solvent for this reaction using 1a (76%, dr = 99:1, 99% ee) (entry 9). Next, the molar ratio of catalyst 1a was examined in this reaction (entries 10–12). The reaction was carried out using 20 mol%, 5 mol% and 2.5 mol% of 1a at −30 °C in toluene, respectively. The use of 20 mol% of 1a afforded almost same results (75%, dr = 97:3, 98% ee) as that of 10 mol% of 1a (entry 10). The reaction using 5 mol% of 1a brought about the significant decrease of chemical yield, but the diastereoselectivity and enantioselectivity were kept a high level of value (45%, dr = 95:5, 94% ee) (entry 11). However, the use of 2.5 mol% of 1a brought about decrease in chemical yield and enantioselectivity except for diastereoselectivity (36%, dr = 93:7, 50% ee) (entry 12). From the above results, it was revealed that the optimum amount of 1a was 10 mol%. The ratio of substrate amounts of 2a and 3a (2a:3a = 1:1 and 2a:3a = 1:2) were examined in the presence of 1a (10 mol%) (entries 13 and 14). However, these ratios brought about a decrease of chemical yield and enantioselectivity, except for diastereoselectivity (38%, dr = 93:7, 52% ee) (56%, dr = 94:6, 90% ee) (entries 13 and 14). Furthermore, the effect of reaction times (24 h and 72 h) also did not afford better results, as the yields and stereoselectivities (42%, dr = 95:5, 88% ee) (60%, dr = 84:16, 95% ee) (entries 15 and 16) were observed to be inferior compared to 48 h. Based on the above results, it was revealed that 10 mol% of catalyst 1a, 2 eq. of 2a, 1 eq. of 3a, toluene as a solvent, 48 h of reaction time and −30 °C temperature are the optimum condition to obtain the Michael adduct [2R,3S]-4 with good chemical yield, excellent diastereoselectivity and enantioselectivity. Next, we examined the effect of substituents on amino organocatalysts 5a-e9 in this addition of 2a with 3a (Scheme 3, Table 3). The reaction of 2a (2 eq.) with 3a (1 eq.) was carried out in toluene at −30 °C for 48 h (Table 3). Catalyst 5a9a with no substitution at the α-position only showed low catalytic activity (40%, dr = 60:40, 31%) and afforded enantiomer adduct 4′ of 4 (entry 1). Furthermore, when 5b9b in which the hydroxyl group was masked by TMS group was used, the chemical yield and, stereoselectivities were significantly less than the result that afforded by 1a with free hydroxyl group (45%, dr = 77:23, racemate) (entry 2). We also carried out the reaction using 5c9c with no substitution for the hydroxyl group at β-position, but this catalyst did not show satisfactory catalytic activity (48%, dr = 66:34, 20%) (entry 3). From these results, the hydroxyl group at the α-position on catalyst 1a may need to promote this reaction with enough enantioselectivity. The reactions using catalysts 5d9d, 5e9e with secondary and tertiary amino groups at the α-position were expected to be more basic than 1a with primary amino group were examined (entries 4 and 5). However, only racemate adduct 4 was obtained with good chemical yield and moderate diastereoselectivity (5d: 75%, dr = 68:32, racemate) (5e: 70%, dr = 55:45, racemate). In the reactions using catalysts 5a–e, no better result was obtained than the result using catalyst 1a. Based on the above results, the utility of β-amino alcohol having a primary amino group at the β-position acting as a base and hydrobonding site, phenyl group at the α-position performing as stereocontrolling site and a hydroxy group forming hydrogen bonds with the substrates was revealed in order to achieve satisfactory chemical yield and stereoselectivities.
Table 2 Optimal condition examination in asymmetric Michael addition of β-keto esters 2a with nitrostyrene 3a using amino alcohol organocatalyst 1a
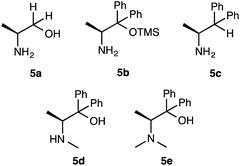 |
| | Scheme 3 Substituted amino alcohol organocatalysts 5a–e. | |
Table 3 Optimal condition examination in asymmetric Michael addition of β-keto ester 2a with nitrostyrene 3a using amino alcohol organo catalysts 5a–e
2.2. Substrate scope
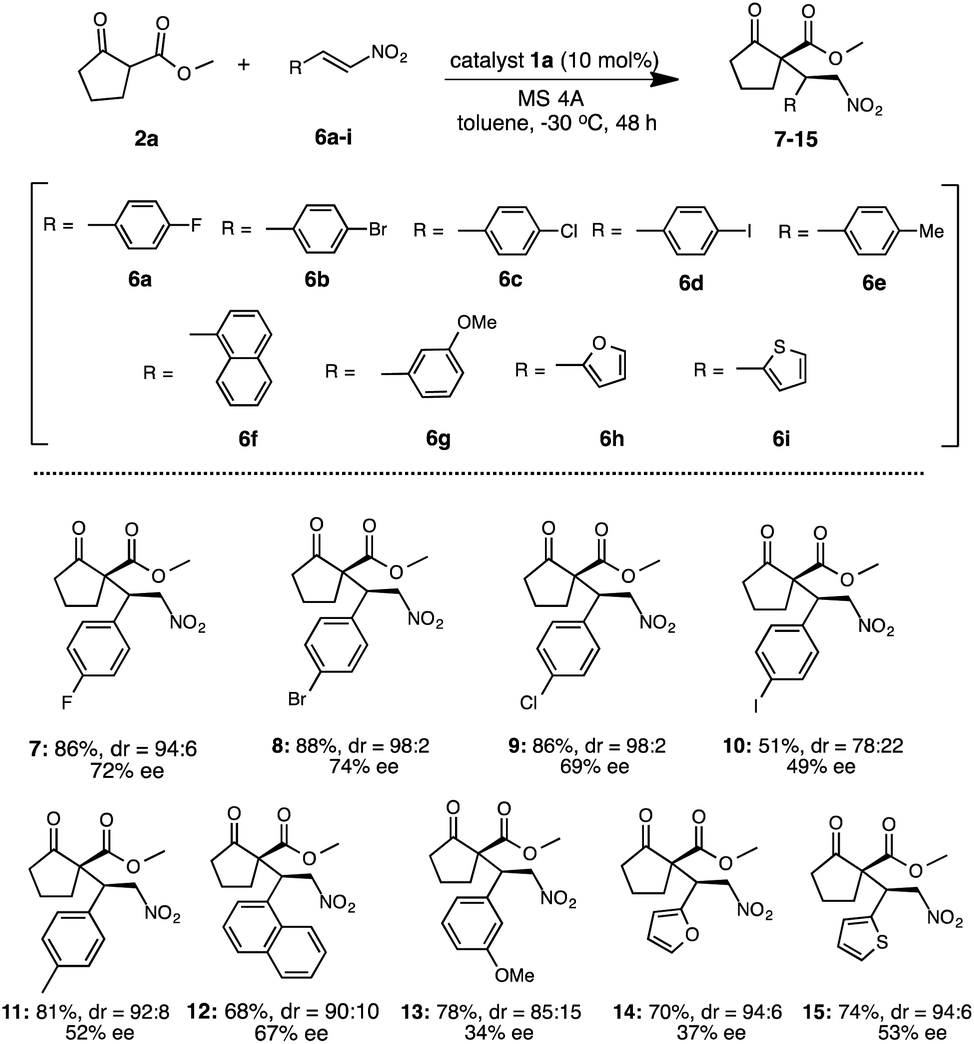
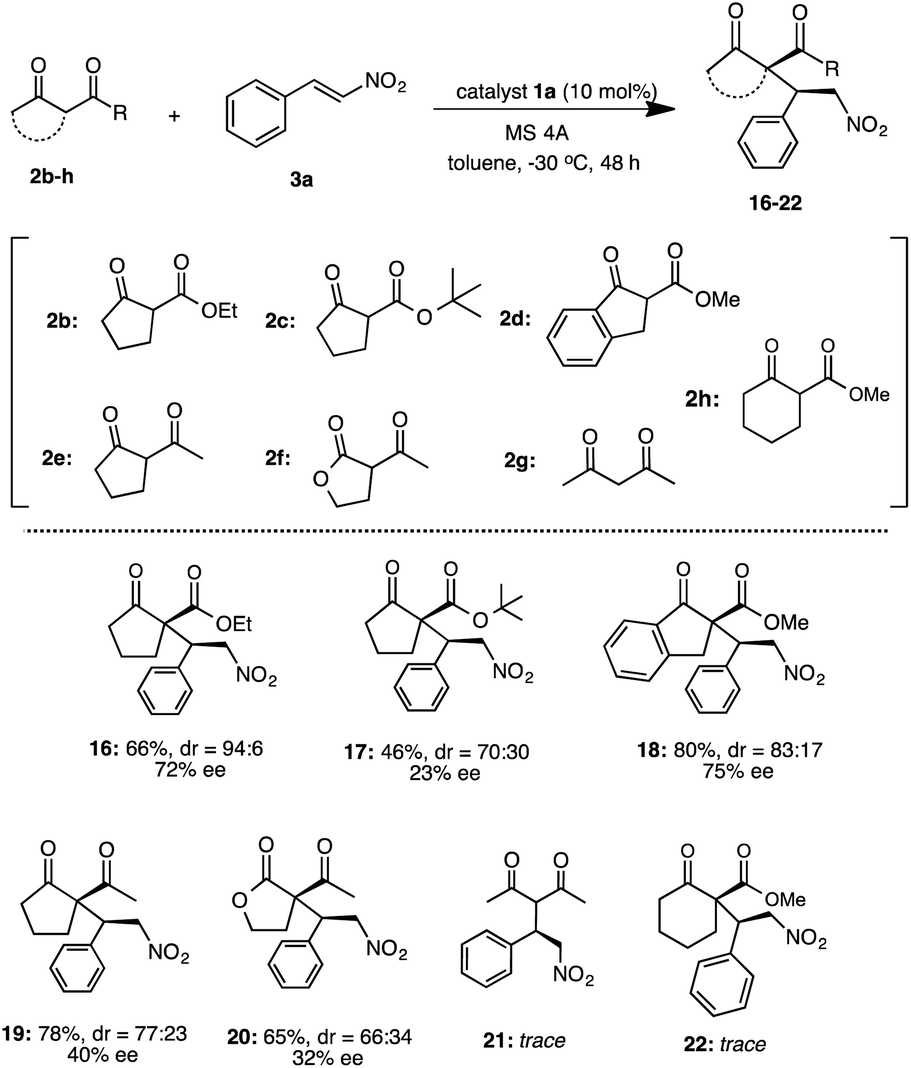
Under the optimized reaction condition using catalyst 1a, the generality of 1a was examined in the asymmetric Michael addition of various β-keto esters with nitroolefins (Schemes 4 and 5). First the reactions of 2a with 6a–i were carried out in toluene at −30 °C for 48 h (Scheme 4) respectively. As summarized in Scheme 4, the desired Michael adducts 7–15 were obtained at moderate to good chemical yields and stereoselectivities. The reaction of β-keto ester 2a with p-halogenated nitrostyrenes 6a–d proceeded with good chemical yield and diastereoselectivities with moderate to good enantioselectivities to afford 7–10 (7, 86%, dr = 94:6, 72% ee) (8, 88%, dr = 98:2, 74% ee) (9, 86%, dr = 98:2, 69% ee) (10, 51%, dr = 78:22, 49% ee). Although the reaction using p-methylated nitrostyrene 6e also afforded the corresponding adduct 11, the enantioselectivity was slightly decreased (11: 81%, dr = 92:8, 52% ee). Furthermore, the use of 1-naphthylnitroolefin 6f also afforded 12 at good chemical yield, moderate diastereoselectivity and enantioselectivity (12: 68%, dr = 90:10, 67% ee). Similarly, 3-methoxynitrostyrene 6g also yielded 13 at good chemical yield and diastereoselectivity, but with low enantioselectivities (13: 78%, dr = 85:15, 34% ee). Moreover, the reaction using heterocyclic 2-1-(2-furyl)-2-nitroethylene 6h was carried out and the corresponding 14 was obtained in good chemical yield and diastereoselectivity with low enantioselectivity (14: 70%, dr = 94:6, 37% ee). Similarly, the use of heterocyclic 2-[(E)-2-nitrovinyl]thiophene 6i also afforded 15 at good chemical yield and diastereoselectivity, but with moderate enantioselectivity (15: 74%, dr = 94:6, 53% ee). In addition, we also examined the reaction of various β-keto esters 2b–d or β-diketones 2e–g with nitrostyrene 3a (Scheme 5). The reactions of β-keto esters 2b–d with 3a respectively, afforded the corresponding Michael adducts 16–18 with moderate to good chemical yields and moderate stereoselectivities. The reaction using cyclopentanone ethyl ester 2b afforded 16 with moderate chemical and good stereoselectivities (16: 66%, dr = 94:6, 72% ee). Moreover, the use of bulky cyclopentanone tert-butyl ester 2c afforded 17, but the reaction brought about the decrease of chemical yield and stereoselectivities (17: 46%, dr = 70:30, 23% ee).11b On the other hand, the reaction using indanone ester 2d proceeded to afford 18 at good chemical yield and stereoselectivities (18: 80%, dr = 83:17, 75% ee).11c Although, the reaction using diketones such as 2-acetyl cyclopentanone 2e or 2-acetyl butyrolactone 2f with 3a, respectively, also afforded the corresponding 19, 20 the enantioselectivities were low to moderate (19: 78%, dr = 77:23, 40% ee)11a (20: 65%, dr = 66:34, 32% ee).11c On the other hand, the use of acycliclic diketone 2g and gave only a trace of 21. Reaction using six membered ring diketo ester 2h was also tried, however the reaction did not proceed. The reaction using a large amount of substrate (3a: 1.0 g, 2a: 1.9 g) was examined to demonstrate the practically utility in the best reaction condition. As a result, the Michael adduct 4 was obtained with 60% chemical yield with good stereoselectivites (dr = 91:9, 86% ee) at 40 h reaction time, although a slight decrease of ee was observed. From this result, it is expected that this Michael addition reaction using simple primary β-Amino alcohol organocatalyst may be useful for practical aspect.
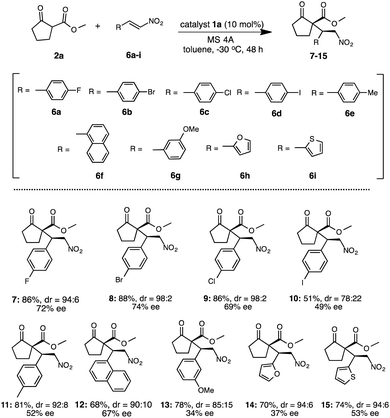 |
| | Scheme 4 Asymmetric Michael addition of 2a with 6a–i. | |
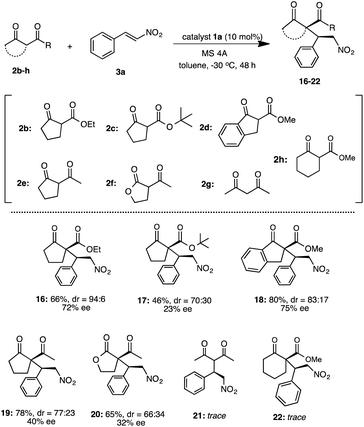 |
| | Scheme 5 Asymmetric Michael addition of 2b–h with 3a. | |
2.3. Reaction mechanism
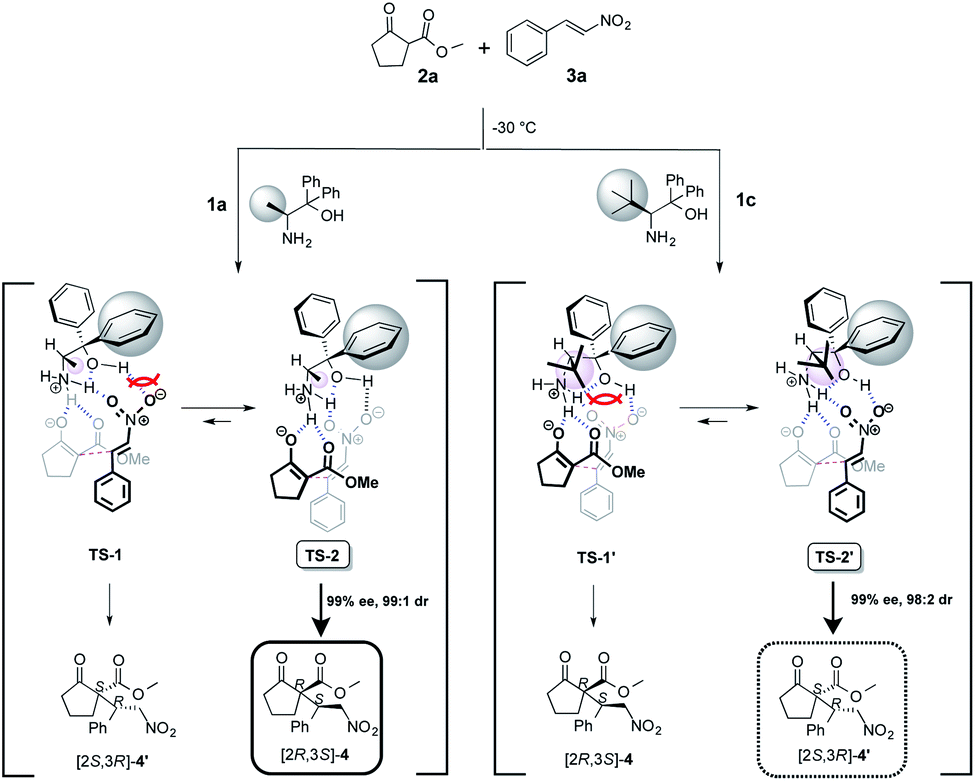
The uses of catalyst 1a with methyl group at α-position afforded the Michael adduct 4a and of 1c with tert-butyl group at α-position afforded enantiomer adduct 4′ of 4 with excellent in the reaction of methyl 2-oxocyclopentanecarboxylate 2a and nitrostyrene 3a at −30 °C, respectively (Table 1). Stereoselectivities (1a: dr = 99:1, 99% ee, 1c: dr = 98:2, 99% ee) Based on the observed excellent stereoselectivities, we proposed the plausible mechanism via a transition state (TS) model to rationalize the stereochemical of Michael addition as shown in Scheme 6. In the mentioned TS, both catalysts 1a and 1c, respectively, act as a base and abstracts a proton on 2a to generate an enolate and then the species is fixed with the ammonium site of catalyst part by hydrogen bonding. In addition, substrate 3a is also fixed with ammonium and hydroxyl group sites on catalyst part by hydrogen bonding. After the fixing of catalyst and substrates, the reaction using catalyst 1a with less bulky methyl group might be assumed to proceed through TS-2 that does not have the steric interaction between phenyl group at α-position on catalyst part and 3a than that of TS-1, to afford [2S,3R]-4 with excellent stereoselectivities.
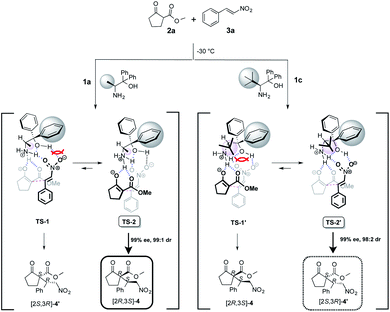 |
| | Scheme 6 Plausible reaction course for Asymmetric Michael addition. | |
On the other hand, the reaction using 1c with bulky tert-butyl group might be assumed to proceed through TS-2′ that does not have the large steric interaction of bulky tert-butyl group on catalyst part and enolate species than that of TS-1′. It may be for a reason that the steric interaction between the bulky tert-butyl group at β-position on catalyst species and enolate species is prioritized in contrast with the steric interaction between phenyl group at α-position on catalyst species and nitrostyrene 3a, to afford [2S,3R]-4′ as a major adduct with high stereoselectivity.
3. Conclusion
We have developed simple primary β-amino alcohols, which act as an efficient organocatalysts in the asymmetric Michael addition of β-keto esters with nitroalkenes, affording highly pure chiral Michael adducts. In particular, simplest β-amino alcohols 1a with methyl group at α-position showed the best catalytic activity and the corresponding Michael adducts having a quaternary chiral carbon center with good to excellent chemical yields (up to 80%), diastereoselectivities (up to 99:1) and enantioselectivities (up to 99% ee). Furthermore, we have found that the both enantiomers of Michael adducts 4, 4′ are separately made by using specific β-amino alcohol organocatalysts such as catalysts 1a with methyl group and 1c with tert-butyl group at β-position, respectively. And also, interestingly, when β-amino alcohols 1b or 1e were used in this reaction, both enantiomers of Michael adducts ([2R,3S]-4 and [2S,3R]-4′) were separately made, depending on the reaction temperature.
4. Experimental
4.1. General information
All reagents and dry solvents were purchased from commercial vendors and used directly without further purification. All reactions were placed in dried sample vials inserted with magnetic beads. Thin-Layer Chromatography (TLC) was performed on Merck silica gel 60 F254 plates and the analytes were identified under UV light. Flash column chromatography was performed using silica gel pore size 60 N (40–100 μm). Infrared (IR) spectra were measured with a JASCO FT/IR-4100 spectrophotometer. 1H and 13C NMR spectroscopic data were recorded using a JEOL JNM-ECA500 instrument with tetramethylsilane as the internal standard. HPLC data were collected using the TOSOH instrument equipped with (UV-8020, DP-8020, and SD-8022) detectors using Daicel CHIRALCEL OD-H column. Optical rotations were measured with a JASCO-DIP-370 digital polarimeter. MS were taken on a JEOL-JMS-700 V spectrometers.
4.2. General procedure for catalytic asymmetric Michael addition of β-keto esters, ketones 2a,2b–g with nitrostyrenes 3a,6a–h using catalyst 1a
To a solution of catalyst 1a (10 mol%) in dry toluene (2 mL) with molecular sieves 4A was added β-keto esters, ketones 2a,2b–g (0.4 mmol) at RT under inert atmosphere and the solution was stirred at same temperature. After 1 h, the reaction was cooled to −30 °C and the respective nitrostyrene 3a,6a–i (0.2 mmol) was added. The reaction was allowed to stir at −30 °C for 48 h. After the completion of the reaction, the solvent was removed under reduced pressure and the residue was purified by flash chromatography on silica gel (n-hexane/AcOEt = 10/1) to give the corresponding chiral Michael adduct. The compounds are the known compounds and the structures were identified by spectral data which were in good agreement with those reported.10,11
Conflicts of interest
There are no conflicts to declare.
Notes and references
-
(a) A. Lattanzi, Org. Lett., 2005, 7, 2579 CrossRef CAS;
(b) Y. Chi and S. H. Gellman, Org. Lett., 2005, 7, 4253 CrossRef CAS;
(c) R. M. De Figueiredo and M. Christmann, Eur. J. Org. Chem., 2007, 16, 2575 CrossRef;
(d) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS;
(e) S. Bertelsen and K. A. Jorgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC;
(f) E. MarquesLopez, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138 RSC;
(g) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef CAS;
(h) P. Kasaplar, C. R. Escrich and M. A. Pericas, Org. Lett., 2013, 15, 3498 CrossRef CAS;
(i) H. X. He and D. M. Du, RSC Adv., 2013, 3, 16349 RSC;
(j) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS;
(k) X. Fang and C. J. Wang, Chem. Commun., 2015, 51, 1185 RSC;
(l) J. Chen, S. Meng, L. Wang, H. Tang and Y. Huang, Chem. Sci., 2015, 6, 4184 RSC;
(m) L. Xu, J. Huang, Y. Liu, Y. Wang, B. Xu, K. Ding, Y. Ding, Q. Xu, L. Yu and Y. Fan, RSC Adv., 2015, 5, 42178 RSC;
(n) T. Sekikawa, T. Kitaguchi, H. Kitaura, T. Minami and Y. Hatanaka, Org. Lett., 2016, 18, 646 CrossRef CAS;
(o) U. Varal, M. Durmaz and A. Sirit, Org. Chem. Front., 2016, 3, 730 RSC;
(p) J. Kaur, P. Chauhan and S. S. Chimni, Org. Biomol. Chem., 2016, 14, 7832 RSC;
(q) H. Zhang, M. Han, T. Chen, L. Xu and L. Yu, RSC Adv., 2017, 7, 48214 RSC;
(r) Y. Zheng, A. Wu, Y. Ke, H. Cao and L. Yu, Chin. Chem. Lett., 2019, 30, 937 CrossRef CAS;
(s) S. H. Xiang and B. Tan, Advances in asymmetric organocatalysis over the last 10 years, Nat. Commun., 2020, 11, 3786 CrossRef CAS.
-
(a) C. Bolm and J. A. Gladysz, Chem. Rev., 2003, 103, 2761 CrossRef CAS;
(b) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2005, 115, 9587 CrossRef;
(c) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH Weinheim, Germany, 2007 CrossRef;
(d) T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863 CrossRef CAS;
(e) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salte, Chem. Rev., 2011, 111, 2626 CrossRef CAS;
(f) J. Gascon, A. Corma, F. Kapteijn and F. X. L. Xamena, ACS Catal., 2014, 4, 361 CrossRef CAS;
(g) D. F. Chen, Z. Y. Han, X. L. Zhou and L. Z. Gong, Acc. Chem. Res., 2014, 47, 2365 CrossRef CAS;
(h) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2015, 115, 9922 CrossRef CAS.
-
(a) J. Kumagai, Y. Kohari, C. Seki, K. Uwai and Y. Okuyama, Heterocycles, 2014, 90, 1124 Search PubMed;
(b) U. V. S. Reddy, M. Chennapuram, C. Seki, E. Kwon, Y. Okuyama and H. Nakano, Eur. J. Org. Chem., 2016, 24, 4124 CrossRef;
(c) H. Nakano, I. A. Owolabi, M. Chennapuram, Y. Okuyama, E. Kwon, C. Seki, M. Tokiwa and M. Takeshita, Heterocycles, 2018, 97, 647 CrossRef CAS.
-
(a) T. Otsuki, J. Kumagai, Y. Kohari, Y. Okuyama, E. Kwon, C. Seki, K. Uwai, Y. Mawatari, N. Kobayashi, T. Iwasa, M. Tokiwa, M. Takeshita, A. Maeda, A. Hashimoto, K. Turuga and H. Nakano, Eur. J. Org. Chem., 2015, 33, 7292 CrossRef;
(b) H. Cui, P. V. Chouthaiwale, F. Yin and F. Tanaka, Asian J. Org. Chem., 2016, 5, 153 CrossRef CAS.
-
(a) Y. Kohari, Y. Okuyama, E. Kwon, T. Furuyama, N. Kobayashi, T. Otuki, J. Kumagai, C. Seki, K. Uwai, G. Dai, T. Iwasa and H. Nakano, J. Org. Chem., 2014, 79, 9500 CrossRef CAS;
(b) J. Kumagai, T. Otsuki, U. V. S. Reddy, Y. Kohari, C. Seki, K. Uwai, Y. Okuyama, E. Kwon, M. Tokiwa, M. Takeshita and H. Nakano, Tetrahedron: Asymmetry, 2015, 26, 1423 CrossRef CAS;
(c) T. Takahashi, U. V. S. Reddy, Y. Kohari, C. Seki, T. Furuyama, N. Kobayashi, Y. Okuyama, E. Kwon, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, Tetrahedron Lett., 2016, 57, 5771 CrossRef CAS;
(d) U. V. S. Reddy, M. Chennapuram, C. Seki, E. Kwon, Y. Okuyama and H. Nakano, Eur. J. Org. Chem., 2016, 24, 4124 CrossRef.
- U. V. S. Reddy, M. Chennapuram, K. Seki, C. Seki, B. Anusha, E. Kwon, Y. Okuyama, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, Eur. J. Org. Chem., 2017, 26, 3874 CrossRef.
-
(a) J. Ye, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 35, 4481 RSC;
(b) R. Manzano, J. M. Andres, M. D. Muruzabal and R. Pedrosa, Adv. Synth. Catal., 2010, 352, 3364 CrossRef CAS;
(c) B. D. Mather, K. Vishwanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487 CrossRef CAS;
(d) J. Luo, L. W. Xu, R. A. S. Hay and Y. Lu, Org. Lett., 2009, 11, 437 CrossRef CAS;
(e) H. Guo, F. Xing, G. F. Du, K. W. Huang, B. Dai and L. He, J. Org. Chem., 2015, 80, 12606 CrossRef CAS;
(f) Y. Zhou, Z. L. Xia, Q. Gu and S. L. You, Org. Lett., 2017, 19, 762 CrossRef CAS;
(g) C. F. Nising and S. Brase, Chem. Soc. Rev., 2008, 37, 1218 RSC;
(h) M. S. Rosello, J. L. Acena, A. S. Fuentes and C. D. Pozo, Chem. Soc. Rev., 2014, 43, 7430 RSC.
-
(a) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS;
(b) Z. H. Zhang, X. Q. Dong, D. Chen and C. J. Wang, Chem.–Eur. J., 2008, 14, 8780 CrossRef CAS;
(c) A. M. Flock, A. Krebs and C. Bolm, Synlett, 2010, 8, 1219 Search PubMed;
(d) K. Murai, S. Fukushima, S. Hayasi, Y. Takahara and H. Fujioka, Org. Lett., 2010, 12, 964 CrossRef CAS;
(e) T. Nemoto, K. Obuchi, S. Tamura, T. Fukuyama and Y. Hamada, Tetrahedron Lett., 2011, 52, 987 CrossRef CAS;
(f) A. Lattanzi, C. D. Fusco, A. Russo, A. Poater and L. Cavallo, Chem. Commun., 2012, 48, 1650 RSC;
(g) D. F. Chen, P. Y. Wu and L. Z. Gong, Org. Lett., 2013, 15, 3958 CrossRef CAS;
(h) P. Vinayagam, M. Vishwanath and V. Kesavan, Tetrahedron: Asymmetry, 2014, 25, 568 CrossRef CAS;
(i) M. Bera, T. K. Ghosh, B. Akhuli and P. Ghosh, J. Mol. Catal. A: Chem., 2015, 408, 287 CrossRef CAS;
(j) M. S. Ullah and S. Itsuno, Chem. Lett., 2018, 47, 1220 CrossRef CAS;
(k) C. S. Afrin and S. Itsuno, J. Catal., 2019, 377, 543 CrossRef;
(l) M. A. Abdelkawy, E. A. Aly, M. A. El-Badawi and S. Itsuno, Catal. Commun., 2020, 146, 106132 CrossRef CAS.
-
(a) C. Tianhao and H. Yanwei, Zhongguo Yiyao Gongye Zazhi, 2001, 32, 322 Search PubMed;
(b) Y. Sakuta, Y. kohari, N. D. M. R. Hutabarat, K. Uwai, E. Kwon, Y. Okuyama, C. Seki, H. Matsuyama, N. Takano, M. Tokiwa, M. Takeshita and H. Nakano, Heterocycles, 2012, 86, 1379 CrossRef CAS;
(c) D. O. Hagan and M. Tavasli, Tetrahedron: Asymmetry, 1983, 10, 1189 CrossRef;
(d) A. Tadatoshi, H. Motoo, Y. Yukio, S. Gohfu, European Patent organisation, EP 75868 A2 1983-04-06, 1983;
(e) M. Gacek and K. Undheim, Acta Chem. Scand., Ser. B, 1975, 29, 206 CrossRef.
-
(a) R. Togashi, M. Chennapuram, C. Seki, Y. Okuyama, E. Kwon, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, Eur. J. Org. Chem., 2019, 24, 3882 CrossRef;
(b) D. Ganesan, M. Chennapuram, Z. Begum, C. Seki, Y. Okuyama, E. Kwon, K. Uwai, M. Tokiwa, S. Tokiwa, M. Takeshita and H. Nakano, Heterocycles, 2019, 98, 1536 CrossRef CAS.
-
(a) C. J. Wang, Z. H. Zhang, X. Q. Dong and X. J. Wu, Chem. Commun., 2008, 1431 RSC;
(b) P. Chauhan and S. S. Chimni, Asian J. Org. Chem., 2012, 1, 138 CrossRef CAS;
(c) N. Horitsugi, K. Kojima, K. Yasui, Y. Sohtome and K. Nagasawa, Asian J. Org. Chem., 2014, 3, 445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and HPLC data. See DOI: 10.1039/d0ra09041g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :
: